")
Back to Archived Journals » Biosimilars » Volume 5
Global regulatory landscape of biosimilars: emerging and established market perspectives
Authors Krishnan A, Mody R, Malhotra H
Received 19 June 2014
Accepted for publication 30 July 2014
Published 17 February 2015 Volume 2015:5 Pages 19—32
DOI https://doi.org/10.2147/BS.S44052
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ajay Singh
Anita Krishnan,1 Rustom Mody,1 Hemant Malhotra2
1Lupin Limited, Biotech Division, Maharashtra, India; 2Division of Medical Oncology, RK Birla Cancer Center, SMS Medical College Hospital, Jaipur, India
Abstract: Biological product development for launch in multiple geographies with varied regulatory expectations would require a planned and focused strategy, involving the selection of the appropriate reference product, defining the extent of process and product characterization and design of nonclinical and clinical studies. The development for established markets like the European Union and the United States, which have precedence in regulatory pathways, may face very different challenges compared to emerging markets, many of which are still in the nascent stages of regulatory guidelines. A clear and concise understanding of the regulatory framework of each region and awareness of the limitations of health care policies, with an added knowledge of the local factors that influence the biosimilar market, would be desirable for a good business strategy. Herein it is attempted to outline the stages of regional guideline implementation in the various global locations and compare the variability in regulatory requirement between them. The factors that could potentially impact biosimilars business in these regions are also outlined. Finally, the prevailing competition between manufacturers of innovative and biosimilar drugs, which could influence the availability of lifesaving off-patent drugs for critical diseases and the advent of more effective, alternate, or next-generation molecules, is also briefly described.
Keywords: guidelines, India, comparability, EMA, US FDA, WHO
Introduction
Biological entities, larger in size but smaller in presence as compared to chemical entities, have made a significant difference to patients around the world by providing targeted solutions to illness rather than symptomatic relief. The advent of newer understanding of the molecular and cellular basis of diseases, such as cancer, arthritis, and multiple sclerosis, through technological advances has led to biotechnology-based therapies finding their importance and preference in the clinical and patient community. The term “biological drug” or “biologics” could encompass a wide variety of molecules, such as therapeutic/supplementary proteins, hormones, monoclonal antibodies, antibody fragments, pegylated proteins, albumin binding domain antibodies, and deoxyribonucleic acid (DNA) vaccines. In terms of sheer size, these molecules are often 100 to 1,000 times bigger than their chemical counterparts, in turn making them complex. All protein biological entities are composed of a string of amino acids that are sometimes disulphide bonded within or with other similar or dissimilar subunits, bent into alpha-helix and beta-sheets typically forming the 1°, 2°, 3° and 4° structure, giving rise to pockets and crevices which are critical for their functionality. Typically, recombinant proteins are produced by manipulating the genetic blueprint of a living cell (prokaryotic or eukaryotic) with the gene of interest and culturing in liquid medium, to tap the expressed protein of interest. This protein which is surrounded by a milieu of other proteins, sugars, and lipids is chromatographically isolated to a purity of more than 98% to form the drug substance. The drug substance is the equivalent of an active pharmaceutical ingredient in a pharma molecule, which is then formulated with an appropriate buffer and excipients to make the final injectable product called the drug product. Due to the complex mode of production involving live organisms in a stringently controlled environment, achieving manufacturing consistency is an indomitable task. Though every batch inherently has product quality differences, for obvious reasons, the variations from batch to batch need to be monitored to ensure conformance within a preset range. The product even in the purest form is still associated with a number of product variants; for example, posttranslational modifications such as glycosylation, oxidation, and deamidation variants. These variants are most often critical quality attributes and have a high impact on the functionality of the molecule. Thorough characterization of these posttranslational modifications and other structural variations that are not present in small molecules is scientifically challenging. Apart from these, there are other process-related impurities that arise from the host, such as host cell proteins, host cell DNA, and endotoxins, which need to be controlled within a safe limit. The product quality differences between batches need to be thoroughly scrutinized in a regulatory application and correlation to clinical trial results need to be drawn, to avoid any safety hazard to the consumers. Therefore, a “one size fits all” approach of regulatory review of a small molecule does not fit a biological drug (Figure 1).
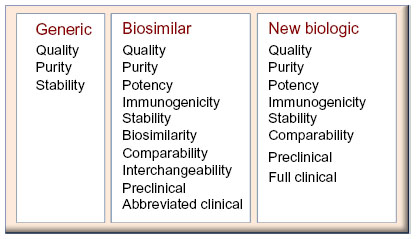 | Figure 1 Comparison of the requirements for biosimilar licensure pathway, with regards to the generic (small molecule) and novel biologics pathways. |
Biosimilars are blockbuster drugs, in the sense that they are affordable copies of the expensive originals, providing much-needed affordable quality health care. A major part of this affordability arises from the reduced clinical trial requirements, the permission for which is granted, when sufficient biosimilarity is demonstrated.1–3 Since the biosimilar is the end product of a biotechnological process, a system wherein genetic manipulation and physiological environment is the key factor, the active ingredient is similar to the original biologic, but never identical. Unlike generics, where an exact identical copy is anticipated, in biosimilars “process is product,” and subtle variations are inherent, since no sponsor has knowledge of the originator’s process or cell lines. The recombinant protein produced will bear the fingerprint of the cell line and process, in terms of product- and process-related impurities, since it is a highly controlled manufacturing environment. Due to this complexity, regulatory requirements of clinical, nonclinical, and analytical comparability of these subtle differences to the originator are mandatory. These regulatory requirements for biosimilar drug applications weigh more than a generic drug application but less than a new biologic drug application. In a generic application, it is sufficient if quality, purity, and bioequivalence evidence is presented. However, with biosimilars, apart from chemistry, manufacturing and controls analytics, the emphasis is on biosimilarity, preclinical, immunogenicity, and limited clinical studies. As molecule exclusivity expires, sponsors all over the globe attempt to produce these recombinant protein drugs. In a business perspective, many factors such as development timeline, cost of goods, drug accessibility, user friendliness, and regulatory compliance are all factors that drive the market in varied degrees in different geographical regions. With multiple prospective manufacturers on the horizon, the need arises for regulatory guidelines that ensure biosimilarity, comparability, and interchangeability with respect to safety and efficacy of the product. The major dictators in this domain are the European Medicines Agency (EMA), the World Health Organization (WHO), and the United States Food and Drug Administration (US FDA), with a number of regional versions of guidelines that have been formulated based on the above mentioned templates. Here we attempt to compare the regulatory scenario in emerging markets with regards to the advanced European Union (EU) and US environment and also to evaluate the driving forces for these two.
