")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
GLIPR1 Protects Against Cigarette Smoke-Induced Airway Inflammation via PLAU/EGFR Signaling
Authors Peng W, Wu Y, Zhang G, Zhu W, Chang M, Rouzi A, Jiang W, Tong L, Wang Q, Liu J, Song Y, Li H, Li K, Zhou J
Received 12 July 2021
Accepted for publication 27 September 2021
Published 8 October 2021 Volume 2021:16 Pages 2817—2832
DOI https://doi.org/10.2147/COPD.S328313
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Min Zhang
Wenjun Peng,1,* Yuanyuan Wu,1,* Ge Zhang,1,* Wensi Zhu,1,* Meijia Chang,1 Ainiwaer Rouzi,1 Weipeng Jiang,1 Lin Tong,1 Qin Wang,1 Jie Liu,1 Yuanlin Song,1– 4 Huayin Li,1 Ka Li,5 Jian Zhou1– 4
1Department of Pulmonary and Critical Care Medicine, Shanghai Respiratory Research Institute, Zhongshan Hospital, Fudan University, Shanghai, 200032, People’s Republic of China; 2Center of Emergency & Intensive Care Unit, Jinshan Hospital, Fudan University, Shanghai, 200540, People’s Republic of China; 3Shanghai Key Laboratory of Lung Inflammation and Injury, Shanghai, 200032, People’s Republic of China; 4Shanghai Engineering Research Center of Internet of Things for Respiratory Medicine, Shanghai, 200032, People’s Republic of China; 5Institute of Clinical Science, Zhongshan Hospital, Fudan University, Shanghai, 200032, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jian Zhou
Department of Pulmonary and Critical Care Medicine, Shanghai Respiratory Research Institute, Zhongshan Hospital, Fudan University, Shanghai, 200032, People’s Republic of China
Email [email protected]
Ka Li
Institute of Clinical Science, Zhongshan Hospital, Fudan University, Shanghai, 200032, People’s Republic of China
Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is a major health problem associated with high mortality worldwide. Cigarette smoke (CS) exposure is the main cause of COPD. Glioma pathogenesis-related protein 1 (GLIPR1) plays a key role in cell growth, proliferation, and invasion; however, the role of GLIPR1 in COPD remains unclear.
Methods: To clarify the involvement of GLIPR1 in COPD pathogenesis, Glipr1 knockout (Glipr1-/-) mice were generated. Wild-type (WT) and Glipr1-/- mice were challenged with CS for 3 months. To illustrate how GLIPR1 regulates CS-induced airway damage, knockdown experiments targeting GLIPR1 and PLAU, as well as overexpression experiments of PLAU, were performed with human bronchial epithelial cells.
Results: Compared with WT mice, Glipr1-/- mice showed exacerbated CS-induced airway damage including lung inflammation, airway wall thickening, and alveolar destruction. After CS exposure, total proteins, total white cells, neutrophils, lymphocytes, IL-6, and matrix metalloproteinase-9 increased significantly in lung of Glipr1-/- mice than those in lung of WT mice. Furthermore, in vivo and in vitro experiments demonstrated that silencing of GLIPR1 inactivated PLAU/EGFR signaling and promoted caspase-1-dependent pyroptosis (a mode of inflammatory cell death) induced by CS and CS extract exposure, respectively. In vitro experiments further revealed the interaction between GLIPR1 and PLAU, and silencing of PLAU blocked EGFR signaling and promoted pyroptosis, while overexpression of PLAU activated EGFR signaling and reversed pyroptosis.
Conclusion: To conclude, GLIPR1 played a pivotal role in COPD pathogenesis and protected against CS-induced inflammatory response and airway damage, including cell pyroptosis, through the PLAU/EGFR signaling. Thus, GLIPR1 may play a potential role in COPD treatment.
Keywords: chronic obstructive pulmonary disease, cigarette smoke, GLIPR1, PLAU, pyroptosis
Introduction
Chronic obstructive pulmonary disease (COPD) is a well-known chronic airway inflammatory disease characterized by progressive and irreversible airflow limitation with airway inflammation and airway remodeling.1 Over the last decade, the incidence of COPD has been increasing consistently and is associated with extremely high mortality; this has made COPD the third leading cause of death worldwide, thereby resulting in a major healthcare burden in both developing and developed countries.1,2 Cigarette smoke (CS) is the main cause of COPD globally, although other risk factors such as air pollution and microbial infections are also responsible.3,4 Exposure to these risk factors, especially smoking, could lead to an imbalance in oxidation and antioxidation and release of inflammatory cytokines, thereby prompting apoptosis of epithelial, endothelial, and other parenchymal cells in the lungs, as evidenced in clinical studies5–7 and animal experiments.8–11 Apoptosis of parenchymal cells further leads to the destruction of lung tissue structure, pulmonary emphysema, and airway remodeling.12,13 Although considerable efforts have been made to target inflammation and oxidative stress, unsuccessful previous therapies have necessitated the urgency of developing novel and effective therapeutic options for COPD.1
Glioma pathogenesis-related protein 1 (GLIPR1) was originally identified and characterized with oncogenic features in human glioblastoma.14 GLIPR1 is related to testes-specific, vespid, and pathogenesis protein-1 (RTVP-1) and is highly expressed in Wilms’ cancer15 and melanoma.16 The transcript level of GLIPR1 is regulated by the degree of promoter methylation.15,17–19 GLIPR1 overexpression promotes the growth, proliferation, and invasion of glioma cells;20,21 whereas, GLIPR1 silencing reduces the proliferation and induces apoptosis in glioma20 and melanoma cells.16 At the same time, silencing GLIPR1 expression also inhibits prostate cancer cell proliferation,22 indicating the necessity of GLIPR1 in cell growth. Thus, GLIPR1 plays an indispensable role in cell growth and proliferation. Nevertheless, its role in COPD pathogenesis has not yet been reported.
Plasminogen activator urokinase (PLAU), also known as urokinase-type plasminogen activator (uPA), plays a key role in tumorigenesis and metastasis of many malignancies such as colon and breast cancers.23,24 Levels of PLAU are strongly correlated with cancer prognosis, including recurrence and metastasis.23–25 Targeting PLAU successfully triggers cell apoptosis and inhibits cell growth, proliferation, invasion, and metastasis both in vivo and in vitro.23,26 In addition, PLAU is critical for various inflammatory processes, such as promoting the activation of inflammatory cells and releasing inflammatory cytokines.27 PLAU-dependent proteolysis destroys basement membranes and extracellular matrix (ECM) proteins, thereby resulting in tissue degradation and remodeling.28–30 PLAU bound to its receptor (PLAUR or uPAR) may also contribute to the peripheral lung-remodeling process in multiple pulmonary diseases,31 and promote cell proliferation through receptor tyrosine kinases, such as EGFR, HER1, and ErbB1.32,33 EGFR is one of the most important components of the PLAUR proliferasome, a macromolecular complex stimulating cell proliferation, and EGFR knockdown leads to cell apoptosis.34
Apoptosis is a form of programmed cell death. However, continuous research has shown that many cell death phenomena could not be explained by apoptosis.35 Pyroptosis has been identified as the other form of programmed necrosis, and is also known as a pro-inflammatory and lytic mode of cell death that influences immunity and diseases.36 Until now, the mechanism of cell pyroptosis has not been completely studied. Inflammatory caspases (caspase-1, murine caspase-11, and human caspase-4/5) mediate cell pyroptosis, and this distinguishes pyroptosis from apoptosis. Caspase-1 is activated by various canonical inflammasomes, including NLRP3, NLRP1, AIM2, and Pyrin;36 whereas, caspase-4/5/11 directly recognize bacterial lipopolysaccharide.37,38 Both of these ways trigger pyroptotic cell death through specific cleavage of gasdermin D (GSDMD) to release a free gasdermin-N domain,39 which forms extensive pores on the plasma membrane that is made of artificial or natural phospholipid mixtures.40 Meanwhile, caspase-1 activation processes pro-IL-1β and pro-IL-18 into biologically mature IL-1β and IL-18, which are released extracellularly via the membrane pores formed by GSDMD-N domains.35,41 The difference between caspase-1 in the canonical inflammasome pathway and caspase-4/5/11 in the non-canonical inflammasome pathway is dependent on the direct and indirect activation of caspase-1, respectively.39 Increasing evidence has demonstrated that CS-induced caspase-1-dependent pyroptosis is present in vein endothelial cells,42 urothelial cells,43 and skeletal muscle.44
In this study, mice and human bronchial epithelial (HBE) cells were treated with CS and CS extract (CSE), respectively, to simulate CS-induced airway damage. GLIPR1 knockout mice and short hairpin RNA (shRNA) targeting GLIPR1 and PLAU were introduced to demonstrate the potential role and underlying mechanism of GLIPR1 in CS-induced airway damage.
