")
Back to Journals » OncoTargets and Therapy » Volume 15
Glioblastoma: Pitfalls and Opportunities of Immunotherapeutic Combinations
Authors Niedbała M , Malarz K , Sharma G , Kramer-Marek G, Kaspera W
Received 26 September 2021
Accepted for publication 5 April 2022
Published 28 April 2022 Volume 2022:15 Pages 437—468
DOI https://doi.org/10.2147/OTT.S215997
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Federico Perche
Marcin Niedbała,1,* Katarzyna Malarz,2,* Gitanjali Sharma,3 Gabriela Kramer-Marek,3 Wojciech Kaspera1
1Department of Neurosurgery, Medical University of Silesia, Regional Hospital, Sosnowiec, Poland; 2A. Chełkowski Institute of Physics and Silesian Centre for Education and Interdisciplinary Research, University of Silesia in Katowice, Chorzów, Poland; 3Division of Radiotherapy and Imaging, The Institute of Cancer Research, London, UK
*These authors contributed equally to this work
Correspondence: Wojciech Kaspera, Department of Neurosurgery, Medical University of Silesia, Regional Hospital, Plac Medykow 1, Sosnowiec, 41-200, Poland, Tel +48 600 301 977, Fax +48 32 368 20 24, Email [email protected] Gabriela Kramer-Marek, Division of Radiotherapy and Imaging, The Institute of Cancer Research, 15 Cotswold Road, Sutton, London, SM2 5NG, UK, Tel +44 208 722 4412, Email [email protected]
Abstract: Glioblastoma multiforme (GBM) is the most common and aggressive primary central nervous system tumour in adults. It has extremely poor prognosis since the current standard of care, comprising of gross total resection and temozolomide (TMZ) chemoradiotherapy, prolongs survival, but does not provide a durable response. To a certain extent, this is due to GBM’s heterogeneous, hostile and cold tumour microenvironment (TME) and the unique ability of GBM to overcome the host’s immune responses. Therefore, there is an urgent need to develop more effective therapeutic approaches. This review provides critical insights from completed and ongoing clinical studies investigating novel immunotherapy strategies for GBM patients, ranging from the use of immune checkpoint inhibitors in different settings of GBM treatment to novel combinatorial therapies. In particular, we discuss how treatment regimens based on single antigen peptide vaccines evolved into fully personalised, polyvalent cell-based vaccines, CAR-T cell, and viral or gene therapies. Furthermore, the results of the most influential clinical trials and a selection of innovative preclinical studies aimed at activating the immunologically cold GBM microenvironment are reviewed.
Keywords: glioblastoma multiforme, targeted therapy, tumour microenvironment, immune checkpoint, vaccine, viral therapy, gene therapy, adoptive cell therapy, CAR-T, CAR-NK
Introduction
Glioblastoma: Definition and Characteristics in Light of the 2021 WHO Classification of Tumors of the Central Nervous System
Glioblastoma multiforme (GBM) is the most common, highly aggressive and invasive primary brain tumour in adults. The current standards of care (SOC) in GBM therapy consist of gross total resection to remove the tumour bulk, followed by radiation therapy (RT) and adjuvant chemotherapy with temozolomide (TMZ).1,2 Promoter methylation of the DNA repair enzyme O6-methylguanine DNA methyltransferase (MGMT) gene is associated with statistically significant improvement in overall survival (OS) in patients receiving RT combined with TMZ.3
In 2009, the Food and Drug Administration (FDA) in the United States approved the inclusion of bevacizumab, a humanised monoclonal antibody against vascular endothelial growth factor (VEGF), for the treatment of patients with recurrent GBM that progressed following prior therapy.4 Several studies clearly demonstrated that the use of bevacizumab in combination with chemotherapy or the use of bevacizumab alone in patients with recurrent GBM shows both radiographic responses and increased progression-free survival (PFS), compared with historical data from patients who received chemotherapy alone.4 Furthermore, in 2015, the FDA approved the use of a tumour-treating fields (TTF) device in the therapy of newly diagnosed GBM based on the promising results of a Phase III study,5 which showed an extension of both the median OS (mOS) and median PFS (mPFS) with a simultaneous low percentage of severe adverse events (AE).
But, despite aggressive therapeutic efforts and a variety of combinatorial treatment regimens, GBM patients still have an extremely poor prognosis with the mOS being 10.2 months and a five-year survival rate of less than 5%.6,7 The recurrence of GBM is inevitable with most patients experiencing it shortly after primary treatment (i.e., mPFS of 1.8 months and mOS of 5 months).6 It is worth noting, that the extent of resection is currently one of the main prognostic factors for the surgically treated patients and has an effect both on mPFS as well as mOS.8
According to the latest 2021 WHO classification of Tumors of the Central Nervous System (CNS), GBM is considered as a grade 4 tumour belonging to the family of adult-type diffuse gliomas (Figure 1).9 GBMs are diagnosed only in IDH wild-type astrocytomas based on histological findings including mitotic activity, necrosis and microvascular proliferation. The presence of one or more of three genetic markers highly specific for aggressive clinical behaviour, namely TERT promoter mutation, EGFR gene amplification, the combined gain of an entire chromosome 7 and loss of an entire chromosome 10 (+7/-10), is required for the diagnosis of GBM in IDH-wildtype astrocytomas without the histological features of glioblastoma.9 On the other hand, all diffuse IDH-mutant astrocytic tumours are considered to be a distinct type (astrocytoma, IDH mutant) and are graded 2, 3 or 4 based on the histological features.9 However, in the case of the presence of other molecular signatures, such as CDKN2A/B homozygous deletion, type IDH-mutant astrocytoma will be considered as grade 4, even in the absence of mitotic activity, increased cellular density, microvascular proliferation and necrosis.9
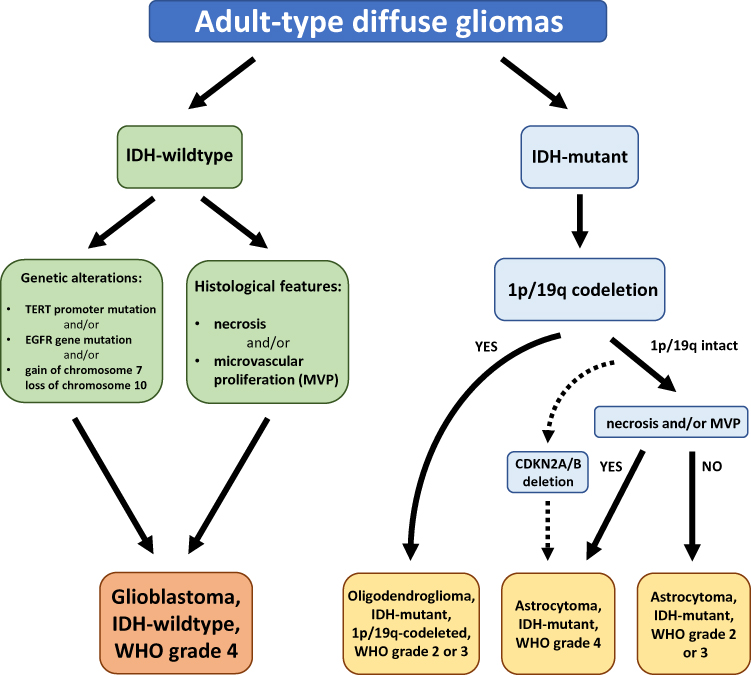 |
Figure 1 The 2021 WHO classification of the major diffuse gliomas in adults. |
Over the last decade, numerous studies have focused on characterising the mutations, transcriptional signatures and signalling alterations towards better understanding of the molecular mechanisms driving GBM formation. One of the most comprehensive and reliable analyses was the study conducted by The Cancer Genome Atlas (TCGA) group in 2008.10 The most frequent genetic alterations found in the analysed GBM tissue samples included receptor tyrosine kinase (RTK)/rat sarcoma (RAS)/PI3K, p53 and RB signalling pathways with the frequency of occurrence of 88%, 78% and 87%, respectively.10 Verhaak et al. have recently identified based on the TCGA data and their own gene expression profile studies, four distinct molecular subtypes of GBM, namely proneural, neural, classical and mesenchymal, characterised by abnormalities in EGFR, NF1, PDGFRA and IDH1.11 Due to their unique molecular signatures, each of the subtypes was found to exhibit a different prognosis. The mesenchymal subtype characterised by high expression of CHI3L1 and MET,11,12 high frequency of NF1 mutation/deletion and low levels of NF1 mRNA expression,11 has the worst prognosis among all subtypes.12 In the classical subtype, the gain of chromosome 7, loss of chromosome 10, homozygous deletion of CDKN2A and EGFR amplification are the most common genetic aberrations.11 The proneural subtype, most common in young adults, has a markedly better prognosis.13 It is characterised by PDGFRA abnormalities, as well as IDH1 and TP53 mutations.11 The presence of the neural subtype has been recently questioned due to the lack of characteristic gene signatures. Several studies have shown that this subtype was misidentified due to the contamination of original samples with non-cancerous cells.14,15
Of note, each of the subtypes has shown a distinct tumour microenvironment (TME), containing different types of non-malignant cells including tumour-associated macrophages (TAMs), dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs) and T lymphocytes.15,16 TAMs are the most numerous group of immune cells found in GBM, reaching 30–40% of the tumour cells.17 Increased tumour infiltration occurs especially in the mesenchymal subtype due to inactivation of NF1.15 TAMs directly suppress T cell function by surface presentation of programmed death-ligand 1 (PD-L1), which activates programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4), respectively.
There is a two-way interaction between tumour cells and the TAMs.18 Tumour cells reprogram immune cells to a pro-tumour phenotype which by releasing factors such as IL-6, IL-1β, EGF, TGF-β, STI1 and PTN, promote tumour progression and may also cause the transcriptomic adaptation of neoplastic cells and shape the mesenchymal phenotype of GBM.18 Thus, the highly immunosuppressive nature of GBM’s microenvironment is a challenge for developing new therapy options.
This paper provides a comprehensive review of the results of clinical trials investigating immunotherapy regimens for GBM patients. We discuss the available approaches including immune checkpoint inhibitors, vaccination, adoptive cell therapies, virotherapy and combinatorial strategies that might change the management of GBM patients.