Definition of biosimilars
Biosimilars, although bearing in essence the same meaning world over, have been defined in different ways and with differences in nomenclature. A brief tabulation has been presented below as a ready reckoner (Table 1).4–10
 | Table 1 Definitions of biological medicines according to regional regulatory agencies |
Market position of biosimilars
Biosimilars are legally approved subsequent versions of innovator biopharmaceutical products following patent and exclusivity expiry. However, the definition and nomenclature of biosimilars differs among the various regulatory agencies across the world. For example, they are known as similar biological medicinal products by the EMA and Korean Food and Drug Administration (KFDA), as follow-on protein products or follow-on biologics by the US FDA and Ministry of Health, Labor, and Welfare (MHLW), and as subsequent entry biologics by Health Canada. Biological drugs constitute one of the largest growing sectors of the pharmaceutical industry. By 2015, sales of biosimilars are expected to reach between US $1.9–$2.6 billion and US $25 billion in 2020, a compound annual growth rate (CAGR) of 7.7%.11 Biosimilar products encompass different therapeutic classes, like erythropoietins, growth hormones, interferons, stimulating factors, low molecular weight heparins, insulins, monoclonal antibodies, and Fc fusion proteins. Of these, monoclonal antibodies have been established as a major product class and occupy a large sector of the biosimilar market space. This is primarily due to the therapeutic success of these molecules and the prevalence of diseases like cancer, arthritis, and psoriasis.
With patents for top-selling monoclonal antibody drugs poised to expire shortly or already expired, there is a clamor among biosimilar manufacturers for the business.12 In a way it is good for health care for two reasons: availability of affordable drugs and research and development focus on novel biologics. For registration of their products, biosimilar sponsors have to prove equivalence or similarity to the reference product. Variations of smaller magnitude, especially risks of immunogenicity, have to be shown as clinically nonsignificant, because of safety and efficacy. These regulatory requirements have now evolved and been included in the guidelines.
Evolution of guidelines for biosimilars
After the first wave of off-patent drugs, EMA was the first to create biosimilar guidelines in 2005, and the first approval came about in 2006. EMA boasts the longest list of approved biosimilars after the implementation of biosimilar guidelines (Table 2).13 The responsibility of implementing regulation (with respect to development, authorization, and manufacturing of biotherapeutics) lies with member states of the EU. EU guidelines to date stand as one of the most stringent regulatory norms for biosimilar product development. The chemistry, manufacturing and controls package and other clinical requirements are extensive, which ensures safety and efficacy of biosimilars produced by the sponsors. Traditionally, sponsors have found it safe to develop simple protein therapeutics like granulocyte-colony stimulating factor, erythropoietin, somatropin, and follitropin. However, recently two biosimilar monoclonal antibodies of infliximab have been approved in a landmark decision, indicating that even complex molecules can gain biosimilar status in the EU if well developed.
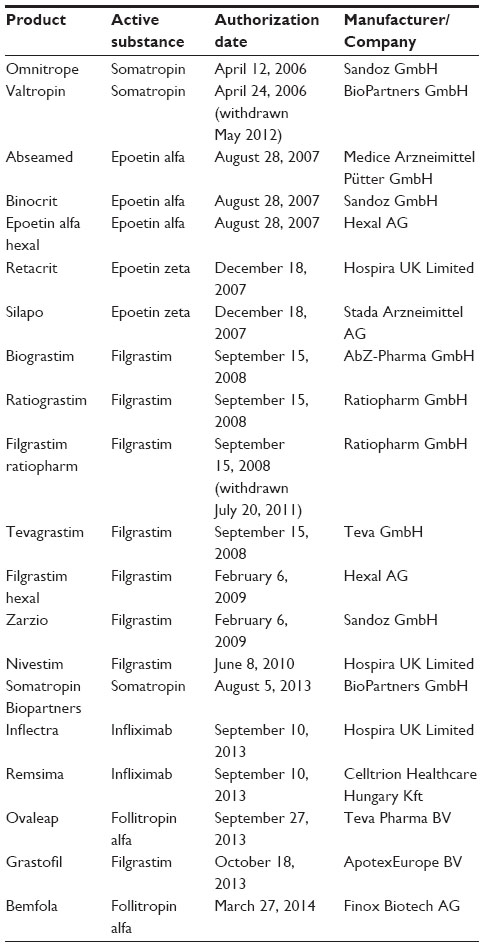 | Table 2 EMA-approved biosimilars |
In 2009, WHO developed a set of globally accepted standards to ensure the safety, efficacy, and quality of similar biotherapeutic products (SBPs). These were mainly targeted to aid and ensure local regulatory authorities to adhere to international standards.2 Ever since there has been a rapid evolution of guidelines, and most countries have adopted the general framework of EMA (Table 3) or WHO, while others have established their individual national guidelines based on these templates. Australia adopted the EU guidelines without any changes, while Singapore and Malaysia amended their guidelines mainly in accordance with the EMA guidelines.14 Brazil and Cuba chose the WHO and Canadian guidelines as the basis for developing regulations for their respective countries.15 However, there are considerable variations in definitions, terminology, reference product for comparability, extent of data requirements, and other aspects. India released official guidelines in June 2012,5 prior to which 20 biosimilars were already approved for use within India under an ad hoc abbreviated process (Figure 2).16
 | Table 3 European Medicines Agency guidelines relevant to biosimilar development and approval |
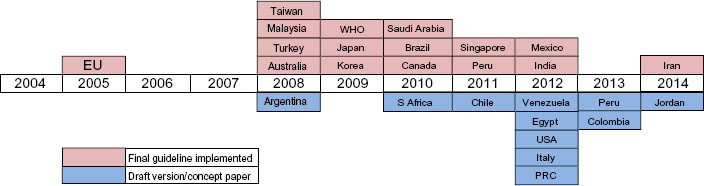 | Figure 2 Evolutionary scale of guidelines in the last decade. |
WHO regulatory framework
To assure safety, efficacy, and quality of biotherapeutic products, WHO guidelines draw certain basic principles which are mandatory for licensure of a biosimilar product. This ensures successful regulatory submissions in most major pharmaceutical geographies around the world.
- Reference product: The chosen reference biotherapeutic product (RBP) should be licensed based on full quality, safety, and efficacy data and should be authorized in the country or region in question. Wherever it may not be feasible, such as countries lacking nationally licensed RBPs, additional criteria (such as the product should be licensed and widely marketed in another jurisdiction) may be applied.