Materials and Methods
Preparation of CSE
CS generated by 20 cigarettes (Daqianmen, tar yield 10 mg per cigarette, nicotine 0.8 mg per cigarette, CO yield 12 mg per cigarette, Shanghai Tobacco Group) were slowly bubbled into 50 mL sterile serum-free cell culture media using a syringe at a flow rate of 60 mL/min. A single cigarette was consumed in about 6 min. CSE solution was filtered (0.22 μM; SLGS033SS, Merck Millipore, MA, USA) and was designated as 100% CSE solution, whose OD value was 3.80 at 320 nm.
Cell Culture
HBE cells were purchased from the American Type Culture Collection (CRL-2741) and cultured in Gibco RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA), and supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and penicillin–streptomycin (100 U/mL). The culture was maintained in 5% CO2 at 37 °C. For CS exposure experiments, HBE cells were treated with CSE for 3 days.
Lentiviral Construction and Infection
shRNA vectors targeting the GLIPR1 gene (shGLIPR1) and PLAU gene (shPLAU) were purchased from The RNAi Consortium (Boston, MA, USA). Lentiviral plasmids were purchased from GeneChem (Shanghai, China). Lentiviruses carrying overexpressing human PLAU (GenBank accession number NM_002658.5) lentiviral vectors (GL125) were from OBiO Technology (Shanghai, China). For viral infections, HBE cells were plated onto 6-well plates and incubated with medium containing 5 μg/mL polybrene and viruses at 37 °C. After 12 hours, the transfection mixture was replaced with normal growth medium.
Animals and CS Exposure
Glipr1 knockout mice (Glipr -/-) of C57BL/6 background were generated with CRISPR-Cas9 gene editing done by Shanghai Model Organisms (Shanghai, China) (Supplementary Figure S1). More details are shown in the Supplemental Materials and Methods. Then, the mice were routinely bred in the animal facility of Zhongshan Hospital at Fudan University (Shanghai, China), together with C57BL/6 mice. All mice were provided free access to water and food at 20–25 °C with the relative humidity of 50–70%. All experimental procedures complied with Guide for Care and Use of Laboratory Animals of USA National Institutes of Health, and Laboratory animal - Guideline for ethical review of animal welfare in China, and were approved by the Animal Care and Use Committee of Zhongshan Hospital at Fudan University. C57BL/6 and Glipr1-/- mice were randomly assigned to wild-type (WT) group, WT CS group, Glipr1-/- group, and Glipr1-/- CS group. Mice of the WT CS group and Glipr1-/- CS group were challenged with smoke generated by burning cigarettes (Daqianmen) for 2 h per day, 6 days a week. After 3 months, the mice were sacrificed under over anesthesia to collect peripheral blood, bronchoalveolar lavage fluids (BALF), and lung tissues. Immediately following the sacrifice, the right upper lobes of the lungs were harvested, fixed in 4% paraformaldehyde overnight, and embedded in paraffin. The sections were then stained with hematoxylin and eosin (HE) and Masson’s staining kit (Sigma-Aldrich, St. Louis, MO, USA) according to conventional protocols for histopathological evaluation.
Total Protein Concentration and Inflammatory Cell Count in BALF
BALF was collected by cannulating the trachea and then centrifuging the BALF at 1,000 rpm for 5 min. The supernatant was aspirated and used for detection of multiple factors and total protein levels. Each cell pellet was resuspended and stained with Wright–Giemsa staining solution (Thermo Fisher Scientific, Waltham, MA, USA). Total protein levels in BALF were measured by Bicinchoninic Acid Protein Assay Kit (#7780, Cell Signaling Technology, Boston, MA, USA) as per the manufacturer’s protocol.
IL-6, IL-1β, and PLAU ELISA
IL-6 (DY406, R&D Systems, Minneapolis, MN, USA), IL-1β (DY401, R&D Systems), and soluble PLAU (ab198512, Abcam, Cambridge, MA, USA) in plasma and BALF were measured using ELISA kits according to the manufacturer’s instructions.
Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from the cells or lung tissues using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), and complementary DNA (cDNA) was synthesized using reverse transcriptase (TOYOBO, Osaka, Japan). mRNA levels were quantified using SYBR Premix EX TaqTM (TaKaRa Bio, Osaka, Japan). Gene-specific primers sequences are provided in Supplementary Table S1.
Western Blotting Analysis
Total protein was extracted using a RIPA Lysis Kit (Beyotime Biotechnology, Shanghai, China) by following the manufacturer’s protocol. Equal amounts of protein from each sample were electrophoretically separated using polyacrylamide gels, and then transferred to polyvinylidene fluoride membranes. The membranes were blocked at 23–25 °C for 1 h and then incubated at 4 °C overnight with primary antibodies against GLIPR1 (ab198215, Abcam), PLAU (ab28230 and ab24121, Abcam), EGFR (ab52894, Abcam), p-EGFR (11862S, Cell Signaling Technology, Boston, MA, USA), caspase-1 (22915-1-AP, Proteintech Group, Chicago, IL, USA), GSDMD (ab209845, Abcam; 97558, Cell Signaling Technology), IL-1β (AF5103, Affinity Biosciences, Cincinnati, OH, USA), and β-actin (4970, Cell Signaling Technology). Then, the membranes were washed three times and incubated with an appropriate horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h. The proteins were visualized using enhanced electro-chemiluminescence reagents (Beyotime Biotechnology), and the bands were analyzed using an imaging system (Bio-Rad, Hercules, CA, USA).
Immunohistochemistry (IHC)
By following the procedure we described,45 anti-Glipr1 antibody (ab198215, Abcam) was incubated with the sections at room temperature for 1 h and overnight at 4 °C. After washing three times with PBS, an HRP-conjugated goat anti-rabbit secondary antibody was incubated with the sections at room temperature for 45 min.
Co-Immunoprecipitation (CO-IP)
Whole-cell total protein was extracted using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific). In brief, 5×106 HBE cells were lysed and centrifuged. Then, the supernatant was collected and diluted to 100 μg/μL. 400 μg of cell extract was incubated with anti-GLIPR1 antibody (SC-517141, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-PLAU antibody (MAB1310-100, R&D Systems), or anti-mouse immunoglobulins (IgG) overnight at 4 °C with gentle rotation. The next day, the mixture of the cell extract and GLIPR1 or PLAU was incubated with Protein A/G PLUS-Agarose (SC- 2003, Santa Cruz Biotechnology) at 4 °C for 1 h. Then, the mixture was washed 6 times and analyzed by Western blotting.
Statistical Analysis
The results are shown as the mean ± SD. Statistical analysis was performed using GraphPad Prism 8.0 software (GraphPad, San Diego, CA, USA). Comparisons between two groups were determined by Student’s t-test; whereas, multiple comparisons groups of more than two groups were determined using two-way ANOVA followed by Tukey post hoc test. P < 0.05 was defined as the threshold for significance.