GBM Immune Microenvironment
Intrinsic molecular and genetic changes of GBM are also influenced by the cells forming a TME, such as endothelial, immune and other parenchymal cells that promote angiogenesis, invasion and proliferation as well as convey immune-suppressive functions.17,19 For decades the central nervous system (CNS) has been perceived as an “immune privileged” organ due to the presence of an intact blood–brain barrier (BBB) and the absence of a conventional lymphatic system that shuttles CNS-specific antigens to peripheral lymph nodes.20,21
Role of a Glymphatic System in Immune Surveillance of the CNS
It has been recently reported that the functional glymphatic system enables exchange of fluids, macromolecules and immune cells between CNS and the immune system.22,23 Louveau et al., discovered functional lymphatic vessels lining the dural venous sinuses that unequivocally express the molecular features (CD31, LYVE-1, podoplanin, VEGFR3, CCL21) of lymphatic endothelial cells.24 These lymphatic vessels are capable of transporting CNS antigens, T cells, B cells, and DCs together with cerebrospinal fluid (CFS) into the peripheral immune system through the foramina of the base of the skull, cribriform plate then to the nasal mucosa to finally reach the cervical lymph nodes.24–26 Furthermore, it is worth emphasising that the migration of immune cells to the CNS is strictly controlled by a complex network of interactions consisting of the BBB, astrocytes and microglia.27 The process of GBM tumorigenesis and the resulting pro-inflammatory state may additionally compromise the integrity of the BBB increasing its permeability and infiltration of circulating monocytes and lymphocytes from the peripheral lymphatic system.28 Besides, the key component of the BBB, astrocyte cells can also enhance pro-inflammatory activity through secretion of cytokines including tumour necrosis factor-α (TNF-α), interferon-α (IFN-α) and transforming growth factor-β (TGF-β).29
Immunosuppressive Landscape of GBM Microenvironment
Indeed, recent studies have provided evidence for the enriched presence of a variety of immune cell types within the GBM TME but with a dominance of immunosuppressive cells including MDSCs, microglia and TAMs, FoxP3+ regulatory T cells (Tregs), and antigen presenting cells (APCs) including DCs and bone marrow-derived macrophages (BMDM).30,31 Moreover, the present CD4+ and CD8+ T cells are frequently functionally deficient, inactivated, or exhausted, often co-expressing the immune checkpoint molecules including programmed cell death receptor 1 (PD-1), lymphocyte activation gene 3 (LAG3) and T cell immunoglobulin mucin 3 (TIM-3).31
TAMs are known to release large amounts of immunosuppressive cytokines including interleukins (e.g., IL-6, IL-10 and TGF-β), which prevent T cells activation directly inhibiting anti-tumoural responses of cytotoxic CD8+ T cells and natural killer (NK) cells.32–34 Similarly, the presence of Tregs within TME may lead to the suppression of immune response by inhibiting the accumulation of CD8+ T cells and simultaneous secretion of pro-inflammatory cytokines, such as IFN-γ, TNF and IL-6.35 MDSCs are immature myeloid cells that are key drivers of immunosuppression. They express chemokine receptors CCR2, CCR5 and CXCR2, which mobilise them to the blood and tumour by chemokine ligands (CCL2, CCL5, CXCL1-8) and other inflammatory mediators such as IL-1β, IL-6, IL-10, VEGF, TGF-β, TNF-α, and prostaglandin E2 (PGE2) secreted by TME. Besides, the presence of CCR2 with CXCR3 is involved in the recruitment of TAMs and the signalling of these chemokine receptors also affects the polarization of TAMs.36 The M1/M2 polarity of TAMs is associated with a different phenotype and distinct production of anti-inflammatory and proinflammatory cytokines. The increase in proinflammatory cytokines such as IL-2, IL-12, IFN-γ and TNF-α is associated with the M1-like phenotype, and IL-4, IL-10 and TGF-β with the M2 phenotype.37 This polarization-dependent phenotypic signature is also characteristic for the microglia cells.38 Numerous reports have shown that microglia cells with the M1-like phenotype have elevated levels of TNF-α, IL-12, IL-23 and IL-1β cytokines. In contrast, high IL-10 expression and protein levels of arginase were found in microglia cells with M2-like phenotype.27,37 Of note, M2-like microglia and macrophage cells are more resistant to the treatment, therefore, their repolarization or reprogramming may result in a significant improvement in GBM prognosis and inhibition of tumour progression.39 Figure 2 visualises the relation between the TME, BBB and its connection to the peripheral immune system.
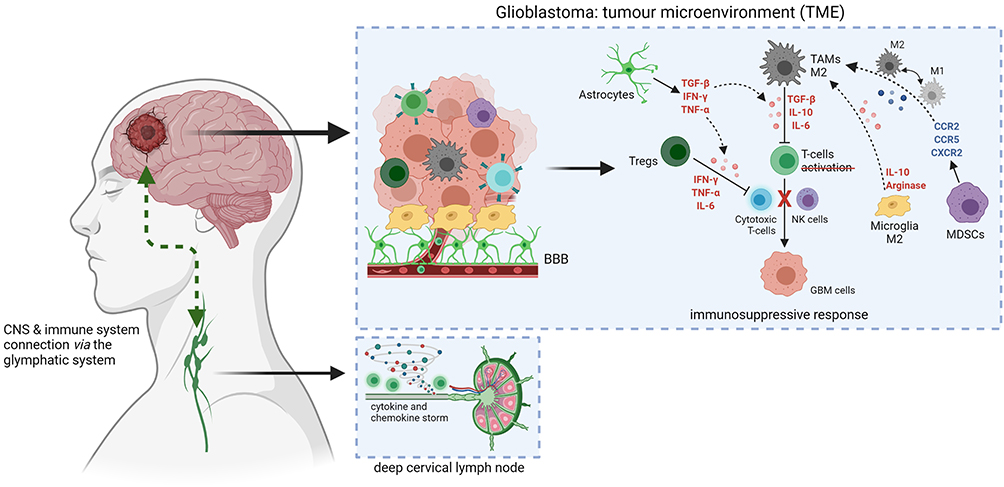 |
Figure 2 Glioblastoma tumour microenvironment alterations and connection with peripheral immune system. Created with BioRender.com. |
Figure 3 presents different immunotherapy approaches for GBM that will be discussed in the following paragraphs.
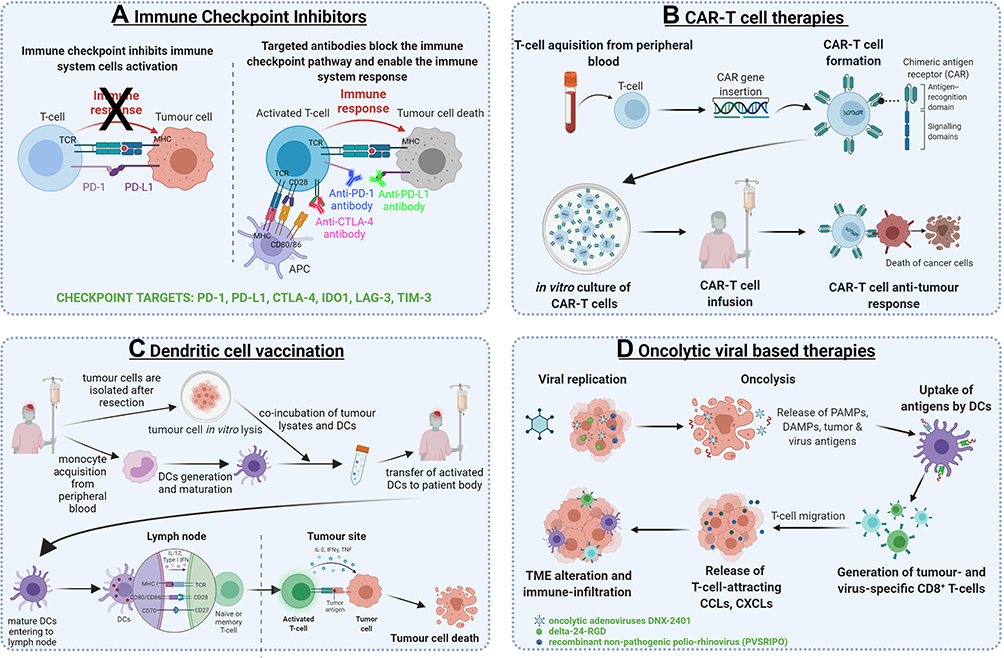 |
Figure 3 The most prominent immunotherapeutic strategies for glioblastoma. (A) The mechanism of immune checkpoint inhibition. (B) The CAR-T therapy from the creation of CAR-T cells to the direct anti-tumour effects. (C) Two different vaccination strategies for glioblastoma: tumour-associated antigen peptide vaccination, autologous dendritic cell transfer following the exposure to tumour lysate. (D) Oncolytic viral therapy mechanisms from oncolysis to alterations of tumour microenvironment. Created with BioRender.com. Abbreviations: PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; CTLA-4, cytotoxic T-lymphocyte antigen 4; IDO1, indoleamine 2,3-dioxygenase; LAG-3, lymphocyte activation gene 3; TIM-3, T cell immunoglobulin mucin 3; TCR, T cell receptor; MHC, major histocompatibility complex; CAR, chimeric antigen receptor; CAR-T, chimeric antigen receptor engineered T cell; IL-12, interleukin 12; IFNγ, interferon gamma; IL-2, interleukin 2; TNF, tumour necrosis factor alpha; DC, dendritic cell; PAMP, pathogen-associated molecular pattern; DAMP, damage-associated molecular pattern; TME, tumour microenvironment. |
Immune Checkpoint Inhibitors and Potential New Checkpoint Blockade Targets
Recently, immunoregulatory agents that block immune checkpoint molecules have emerged as a promising treatment strategy against a variety of cancers including melanoma, lung cancers and head and neck cancers.40 However, the immune checkpoint inhibitors (ICPIs) have been shown to demonstrate only limited therapeutic efficacy in GBM patients due to a highly immunosuppressive TME of these tumours.
PD-1/PD-L1 Axis Inhibitors
One of the most important immunosuppressive molecules is PD-1, an immunoglobulin receptor expressed by B cells, APCs, myeloid cells, NK and T cells.41 PD-1 has two ligands, PD-L1 (CD274, B7-H1) and PD-L2 (CD273, B7-DC). Upon ligand binding to PD-1, an inhibitory signal is transmitted into the T cell that leads to the inhibition of cytokine production, suppression of T cell proliferation and inhibition of their cytotoxic and memory potential.42,43 The activated PD-1/PD-L1 signalling promotes cancer immune escape by increasing proliferation of Treg cells and mediating the suppression of NK and B cells.44,45
In GBM elevated levels of PD-L1 have been found in patients with primary and recurrent disease.46,47 Increased PD-L1 levels have also been associated with worse OS amongst patients,48 but its role as a prognostic factor still remains controversial.49
Recently, several anti-PD-1 (e.g., nivolumab, pembrolizumab, cementiplimab) and anti-PD-L1 (e.g., atezolizumab, avelumab, durvalumab) monoclonal antibodies (mAb) have been approved for the treatment of solid tumours.50,51 However, there are still challenges that need to be overcome to routinely use these drugs for the treatment of GBM. Even though a number of studies investigating ICPIs have demonstrated promising results,52–54 further clinical and preclinical trials are required to determine whether immunotherapy can be implemented as part of standard therapy for GBM.