- Quality: All aspects of quality and heterogeneity should be assessed, including head-to-head comparisons with the reference product. Due to unavailability of the drug substance of RBP, comparison is performed mostly with the commercial product. It is important to show that the excipients do not pose interference in the analytical test methods. Wherever this is not possible, extraction of drug substance needs to be performed. In that case demonstration that product heterogeneity and relevant attributes of the active moiety are not affected by the extraction procedure is required. Where possible, the product should be tested with and without manipulation.
- Nonclinical data: Data should include pharmacodynamic, pharmacokinetic (PK), and comparative repeat-dose toxicity studies in a relevant species. The selection of PK study design, which could be single-dose studies, steady-state studies, or repeated determination of PK parameters, needs to be adequately justified by the sponsor. The pharmacokinetics of the SBP and RBP are compared in terms of absorption, bioavailability, and elimination characteristics. Clinically relevant pharmacodynamic (PD) markers should be selected and may be investigated in the context of combined PK/PD studies.
- Clinical studies: Similarity of the efficacy of the SBP and the RBP will usually have to be demonstrated in adequately powered, randomized, and controlled clinical trial(s). Although equivalence designs are the preferred choice for the comparison of efficacy and safety of the SBP with the RBP, noninferiority designs may also be considered if appropriately justified. Immunogenicity should always be investigated in humans before authorization.
- Pharmacovigilance: Monitoring, or a pharmacovigilance plan, at the postmarketing phase is included in the guideline to supplement the limited clinical data that is present during authorization. In some cases an associated risk management plan is also advised.
WHO guidelines, although they followed the EMA guidelines chronologically, are not geography specific and thus were a template for many other national regulatory agencies (NRA). We scrutinized the regulatory landscape in all these different global locations.
European Union (EMA)
The EU is the forerunner for the biosimilar market, as is evident from the number of approved drugs, market size, promulgation of the guidelines, and so forth. The EMA/European Commission were the first to implement a well-documented legal and regulatory pathway for the approval of biosimilar products that is distinct from the generic pathway. Clear definitions in terms of the analytical, preclinical, and clinical data requirements have been specified in the guidelines, which are more detailed than those for generics. The EMA requirements for comprehensive comparability studies between the biosimilar and reference product are elaborately defined in the EMA guidelines. The EMA guidelines are some of the most stringent, and recommend a case-by-case review of submissions, considering the diversity and complexity of biological products.17
EMA first released general guidelines for quality issues and nonclinical and clinical issues which encompass quality, consistency, the manufacturing process, safety, and efficacy considerations. This was followed by far more detailed product-specific guidelines, by EMA/Committee for Medicinal Products for Human Use (CHMP), for products like erythropoietin, growth hormone, granulocyte-colony stimulating factor (G-CSF), insulin, interferon beta, low-molecular weight heparins, and monoclonal antibodies.18 The list of guidelines/concept papers are listed in Table 4. The EMA/CHMP guidelines are adopted standards in countries like Australia, Canada, Japan, Korea, and South Africa.1,15
 | Table 4 European Medicines Agency guidelines relevant to product-specific biosimilar development and approval |
United States of America (US FDA)
The market in the United States for recombinant therapeutics reached $507 million in 2010 and $1.1 billion in 2011. The market is expected to reach $1.3 billion by 2016, a CAGR of 4.1%.19
In the US, approval for biological products is made through the Public Health Service Act and for small molecule drugs through the Federal Food, Drug, and Cosmetics Act. Unlike EMA, the US FDA was a late entrant to the biosimilars regulatory pathway. On March 23, 2010, the Biologics Price Competition and Innovation Act (BPCIA) was signed as part of the Patient Protection and Affordable Care Act, which created a new licensure pathway for biosimilars within the auspices of the US FDA. Biosimilar applications can be submitted under section 351(k) of the Public Health Service Act. The BPCIA was considered equivalent to the Drug Price Competition and Patent Term Restoration Act of 1984 or the Hatch–Waxman Act of generics, which enabled the availability of affordable drugs for public health and fostered innovation by the originator companies. However, the generic manufacturers had to only establish the sameness or exactness of their molecules to the approved originator drug supported by bioequivalence studies and could rely to a large extent on the safety data of the originator while seeking approval. Since the biological products cannot be the same or exact, provisions for demonstrating “similarity” had to be included in the BPCIA. The Act defined the basic requirement of a demonstration of biosimilarity between reference and biosimilars that was proposed with the same mechanism of action and same indications. The Act also proposes that the sponsor’s product should be shown as interchangeable with the originator’s product. Given the environment of major resistance from originators, the Act also provided exclusivity periods for reference biologics and some already approved follow-on biologics. A complex framework addressing the procedure for resolving patent disputes between the biologic innovator and the biosimilar’s sponsor were also defined. However, specific criteria for biosimilarity or interchangeability, such as the extent of analytical data, animal studies, or clinical trial designs, were not defined. Until now, the BPCIA has provided case-specific determinations to the sponsor’s individual molecules rather than providing any criteria for a class of molecules.20
The US FDA finally issued three draft guidance documents in 2012, recommending a stepwise approach to demonstrate biosimilarity, laying importance on “totality of evidence”:6,21,22
- Quality Considerations in Demonstrating Biosimilarity to a Reference Protein Product
- Scientific Considerations in Demonstrating Biosimilarity to a Reference Product
- Biosimilars: Questions and Answers Regarding Implementation of the BPCIA of 2009.
The Scientific Considerations guidance calls for the demonstration of biosimilarity between the reference and biosimilar in terms of safety, potency, and purity, with no clinically meaningful differences. The guidance insists on the following main points:
- Analytical studies: Demonstration of the product as highly similar to the reference, notwithstanding minor clinically irrelevant differences.
- Animal studies: Including toxicity studies, PK and PD measurements, and immunogenicity studies.
- Human clinical studies: Including PK and PD measurements, immunogenicity results, and safety and efficacy data, plus equivalence studies showing comparability on both upper and lower margins. It also recommends sponsors to establish an early meeting with US FDA officials with preliminary data and product development plans and understand the requirements of submission, while also drawing a schedule of milestones/meetings for future work.