Results
Glipr1 Levels Were Increased in the Lung Tissues of Mice After CS Exposure
To investigate the potential role of Glipr1 in COPD, we compared the expression levels of Glipr1 in the lung tissues of mice in response to CS exposure. The mRNA expression of Glipr1 was significantly higher in the lung tissues after CS exposure (Figure 1A). Consistently, Western blotting analysis revealed elevated protein levels of Glipr1 in lung tissues after CS exposure as well (Figure 1B). These results suggested a conceivable correlation between GLIPR1 and CS-induced airway damage.
 |
Figure 1 Glipr1 knockout exacerbated CS-induced airway damage. (A) mRNA and (B) protein levels of Glipr1 in lung tissues of mice were increased after CS exposure. (C) Weight of mice (in grams) after CS exposure. (D) Hematoxylin and eosin (HE) staining. (E–H) Quantitative analysis of lung damage as assessed histopathologically. (E) Lung injury scores. (F) Mean linear intercept (MLI). (G) Mean alveolar septal thickness (MAST). (H) Destructive index (DI). Ten fields were randomly selected for scoring. (I) Masson’s staining. Error bars represent SD. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviations: WT, wild-type; CS, cigarette smoke; WT CS, wild-type cigarette smoke. |
Glipr1 Knockout Exacerbated CS-Induced Airway Damage
To further clarify the role of Glipr1 in COPD pathogenesis, Glipr1 knockout mice were generated. The initial weight of each mice was between 20 and 23 g. Weight loss was induced by CS exposure for 3 months in both WT and Glipr1-/- mice; however, it was more pronounced in the Glipr1-/- mice (Figure 1C). HE staining revealed no obvious lung damage in Glipr1-/- mice in comparison with that in WT mice; however, COPD phenotypes, such as lung inflammation, airway wall thickening, alveolar destruction, and airway remodeling, were detected in response to CS exposure in WT mice, and were apparently aggravated in CS-exposed Glipr1-/- mice (Figure 1D). In accordance with this, lung pathology indicated augmented inflammatory cell infiltration around the airway in CS-exposed Glipr1-/- mice than in CS-exposed WT mice (Figure 1D). Lung injury score, a relatively quantitative indicator, of WT and Glipr1-/- mice was significantly increased after CS exposure, and the score of CS-exposed Glipr1-/- mice was much higher than that of CS-exposed WT mice (Figure 1E). Quantitative analysis also suggested that alveolar-space broadening, alveolar septum thickening, and alveolar wall destruction were significantly exacerbated in CS-exposed Glipr1-/- mice than in CS-exposed WT mice (Figure 1F–H).
Furthermore, Masson’s staining demonstrated that both WT and Glipr1-/- mice presented with fibrous deposition around the airway after CS exposure (Figure 1I). Compared with CS-exposed WT mice, fibrosis level of lung tissues in CS-exposed Glipr1-/- mice was significantly exacerbated (Figure 1I).
Glipr1 Knockout Caused More Severe Inflammatory Infiltration After CS Exposure
Chronic inflammation was implicated in response to CS exposure in COPD pathogenesis. Therefore, in this study, BALF was used for total protein detection and cell counting to evaluate inflammatory infiltrates in the lungs. Chronic CS exposure caused more severe protein accumulation in Glipr1-/- mice than in WT mice (Figure 2A). CS exposure led to significant increase in total cells, neutrophils, and macrophages in the BALF in both WT and Glipr1-/- mice (Figure 2B–D). Further, the counts of total cells, neutrophils, and lymphocytes in the BALF were significantly higher in CS-exposed Glipr1-/- mice than in CS-exposed WT mice (Figure 2B, C, and E). Furthermore, IL-6, a proinflammatory cytokine, was noticed in the BALF and plasma of both WT and Glipr1-/- mice following CS exposure (Figure 2F and G). Compared to the CS-exposed WT mice, IL-6 levels in the BALF and plasma were significantly increased in the CS-exposed Glipr1-/- mice (Figure 2F and G).
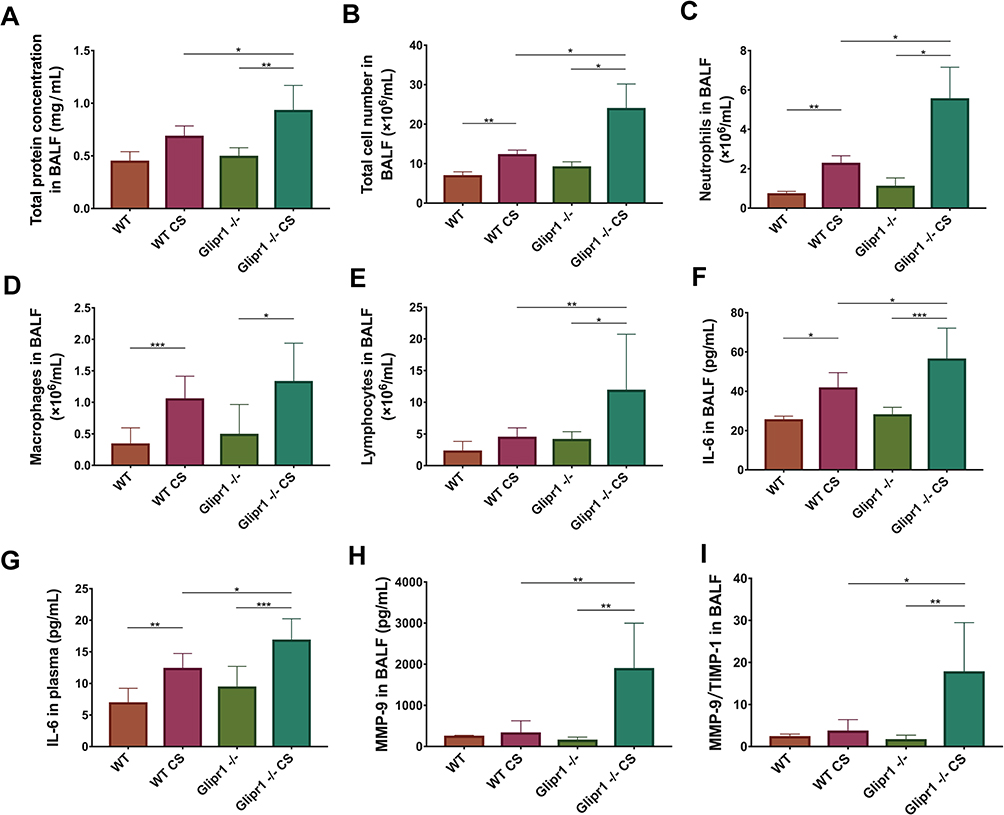 |
Figure 2 Glipr1 knockout caused more severe inflammatory infiltration after cigarette smoke exposure. (A) Total protein levels, (B) total cell counts, (C) neutrophils, (D) macrophages, and (E) lymphocytes in bronchoalveolar lavage fluid (BALF) were assessed. IL-6 concentrations in (F) BALF and (G) plasma samples were measured by ELISA. (H) MMP-9 concentration in BALF were measured by ELISA. (I) Ratio of MMP-9/TIMP-1 in the BALF. Error bars represent SD. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviations: WT, wild-type; CS, cigarette smoke; WT CS, wild-type cigarette smoke; MMP-9, matrix metalloproteinase; TIMP-1, tissue inhibitor of metalloproteinase. |
Matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinase (TIMP) are two important factors in the regulation of ECM synthesis and degradation, which is in a dynamic equilibrium under normal physiological circumstances.46,47 The increase in MMP-9 and imbalance between MMP-9 and TIMP-1 causes abnormal degradation or accumulation of ECM in alveolar and small airway walls, thereby contributing to COPD pathogenesis, and were associated with acute exacerbation of COPD.47–49 Here, we evaluated the levels of MMP-9 and TIMP-1 in the BALF. ELISA detection showed a sharp rise in MMP-9 and ratio of MMP-9/TIMP-1 in CS-exposed Glipr1-/- mice than in CS-exposed WT mice (Figure 2H and I).
Glipr1 Knockout Aggravated Caspase-1-Dependent Pyroptosis in CS-Induced Airway Damage
To evaluate the role of Glipr1 in pyroptosis, pro-caspase-1, cleaved-caspase-1, GSDMD, GSDMD N-terminal domain, pro-IL-1β, and cleaved-IL-1β were examined by Western blotting analysis and ELISA. The expression of cleaved-caspase-1, GSDMD N-terminal domain, and mature IL-1β were significantly increased after CS exposure in both WT and Glipr1-/- mice (Figure 3A–F). In comparison with the CS-exposed WT mice, activation of caspase-1, separation of GSDMD N-terminal domain, and maturation and secretion of IL-1β exhibited a significant increase in CS-exposed Glipr1-/- mice (Figure 3A–F). These data illustrated that Glipr1 knockout exacerbated CS-induced pyroptosis, thereby suggesting potential anti-pyroptosis effect of Glipr1 when challenged with CS exposure.