In one of the first studies, Chamberlain et al. showed that nivolumab monotherapy in patients (n=16) with recurrent GBM who did not respond to bevacizumab, a VEGF-A inhibitor, did not prolong the OS.55 On the other hand, a phase II clinical trial (NCT02550249) with nivolumab as a neoadjuvant administered before and after surgical resection of the recurrent GBM (n= 30 patients) showed no significant clinical benefit, with median PFS and OS at 4.1 and 7.3 months respectively. However, two of three patients in the group treated with nivolumab before and after surgery remained alive for 33 and 28 months. Additionally, molecular analyses of tumour tissue samples obtained post-nivolumab treatment revealed an enhanced expression of chemokine transcripts, higher immune cell infiltration and augmented T cell receptor (TCR) clonal diversity among tumour-infiltrating lymphocytes (TILs), supporting a local immunomodulatory effect of the treatment.56 The effects in the immune TME were observed in a phase II clinical trial (NCT02337686) of neoadjuvant therapy with pembrolizumab against recurrent GBM. This study showed improved survival benefit in the group treated with neoadjuvant pembrolizumab (n=16), where the mOS was 13.7 months in comparison to 7.5 months in the group receiving the mAb as adjuvant treatment (n=19). The mPFS was 3.3 months in the neoadjuvant arm and 2.4 months in the adjuvant group. Additionally, the molecular study revealed that pembrolizumab treatment before surgery activated TILs, producing an IFN-γ response within the TME and increasing the expression of T cells and IFN-γ-related genes while decreasing the expression of cell cycle-related genes. Surprisingly, such changes were not observed with adjuvant treatment. Importantly, decreased PD-1 expression on peripheral blood T lymphocytes, decreased monocyte population, and enhanced T cell clonal expansion were significantly more frequently observed after pembrolizumab as a neoadjuvant treatment.57 Interestingly, the first report from a phase II study (NCT02337686) in patients with recurrent and resectable GBM (n = 15) failed to show a statistically significant increase in CD8+ T cells under pembrolizumab neoadjuvant treatment. The authors suggested it may be due to the very low number of T cells present in the TME and an excessive number of CD68+ macrophages.58
The CheckMate 143 study (NCT02017717) was the first multi-centre randomised phase III clinical trial which tested the safety and efficacy of treatment with nivolumab compared with bevacizumab, in a cohort of 369 patients following recurrence of GBM after RT and TMZ therapy.59 The study revealed no significant difference in patient OS between these groups. The mOS was 9.8 months for the nivolumab arm and 10 months in the bevacizumab group. Moreover, the overall response rate was lower in the nivolumab-treated group (8%) compared with the bevacizumab cohort (23%). Nevertheless, the response to treatment with nivolumab was twice as long (more durable). In addition, high grade (3–4) treatment-related adverse events (TRAEs) were similar in both groups. They occurred in 18% of patients in nivolumab arm and 15% of patients in bevacizumab arm. Importantly, there were no unexpected neurological TRAEs or TRAE-related deaths in this study.59,60 The CheckMate143 study also assessed the safety profile and tolerance of combination treatment of nivolumab plus RT and TMZ in two cohorts of patients (1c and 1d) with newly diagnosed GBM. The results showed that TRAEs were observed in as many as 67% (1c) and 70% (1d) of patients, with the majority involving fatigue, headache and increased alanine aminotransferase (ALT). Grade 3–4 treatment-related adverse events were demonstrated in >2 patients in 1c and 1d cohorts, which included increased ALT and lipase activity.59,61 Moreover, another randomised phase II clinical trial (NCT03452579) in recurrent GBM showed no difference in PFS and OS rates with nivolumab along with bevacizumab administered at two doses: standard (10 mg/kg) and low (3 mg/kg).62 Phase III clinical studies of nivolumab combined with RT with or without TMZ in patients with MGMT-unmethylated and -methylated newly diagnosed GBM are currently underway (NCT02667587 – CheckMate 548 study, NCT02617589 - CheckMate 498 study). The first published report of the CheckMate 498 trial (n=560) showed no difference in patient survival after treatment with nivolumab combined with RT compared with TMZ plus RT, where the mOS for both combinations was approximately 14 months.63,64 Similar preliminary results were obtained for the CheckMate 548 trial (n=693), which showed no significant statistical improvement in OS or PFS after combination treatment with nivolumab and TMZ plus RT.63,65 Furthermore, ongoing phase II study (NUTMEG) evaluates whether nivolumab combined with TMZ improves OS in newly diagnosed GBM in elderly patients (65 years of age and/or older) (NCT04195139). Currently, there is recruitment of patients for a phase II study designed to determine the potential of increased therapeutic effect of nivolumab combined with RT and bevacizumab in recurrent MGMT-methylated GBM (NCT03743662).
The Keynote-028 (NCT02054806) phase I clinical trial provided data on the efficacy and safety of pembrolizumab in various advanced solid tumours, including patients (n = 26) with recurrent GBM, who were naïve to bevacizumab and had PD-L1 expression on >1% of tumour and stromal cells.66 Results of this study showed that pembrolizumab was generally safe, as only 5 patients experienced grade 3–4 toxicities. In addition, two partial responses to the treatment were noted during the study lasting 8.3 and 22.8 months, respectively, while the 16 patients experienced stable disease. Partial response is defined as at least a 30% decrease of the sum of the longest diameter of assessed tumours.67 The mOS for the whole cohort was 13.1 months and the mPFS was 2.8 months.66,68 In another phase II clinical trial (NCT02337491) the safety and response to pembrolizumab combined with bevacizumab was evaluated in patients with recurrent GBM. Despite the fact that the majority of patients showed good tolerance to this regimen, the addition of bevacizumab did not improve efficacy.69 In addition, the safety and efficacy of pembrolizumab administered in combination with bevacizumab and hypo-fractionated stereotactic irradiation was investigated in patients with recurrent GBM (NCT02313272). The early encouraging reports showed that a complete or partial response lasting more than 6 months was registered in 53% of the study cohort. Furthermore, at 6 and 12 months the OS was 94% (16/17 patients) and 64% (7/11), respectively.70
In turn, a phase I/II clinical study of pembrolizumab combined with RT and TMZ in newly diagnosed GBM was terminated after phase I but the results and reasons for the discontinuation have not been disclosed so far (NCT02530502).
Also, the results of a phase II study (NCT03661723) completed in mid-2021 at the Dana-Farber Cancer Institute, where the combination of pembrolizumab and re-irradiation in resistant or non-resistant to bevacizumab recurrent GBM, were investigated, have not been released yet.
Furthermore, another anti-PD-1 antibody, spartalizumab, is currently undergoing a phase I/II clinical trial in combination therapy with BLZ945, an oral inhibitor of colony-stimulating factor 1 receptor (CSF1R) in several solid tumours, including GBM (NCT02829723).
There was also a phase II trial evaluating the safety and response to the treatment of durvalumab and/or bevacizumab in combination with radiotherapy in five cohorts of patients (n=158) with newly diagnosed MGMT non-methylated and recurrent GBM (NCT02336165). Preliminary data from cohort A (n=40, newly diagnosed GBM) revealed that durvalumab treatment with RT was well-tolerated. Adverse events (AEs) with maximum CTCAE grade ≥3 occurred in 14 patients, with the most common being asymptomatic lipase elevation (n=6) and amylase elevation (n=2). At the time the report was published, the mOS was 15.1 months, and 8 patients were still alive (with survival ranging from 15.7–34.9 months).71 In contrast, the report for cohort B (n=30, recurrent bevacizumab-naïve GBM patients) that received durvalumab as a single agent showed that 6 patients (20%) remained free of disease progression at 6 months and OS were 59.0% and 44.4% for 6, and 12 months, respectively. Post-treatment there was a partial response in 13.3% of the cohort population (n=4) and none of the patients experienced high-grade treatment-related adverse events (grade 4). The longest OS was 86 weeks for one patient.72 The following reports discussed data from arms B2, B3 and C, which used combination treatment of durvalumab with bevacizumab in recurrent bevacizumab-naïve or refractory GBM.73,74 Overall, 66 patients were enrolled in the B2 and B3 arms, which received the combination of these two drugs at two doses: standard (B2 arm – 10 mg/kg) and low (B3 arm – 3 mg/kg). Unfortunately, there was no improvement in patient outcome in both cohorts compared with durvalumab monotherapy. PFS rates at 6 months were 15.2% and 21.1% for B2 and B3, respectively.73 On the other hand, preliminary results in cohort C with bevacizumab-resistant GBM were promising, with most patients tolerating therapy well, 36% of the population having OS ≥22 weeks, 50% having PFS ≥8 weeks, and 3 patients still alive at the time of reporting.74 Besides, durvalumab is currently being studied in a phase I/II trial called STERIMGLI in combination with hypo-fractionated stereotactic radiotherapy in patients with recurrent GBM (NCT02866747).