The Quality Considerations guidance dwells more on the physicochemical characterization needs of the application. The US FDA considers certain important underlying factors while evaluating applications, like manufacturing process consistency, structural similarity, potency assays relevant to mechanism of action, PD studies extending the understanding of the mechanism of action, comparative PK, and immunogenicity. In physicochemical characterization, the emphasis is on display of primary and higher order structure and post-translational and chemical modifications, whereas in biological characterization, comparative potency and receptor binding assays are the expectations. The commercial history and experience of the sponsor could also be added valid points. Another major consideration of the BPCIA is the interchangeability of biosimilars. A sponsor must demonstrate that the biosimilar drug produces the same clinical effects as the originator in patient studies. This ensures automatic interchangeability of the biosimilars with the reference originator product, without the need for clinician’s discretion. Once approved or licensed for marketing, the clinician will not have to intervene regarding switching/swapping these drugs.6
The Questions and Answers Guidance provides a set of answers that biosimilar sponsors require early on, like apprehensions regarding differences in formulation, exclusivity issues, and meeting schedules. This guidance also provides a list of definitions distinguishing terms such as “chemically synthesized polypeptide,” “biological product,” and “protein.”
Japan (Pharmaceuticals and Medical Devices Agency)
Japan’s national regulatory agency is the Pharmaceuticals and Medical Devices Agency (PMDA); it is one of the countries that had defined regulatory pathway for biosimilars early on in 2009.22,23 The pathway is very similar and in line with the EU in terms of comparability data requirements. Also in Japan, the regulation of biosimilars principally follows the guidelines of the International Committee on Harmonization, especially guideline Q5E, which deals with changes in manufacturing process and the evaluation of comparability, and guideline Q6B, which concerns the comparability of products. The PMDA has been supporting biosimilar development in Japan by consulting with pharmaceutical companies.
Recombinant plasma proteins, recombinant vaccines, PEGylated recombinant proteins, and nonrecombinant proteins that are highly purified and characterized are defined in the scope of the guideline. However, some inclusions of EU, like polyglycans (low molecular weight heparin) and synthetic peptides have been excluded since extensive characterization is not required for simple molecules without higher order structures.15 Comparative stability studies with reference biologics is not mandatory in Japan.
The detailed guidelines are outlined in:22,23
- “Guidelines for the Quality, Safety, and Efficacy Assurance of Follow-on Biologics” (Yakushokushinsahatu 0304007 by MHLW/March 4, 2009)
- “Nonproprietary Name and Brand Name of Follow-on Biologics” (Yakushokushinsahatu 0304011 by MHLW/March 4, 2009)
- “Revision of Marketing Approval Application” (Yakushokushinsahatu 0331015 by MHLW/March 4, 2009).
Although EMA guidelines are the template for MHLW, the requirements of comparative stability, which may not be needed always in Japan, pose as differentiators. So is the case with toxicology studies, wherein impurities need not be evaluated through nonclinical studies. Somatropin and epoietin alfa have been authorized for marketing in Japan following the abovementioned route. The PMDA has been supporting biosimilar development in Japan by consulting with pharmaceutical companies before any formal guidelines were drawn. Recently PMDA has made great efforts to reduce the time lag of the approval process. The time period required for the approval of standard products has reduced by almost half, from 21 to 12 months, and priority products from 12 months to 9 months. The application lag of PMDA when compared with the US FDA is large, about a year, whereas the postapplication lag is much less, about a month.24,25
A survey interview of representatives from various types of companies, including contract manufacturing companies, bioventures, generic manufacturers, biotechnology companies, and pharmaceutical companies having a high level of activity in the field of biopharmaceutical development, was conducted to derive opinions from key leaders. Among the regular discussions, it was also stated that incentives for development of innovator biopharmaceuticals should not suffer in the competitive race with biosimilars, since companies developing innovator products are taking a huge risk given the number of unpredictable events that may occur during nonclinical and clinical studies. The survey discussions stated that only one to two companies had a biosimilar product under development, using the company’s own unique manufacturing technologies. The other survey participants revealed that their companies were not engaged in biosimilar development, nor did they have any plans to do so. Four respondents reflected their company’s vision to focus on new- or second-generation biopharmaceuticals than development of biosimilars. Some opinions that could be worth noting were the possibility of using offshore manufacturing contractors in India or the People’s Republic of China.26
Korea (KFDA)
The market for biosimilars in Korea is promising, as it has been in other parts of the world. Samsung, the electronics giant in Korea, announced its entry to the biosimilar arena with an investment of $389 million for an anticipated 5-year period in 2009. The regulatory framework of biosimilar products in Korea had been established by 2009. It is governed by Pharmaceutical Affairs Act, Notification of the Regulation on Review and Authorization of Biological Products, and the Guideline on Evaluation of Biosimilar Products. The Pharmaceutical Affairs Act is the supreme regulatory authority for all licensure of biologic products. The KFDA operates within the confines of the former. The Notification of the Regulation on Review and Authorization of Biological Products serves as the guiding document for all biosimilars.15,27
Since the Korean guideline for biosimilar products coincided with the timeline of the WHO regulatory guidelines, most of the requirements are similar except for that of the clinical evaluation to demonstrate similarity. Equivalence testing, where the upper and lower margins need to be specified with justified preset limits, is mandatory with KFDA, whereas in WHO it is preferred to the noninferior mode of testing where only the lower margin is specified. This opens the possibility for extrapolation of efficacy data to other indications of the reference product.3,27
Canada (Health Canada)
The federal regulations that evaluate the safety, efficacy, and quality of subsequent entry biologics that are developed on expiry of patents for biological drugs in Canada is Health Canada. “Guidance for Sponsors: Information and Submission Requirements for Subsequent Entry Biologics (SEBs)” was issued by Health Canada in 2010.4
Similar to WHO, the Canadian guidelines states that the biologic drug that was authorized based on a complete clinical data package with sufficient safety and efficacy data could qualify as a suitable reference. Additionally, the product should be subjected to advanced analytical testing for extensive characterization and should meet appropriately defined criteria/limits. Health Canada insists on the physicochemical and biological characterization, extensive in-process analysis of all critical intermediates, full scale stability data, ranging of impurities, ranging studies of reference and product to plot the differences in variability, nonclinical and clinical, and safety and efficacy data. Postmarketing requirements like adverse drug reaction report, periodic safety update reports, suspension or revocation of notice of compliance (NOC) of authorization in the event of any compromise of the safety of drug have all been defined in the guidance.4
The only biosimilar that was authorized in Canada was somatropin. The recent authorization of monoclonal antibodies, remsima and inflectra, in January 2014 is an indication of acceptance of good quality, affordable “subsequent entry biologics.” This may induce the much-awaited first 351k biosimilar filing with the US FDA this year. This may also have a cross-border influence and increase the public outcry for affordable drugs in the US, where biosimilars are still not recognized. There have been incidents of people organizing bus trips to Canada from the US as part of medical tourism to buy affordable copies.28,29
The People’s Republic of China (State Food and Drug Administration)
The People’s Republic of China and India are always compared in every sphere due to their large economies. The People’s Republic of China has a major presence in manufacturing chemicals, intermediates, and large-volume active substances, while India’s strengths have been sourcing active substances and finished dose products. At the CPhI worldwide preconnect conference, panelists Ina Lennon (Chiral Quest) and Gurpreet Sandhu (Reva Pharmachem) compared the pharmaceutical scenarios in the two countries.30 Both have had their share of challenges, like devaluation of the rupee in India and retention of metropolitan talent in the People’s Republic of China. India has an edge over the People’s Republic of China in language skills and experience of regulatory authorities. India boasts the largest number of US FDA-approved facilities outside the US. The pace is picking up in the People’s Republic of China, but most of the players are focusing on the huge internal market, which is investment friendly.30
Over 40 biopharmaceutical products have been already approved by the State Food and Drug Administration in the People’s Republic of China, which include a majority of biosimilars. Guidance for Review and Approval of Biosimilar Products guidelines was announced by the Department of Health in 2008, and is mainly based on EMA guidelines with consideration of local regulatory environment:22
- Guidelines for Registration of Drugs (2010)
- Guidance for Review and Approval of Recombinant Protein Drugs (2002)
- Guidance for Review and Approval of Biosimilar Product (2008)
- Points to Consider for Common Technical Documents (CTD) in Review and Approval of Biosimilar Product (2010).