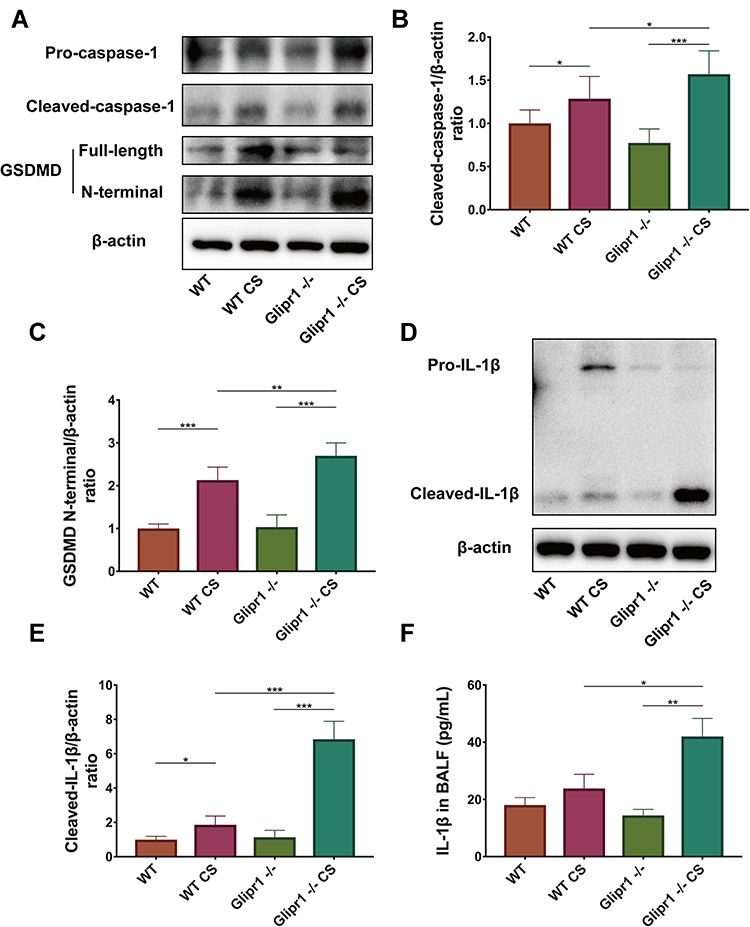 |
Figure 3 Glipr1 knockout aggravated caspase-1-dependent pyroptosis in CS-induced airway damage. (A) Levels of pro-caspase-1, cleaved-caspase-1, gasdermin D (GSDMD), GSDMD N-terminal domain, and β-actin proteins in the lung tissues of mice were measured by Western blotting analysis. Quantitative analysis of (B) cleaved-caspase-1 and (C) GSDMD N-terminal domain. (D) Levels of pro-IL-1β and cleaved- IL-1β in the lung tissues of mice were measured by Western blotting analysis. (E) Quantitative analysis of cleaved-IL-1β expression. (F) IL-1β concentration in bronchoalveolar lavage fluid (BALF) were measured by ELISA. Error bars represent SD. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviations: WT, wild-type; CS, cigarette smoke; WT CS, wild-type cigarette smoke; MMP-9, matrix metalloproteinase; TIMP-1, tissue inhibitor of metalloproteinase. |
Glipr1 Protected CS-Induced Airway Damage via PLAU/EGFR Signaling
In CS-exposed Glipr1-/- mice, the increase of Glipr1 was much lower than that in the CS-exposed WT mice (Figure 4A and B). As previous studies have indicated the association between PLAU and COPD,46,50 we determined whether the expression of Plau was affected by CS exposure and Glipr1 knockout. To examine the expression level of Plau, Western blotting analysis, qRT-PCR, and ELISA were performed. The mRNA and protein levels of Plau were significantly increased in mice following CS exposure (Figure 4A, C, F and G). Glipr1 knockout significantly reduced Plau expression in both the absence and presence of CS exposure (Figure 4A, C, F and G). In consistent with this, IHC showed that Glipr1 expression increased after CS exposure in both WT and Glipr1-/- mice, while Glipr1 knockout significantly decreased Glipr1 expression in Glipr1-/- mice with and without CS exposure compared with WT and CS-exposed WT mice (Figure 4H). Moreover, Glipr1 was highly expressed in bronchial epithelial cells and alveolar epithelial cells (Figure 4H).
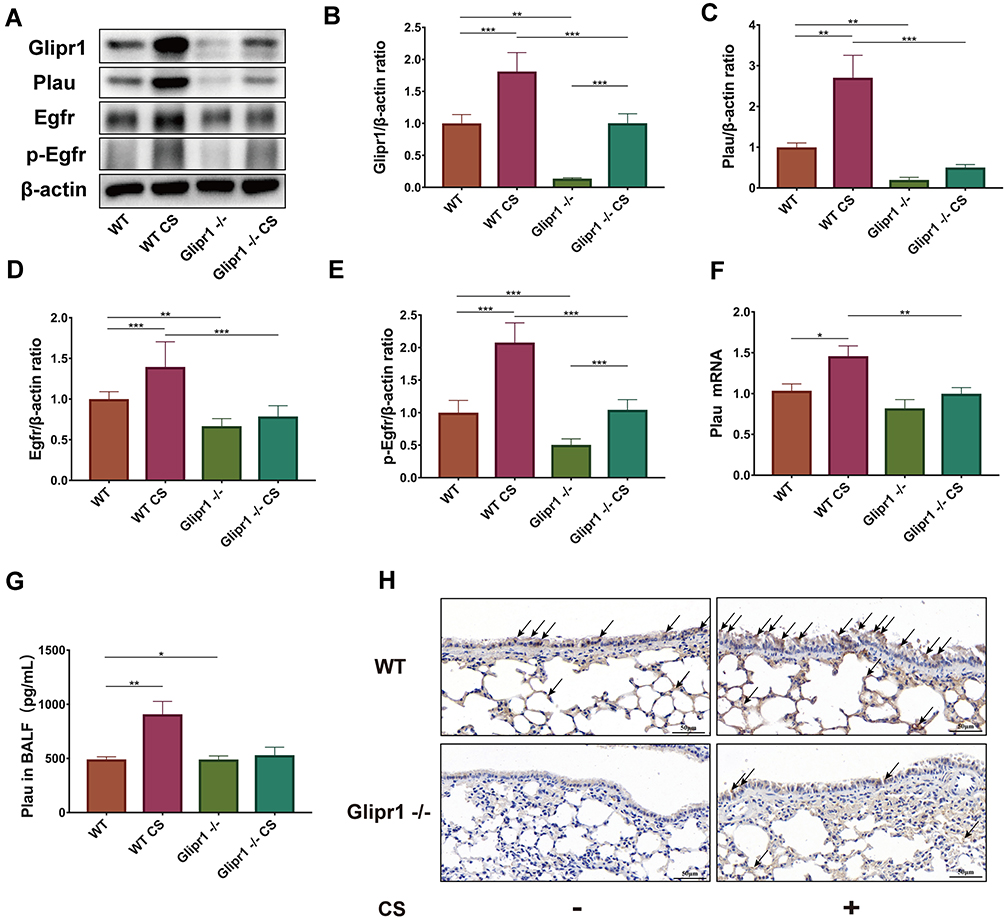 |
Figure 4 Glipr1 protected CS-induced airway damage through PLAU/EGFR signaling. (A) Levels of Glipr1, Plau, Egfr, p-Egfr, and β-actin proteins in the lung tissues of mice were measured by Western blotting analysis. Quantitative analysis of (B) Glipr1, (C) Plau, (D) Egfr, and (E) p-Egfr. (F) mRNA levels and (G) protein expression in bronchoalveolar lavage fluid (BALF) of Plau. (H) IHC. Error bars represent SD. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviations: WT, wild-type; CS, cigarette smoke; WT CS, wild-type cigarette smoke. |
PLAU also activates EGFR and induces phosphorylation of EGFR.51 Therefore, we detected the levels of EGFR and p-EGFR. Compared with WT mice, CS-induced increase in EGFR expression and its activation were largely hindered by Glipr1 knockout (Figure 4A, D, and E). These results indicated that Glipr1 was crucial for activating the PLAU/EGFR signaling against CS-induced airway damage.
GLIPR1 Knockdown Exacerbated CSE-Induced Pyroptosis in vitro
As the results shown in IHC staining (Figure 4H), Glipr1 was highly expressed in bronchial epithelial cells. Thus, to confirm the role of GLIPR1 in CS-induced damage, shGLIPR1 was used to silence GLIPR1 in human bronchial epithelial (HBE) cells. CSE significantly led to the activation of caspase-1, cleavage of GSDMD, and maturation of IL-1β, which were apparently exacerbated by GLIPR1 knockdown (Figure 5A and B). These data demonstrated that GLIPR1 knockdown exacerbated CSE-induced pyroptosis in vitro.