In addition, atezolizumab was evaluated in a phase I clinical study, in a small group of patients (n = 16) with recurrent GBM (NCT01375842). The published report showed a safe profile for the therapy used, with none of the patients experiencing AEs of the highest grades (4–5). There was only 1 patient who achieved a partial response (5.3 months), while the mOS in the whole cohort was 4.2 months (range 1.2–18.8 months). Interestingly, the highest survival rate of more than 16 months was recorded in two patients with tumour harbouring mutation in IDH1 gene and one with a DNA polymerase epsilon (POLE) mutation. Besides, a correlation between low baseline levels of circulating CD4+ T and B lymphocytes (<0.40×106 cells/mL and <0.19×106 cells/mL, respectively) and low PFS and OS rates in patients was reported.75 Currently, M.D. Anderson Cancer Center is conducting a phase I/II clinical trial using atezolizumab in combination with TMZ and RT in newly diagnosed GBM (NCT03174197). Initial data (n = 60 patients) revealed promising efficacy, with mPFS rates of 9.7 months and a mOS of 17.1 months. Of note, the mPFS was 16.7 months in patients with MGMT promoter methylation status and only 7.9 months in MGMT unmethylated patients.76
Furthermore, phase II clinical trials have investigated the use of avelumab as a monotherapy (ongoing) in a group of patients with recurrent IDH-mutated HGG (NCT03047473). The study showed good tolerability to the treatment (n = 6 patients) with no dose-limiting toxicities. Median PFS and OS rates were 4.2 and 10.1 months, respectively.77
Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) Inhibitors
CTLA-4 is a surface receptor expressed by CD4+ and CD8+ T cells and it binds the ligands CD80 and CD86 present on the surface of APCs. Ligand binding to the receptor leads to the suppression of T cell activation. Both ligands can also bind to CD28 activating T lymphocytes.78 However, CTLA-4 has greater affinity for CD80 and CD86 than CD28. In addition, CTLA-4 can remove both ligands from the surface of APCs via trans-endocytosis, leading to impaired co-stimulation via CD28.79 Constitutive expression of CTLA-4 is also related to the development and activity of Treg cells.80
In a preclinical study, Fecci et al. showed that a CTLA-4 blockade by an experimental antibody (clone 9H10) ensured long-term survival in 80% of treated mice while concomitantly restoring T cell proliferation and activity. Notably, this phenomenon only affects the CD4+ CD25- T cell population.81 In turn, Reardon et al. demonstrated that a combination of anti-CTLA-4 (9D9 mouse IgG2b, K) and anti-PD-1 (332.8H3 mouse IgG1, K) agents assured long-term survival in 75% of treated mice, while the use of these agents as a monotherapy showed much lower effect, with survival rates of 50% for anti–PD-1 treatment and 15% for anti–CTLA-4.53 Ipilimumab was the first humanised anti–CTLA-4 antibody approved for the treatment of patients with metastatic melanoma.82 Despite many reports demonstrating its efficacy in patients with melanoma metastasised to the brain, ipilimumab has not been used so far as a monotherapy for the treatment of GBM.83,84
In the Checkmate-143 clinical trial (NCT02017717) ipilimumab was assessed in combination with nivolumab in a recurrent GBM. Unfortunately, this study revealed a poor safety profile for this combination as almost the entire cohort of patients experienced high-grade treatment-related adverse events (9 out of 10) compared with the monotherapy arms, where there were no grade 3–4 toxicities. In addition, the study showed only two partial responses to combination therapy.85 Additionally, there are also two additional ongoing phase I/II clinical studies investigating the effects of combining ipilimumab with nivolumab in patients with recurrent GBM with an elevated mutational burden as well as after the resection of a recurrent GBM (NCT04145115, NCT03233152). A report from the second study has demonstrated the safety of the combination of ipilimumab and nivolumab after intratumoural and intracavitary administration. The reported mOS was 71 weeks, with 51% and 34% at one and two years, respectively.86
There is an ongoing open-label trial with ipilimumab and/or nivolumab in combination with TMZ in patients with newly diagnosed GBM (NCT02311920). The safety profile study results have showed that the treatment has been well tolerated, with none of the patients experiencing high-grade (5) AEs so far. The grade 4 toxicities occurred in 16% of patients.87 Furthermore, a phase I clinical trial on safety and survival profiles of dual therapy (nivolumab and ipilimumab) before and after surgery in patients with recurrent GBM was initiated in October 2020 (NCT04323046). In addition, participants are being recruited for a phase II study and a randomised phase II/III study that will determine the efficacy of the combination therapy of ipilimumab, nivolumab and RT for newly diagnosed MGMT-unmethylated GBM (NCT04396860, NCT03367715).
Last but not least, an anti–CTLA-4 mAb (tremelimumab) is currently being tested as monotherapy and in combination with durvalumab in patients with recurrent GBM (NCT02794883).
Novel Immune Checkpoint Targets
Due to the fact that some patients do not respond to drugs targeting PD-1/PD-L1 axis or CTLA-4 checkpoint, other immune checkpoint targets have been recently identified including indoleamine 2,3-dioxygenase (IDO1), lymphocyte activation gene 3 (LAG-3) and T cell immunoglobulin mucin 3 (TIM-3).88
IDO1 Inhibitors
IDO1 is the rate-limiting enzyme involved in the conversion of tryptophan to kynurenine and its by-products. Depleting tryptophan may cause T cell deactivation, the induction of T cell apoptosis and an increase in Treg cell production.89 Interestingly, the high expression of IDO1 mRNA and protein levels has been found to correlate with poor survival and clinical outcome in GBM.90,91 Currently, there are ongoing phase I/II clinical trials with two IDO1 inhibitors – epacadostat and INT230-6 – in combination with nivolumab in patients with advanced solid tumours, including recurrent GBM (NCT02327078, NCT03058289). Additionally, participants are being recruited for an open-label phase I study aiming to determine the potential side effects of combination therapy including nivolumab, an irreversible inhibitor of IDO1 (BMS-986205), and standard RT with or without TMZ for newly diagnosed GBM (NCT04047706).
LAG-3 Inhibitors
Another immune checkpoint, the LAG-3 receptor, is expressed on the surface of activated T cells, B cells and NK cells.92 It mainly binds to MHC II on APCs and negatively modulates the function of T cells by promoting apoptosis and decreasing proliferation.93 Human GBM samples revealed the presence of LAG-3 on TILs and lymphocytes of the perivascular niche of the tumour.
The preclinical studies showed that blocking LAG-3 using a single agent or in combination with anti–PD-1 may lead to the eradication of GBM in mice, especially if anti–LAG-3 is administered at an early stage of treatment.94 Excitingly, a phase I clinical trial of anti–LAG-3 mAb (BMS-986016) as monotherapy or in combination with nivolumab in patients with recurrent GBM is currently underway (NCT02658981). The maximum tolerated dose of anti–LAG-3 is currently considered to be 800 mg in monotherapy arm and 160 mg in combination therapy arm with nivolumab (240 mg).95 The same combination of mAbs is currently being tested for clinical response after cytokine microdialysis and tumour resection in patients with recurrent GBM (NCT03493932).96 Although anti-LAG-3 therapies have shown efficacy in preclinical studies, a downside includes the fact that LAG-3 is only expressed in a small percentage of TILs, suggesting it may only be useful for a small subset of patients.
TIM-3 Inhibitors
The final checkpoint target to be explored is TIM-3, a surface receptor expressed by CD4+ T-helper and cytotoxic CD8+ T cells, which secrete IFN-γ. After binding to its ligand, the galectin-9/TIM-3 axis participates in suppressing the immune response, finally leading to an exhaustion of T cells.97 This phenomenon is associated with the loss of T cell effector functions, the expression of multiple inhibitory receptors and an altered transcriptional programme.98 Interestingly, Li et al. indicated that the expression of galectin-9/TIM-3 is significantly higher in GBM than in lower grade gliomas. Therefore, the high level of TIM-3 may indicate a worse prognosis.99 In addition, the level of TIM-3 expression has been shown to correlate with PD-1 expression in both in vitro and in vivo models.100 Koyama et al. indicated that TIM-3 upregulation may influence the mechanism of resistance to the anti-PD-1 blocking in a murine model of lung cancer.101 In turn, later studies on murine glioma have reported that dual therapy with anti-PD-1 and anti-TIM-3 mAbs significantly increased survival in comparison to monotherapy.102 The TIM-3 antibodies (MBG453, TSR-022 and Sym023) are currently tested as monotherapy and in combination with anti-PD-1 in phase I/II clinical trials in patients with solid tumours (melanoma, NSCLC and RCC) and haematological malignancies (NCT02608268, NCT02817633 and NCT03489343). Recently, a phase I clinical trial combining the anti-TIM-3 inhibitor MBG453 with the PD-1 inhibitor spartalizumab and stereotactic radiosurgery has been initiated in patients with GBM (NCT03961971).
Vaccination in Glioblastoma
Currently, a plethora of methods are available for achieving positive immunization against a tumour, from relatively simple vaccines targeting single antigens to polyvalent cell-based vaccines that incorporate additional adjuvants. Therefore, vaccines have gained a lot of attention in the last decade for the treatment of GBM.
The specific targets of vaccines can be divided into two distinct groups: tumour-associated antigens (TAA) and tumour-specific antigens (TSA). The former group represent those antigens which can be found on healthy cells across the human body, but which can also be overexpressed on cancer cells. The latter group is prevalent only on malignant cells, potentially making them a better and more selective target. In highly heterogeneous tumours, such as GBMs, it is impossible to find the target that would selectively appear in all tumour cells, without being present in other, healthy cells. Therefore, it is of paramount importance to develop therapies that would affect the whole population of GBM cells, without causing off-target effects in healthy brain and surrounding tissues. Such measures must be taken to elicit a strong and widespread anti-tumour response and to minimise the adverse effects outside of the tumour.
Peptide Vaccination
Targeting EGFRvIII
The most investigated type of vaccines targeting GBM are the peptide vaccines. Rindopepimut (PEP-3-KLH, CDX-110) was the first vaccine to specifically target a splice variant of EGFR (EGFRvIII) and was designed to generate humoral and cytotoxic T cell response against tumours expressing this mutation. That vaccine incorporates a 14-mer peptide (PEPvIII) that has an amino acid structure corresponding to that of EGFRvIII and is conjugated to an immunogenic metalloproteinase called keyhole limpet hemocyanin (KLH).103,104 Several studies have been conducted using this vaccine. The phase II ACTIVATE and ACT II trials both showed that rindopepimut may confer a substantial improvement in PFS and OS over the matched controls, along with minimal toxicity.105,106 Moreover, these studies reported the loss of EGFRvIII on recurrence in about 90% of collected samples. The next complex clinical trial was the ACT III trial, which was a multi-centre, single-arm study that ultimately recruited 65 patients and reported PFS and OS values consistent with the previous data.107 On the other hand, the later ACT IV study, which was a randomised, double-blinded phase 3 trial, involving 745 patients from 165 hospitals108 was terminated when no significant mOS difference was demonstrated between patients who received a standard of care (SoC) and either rindopepimut or a placebo.109 The study did show that EGFRvIII was lost on recurrence in almost 60% of the patients in both groups and that the elimination of EGFRvIII did not correlate with PFS or OS. The postulated reasons for rindopepimut’s failure were: the selection pressure leading to an antigen loss and immune escape, the relatively low and heterogeneous prevalence of EGFRvIII in GBM samples and the negative effect of TMZ SoC treatment, which might have hindered an immunological reaction in the study group. Recently, the results of the double-blinded, randomised phase II ReACT study involving 73 patients were published. The studies showed a positive PFS signal in the study group receiving combined bevacizumab and rindopepimut treatment on GBM recurrence.110 However, these findings should be verified in a larger study cohort.