India (Central Drugs Standard Control Organization)
Mohal Sarabhai (Asenco) in the 2013 CPhI meeting explored the strengths of India, including regulatory compliance, a large talent pool, the ability to develop new and differentiating technologies, and low manufacturing costs. India is often called the “Pharmacy of the World” due to all these factors.30
The apex regulatory bodies22 under the government of India that are involved in the approval process of SBPs are:
- Central Drugs Standard Control Organization
- The office of Drug Controller General of India
- Review Committee on Genetic Manipulation
- Genetic Engineering Approval Committee.
As far as the products are concerned, various biosimilar products being marketed currently include erythropoietin, human growth hormone, recombinant human insulin, G-CSF, and interferon. Industry statistics indicate that in 2010, epoetin alfa (erythropoietin biosimilar) occupied more than 40% of the market share, followed by filgrastim (G-CSF biosimilar) with a 33% market share and somatropin (human growth hormone biosimilar) with a 25% market share.
The Indian biosimilar industry is estimated to be a US $338 million industry that has been growing at a CAGR of 30% since 2008. There are around 25 Indian companies operating in the biosimilar space, marketing close to 50 products in the Indian market and a few of these products in some of the unregulated markets.
Islamic Republic of Iran (National Regulatory Authority)
The National Regulatory Authority in the Islamic Republic of Iran is the Ministry of Health and Medical Education, which is the official body for establishing quality standards for the regulation of medicinal products. The Division of Pharmaceuticals and Narcotic Affairs is the principal governing agency; the other divisions which share some overlapping responsibilities are the Drug Control Laboratory (FDCL), the Adverse Drug Reaction Centre, Clinical Trials Evaluation Committee, and the Centre for Diseases Control. The office of biologics, in collaboration with FDCL and the Adverse Drug Reaction Centre, is responsible for the marketing authorization and licensing, lot release, and regulatory inspections of biologics products.31 A wide list of locally manufactured biopharmaceuticals such as interferons, pegylated interferons, somatropin, follitropin, filgrastim, and erythropoietin are available in the Iranian market. Others, such as B-mab, T-mab, R-mab and E-cept, are in registration phases.32
The Iran NRA prepared a draft guideline on the registration of biosimilars based on the WHO draft guideline of 2009. After some amendments, the final draft has been finalized and approved in September 2010 by the Iran expert committee on biologicals, followed by the approval from the Head of the Iran US FDA in February 2011. Since the guideline has been drawn per WHO guidelines, the framework includes similar requirements.31
The Iranian guidelines are similar to the WHO guidelines in several areas, including head-to-head comparison of an SBP to a reference product in quality, requirement of extensive characterization beyond typical monograph specified test methods, demonstration of similarity at the levels of both drug substance and drug product between the RBP and SBP, equivalence in dosage form, clinical trial with a small sample size, and route of administration.31 The major difference that emerges on a comparison is that WHO does not recommend an SBP as a choice for RBP. However, in Iran, an SBP with US FDA or European Medicines Agency approval and accessible periodic safety update reports which is licensed in Iran and has a good marketing history can be considered an RBP. This helps in cases where the original drug is not registered. Also, Iran specifically recommends that specifications for an SBP should be the same as for the reference product, but does not consider a head-to-head comparative accelerated stability study as mandatory.31 Presently, the Iranian NRA does not insist on comprehensive clinical trials; it relies more on national postmarketing surveillance data for drug safety. Also, Iran imposes an import tariff as high as 65% on imported drugs in a bid to promote locally manufactured drugs, which as per EMA/US FDA/WHO regulatory norms may not be called “highly similar.” However, since the national adverse drug reaction reporting system is well established and implemented, there is no safety concern.32
Jordan (Jordan Food and Drug Administration)
The Jordan Food and Drug Administration (JFDA) comprises two main directorates, the Drug Directorate and Food Directorate, for drug and food safety, respectively. An individual registration department that comes under the auspices of the drug directorate performs regular registering of drugs for approval. The Technical Committee for the Registration of New Drugs is the committee responsible for registration of originator, new drugs, and biological and biosimilar products. The chairman of this committee is the secretary general of the Minister of Health, and the vice chairman is the director of the drug directorate.33
The committee decides on registration of new drugs and drugs that have a registered equivalent. It does this within a maximum period of 180 days from the date of the submission of the application of completed documents. In addition, the committee also ensures protection of the information in this application.