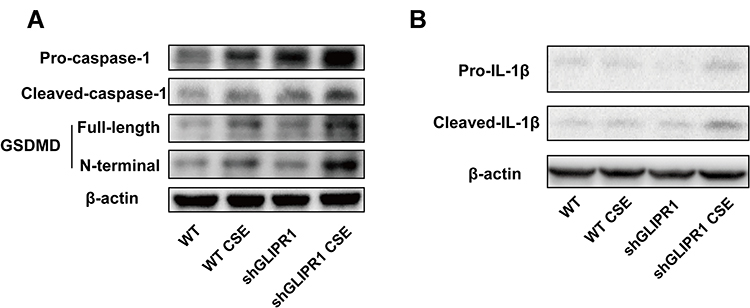 |
Figure 5 GLIPR1 knockdown exacerbated CSE-induced pyroptosis in vitro. (A) Levels of pro-caspase-1, cleaved-caspase-1, gasdermin D (GSDMD), GSDMD N-terminal domain, and β-actin proteins in human bronchial epithelial (HBE) cells were measured by Western blotting analysis. (B) Levels of pro-IL-1β and cleaved-IL-1β in HBE cells were measured by Western blotting analysis. shRNA vectors targeting GLIPR1. Abbreviations: WT, wild-type; CSE, cigarette smoke extract; WT CSE, wild-type cigarette smoke extract. |
GLIPR1 Knockdown Reduced the PLAU/EGFR Signaling in vitro
GLIPR1 expression, on both the gene and protein levels, was significantly promoted by CSE stimulation (Figure 6A, B, and F). Protein levels of PLAU, EGFR, and p-EGFR were increased after the CSE challenge (Figure 6A, C–E). Consistently, qRT-PCR assays showed that the mRNA levels of PLAU and EGFR were much higher after CSE exposure (Figure 6G and H). GLIPR1 silencing considerably reduced the levels of PLAU, EGFR, and p-EGFR with or without CSE exposure (Figure 6A–H), thereby suggesting a pivotal role of GLIPR1 in activating the PLAU/EGFR signaling.
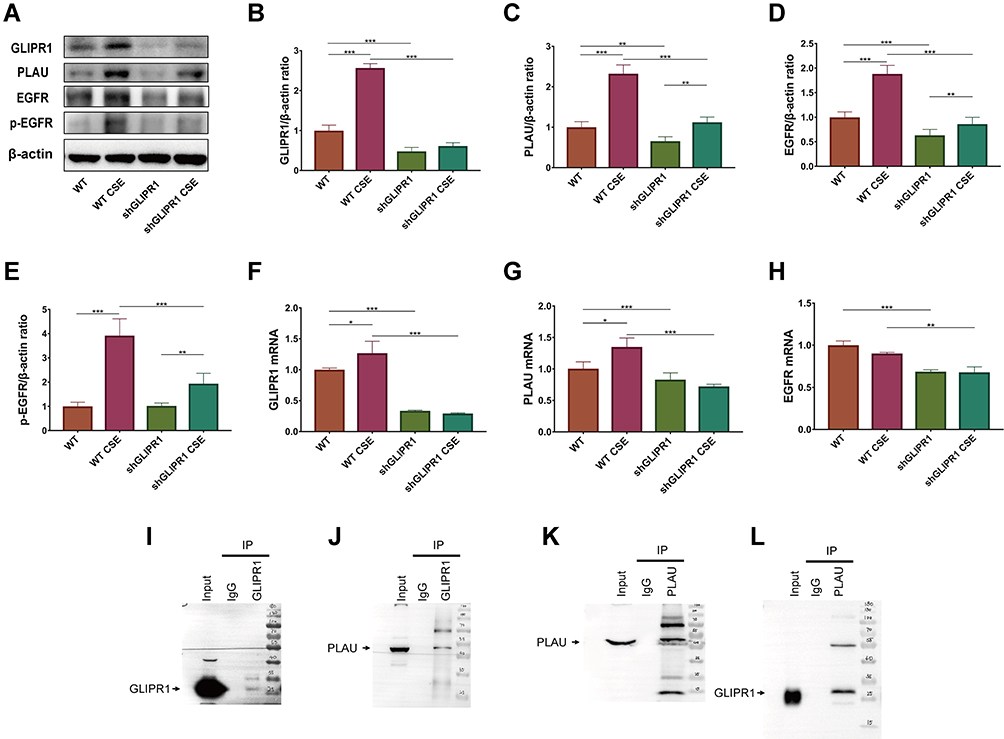 |
Figure 6 GLIPR1 knockdown reduced PLAU/EGFR signaling in vitro. (A) Levels of GLIPR1, PLAU, EGFR, p-EGFR, and β-actin proteins in human bronchial epithelial (HBE) cells were measured by Western blotting analysis. Quantitative analysis of (B) GLIPR1, (C) PLAU, (D) EGFR, and (E) p-EGFR. mRNA levels of (F) GLIPR1, (G) PLAU, (H) EGFR. (I) GLIPR1 was pull downed by Western blotting using anti- GLIPR1 antibody. (J) Immunoprecipitation assay showed that PLAU was pull downed by GLIPR1. (K) PLAU was pull downed by Western blotting using anti- PLAU antibody. (L) Immunoprecipitation assay showed that GLIPR1 was pull downed by PLAU. Error bars represent SD. *P < 0.05; **P < 0.01; ***P < 0.001. shRNA vectors targeting GLIPR1. Abbreviations: WT, wild-type; CSE, cigarette smoke extract; WT CSE, wild-type cigarette smoke extract. |
GLIPR1 Formed Complex with PLAU
To determine whether GLIPR1 interacts with PLAU, a CO-IP assay was performed. Firstly, GLIPR1 was combined with protein A/G PLUS-agarose and then was pull downed by anti-GLIPR1 antibody and tested using Western blotting (Figure 6I). Then, the immunocomplexes with GLIPR1 were subjected to Western blotting analysis and PLAU expression was clearly detected in HBE cells (Figure 6J). In consistent with this, Western blotting by anti-PLAU antibody was used to analyze the immunocomplexes with PLAU, in which GLIPR1 expression was clearly detected using anti-GLIPR1 antibody (Figure 6K and L). Our results demonstrated the interaction between endogenous GLIPR1 and endogenous PLAU in HBE cells, suggesting the potential biological function of GLIPR1 in regulation of PLAU.
PLAU Knockdown Exacerbated CSE-Induced Pyroptosis in vitro
To clarify the role of PLAU in CS-induced damage, shPLAU was used to silence PLAU in HBE cells. CSE-induced activation of caspase-1, cleavage of GSDMD, and maturation of IL-1β were significantly exacerbated by PLAU knockdown (Figure 7A and B), indicating that PLAU knockdown exacerbated CSE-induced pyroptosis in vitro.
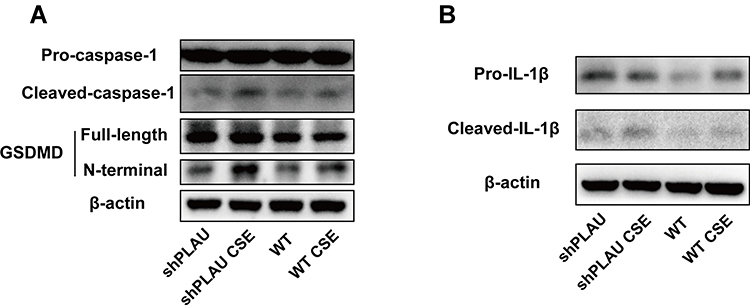 |
Figure 7 PLAU knockdown exacerbated CSE-induced pyroptosis in vitro. (A) Levels of pro-caspase-1, cleaved-caspase-1, gasdermin D (GSDMD), GSDMD N-terminal domain, and β-actin proteins in human bronchial epithelial (HBE) cells were measured by Western blotting analysis. (B) Levels of pro-IL-1β and cleaved-IL-1β in HBE cells were measured by Western blotting analysis. shRNA vectors targeting PLAU. Abbreviations: WT, wild-type; CSE, cigarette smoke extract; WT CSE, wild-type cigarette smoke extract. |
PLAU Knockdown Reduced EGFR Signaling in vitro
PLAU knockdown significantly blocked the CSE-induced increase in protein expression of PLAU, EGFR, and p-EGFR (Figure 8A–D). qRT-PCR assays also verified that PLAU knockdown significantly reduced the mRNA levels of PLAU and EGFR with or without CSE exposure (Figure 8E and F).