As was made evident with anti-EGFRvIII vaccines, the hallmark of anti-single antigen therapies should be the discovery of a stable target that is homogenously expressed among almost all of the malignant cells. Furthermore, to make vaccines a more successful treatment regimen for GBM patients multiple, immunogenic antigens and more potent vaccine vectors should be tested in combination with checkpoint modulators and other new drugs that reverse GBM immunosuppression.
Mutated IDH1 – A Potential Target in Astrocytoma Grade 4
In the light of the 2021 WHO classification, GBMs are diagnosed only in case of IDH wild-type astrocytomas, therefore the mutated IDH1 (isocitrate dehydrogenase type 1) could be a viable target only in the case of IDH1-mutated grade 4 astrocytomas which are considered as high-grade gliomas (HGG).
Moreover, the mutated IDH1 protein can act as a neoantigen that is not present in healthy cells of the human body. Therefore, the widespread prevalence of this mutation in IDH1-mutated gliomas makes it a perfect candidate as a vaccine target.111,112 Only one clinical trial so far has investigated the use of an IDH1R132H-targeted vaccine in gliomas, including GBM. The IDH1R132H is a mutation with the amino acid exchanged to the glutamine from an arginine at a specific position of 1DH1. The NOA-16 trial, a phase I multi-centre study (NCT02454634), enrolled a total of 32 patients, who were injected with a 20-mer peptide vaccine subcutaneously and were topically administered imiquimod, an immune response modifier. According to the initial report, the trial demonstrated safety in combining the peptide vaccine alongside the SoC, an absence of dose-limiting toxicities as well as good humoral and cellular immunogenicity.113 Moreover, recently published data have shown a 3-year PFS rate of 0.63 and a 3-year death-free rate of 0.84 after administration of this vaccine. Only 2 patients did not elicit a vaccine-induced immune response.114 Yet another trial, NOA-21 or AMPLIFY-NEOVAC (NCT03893903), could be treated as a more complex successor to NOA-16. It is an open-label, multi-centre, randomised phase I trial that is recruiting patients suffering from recurring grade 2, 3 and 4 IDH1R132H-mutated gliomas. However, this time the treatment protocol involves the concurrent administration of a peptide vaccine as well as avelumab, the anti-PD-L1 mAb in one of the study arms. Any initial reports are anticipated. Furthermore, there is another ongoing study that is exploring the IDH1 vaccine on patients with grade 2 recurrent gliomas. The PEPIDH1M vaccine is being administered with a specific growth factor, Granulocyte Macrophage Colony Stimulating Factor (GM-CSF) and tetanus-diphtheria toxoid (Td) in the RESIST trial (NCT02193347).
Polyvalent Peptide Vaccines
To avoid some of the disadvantages of the single-antigen treatment options, multi-antigen targeting modalities are being pursued. An example of this is IMA950, a vaccine containing 11 tumour-associated peptides (TUMAPs), designed to activate TUMAP-specific T cells. It combines 9 HLA-A2–restricted peptides that are eluted from the surface of GBM tumour samples and 2 HLA-DR–binding peptides that aid the CD4+ T cell response. These antigens were identified as immunogenic TAAs on multiple GBM samples in vitro. The vaccine is combined with polyinosinic-polycytidylic acid stabilised with polylysine and carboxymethylcellulose (poly-ICLC), an immunogenic molecule that aids in inducing the CD8+ T cell response (NCT01920191). Intriguingly, the vaccine formulation and administration had to be optimised in order to elicit meaningful T cell responses. Moreover, a post hoc analysis revealed no synergy in the patients treated concomitantly with bevacizumab.115,116 The results of another clinical trial (NCT01222221) investigating SoC treatment with IMA950 combined with GM-CSF instead of poly-ICLC as an adjuvant, revealed that 50% of the patients presented an immune response to more than one of the tumour-associated peptides regardless of steroid use. The PFS was analysed at 6 and 9 months and was observed to be 74% and 31% respectively.117 One single-centre, randomised phase I/II trial, IMA950-106 (NCT03665545), is currently recruiting 24 patients with recurrent GBM that will undergo IMA950/Poly-ICLC treatment or a combination regimen that utilises IMA950/Poly-ICLC along with pembrolizumab anti-PD-1 mAb. The main characteristic of the IMA950 vaccine is its pre-set antigen target range. The IMA950 is not a truly personalised vaccine, though its broad spectrum of targeted antigens may increase its effectiveness. On the other hand, the use of pre-set antigens means that the vaccine could potentially be used even in a neoadjuvant manner, before there is an access to tumour samples (by biopsy or after resection).118
Another attempt to create a personalised, polyvalent peptide vaccine has been made with the use of heat shock proteins (HSPs). HSPs belong to the wide family of complex peptide molecules, which are known for their chaperone activity, including assistance in protein folding. Usually, their expression arises as a response to cell stress, regardless of its source. It has been noted that the constitutive upregulation of HSP27, HSP72, HSP73 and HSP90 is a characteristic feature of GBM.119,120 HSPs are constitutively expressed and function as molecular chaperones that aid in the synthesis and folding of a variety of peptides, including antigens and neoantigens, which subsequently have the ability to specifically bind to APCs and invoke both the CD4+ and CD8+ T cell immune responses. This ability was a driving factor for the development of the Prophage heat-shock peptide protein complex–96 (HSPPC-96) autologous vaccine. HSPPC-96 formulation starts with GBM resection and collection of an appropriate amount of tumour tissue (at least 7 g). The samples are then moved to the laboratory, where the process of HSPPC-96 extraction and purification occurs. There have been several clinical trials of HSPPC-96 during the past decade, starting with a phase I study (NCT00293423) which proved the safety profile and a positive immunogenicity in patients with recurrent GBM.121 Moreover, it was demonstrated that the immune responders had vastly better mOS than the solitary non-responder (47 vs 16 weeks). The following Phase II, single-arm part of this study (NCT00293423),122,123 involving 41 cases of recurring GBM, reported an mOS of 42.6 weeks (95% CI, 34.7–50.5). However, one of the main findings was the fact that pre-treatment lymphopenia (an absolute lymphocyte count of <1.0×109 cells/L) resulted in a significantly lower mOS (49.1 vs 37.1 weeks, P = 0.039). In the HeatShock trial (NCT00905060) it was demonstrated that the high level of PD-L1 expression on circulating myeloid cells may be a prognostic factor for the efficacy of the HSPPC-96 vaccine. Among the patients with a high level of PD-L1 expression (n = 15), the mOS was 18.0 months (95% CI, 10.0–23.3); in the group of patients expressing low levels of PD-L1 (n = 17), the mOS was 44.7 months (95% CI, incalculable). This difference was highly significant (hazard ratio, 3.3; 95% CI, 1.4–8.6; P = 0.007).124 This has led to the preparation of a randomised, double-blinded phase II trial (NCT03018288) that is actively recruiting 108 patients with newly diagnosed GBM. The patients of the experimental arm will undergo SoC treatment, including tumour resection, and then will be administered pembrolizumab and the HSPPC-96 vaccine. Unfortunately, another clinical trial investigating this vaccine in combination with bevacizumab in recurrent GBM (NCT01814813)125 showed no survival benefit in any of the scenarios and therefore was terminated.126 There is a relatively large-scale, randomised study being organised in Beijing that will recruit up to 150 patients with newly diagnosed supratentorial gliomas (NCT03650257).127 More evidence on HSPPC-96 is needed in order to prove its efficacy. The most awaited results will be from the trial NCT03018288 that is combining vaccine therapy with pembrolizumab. This innovative combinatorial approach might help generating the robust and stable immune response that could potentially overcome the highly immunosuppressive GBM TME. Furthermore, as HSPPC-96 generates a cocktail of different antigens and neoantigens in each patient who undergoes vaccination, it might lead to a highly personalised therapy. On the other hand, it might be difficult in some patients to secure enough tumour material for generation of the vaccine. Another evident downside of this approach, which is true for HSPPC-96 and other personalised, autologous vaccines, is the fact that they require a specialised manufacturing process in the laboratory for each individual, resulting in expensive final costs and in turn expensive prices for patients.
Role of Whole Exome Sequencing in GBM Vaccination
The effort to create the most personalised, polyvalent and immunogenic GBM vaccines has led to another therapeutic strategy that is currently being investigated. These are ambitious peptide vaccine projects that aim to choose suitable targets among TAAs and TSAs by deeply analysing individual patients’ tumoural genomic and transcriptomic features.
The first of these trials started in 2014 and has only recently been published.128 The GAPVAC-101 phase I trial (NCT02149225)129 enrolled 15 patients who underwent a two-phase vaccination treatment alongside the SoC. This protocol starts with the administration of the APVAC1 vaccine, which consists of 9 HLA class I and HLA-DR peptides chosen from the premanufactured library of candidate synthetic peptides. In the second stage of treatment, patients received the APVAC2 vaccine, formulated from premanufactured or de novo synthetised peptides according to the results of each tumour’s transcriptome and immunopeptidome. Both of the vaccines used poly-ICLC and GM-CSF as adjuvants. The median ingredient formulation time for APVAC2 was 96 days, though even more time was required for the manufacturing (a median of 86 days). The trial has proven that this approach results in a highly immunogenic response targeting the projected antigens and is tolerable, albeit with some related toxicity – including an anaphylactic reaction and a brain oedema. The 15 patients that received at least the APVAC1 had a mOS of 29 months since diagnosis and mPFS of 14.2 months. The biggest advantage of this trial was the establishment of a whole manufacturing process that involves the recognition of feasible therapeutic targets and the production of de novo synthetic peptides. This whole process took place without disrupting the SoC treatment protocol.
Another important trial, currently in its final stage also uses a highly personalised vaccine, although mostly among patients with MGMT-unmethylated GBMs. The trial NCT02287428130 investigates the use of the vaccine in parallel to the SoC regime, but new cohorts were added that additionally received pembrolizumab along the investigated vaccine. Moreover, the trial presumed to enrol patients with MGMT-methylated GBMs in later stages. The 10 enrolled patients were subjected to the SoC surgery and RT.131 Following the collection of tumour tissue during resection, the samples underwent whole-exome sequencing and RNA-sequencing analysis, which led to the identification of target epitopes. These data enabled the formulation of individual vaccines based on approx. 4 peptides (20–30 amino acids in length), each with the addition of a poly-ICLC adjuvant. The reported data demonstrated that in this case a median time of 19.9 weeks between resection and the first vaccine was achieved. Eight patients received the vaccination protocol and they experienced tolerable AEs. The mPFS in this group was 7.6 months and the mOS was 16.8 months. The most important finding from this study is the evidence that dexamethasone (DEX), a highly potent corticosteroid frequently given to treat GBM patients with cerebral oedema, interfered with the process of generating a meaningful immune response. Only the 2 patients who did not receive DEX generated the polyvalent, circulating CD4+ and CD8+ T cell responses.