Since 2008, the JFDA had started to review applications for biosimilars on the basis of EMA guidelines. Emphasis has been laid on postmarketing surveillance, since to date no clinical studies have been conducted in Jordan. There is an ongoing debate regarding interchangeability and substitutability of the biosimilar product with the RBP, just as anywhere else in the world. The draft guidelines had been formulated and were available for comments till January 2014. The basis for these guidelines has been the EMA, the International Conference on Harmonisation, WHO, and also the lessons learned from submitted applications since 2008. Following the scope for customization of guidelines to be country specific as per WHO, the choice of RBP has been specifically defined as the first product registered internationally with a particular active ingredient.33,34
Middle East
A group of physicians and neurologists interested in multiple sclerosis, endocrinology, and pharmacology from across Middle Eastern countries convened to discuss the appropriate procedure for approval of biosimilars. In the absence of regional guidelines, they recommended the implementation of EMA guidelines for the requirements of safety, efficacy, and pharmacovigilance. The meeting minutes were published in 2008. The expert group recommended demonstration of consistent process with adequate quality control methods and comparable PK and PD of biosimilar and reference drugs. Double arm direct comparative clinical trials and postmarketing surveillance were also mandatory as per the recommendation. Efforts need to be initiated to have regional guidelines according to the group of expert physicians.35
The business of biologics in emerging markets
Biopharmaceuticals are different from small molecule chemical drugs in terms of the complexity which makes characterization difficult as well as a regulatory requirement. Generic drugs may not have the same elaborate and stringent approval processes, but face similar marketing concerns as biosimilars. Both generics and biosimilar markets share the same concerns, which are varied between established markets like EU/US and other emerging markets. According to Hordur Thoralissonn (Actavis Inc.), the term “emerging” is used to define the level of development of generics rather than development of the country per se. Extrapolation of such general issues apart from regulatory hurdles could be done from generics to biosimilars quite seamlessly. Peter Wittner (Interpharm Consultancy) suggested that a Darwinian survival of the fittest theory is almost invisibly present in the generics market to provide affordable drugs, which we may, in all probability, extend to biosimilars as well. According to him the European markets are attractive but need extensive and careful preparation before entry. Spain and Italy were good markets according to him, considering the underuse of generics in these countries. The “pharmerging” countries like the BRIC nations (Brazil, Russia, India, and the People’s Republic of China) according to him can prove to be attractive and huge, but their poor infrastructure, reduced health insurance coverage, and so forth can be intimidating.36
Bill Haddad (Biogenerics Pharma GmbH) and Richard Dicicco (Harvest Moon Pharmaceuticals USA Inc.) discussed the prospects of biosimilars in the SMi’s 13th Annual Meeting (2010). They were of the opinion that the “emerging” markets could be a better investment proposition than the US and EU markets. The former is reportedly biosimilar unfriendly, as it favors everlasting and perpetual patents, and the latter has such stringent guidelines that it does not make it any cheaper to develop biosimilars than original drugs; also, 15 of the member countries in the EU have laws to prevent substitution. Therefore, the private sector of pharmerging markets is attractive, since the cost of drugs is only about 25% cheaper than the developed counterparts. Also, government supply of these drugs through tenders could be good options in Latin America and the Middle East. Other factors which should potentially be evaluated before market penetration include protectionism (favoring of local players); the presence of import bans, limitations, or tariffs; reduced access to health care in challenging terrains; cold chain management; and a preference for dosage regimens that prevent frequent visits to the health care practitioner. Most generic sponsors enter into strategic partnerships with local manufacturers to gain access in these niche markets. This could be in the form of shifting production to local manufacturing units, support of science and technology needs, providing research and development support, helping to build quality systems, or providing innovative delivery devices that are better than those of the originator for self-administration. Supergenerics or biobetters also offer higher pricing margins and exclusivity in the market.36
Competitive biosimilar space
Contrasting emerging and established markets for the launch of affordable drugs is one side of the biosimilar coin. The other side is the emerging resistance of the global biotech giants against the industry of biosimilars. Originator companies have been devising newer strategies to combat the biosimilar competition. The originators frequently change delivery devices (switching from vials to prefilled syringes to autoinjectors) and changing of presentations (from liquid to lyophilized) combined with the withdrawal of older presentation/device. This is a matter of concern for biosimilar sponsors, who have to adapt their biosimilar development to a different presentation/device with the discontinuation of the original marketed reference standard. Some other strategies, like patenting of analytical methods by the innovator to evaluate critical quality attributes, limits the biosimilar sponsor in accurately determining the potency of their product, since the method is patented beyond the molecule’s patent life. The same also applies to formulation patents, which often have patent life beyond the molecule’s patent life. Frequent process changes by the originator gives rise to reference standards that are considerably different, and the biosimilar sponsor is left in the lurch with a near-to-completion process but without a comparable reference. The case of postapproval process changes for Enbrel, Rituxan, Mabthera, and Aranesp37 is a well-known example. The changes in physicochemical profile correlated with an altered potency profile. Nevertheless, they were available in the market with the same unaltered labels, indicating a comparable clinical profile that was acceptable to the regulatory authorities. On the contrary, WHO requires a suffix for glycosylated molecules which do not fall in the “highly similar” category, leading to molecules with extended names like epoietin zeta.38
Nomenclature of biosimilars
The WHO established the “International Nonproprietary Names” (INN) mode of nomenclature for biological and biotechnological substances which are indicative of the active ingredient and a classified group. The WHO has classified families of molecules and has provided appropriate stems and prestems that follow the typical INN schemes with prefixes and suffixes. This scheme has been recognized by all regulatory agencies the world over and sponsors use a brand name apart from this “generic” name. This avoids conflicts with existing names and instills confidence in the prescriber and user. The INN mode has a provision for glycosylated proteins to use different suffixes if glycosylation variations are extensive. However, in a recent WHO meeting there was lobbying from the innovators to consider a two-part name for biosimilars, with an INN first half and a second part indicating that this is a biosimilar.39 The originator companies, even after process changes of a glycosylated molecule, still retained the same primary INN on their label. This is one of the contentious issues with the biosimilar sponsors, as there will be no uniformity in the concept of naming and will eventually lead to apprehension of prescriber and user about a possibility of inferior quality in a biosimilar due to a different name. This could become contradictive, with the regulatory approval indicating a highly similar product but yet with a different name. The Pharmaceutical Research and Manufacturers Association (PhRMA) and the Generic Pharmaceutical Association (GPhA) in the US are still at loggerheads on this issue. The PhRMA has put forth the requirement of “Distinguishable Nonproprietary Names,” which will facilitate accurate attribution of adverse drug reactions to the right product during the tracking process, relieving the public of doubts. This, they predict, would alleviate confusions in a market that is anticipated to be flooded with a combination of biosimilars, interchangeable biosimilars, and multiple innovator biologics very soon, resulting in effective pharmacovigilance. However, the biosimilars sponsors quote that every adverse drug reaction tracking is through batch numbers, brand name, and manufacturer and is never general.40
Interchangeability
Since biological drugs can never be exact copies, the question whether they can be substitutes of original biologics remains unclear. The major concern about interchangeability is that repeated switches between the biosimilars and the reference biological may increase immunogenicity, leading to adverse reactions. Some inherent differences arising due to a postapproval process or a formulation change could lead to differential immunogenicity that may not necessarily be assessed through characterization or clinical trials during the time of application/approval, but may become evident during postapproval surveillance/pharmacovigilance. But this would be the case even for an originator drug that has undergone a process change but is marketed with the same name. Even during pharmacovigilance the studies are typically designed on patient population and never follow a single patient, making it very difficult to track the status of interchangeability issues. Therefore, it would be difficult for regulatory bodies to certify that the drug is truly interchangeable without adequate data. There has been considerable debate over this issue in the EU and US.20,41 According to EMA, approved biosimilar status signifies that the biosimilar can be used interchangeably with reference drugs. However, automatic substitution is not possible according to EU pharmaceutical law governing similar biological medicinal products. The EMA in a public consultation for the revision of the 2005 Guideline has included a new element wherein a statement mentions that a biosimilar application when assessed for marketing authorization does not certify the interchangeability status. Since these provisions come under the national laws of the EU Member States, the EMA does not have the power to make such a determination. According to the EMA, automatic substitution does not yet prevail for any approved biosimilar.42 The EU Generics Association also claims that more than 12 countries have rules against automatic substitution. However, France has now permitted the switching of biosimilars and generics with the originals as part of a new law concerning the social security budget (Article 47 of the Law of 23 December 2013), which came into effect on January 1, 2014.43 In the US, the US FDA has the power to make such announcements, although no such proclamation has been made so far. The guidances issued in 2012 do not discreetly address this. A survey of prescribers by the Alliance for Safe Biologic Medicines in 2012 indicated that they prefer different names so that they do not have to use their discretion for substitution/switching.44,45 Even as new guidelines are being drawn globally, in both established and emerging markets, this whole cloud of confusion prevails regarding the three facets of interchangeability:
- Automatic substitution of originator with biosimilars
- Switching of originator drug with biosimilar drug and vice versa
- Naming of biosimilar drugs.