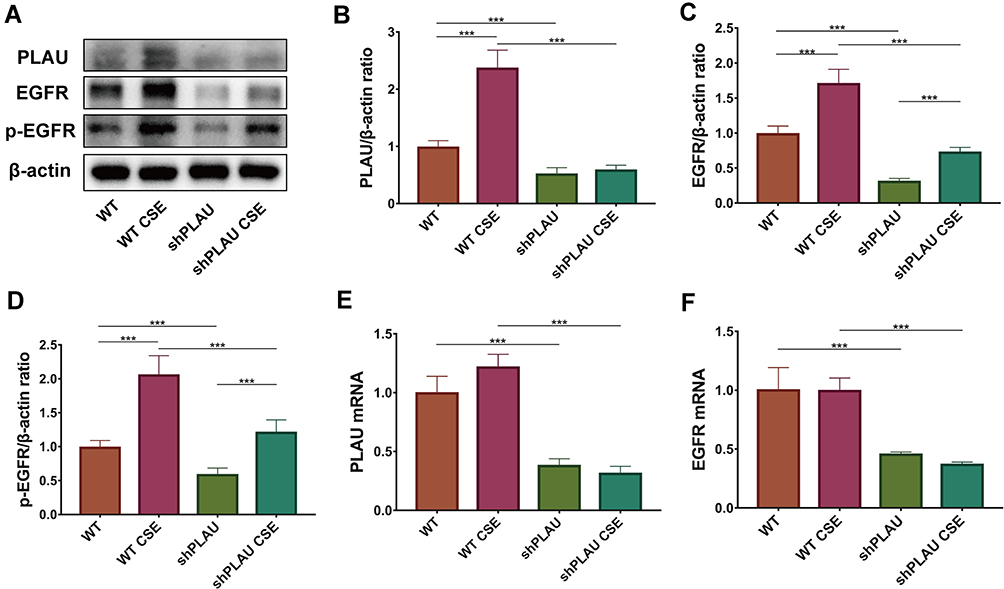 |
Figure 8 PLAU knockdown reduced EGFR signaling in vitro. (A) Levels of PLAU, EGFR, p-EGFR, and β-actin proteins in human bronchial epithelial (HBE) cells were measured by Western blotting analysis. Quantitative analysis of (B) PLAU, (C) EGFR, and (D) p-EGFR. mRNA levels of (E) PLAU, (F) EGFR. Error bars represent SD. *P < 0.05; **P < 0.01; ***P < 0.001. shRNA vectors targeting PLAU. Abbreviations: WT, wild-type; CSE, cigarette smoke extract; WT CSE, wild-type cigarette smoke extract. |
PLAU Overexpression Reversed CSE-Induced Pyroptosis in vitro
To further prove the role of PLAU, we examined whether PLAU overexpression via transfection with the lentivirus-mediated PLAU-OE vector inhibited CSE-induced pyroptosis. Western blotting analysis revealed that CSE-induced activation of caspase-1, cleavage of GSDMD, and maturation of IL-1β were significantly reduced by PLAU overexpression (Figure 9A–E).
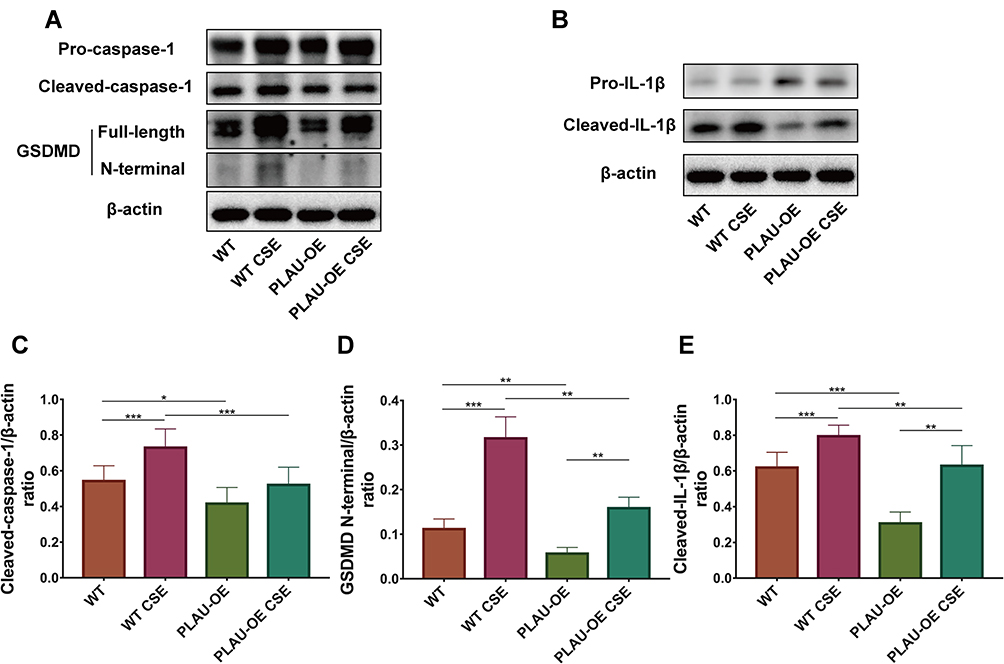 |
Figure 9 PLAU overexpression reversed CSE-induced pyroptosis in vitro. (A) Levels of pro-caspase-1, cleaved-caspase-1, gasdermin D (GSDMD), GSDMD N-terminal domain, and β-actin proteins in human bronchial epithelial (HBE) cells were measured by Western blotting analysis. (B) Levels of pro-IL-1β and cleaved-IL-1β in HBE cells were measured by Western blotting analysis. Quantitative analysis of (C) cleaved-caspase-1, (D) GSDMD N-terminal domain, and (E) cleaved- IL-1β. Error bars represent SD. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviations: WT, wild-type; CSE, cigarette smoke extract; WT CSE, wild-type cigarette smoke extract; PLAU-OE, PLAU overexpression. |
PLAU Overexpression Increased EGFR Signaling in vitro
Western blotting analysis showed that PLAU was significantly overexpressed after the transfection with the lentivirus-mediated PLAU-OE vector in HBE cells with or without CSE exposure (Figure 10A and B). After CSE challenge, the protein levels of PLAU, EGFR and p-EGFR were increased in cells treated with or without PLAU-OE transfection. PLAU overexpression increased the protein expression of PLAU, EGFR and p-EGFR in cells with or without CSE exposure (Figure 10A–D), indicating that PLAU overexpression increased EGFR signaling in vitro.
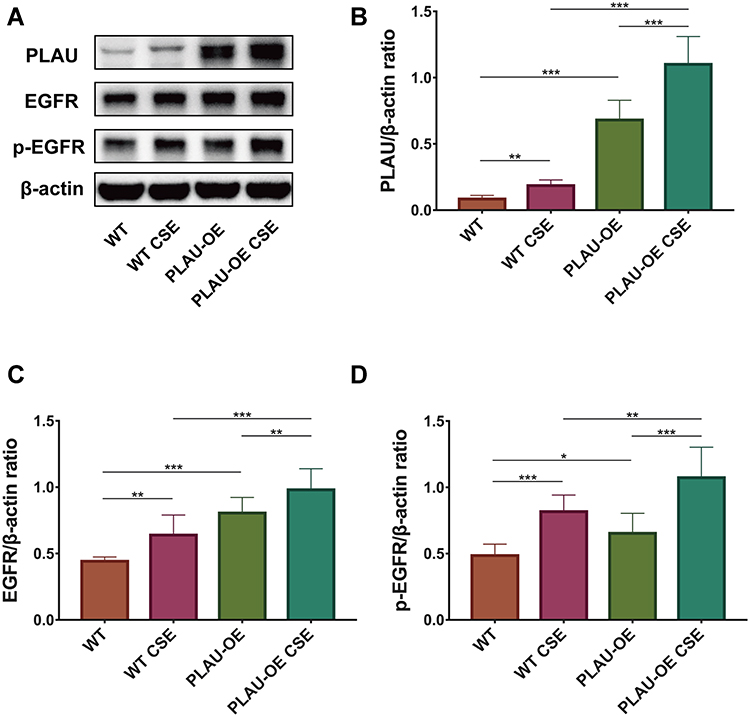 |
Figure 10 PLAU overexpression increased EGFR signaling in vitro. (A) Levels of PLAU, EGFR, p-EGFR, and β-actin proteins in human bronchial epithelial (HBE) cells were measured by Western blotting analysis. Quantitative analysis of (B) PLAU, (C) EGFR, and (D) p-EGFR. Error bars represent SD. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviations: WT, wild-type; CSE, cigarette smoke extract; WT CSE, wild-type cigarette smoke extract; PLAU-OE, PLAU overexpression. |
Discussion
Accumulating data suggested the key role of GLIPR1 as an oncoprotein that promotes cell growth, proliferation, invasion, and metastasis in many tumors, including gliomas, melanoma, and Wilms’ cancer.15,16 GLIPR1 is also vital for promoting stemness of glioblastoma stem cells, which is related to resistance against antitumor drugs and tumor recurrence.52 Furthermore, GLIPR1 is critical for the response of cisplatin-resistant lung cancer cells to cisplatin.53 On the contrary, GLIPR1 functions as a tumor suppressor in prostate and lung cancers.19,22 Although the specific mechanism underlying the dual function of GLIPR1 is still unclear, it has been recently demonstrated that low level of GLIPR1 is necessary for the growth of prostate cancer cells, whereas silencing of GLIPR1 inhibits proliferation of prostate cancer cells.22 In this study, Glipr1 knockout not only significantly increased CS-induced release of proinflammatory cytokines, such as IL-6 and IL-1β, and recruitment of inflammatory cells but also aggravated CS-induced airway damage, such as pulmonary inflammation and airway remodeling. This suggests the protective effect of GLIPR1 against CS-induced inflammation and airway damage.