Both of these studies laid the foundations for more complex therapeutic strategies potentially viable from a manufacturing point of view. On the other hand, this kind of approach seems to be prohibitively costly, both in terms of monetary expenses and the time required to prepare the personalised vaccines. Moreover, a study by Keskin et al. provided further proof of a negative impact of DEX administration on immune system functioning in patients suffering from GBM. Even relatively small doses of DEX have a suppressive effect on immune system interactions that are crucial for the novel therapies.131
Cell Vaccination
The next step in designing more complex and ultimately more polyvalent vaccines is to use immunised cells as a treatment modality. In contrast to peptide vaccines, which are a more passive measure used to stimulate the host’s immune system, cell vaccines could be distinguished as active measures whose role is to actively mediate the immune response by presenting specific antigens.132
Dendritic Cell Vaccination
Dendritic cell vaccination (DCV) is an active immunotherapy that uses the patients’ own dendritic cells (DCs), which are exposed ex vivo to a variety of TAAs and TSAs, with the aid of additional immunostimulatory molecules, and subsequently injected back into the patient’s body. DCs are strong candidates for the development of highly immunogenic and personalised vaccines as they are among the most potent APCs and play a crucial role in the development of acquired immunity.133 The proposed DC vaccines have made much progress since the first phase I and II studies during the past few years. These efforts were met with the huge success of having the first DC vaccine – Sipuleucel-T (Provenge) – registered by the FDA in 2010 as a treatment for metastatic hormone refractory prostate cancer.134
The results of one such early-phase, proof-of-concept study were published recently.135 The patients suffering from newly diagnosed GBM (NCT02709616 – PerCellVac), recurrent GBM (NCT02808364 – PerCellVac2) or lung cancer with brain metastasis (NCT02808416) had their DCs harvested with leukapheresis. Following tumour resection or biopsy, the overexpressed TAAs were identified, which guided the choice of appropriate plasmids from the library that encode 87 different TAAs. In order to personalise the therapy, the patients’ DCs were subject to TAA plasmid transfection. The number of TAAs used varied between 3 and 13 each time and was guided by a number of specific criteria. The patients were then treated with a series of DC infusions which were enhanced by low-dose cyclophosphamide therapy (for regulatory T cell depletion), the administration of poly I:C (for enhancing anti-tumour immunity), and imiquimod (for increasing DC function) or combined with anti-PD-1 antibody therapy in the case of lung cancer. The obvious downside of these trials is the small sample (a total of 10 patients), which was further split into 3 groups of completely distinct disorders. Regardless of this flaw, the researchers found that the therapy induced the antigen-specific T cell response. In addition, it was quite safe with a lack of grade 3–4 adverse events and was associated with better outcomes than those found in regular patients treated in the same hospital with the current SoC.
Among the multiple studies of DC vaccine therapies for GBM, one of the proposed vaccines – DCVax-L – has made it to a phase III clinical trial (NCT00045968).136 The initial results of this trial have been published,137 but the apparent lack of precise primary endpoint information has raised important questions among the scientific community.138 DCVax-L is an autologous, tumour lysate-pulsed DC vaccine that was evaluated in cases of newly diagnosed GBM. In the aforementioned study, a total of 331 patients were randomised and added to the “intention to treat” (ITT) study group. Noteworthy, the initial study group consisted of 1599 screened patients. This group was reduced due to very restrictive inclusion and exclusion criteria as well as by other undesirable circumstances, such as unsuccessful leukapheresis or an inadequate amount of tumour tissue collected for vaccine generation. Of note, all of the patients during recurrence were given the choice of being administered DCVax-L, even if they initially belonged to the control group. At the time of publication, almost 90% of the ITT patients received the vaccine thanks to this apparent cross-over. Although the primary endpoint in this case was the PFS, the authors have avoided providing any information regarding this indicator. Instead, they announced that the mOS of the entire blinded study population was 23.1 months from the time of surgery and concluded that it might be favourable in comparison to the SoC. Moreover, the researchers had derived a subgroup of extended survivors (n = 100), who achieved a mOS of 40.5 months. According to the report, such a high mOS is the result of a selection bias that might accumulate favourable prognostic factors. A more detailed endpoint analysis of the study arms is yet to be published. Another trial of the DCVax-L vaccine is underway among the patients who were excluded from the previous study (NCT02146066).139
A further vaccine that has advanced to a phase III trial is ICT-107, an autologous, pulsed vaccine that is produced by exposing patients’ DCs to the pre-selected cocktail of 6 GBM TAAs.140 The first significant clinical trial to investigate this vaccine (NCT01280552)141 was a randomised, double-blinded, placebo-controlled phase II study that enrolled 124 patients with newly diagnosed GBM, with no prior chemoradiation. It concluded that ICT-107 was well tolerated and reported that the PFS in the ITT population was significantly higher – by 2.2 months (P = 0.011).142 To confirm the promising reports, the NCT02546102 phase III randomised, double-blinded, controlled trial was started in 2015. Unfortunately, the study was suspended in 2018 due to a lack of sufficient funding.143
There are at least two more phase III clinical trials that are investigating the role of DC autologous pulsed vaccines: DEN-STEM (NCT03548571)144 and ADCTA-SSI-G1 (NCT04277221),145,146 which are currently recruiting patients with newly diagnosed and recurrent GBMs, respectively.
Apart from the inherent immunogenicity of DCs and vaccines based on these cells, there are other measures being taken to improve their immunogenic potential even further. There were several early-phase studies that added immunomodulatory molecules to the DC vaccines administered to the patients. A series of such studies were carried out by Duke University Medical Center and the University of Florida, which covered an innovative CMV pp65-LAMP mRNA-loaded DC vaccine in different setups. This involves a vaccine targeting the cytomegalovirus pp65, a protein expressed in more than 90% of GBM glioma cells, but not in the surrounding brain cells. Among the most notable trials were ATTAC, ATTAC-GM (NCT00639639) and ELEVATE (NCT02366728).147,148 These complementary trials evaluated several parameters: the efficacy of vaccination alongside SoC and the effect of preconditioning the vaccination site with Tetanus-Diphtheria (Td) toxoid or GM-CSF. The ATTAC trial describes a cohort of 6 patients who were analysed in terms of change in DC migration and infiltration. There is evidence showing more efficient accumulation of DCs in the vaccine site-draining lymph nodes. According to the murine model, which was investigated in parallel, this mechanism seems to be dependent on the chemokine CCL3. Moreover, in contrast to the solitary DC-vaccine group, which had an mOS and a PFS comparable to those of the SoC treatment group, the group that merged the DC vaccination with Td preconditioning had significantly higher mOS and PFS. These findings were further confirmed in a continuing ATTAC-GM trial, where a group of 11 patients were enrolled. Although the PFS and OS endpoints were not the priority in such small trials, there was an evident cohort of long-term survivors who comprised approximately 35% of the study group patients. Such patients exceeded 60 months of follow-up. Moreover, the mOS in both of these trials fluctuated at around 38 months, which is far longer than in the historical controls.149,150
To improve the quality of the collected data, another study was designed by the same research group. The phase II ELEVATE study, a double-blinded, randomised trial (NCT02366728)147 aims to recruit 100 patients with newly diagnosed GBM in order to investigate the same autologous DC vaccine with the vaccination site preconditioned with Td toxoid. We are still awaiting the mOS and PRF results, but the first report has been published confirming the increased lymph node uptake of DC vaccine cells in the arm treated with Td preconditioning.149 The effort to evaluate the efficacy of this vaccine in a larger group and more controlled environment is currently in progress in the randomised, blinded, placebo-controlled phase II ATTAC-II trial.151,152
Adoptive Cell Therapies
CAR-T Cell Therapies for GBM
Another broad range of strategies that utilise the administration of live cells are known as adoptive cell therapies (ACT). One such promising approach uses chimeric antigen receptor engineered T cells (CAR-Ts) to invoke a direct cytotoxic effect on the tumour cells in a highly specific manner.153 CAR-Ts are artificially constructed T cells that originate from the patient’s own T cells, which are harvested and then genetically modified with the addition of chimeric antigen receptors (CARs) as well as other immunomodulatory molecules.153 The rationale behind this approach is to provide the T cells with the ability to recognise specific antigens independently of the MHC signalling. The CARs themselves are molecules that consist of an extracellular domain which is usually structurally similar to the variable parts of antibody light and heavy chains. This extracellular domain is fused to the intracellular molecule that typically mediates T cell co-stimulation or activation (e.g., CD28 or CD3). CAR-Ts can be targeted against the tumour-specific antigens of choice in order to invoke cytotoxic effects specifically on the tumour cells without the disadvantages of MHC restriction.154,155 This avant-garde approach has been highly successful in the treatment of disseminated cancers, such as B cell precursor acute lymphoblastic leukaemia (ALL), which led to its FDA approval in 2017.156 On the other hand, the CAR-T therapies are yet to show efficacy in the case of solid tumours, such as GBM. Among the most tested targets for CAR-T therapies in GBM are EGFRvIII, human epidermal growth factor receptor 2 (HER2) or IL receptor 13Rα2 (IL-13Rα2).
One of the first reports of CAR-T therapy in GBM was published in 2015 by Brown et al. as a result of the clinical trial NCT00730613.157,158 In this study, 3 patients with recurrent GBMs were treated with an autologous CAR-T therapy specific against IL-13Rα2.159 It has proven that the intracranial administration of CAR-Ts is tolerable, safe and can evoke the immune-mediated anti-GBM response. This led to another phase I clinical trial (NCT02208362)160 that has provided one of the most compelling case reports so far.161,162 The study describes the clinical history of a 50-year-old man suffering from a multifocal, recurrent GBM who was treated with repeated intracranial infusions of IL-13Rα2 targeting CAR-T cells (both into the resection cavity and intraventricularly). This approach resulted in a complete regression of the intracranial and spinal lesions, but sadly the patient had disease relapse after 7.5 months. Noteworthy, this trial has used the optimised, second-generation CAR-Ts that have an integrated 4–1BB (CD137) co-stimulatory domain. The results regarding a larger cohort of up to 92 enrolled patients are still to come.