Conclusion
The generic companies and biosimilar associations have united to counter misconceptions regarding their drugs. They have been applying continuous pressure on the regulatory agencies to favor the manufacture of generic/biosimilar drugs for the benefit of the patient population with low-cost alternatives. Unifying the approval pathway globally will abolish the need for bridging studies, which could make biosimilar development cost effective (since the sponsors will then have a single product development cycle for all geographies) but with the same standards of safety and efficacy. The European Generic Medicines Association and the GPhA, together in the Transatlantic Trade and Investment Partnership negotiations, have put forward their support for regulatory convergence of pharmaceuticals in the US and EU. They recommend combined regulatory approval pathway and compliance inspections. The combined development costs could be about $200–$300 million less per product than the individual route, leading to a huge cost difference that can be passed on to the patients in terms of low-cost medicines. There is a great deal of importance being laid on a unified International Committee on Harmonization authorized reference standard for biosimilar development, which would be acceptable to all regulatory agencies. This will bring down the development costs and fulfill the global aspirations of biosimilar companies. The public looks toward the biosimilar companies for lowering of costs of older drugs and in parallel are looking up to the innovator drug companies for new and more effective therapeutics. The war between “originator” and “biosimilar” sponsors is being closely watched, and the public hopes that there will be harmonization of regulatory expectations that are important to ensure safety and efficacy of biosimilars across regulatory agencies while also ensuring that patients have access to biosimilars.
Disclosure
The authors report no conflicts of interest in this work.
References
Weise M, Bielsky MC, De Smet K, et al. Biosimilars: what clinicians should know. Blood. 2012;120(26):5111–5117. | |
Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars. Ann Oncol. 2008;19:411–419. | |
US FDA. How drugs are developed and approved [webpage on the Internet]. Silver Spring, MD: US FDA; 2014. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/default.htm. Accessed August 25, 2014. | |
EMA. Guideline on similar biological medicinal products [webpage on the Internet]. Canary Wharf, London: EMA; 2005. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf. Accessed August 25, 2014. | |
Expert Committee on Biological Standardization. Guidelines on evaluation of similar biotherapeutic products (SBPs) [webpage on the Internet]. Geneva, Switzerland: WHO; 2009. Available at: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf?ua=1. Accessed August 25, 2014. | |
Korean Food and Drug Administration. Evaluation Guidelines for Biosimilars [webpage on the Internet]. KFDA; 2009. Available at: http://www.biosimilars.ca/docs/Evalutation_Guidelines_for_Biosimilars.pdf. Accessed August 25, 2014. | |
Health Canada. Guidance for sponsors: information and submission requirements for subsequent entry biologics (SEBs) [webpage on the Internet]. Ottawa, Ontario, Canada: Health Canada; 2010. Available at: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/seb-pbu/seb-pbu-2010-eng.pdf. Accessed August 25, 2014. | |
Government of India. Guideline on similar biologics: Regulatory requirements for marketing authorization in India [webpage on the Internet]. Government of India; 2012. Available at: http://dbtbiosafety.nic.in/Files%5CCDSCO-DBTSimilarBiologicsfinal.pdf. Accessed August 25, 2014. | |
US Department of Health and Human Services. Guidance for industry. Scientific considerations in demonstrating biosimilarity to a reference product [webpage on the Internet]. Rockville, MD: Food and Drug Administration; 2012. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed August 25, 2014. | |
US Department of Health and Human Services. Guidance for industry. Quality considerations in demonstrating biosimilarity to a reference product [webpage on the Internet]. Rockville, MD: Food and Drug Administration; 2012. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf. Accessed August 25, 2014. | |
IMS Health. Shaping the biosimilars opportunity: A global perspective on the evolving biosimilars landscape [webpage on the Internet]. London, UK: IMS Health; 2011. Available at: http://www.imshealth.com/ims/Global/Content/Home%20Page%20Content/IMS%20News/Biosimilars_Whitepaper.pdf. Accessed August 25, 2014. | |
Genetics and Biosimilars Initiative. US $ 54 billion worth of biosimilar patents expiring before 2020 [webpage on the Internet]. GaBI; 2011. Available at: http://www.gabionline.net/Biosimilars/Research/US-54-billion-worth-of-biosimilar-patents-expiring-before-2020. Accessed August 25, 2014. | |
European Medicines Agency. Biosimilar medicines – Biosimilar medicines authorised via the Agency [webpage on the Internet]. London, UK: European Medicines Agency. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/document_listing/document_listing_000318.jsp&murl=menus/special_topics/special_topics.jsp&mid=WC0b01ac0580281bf0. Accessed August 25, 2014. | |
Guidance on registration of similar biological products in Singapore [webpage on the Internet]. 2011. Available at: http://www.hsa.gov.sg/content/dam/HSA/HPRG/Western_Medicine/Overview_Framework_Policies/Guidelines_on_Drug_Registration/Appendix%2017_Guidance%20on%20Registration%20of%20Similar%20Biological%20Products%20in%20Singapore%202011.pdf. Accessed January 20, 2015. | |