Pyroptosis is a newly discovered mode of programmed and inflammatory cell death, which is mainly determined by the activation of many inflammatory caspases, such as caspase-1, caspase-4, caspase-5, and caspase-11, resulting in the cleavage of gasdermin family members including GSDMD.39 Previous studies have presented that long-term exposure to CS causes pyroptosis activation both in vivo54 and in vitro.42,43 In this study, we observed that a 3-month CS exposure triggered pyroptosis dependent on caspase-1 in lung tissues, which was notably augmented by Glipr1 knockout. Interestingly, Glipr1 knockout itself did not induce the expression of pyroptosis-related components in the lung tissues of mice. Nevertheless, after CS exposure, the expression of several signaling molecules associated with caspase-1-induced pyroptosis, such as cleaved-caspase-1, free N-terminal peptide from GSDMD, and mature IL-1β, were significantly augmented in Glipr1-/- mice. To further confirm the role of GLIPR1 as a protective effector against CS exposure, we found that GLIPR1 knockdown in HBE cells resulted in the aggravation of caspase-1-dependent cell pyroptosis induced by CSE. These results revealed that silencing Glipr1 had a tendency to accelerate cell pyroptosis when challenged with CS exposure, thereby suggesting that GLIPR1 tended to protect the epithelial cells and lung tissues against CS-induced caspase-1-dependent cell pyroptosis.
In vivo, CS-exposed Glipr1-/- mice showed distinct downregulation of the PLAU/EGFR signaling pathway, which was highly activated in the CS-exposed WT mice. In vitro, GLIPR1 knockdown also resulted in the blockade of PLAU/EGFR signaling with or without CSE challenge. PLAU is critical for ECM degradation, cell proliferation, adhesion, invasion, migration, and metastasis.28,55 The ability of PLAU to promote proliferation occurs not only in lung carcinoma-derived cells and malignant mesothelioma cells but also in nonmalignant lung epithelial cells.31,56 Moreover, PLAU plays a key role in the development and persistence of inflammatory responses and immune responses.57 In the human lung, PLAU is strongly expressed in bronchiolar epithelial cells, alveolar epithelial cells, and alveolar macrophages.50 In patients with COPD, the expression of PLAU increases in tissues surrounding the small conducting airways, and is linked with tissue degradation and remodeling.46 Consistent with the aforementioned studies, in the current study, long-term CS exposure caused a significant increase in the expression of PLAU in both WT and Glipr1-/- mice. However, Glipr1 knockout significantly blocked the PLAU/EGFR signaling and exacerbated CS-induced airway damage. A previous study has stated that targeting PLAU can inhibit cell survival and proliferation.23,26 PLAU-associated EGFR also produces signals that promote cell survival and proliferation.58,59 EGFR-induced cell survival and proliferation is dependent on PLAU, since EGFR-dependent cell proliferation is blocked in the absence of PLAU receptor expression.32–34 In this study, both in vivo and in vitro experiments demonstrated the indispensable role of GLIPR1 in activating the PLAU/EGFR signaling. Furthermore, in vitro study revealed that GLIPR1 forms complex with PLAU. Silencing PLAU exacerbated CSE-induced pyroptosis and inhibited EGFR signaling while overexpressing PLAU significantly reversed CSE-induced pyroptosis and activated EGFR signaling in vitro. As PLAU/EGFR interaction was critical for cell survival, growth, and proliferation, we speculated that GLIPR1 protected CS-induced airway damage, including inhibiting pyroptosis, through the PLAU/EGFR signaling pathway. To the best of our knowledge, this is the first study to elucidate the role of GLIPR1 in COPD pathogenesis.
In this study, Glipr1 knockout did not result in death or visible developmental defects in the mice. Although total protein, total cell number, macrophages and lymphocytes tended to increase in BALF of Glipr1-/- mice compared with WT mice, the differences were not statistically significant (Figure 2A, B, D and E). Besides, in Glipr1-/- mice compared with WT mice, no obvious changes in lung tissue structure was shown by HE staining (Figure 1D), and we did not find a significant increase in inflammatory factors, such as IL-6 and IL-1β, in plasma and BALF (Figures 2F, G and 3F). We speculate that there may be compensation and regulation mechanisms in mice to protect lung against the defect of Glipr1 knockout under physiological conditions, while the compensation mechanisms may be disturbed and weakened when mice are exposed to cigarette smoke, which means the effect of Glipr1 knockout is dominant and CS damage phenotype is manifested. Coincidentally, compared to WT mice, prkn-/- mice showed no obvious lung damage in the absence of CS and presented more severe COPD phenotype in the presence of CS through HE staining of lung sections.60 Nevertheless, the specific mechanism of compensation still needs further study.
Conclusion
In summary, silencing GLIPR1 aggravated CS-induced airway damage, including inflammatory infiltration, alveolar damage, airway remodeling, and caspase-1 dependent cell pyroptosis. GLIPR1 functioned as a protective factor in COPD pathogenesis. The protective effect of GLIPR1 against CS-induced inflammatory response and airway damage was related to the activation of the PLAU/EGFR signaling and inhibition of pyroptosis. Thus, GLIPR1 may play a potential role in the treatment of COPD, and this warrants further investigation.
Acknowledgments
We gratefully acknowledge all members of the Jian Zhou laboratory for valuable discussion.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article was submitted, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Funding
Grants: this work was supported by National Natural Science Foundation of China (81770039; 82070045), Science and Technology Innovation Action Plan of Shanghai Science and Technology Commission (18140904002), National key R&D plan (2016YFC1304104), Science and Technology Commission of Shanghai Municipality (20DZ2261200), Zhongshan Hospital Clinical Research Foundation (2019ZSGG15), Shanghai Municipal Key Clinical Specialty (shslczdzk02201), and Science and Technology Commission of Shanghai Municipality (20DZ2254400; 20XD1401200).
Disclosure
All authors have no conflicts of interest for this work to declare, financial or otherwise.
References
1. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389(10082):1931–1940. doi:10.1016/S0140-6736(17)31222-9
2. Barnes PJ, Burney PG, Silverman EK, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers. 2015;1:15076.
3. Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370(9589):765–773.
4. Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87(3):1047–1082.
5. Yokohori N, Aoshiba K, Nagai A; Respiratory Failure Research Group in J. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest. 2004;125(2):626–632.
6. Calabrese F, Giacometti C, Beghe B, et al. Marked alveolar apoptosis/proliferation imbalance in end-stage emphysema. Respir Res. 2005;6:14.
7. Imai K, Mercer BA, Schulman LL, Sonett JR, D’Armiento JM. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J. 2005;25(2):250–258.
8. Lucey EC, Keane J, Kuang PP, Snider GL, Goldstein RH. Severity of elastase-induced emphysema is decreased in tumor necrosis factor-alpha and interleukin-1 beta receptor-deficient mice. Lab Invest. 2002;82(1):79–85.
9. Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol. 2003;28(5):555–562.
10. Bartalesi B, Cavarra E, Fineschi S, et al. Different lung responses to cigarette smoke in two strains of mice sensitive to oxidants. Eur Respir J. 2005;25(1):15–22.
11. Kuo WH, Chen JH, Lin HH, Chen BC, Hsu JD, Wang CJ. Induction of apoptosis in the lung tissue from rats exposed to cigarette smoke involves p38/JNK MAPK pathway. Chem Biol Interact. 2005;155(1–2):31–42.