Another phase I dose-escalation clinical trial (NCT01454596) investigated the feasibility of CAR-T-EGFRvIII therapy (18 patients with recurrent GBM).155,163 Among 18 patients two severe hypoxia events were observed, including one that resulted in a CAR-T treatment mortality. It is worthy to note that the fatal case was linked to the highest administered CAR-T cells dose.164 In another phase I trial targeting EGFRvIII (NCT02209376) enrolling 10 patients, 7 underwent neurosurgical resection at some point after being subject to CAR-T infusion. The data confirmed that the therapy was safe and led to EGFRvIII antigen loss in 5 out of 7 cases. Moreover, the resistance mechanisms due to IDO1 and PD-L1 upregulation alongside the FoxP3+ Treg cells were observed.165 This indicates that combining CAR-T cells with an immune checkpoint inhibitor therapy might be beneficial.
Furthermore, there was a dose-escalation trial of HER2-specific CAR-T cells (NCT01109095).166,167 Patients (n = 16) received intravenous infusions of second-generation, autologous CAR-T virus-specific T cells (VSTs). Apart from targeting HER2 via the CAR, these cells were designed to be specific for cytomegalovirus, Epstein–Barr virus or adenovirus antigens to improve their persistence in vivo due to additional stimulation by APCs. Unfortunately, the study revealed that the CAR-Ts lasted for only 6 weeks in 7 patients and reached a maximum persistence of 12 months in 2 patients. It was found that only 1 patient with an unresectable thalamic GBM had a partial response lasting 9.2 months that transformed into stable disease after the second administration of CAR-T cells. Ultimately, he survived 26.9 months after the first infusion. Moreover, 7 out of 16 patients had stable disease for 8 weeks up to 29 months. The cohort was characterised by a mOS of 24.5 months since diagnosis. The results from these early-phase studies reveal that the CAR-T therapies in GBM share similar disadvantages with the other highly targeted therapies: a loss of the single antigens, an unsatisfactory viability of CAR-T cells in vivo and the triggering of immunosuppressing compensatory responses.
In the case of HER2-targeting CAR-T cells, a path similar to the previous studies161 of IL-13Rα2–targeting CAR-T cells is investigated. The addition of memory-enriching 41-BB costimulant to the HER2-targeted CAR-T cells is being evaluated in a phase I clinical trial (NCT03389230). The therapy will involve the intratumoural or intracavitary administration of CAR-T cells in 42 participants.168
Interestingly, also other antigens are being evaluated as potential targets for CAR-T cells, including but not limited to EphA2, known as ephrin A2 or erythropoietin-producing human hepatocellular carcinoma receptor. This tyrosine kinase receptor might be overexpressed in GBM cells. Patients with recurrent EphA2-positive glioblastoma are currently recruiting for a dose-escalation phase I trial. They will undergo intravenous injection of EphA2-redirected CAR-T cells alongside a lymphodepletion protocol with the use of fludarabine and cyclophosphamide. Interestingly, the same research group has recently published a preliminary report of 3 cases that prove the feasibility of this approach. The experimental therapy was tolerated well and the patients did not present neurotoxicity. One patient was reported to have stable disease.169
Another strategy being evaluated at this moment is the use of bivalent170 and trivalent CAR-Ts. The promising in vitro study of a single population of CAR-Ts targeting HER2, IL13Rα2 and EphA2 has shown that these CAR-Ts have cytolytic properties against the cells that express any of these three targets. Moreover, the trivalent CAR-Ts were proven to target the experimental cohorts of GBM cells more efficiently than any of the bivalent CAR-Ts populations that were targeted against any of the three aforementioned targets.171
Regardless of these advances, other strategies are also being investigated, including a combination of CAR-T cell therapy with an oncolytic adenovirus secreting an EGFR bispecific T cell engager (OAd-BiTE). An in vitro study demonstrated that the OAd-BiTE managed to retarget folate receptor alpha (FR-α)–specific CAR-T cells to a secondary target, a population of FR-α–negative and EGFR-positive tumour cell lines. Importantly in vivo, in Panc-1 tumour-bearing xenografts, tumour regression was observed.172 In addition, the use of EGFRvIII-targeting CAR-T cells alongside EGFRwt-targeted BiTEs resulted in the regression of U251 orthotopic xenografts following the intraventricular administration of CART-EGFRvIII.BiTE-EGFR.173 It suggests that even in the case of heterogeneous tumours such as GBMs, a wider array of BiTEs could be used alongside a single population of CAR-Ts.
Another preclinical study assessed novel CAR-T cells that use an artificial synNotch priming receptor, that is used to activate CAR expression in the presence of synNotch ligand.174 This was described as a “prime-and-kill” concept. The researchers have developed two separate strategies for this tumour recognition circuit. The first uses a synNotch receptor activated by an antigen specific for CNS tissue, the second one involves a highly tumour-specific antigen for priming, such as EGFRvIII. These approaches, combined with CAR targeting of less specific tumour antigens (such as EphA2 and IL13Rα2), might lead to higher efficacy in overcoming tumour heterogeneity by local, in-tumour multi-antigen reactivity. It has been proven in a preclinical mouse model that such synNotch-CAR-T cells were more effective tumour killers and were more persistent and durable.174
Other strategies that involve the possibility of making CAR-T cells more resistant to the GBM immunosuppressive microenvironment are also being investigated. For example, so-called “armoured” CAR-Ts175 that are characterised by a constitutive secretion of IL-12, are investigated in mice. Preclinical studies have shown that these cells can overcome the PD-L1–induced immunosuppressive mechanisms, which enabled them to remain effective despite the hostile TME. Furthermore, Kuhn et al. reported that CAR-Ts can be modified to constitutively express CD40L.176 This is very beneficial as these CD40L+ CAR-Ts activate the host APCs, leading to the recruitment of CD4+ and CD8+ tumour-specific T cells. Moreover, in the scenario of primary target antigen loss and the expression of CD40 on tumour cells, the cytotoxicity of CAR-Ts can be maintained.
Emerging CAR-NK Cell Therapies
As treatment with CAR-T cells has demonstrated only limited efficacy against solid tumours, new CAR-based approaches are currently being investigated. There is growing interest in developing CAR-engineered NK (CAR-NK) cells that compared with CAR-T cells could offer some significant advantages: (i) can be prepared from allogeneic donors; (ii) have a lower chance of cytokine release syndrome and graft-versus-host disease; (iii) target tumours without pre-sensitisation or HLA-matching mechanisms; and (iv) have high feasibility for off-the-shelf production.
NK cells are able to recognise tumour cells via an interplay between the activating and inhibitory receptors, without the involvement of MHC class I molecules.177 The stimulation of activating receptors can be evoked by a range of factors such as stress ligand (MICA, MICB) expression, restriction of MHC class I receptors or even antibody-dependent cellular cytotoxicity (ADCC). Upon their activation, NK cells may respond via secretion of granzymes and perforin which are responsible for a direct cytotoxicity, caspase induced apoptosis or cytokine release. The wide spectrum of mechanisms that have an impact on NK cells’ activation and a broad range of effector functions make them less prone to single points of failure and therefore could be utilised by tumour cells to resist the immune response.
Earlier studies on CAR-T cells formed the foundation for the development of CAR-NK cells. Such cells preserve their intrinsic killing abilities but gain an additional targeting mechanism through the CAR-mediated pathway. Early CAR-NK cells initially used CARs that included additional co-stimulatory, intracellular domains which were originally optimised for CAR-T cells, such as 4–1BB or CD28.178 In turn, these were exchanged for intracellular domains normally found in NK cells (e.g., DAP10, DAP-12 or 2B4). These modifications proved greater efficacy in vitro.178,179
In most scenarios due to the manufacturing processes, such as irradiation, NK cells and CAR-NK cells present no relevant expansion capability following their transfer to a recipient. This means that potential patients will require repeated administration, as the cells will eventually deplete during the therapy. One of the possible ways is intravenous administration, however the desire to achieve the maximal concentration of killer cells in the tumour cell vicinity suggests that the most desirable administration method is a direct injection into the tumour cavity or even tumour margins during surgery. In case of neurosurgical pathologies, repeated, local administration is possible through the use of Ommaya or Rickham reservoirs, which are subcutaneous implants equipped with a drain system, which can be placed inside the resection cavity.
As the research on CAR-NKs is not as advanced as on CAR-Ts, only a handful of NK-based ACTs converted to early phase clinical trials. The best known phase I CAR-NK clinical trial is the CAR2BRAIN study (NCT03383978) that began in 2017 in Germany.180 The aim of this trial was to recruit 30 patients diagnosed with recurrent HER2-positive glioblastoma, who were treated with HER2 specific NK-92/5.28.z cells.177 According to the preliminary data from the parallel preclinical studies held by the same research team, the proposed NK-92/5.28.z cell therapy has shown a promising synergistic effect when combined with PD-1 checkpoint inhibition in murine models.181,182 This combinatorial approach enhanced the cytotoxic CAR-NK effect and enabled the treatment of gliomas refractory to the monotherapy and modulated the GBM TME from immunosuppressive to more cytotoxic settings. These results led to adding an additional cohort of patients in the CAR2BRAIN study, who would undergo a combinatorial regimen that will use the aforementioned CAR-NK cells alongside the PD-1 checkpoint inhibition. This study is due to finish in the end of 2022, however it is unknown if the addition of PD-1 inhibitor cohort is going to delay its results. The reports from this study are highly anticipated.
Another new clinical trial started in 2021 in Texas, USA (NCT04991870).183 It aims to recruit 25 participants with recurrent supratentorial GBM who will be subjected to a treatment regimen consisting of the intratumoral administration of allogeneic umbilical cord blood NK cells with a subsequent resection. Although this study does not investigate CAR-NK cells specifically, these genetically modified NK cells may open up new perspectives for further CAR-NK cells’ augmentation and therefore deserve to be mentioned. The newly developed CB-NK-TGF-betaR2-/NR3C1- cells are lacking TGFβR2 as well as NR3C1 glucocorticoid receptors, which should harden these cells against the effects of dexamethasone administration as well as the immunosuppressive GBM TME. Moreover, these cord blood-derived NK cells should retain their CD16 receptors, as opposed to the NK92 cells used in the previous trial.178 It is estimated that this trial will be completed by January 2024 and hopefully some of the preliminary data will be unveiled soon.