Wang J, Chow SC. On the regulatory approval pathway of biosimilar products. Pharmaceuticals (Basel). 2012;5(4):353–368. | |
Guise JW, Carson BM. Biogeneric regulatory policies in China and India: a comparison study. Drug Information Journal. 2010;44:55–67. | |
Tzeng L. Follow-on biologics, data exclusivity, and the FDA. Berkeley Technology Law Journal. 2010;25(1):135–158. | |
Genetics and Biosimilars Initiative. Biosimilars marketed in Europe [webpage on the Internet]. GaBI; 2012. Available at: http://www.gabionline.net/Reports/Biosimilars-marketed-in-Europe. Accessed August 25, 2014. | |
BCC Research. Biosimilars: Global Markets. [webpage on the Internet]. Wellesley, MA: BCC Research; 2012. Available at: http://www.bccresearch.com/market-research/biotechnology/biosimilars-global-market-bio090a.html#gsc.tab=0. Accessed August 25, 2014. | |
Pharmatching Portrays. Pharma & Outsourcing Practice. Biosimilars|Guidelines|Players|Providers [webpage on the Internet]. Pharmatching; 2012. Available at: https://www.pharmatching.com/inforena/pharma-outsourcing-practice-biosimilars-guidelines-players-providers/download/91146/Pharmatching_Portrays_Biosimilars.pdf. Accessed August 25, 2014. | |
Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov. 2012;11(7):527–540. | |
Reddy S, Balamuralidhara V, Pramod Kumar TM, et al. Regulatory strategies for biosimilars in regulated and emerging markets. Pharma Times. 2013;45(1):11–14. | |
PMDA. Recent regulations of biosimilars in Japan. [webpage on the Internet] PMDA; 2011. Available at: http://www.pmda.go.jp/regulatory/file/english_presentation/biologics/B-E1arato.pdf. Accessed August 25, 2014. | |
Guidelines for the Quality, Safety and Efficacy Assurance of Follow-On Biologics [webpage on the Internet]; MHLW: Japan, 2009. Available at: http://www.pmda.go.jp/english/service/pdf/notifications/PFSB-ELD-0304007.pdf. Accessed August 25, 2014. | |
Ando Y, Tominaga T, Kondo T. PMDA update: the current situation and future directions. GaBI J. 2013;2(1):41–44. | |
Horikawa H, Tsubouchi M, Kawakami K. Industry views of biosimilar development in Japan. Health Policy. 2009;91(2):189–194. | |
Suh SK, Park Y. Regulatory guideline for biosimilar products in Korea. Biologicals. 2011;39(5):336–338. | |
Werble C. Canadian Biosimilar Approvals for Remicade: Time to Restart the Bus [webpage on the Internet]? 2014. Available at: http://pharmamedtechbi.com/publications/rpm-report/first-take/2014/1/canadian-biosimilar-approvals-for-remicade. Accessed August 25, 2014. | |
Health Canada. Subsequent entry biologics approved in Canada [webpage on the Internet]. Health Canada; 2014. Available at: http://www.gabionline.net/Biosimilars/General/Subsequent-entry-biologics-approved-in-Canada. Accessed August 25, 2014. | |
Kuhrt K, Gilpatrick J. A report from CPhI Worldwide 2013, Fifth Annual Pre-Connect Conference (October 22–24, 2013 – Frankfurt, Germany). Drugs Today (Barc). 2013;49(11):745–750. | |
Hadavand N, Valadkhani M, Zarbakhsh A. Current regulatory and scientific considerations for approving biosimilars in Iran. Biologicals. 2011;39(5):325–327. | |
Cheragalli AM. Current status of biopharmaceuticals in Iran’s pharmaceutical market. GaBI J. 2013;2(1):26–29. | |
Haddadin RD. Concept of biosimilar products in Jordan. Biologicals. 2011;39(5):333–335. | |
Jordan Food and Drug Administration. Guidance for registration of biosimilars [webpage on the Internet]. Available at: http://www.jfda.jo/Download/News/239_530.pdf. Accessed August 25, 2014. | |
Bohlega S, Al-Shammri S, Al Sharoqi I, et al. Biosimilars: opinion of an expert panel in the Middle East. Curr Med Res Opin. 2008; 24(10):2897–2903. | |
Edwards C. Generics, Supergenerics and Patent Strategies – SMi’s 13th Annual Meeting. IDrugs. 2010;13(7):433–436. | |
Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29(4):310–312. | |
International nonproprietary names (INN) for biological and biotechnological substances [webpage on the Internet]. 2006. Available at: http://www.who.int/medicines/services/inn/BioRevforweb.pdf. Accessed August 25, 2014. | |
The INN crowd. Nature biotechnology. 2013;31(12):1055. | |
PhRMA Comments to Generic Pharmaceutical Association Citizen. Petition Docket No FDA-2013-P-1153 [webpage on the Internet]. 2014. Food and Drug Administration. Available at: http://www.fdanews.com/ext/resources/files/02/02-03-14-PhRMA-Comments.pdf. Accessed August 25, 2014. | |
Amgen Inc. Amgen supports World Health Organization’s proposed distinguishable naming policy for biosimilars [press release]. Thousand Oaks, CA: Amgen Inc.; 2013 [March 24]. Available from: http://www.amgen.com/media/amgen_statement_WHO_biosimilar_naming.html. Accessed August 25, 2014. | |
Ebbers HC, Chamberlain P. Interchangeability An insurmountable fifth hurdle? GaBI J. 2014;3(2):88–94. | |
France to allow biosimilars substitution [webpage on the Internet]. 2014. Available at: http://www.gabionline.net/Policies-Legislation/France-to-allow-biosimilars-substitution/(highlight)/france%20substitution. Accessed August 25, 2014. | |
Naming and interchangeability of biosimilars raised in new survey [webpage on the Internet]. 2012. Available at: http://www.gabionline.net/Biosimilars/General/Naming-and-interchangeability-of-biosimilars-raised-in-new-survey. Accessed August 25, 2014. | |
Dolinar R. It’s all about the name: what is the imperative of adopting a unique names for biologic and biosimilar therapeutics [webpage on the Internet]? Food Drug Pol Forum. 2012;2(22). Available at: http://safebiologics.org/pdf/fdli-asbm-its-all-about-the-name.pdf. Accessed August 25, 2014. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.