12. Zheng T, Kang MJ, Crothers K, et al. Role of cathepsin S-dependent epithelial cell apoptosis in IFN-gamma-induced alveolar remodeling and pulmonary emphysema. J Immunol. 2005;174(12):8106–8115.
13. Brass DM, Hollingsworth JW, Cinque M, et al. Chronic LPS inhalation causes emphysema-like changes in mouse lung that are associated with apoptosis. Am J Respir Cell Mol Biol. 2008;39(5):584–590.
14. Murphy EV, Zhang Y, Zhu W, Biggs J. The human glioma pathogenesis-related protein is structurally related to plant pathogenesis-related proteins and its gene is expressed specifically in brain tumors. Gene. 1995;159(1):131–135.
15. Chilukamarri L, Hancock AL, Malik S, et al. Hypomethylation and aberrant expression of the glioma pathogenesis-related 1 gene in Wilms tumors. Neoplasia. 2007;9(11):970–978.
16. Tyndall J, Awasthi A, Woolley AG, Wilbanks SM, Jeffs A. Variable expression of GLIPR1 correlates with promoter methylation and invasive potential in melanoma cells. Eur J Cancer. 2012;48:63.
17. Jacoby E, Yalon M, Leitner M, et al. Related to testes-specific, vespid and pathogenesis protein-1 is regulated by methylation in glioblastoma. Oncol Lett. 2014;7(4):1209–1212.
18. Awasthi A, Woolley AG, Lecomte FJ, et al. Variable expression of GLIPR1 correlates with invasive potential in melanoma cells. Front Oncol. 2013;3:225.
19. Ren C, Li L, Yang G, et al. RTVP-1, a tumor suppressor inactivated by methylation in prostate cancer. Cancer Res. 2004;64(3):969–976.
20. Rosenzweig T, Ziv-Av A, Xiang C, et al. Related to testes-specific, vespid, and pathogenesis protein-1 (RTVP-1) is overexpressed in gliomas and regulates the growth, survival, and invasion of glioma cells. Cancer Res. 2006;66(8):4139–4148.
21. Xiang C, Sarid R, Cazacu S, et al. Cloning and characterization of human RTVP-1b, a novel splice variant of RTVP-1 in glioma cells. Biochem Biophys Res Commun. 2007;362(3):612–618.
22. Sheng X, Bowen N, Wang Z. GLI pathogenesis-related 1 functions as a tumor-suppressor in lung cancer. Mol Cancer. 2016;15:25.
23. Wang Y. The role and regulation of urokinase-type plasminogen activator receptor gene expression in cancer invasion and metastasis. Med Res Rev. 2001;21(2):146–170.
24. Eppenberger U, Kueng W, Schlaeppi JM, et al. Markers of tumor angiogenesis and proteolysis independently define high- and low-risk subsets of node-negative breast cancer patients. J Clin Oncol. 1998;16(9):3129–3136.
25. Harbeck N, Kates RE, Schmitt M. Clinical relevance of invasion factors urokinase-type plasminogen activator and plasminogen activator inhibitor type 1 for individualized therapy decisions in primary breast cancer is greatest when used in combination. J Clin Oncol. 2002;20(4):1000–1007.
26. Pillay V, Dass CR, Choong PF. The urokinase plasminogen activator receptor as a gene therapy target for cancer. Trends Biotechnol. 2007;25(1):33–39.
27. Dinesh P, Rasool M. uPA/uPAR signaling in rheumatoid arthritis: shedding light on its mechanism of action. Pharmacol Res. 2018;134:31–39.
28. Dano K, Andreasen PA, Grondahl-Hansen J, Kristensen P, Nielsen LS, Skriver L. Plasminogen activators, tissue degradation, and cancer. Adv Cancer Res. 1985;44:139–266.
29. Mignatti P, Rifkin DB. Biology and biochemistry of proteinases in tumor invasion. Physiol Rev. 1993;73(1):161–195.
30. Vassalli JD, Sappino AP, Belin D. The plasminogen activator/plasmin system. J Clin Invest. 1991;88(4):1067–1072.
31. Shetty S, Bdeir K, Cines DB, Idell S. Induction of plasminogen activator inhibitor-1 by urokinase in lung epithelial cells. J Biol Chem. 2003;278(20):18124–18131.
32. LaRusch GA, Mahdi F, Shariat-Madar Z, et al. Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood. 2010;115(24):5111–5120.
33. Liu D, Aguirre Ghiso J, Estrada Y, Ossowski L. EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell. 2002;1(5):445–457.
34. Eden G, Archinti M, Arnaudova R, et al. D2A sequence of the urokinase receptor induces cell growth through alphavbeta3 integrin and EGFR. Cell Mol Life Sci. 2018;75(10):1889–1907.
35. Shi J, Gao W, Pyroptosis: SF. Gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254.
36. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157(5):1013–1022.
37. Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–192.
38. Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol. 2015;32:78–83.
39. Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665.
40. Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–116.
41. Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–158.
42. Wang X, Bian Y, Zhang R, et al. Melatonin alleviates cigarette smoke-induced endothelial cell pyroptosis through inhibiting ROS/NLRP3 axis. Biochem Biophys Res Commun. 2019;519(2):402–408.
43. Wu Z, Liu Q, Zhu K, et al. Cigarette smoke induces the pyroptosis of urothelial cells through ROS/NLRP3/caspase-1 signaling pathway. Neurourol Urodyn. 2020;39(2):613–624.
44. Ding J, Li F, Cong Y, et al. Trichostatin A inhibits skeletal muscle atrophy induced by cigarette smoke exposure in mice. Life Sci. 2019;235:116800.
45. Peng W, Chang M, Wu Y, et al. Lyophilized powder of mesenchymal stem cell supernatant attenuates acute lung injury through the IL-6-p-STAT3-p63-JAG2 pathway. Stem Cell Res Ther. 2021;12(1):216.
46. Gosselink JV, Hayashi S, Elliott WM, et al. Differential expression of tissue repair genes in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(12):1329–1335.
47. Mercer PF, Shute JK, Bhowmik A, Donaldson GC, Wedzicha JA, Warner JA. MMP-9, TIMP-1 and inflammatory cells in sputum from COPD patients during exacerbation. Respir Res. 2005;6:151.
48. Hao W, Li M, Zhang C, Zhang Y, Wang P. Inflammatory mediators in exhaled breath condensate and peripheral blood of healthy donors and stable COPD patients. Immunopharmacol Immunotoxicol. 2019;41(2):224–230.
49. Uysal P, Uzun H. Relationship between circulating serpina3g, matrix metalloproteinase-9, and tissue inhibitor of metalloproteinase-1 and −2 with chronic obstructive pulmonary disease severity. Biomolecules. 2019;9:2.
50. Wang IM, Stepaniants S, Boie Y, et al. Gene expression profiling in patients with chronic obstructive pulmonary disease and lung cancer. Am J Respir Crit Care Med. 2008;177(4):402–411.
51. Zhang LC, Shao Y, Gao LH, et al. TEM8 functions as a receptor for uPA and mediates uPA-stimulated EGFR phosphorylation. Cell Commun Signal. 2018;16(1):62.
52. Bier A, Giladi N, Kronfeld N, et al. MicroRNA-137 is downregulated in glioblastoma and inhibits the stemness of glioma stem cells by targeting RTVP-1. Oncotarget. 2013;4(5):665–676.
53. Gong X, Liu J, Zhang D, et al. GLIPR1 modulates the response of cisplatin-resistant human lung cancer cells to cisplatin. PLoS One. 2017;12(8):e0182410.
54. Li F, Ding J, Cong Y, et al. Trichostatin A alleviated ovarian tissue damage caused by cigarette smoke exposure. Reprod Toxicol. 2020;93:89–98.
55. Mondino A, Blasi F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol. 2004;25(8):450–455.
56. Shetty S, Kumar A, Johnson A, Pueblitz S, Idell S. Urokinase receptor in human malignant mesothelioma cells: role in tumor cell mitogenesis and proteolysis. Am J Physiol. 1995;268(6 Pt 1):L972–82.
57. Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3(12):932–943.
58. Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11(1):23–36.
59. Evangelopoulos ME, Weis J, Kruttgen A. Signalling pathways leading to neuroblastoma differentiation after serum withdrawal: HDL blocks neuroblastoma differentiation by inhibition of EGFR. Oncogene. 2005;24(20):3309–3318.
60. Araya J, Tsubouchi K, Sato N, et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy. 2019;15(3):510–526.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.