CAR-NK cell therapies open up new perspectives on solid tumour treatment, including GBM. The lack of expansion of CAR-NK cells in patients could be mitigated by repeated, high concentration injections into the tumour resection cavity or even to the tumour-surrounding brain tissue. Local delivery along the low risk of GvHD or CRS will be helpful in reducing any systemic adverse events which will help in creating combinatorial regimens with other, more aggressive immunotherapies. Moreover, the next generation of CAR-NK cells could be tailored with additional co-stimulatory domains typical for natural NK cells and could be modified to produce stimulating cytokines in order to reinvigorate the cold GBM TME. The future modifications to the CAR-NKs could include the knockdown of receptors responsible for immune cell exhaustion, such as TGFβR or IL-10RA, as well as the expression of innovative bi-/multi-specific CARs.184–187
Non-Viral Vector Mediated Gene Therapy
The 21st century marks the onset of emerging gene therapies that might become a compelling option for the uneven fight against glioblastoma. Cancer gene therapies as a whole rely on altering the tumour genetic repertoire in order to affect the cancer cells in a number of ways. Gene therapies might lead to direct cytotoxicity; they might interfere with tumour resistance pathways; or they might lead to a modification of the microenvironment. There is a substantial number of distinct approaches to tumour gene therapy. Over the past 20 years they have included the transfer of tumour suppressor genes, genes for immunomodulatory molecules or the direct interference of small interfering RNA (siRNA), microRNA (miRNA) or anti-sense cDNA with tumour progression pathway antigens.188–191 The successful delivery of such genetic information carriers necessitated the development of efficient vector systems that enable the BBB to be crossed, the passive or active targeting of cancer cells as well as good penetration of a hostile, solid TME. Currently, there are vectors at different stages of evolution that belong to various nanoparticle classes: spherical nucleic acid gold nanoparticles, liposomes, polymers and, finally, the viral vectors that will be discussed later on. Most of these carrier systems have been examined in preclinical studies, though there are some early-phase clinical trials that are worth mentioning.
The phase II study NCT02340156 was established in 2015 to combine the standard TMZ therapy with a novel targeted p53 gene therapy in patients with recurrent glioblastoma. This was preceded by encouraging preclinical study results.192 Both of these studies used the liposome SGT-53 nanocomplex to deliver a human wild-type p53 suppressor gene plasmid. Antitumour cell targeting in this case was mediated by an anti-transferrin receptor (TfR) antibody fragment. In the in vitro and in vivo study, SGT-53 successfully sensitised human glioblastoma cell lines to TMZ treatment, prolonged its antitumour effect and increased the survival in murine models. The most significant increase was found in mice treated with TMZ and two cycles of systemic SGT-53 administration, which resulted in an increase in mOS of 258% compared with TMZ alone. Despite these positive outcomes, the aforementioned NCT02340156 clinical trial has not produced any published results. According to the available data, the original intent was to enrol 26 patients into a single arm. Unfortunately, the trial was terminated following the enrolment of at least one patient. No report has been published that would provide some insight into this unfortunate occurrence.
Another phase 0 clinical study that used a novel nanoparticle vector had its results published recently.193 The clinical trial NCT03020017 aimed to assess the safety profile and efficacy of NU-0129 nanoparticle trafficking to glioblastoma cells. NU-0129 is a spherical siRNA nucleic acid molecule with a gold particle nanocore that specifically targets the Bcl2L12 GBM oncogene. This study confirmed that administering a microdose of NU-0129 in 8 patients was safe. The drug was positively identified in the tumour, immune and endothelial cells and a BBB crossing was further confirmed. Moreover, an evident downregulation of Bcl2L12 expression was observed in the tumour cells, which demonstrates the direct biological effect of this therapy. On the other hand, this ambitious study mostly yielded data on pharmacokinetics and safety assessment, while it is lacking information about the potential biological effect and clinical response, which should be examined in successive studies.
The discovery of new, innovative nanocarriers should be continued with subsequent clinical studies. Their potential lies not only in the ability to become a vector for gene therapeutics, but also for the specific delivery of other drugs, such as chemotherapeutics, sensitisers or contrasting agents. Additionally, they can improve the pharmacodynamics and pharmacokinetics of more conventional drugs and can aid in crossing the BTB or blood–tumour barrier.
Viral-Based Therapies
As already mentioned, another family of novel oncology therapies that are being developed involve virus vectors in a variety of ways. One of these modalities includes the subset of oncolytic viruses, which are capable of inducing direct cytotoxic effects in tumour cells via their selective replication. Viruses can also be used in a non-lytic manner, where they serve as potential vectors for various gene therapy agents. The genes delivered to the tumour cells might then interfere with tumour resistance pathways, leading to a modification of the microenvironment or sensitising the tumour cells to anticancer drugs.
One of the first oncolytic adenoviruses developed to fight GBM was DNX-2401, or Delta-24-RGD (tasadenoturev). It is selective for tumour cells which are characterised by a dysfunctional Rb pathway, and it is capable of inducing direct oncolysis as well as enhancing antitumour host immune responses. It was recently evaluated in a phase I dose-escalation trial (NCT00805376),194 which engaged the recurrent GBM in 37 patients with an intratumoural administration via catheter. In a treated arm it achieved a survival rate of more than 3 years in 20% of the patients, and in 3 cases a PFS of over 3 years was achieved with an almost complete regression of enhancing tumours. Moreover, the release of damage-associated molecular patterns (DAMPs) and immunogenic cell death was observed in the post-treatment tumour samples.195 Another phase II multi-centre study has just completed the recruitment stage and is currently active (NCT02798406).196 The CAPTIVE/KEYNOTE-192 study enrolled 49 participants with recurrent HGG who underwent intratumoural administration of DNX-2401 and sequential, adjuvant pembrolizumab therapy. The full results from this phase II dose-escalation study have not been published yet, but a short SNO2019 conference report was made available.197 It demonstrates that this combination therapy remained safe and indicated that the researchers have not encountered any dose-limiting conditions. The most common AEs were headache and vasogenic oedema. The most prominent cases are those of 2 patients who responded well, with a tumour regression of >94%, despite not being subjected to surgery. The mOS presented by the cohort of 48 patients was 12 months counting from intratumoural injection, which seems to be a promising result for recurrent GBM. The final results are still to come.
Not only adenoviruses can be used as oncolytic modalities. Other promising results were achieved with the use of PVSRIPO – a live, recombinant polio-rhinovirus chimera specific to the CD155 poliovirus receptor. In a dose-escalation trial (NCT01491893), PVSRIPO was administered intratumourally with convection enhancement into the bulk of WHO grade 4 recurrent tumours.198 It was reported that the survival rate of the study group reached 21% at 24 months, a proportion which was maintained up to 36 months post-treatment. PVSRIPO is currently undergoing further investigation in a multi-centre phase II trial (NCT02986178) that again is using the convection-enhanced intratumoural delivery of this viral vector.199 Moreover, a combination therapy with the parallel administration of atezolizumab was planned for another study (NCT03973879). These bold plans appear to have been postponed, as the study was withdrawn and is currently awaiting resubmission.200
Another noteworthy strategy for viral therapies is to use retroviral replicating vectors (RRVs), which trigger anti-tumour responses in a non-oncolytic manner with the use of a suicide gene . This strategy has been pursued by Tocagen Inc. in a variety of clinical trials in the past decade. This approach uses Toca 511 or vocimagene amiretrorepvec, which is an RRV responsible for the delivery of cytosine deaminase to malignant glioma cells. The administration of Toca FC follows this alteration of tumour cells. Toca FC is a prodrug 5-fluorocytosine which is converted in these malignant cells into a chemotherapeutic, 5-fluorouracil. A series of phase I trials, NCT01156584 and NCT01470794, have resulted in the publication of promising results.201,202 The initial reports have shown a feasible safety profile of Toca 511 + Toca FC therapy administered via injection into the resection cavity walls, and have reported amOS of 13.6 months in patients with recurrent or progressive HGG.203 Moreover, another report was published more recently,204 stating that Toca 511 + Toca FC managed to achieve an OS of 34.8% after 24 months in a phase III dose regimen subgroup of patients. However, the most promising results were linked to the subgroup displaying complete responses (11.3% of the 53 analysed patients, or 21.7% in the Phase III-eligible subgroup). At the time of publication, these patients had survived 33.9–52.2 months post-treatment, with a stable response. This has led to a phase II/III, randomised, multi-centre clinical trial, Toca 5 (NCT02414165),205 though it was unfortunately terminated by the sponsor. According to Tocagen Inc. press releases, the OS endpoint among patients with resectable, recurring HGGs was not achieved and was unsatisfactory compared with the SoC group (an mOS of 11.1 vs 12.2 months).206,207 However, a deep subgroup analysis has revealed that this treatment might be viable among patients with a second recurrence.
In a multitude of different strategies examined in preclinical studies, there have been several notable combinatorial approaches. One interesting concept involves the use of an oncolytic virus in synergy with CAR-T cells. In this case, a chimeric orthopox oncolytic virus (OV19t) is used as a carrier for a CD19t transgene. This virus, which has the ability to invade glioblastoma cells (as verified in a mouse model bearing a U251T subcutaneous tumour), has direct oncolytic activity. Moreover, it forces the dying tumour cells into the production and presentation of truncated CD19, which is selectively recognised by the CD19-CAR T cells administered afterwards. This combination approach utilises two distinct killing mechanisms that aid each other. The OV helps to invade the solid TME, while the direct cytotoxic effect of the CAR-T cells helps with the release of further intact OVs which effectively multiplies the number of tumour cells infected later on. According to a paper published in 2020, this unique therapy has delayed the development of different tumours in mice and has effectively prolonged life.208 This early-phase study should be treated as a proof of concept that gives hope of tailoring a specific combination of synergistic modalities that could overcome glioblastoma’s resistance to single therapies.
Summary
Unsatisfactory results of the SoC treatment for high-grade gliomas have recently led to the development of multiple, highly specific and personalised therapies. Still, however, the majority of the clinical trials have failed to demonstrate efficacy by an improvement in OS. The hostile and cold GBM TME, coupled with the innate heterogeneity of these tumours, effectively hinders most of the immunotherapeutic strategies. Increasing knowledge about different subpopulations of immune cells that either activate or suppress GBM progression could allow the design of more specific therapeutic approaches.
It seems evident that the use of several ICPIs administered in parallel should be investigated and the time when the ICPIs are given re-considered as several trials have shown this has a great effect on the therapy outcome.209,210 For example, the neoadjuvant administration of pembrolizumab primed the host’s immune response to a much higher extent than when the drug was added to SoC post-surgery. Furthermore, using polyvalent vaccines (e.g., IMA950, HSPCC-96 or APVAC1) could be more beneficial as the monovalent vaccines are more prone to causing a mechanism of escape via antigen loss. The fact that these vaccines are not truly personalised for each patient remains one of their advantages as they utilise the variety of pre-set peptide antigens, suggesting a more effective formulation and production process.
The aspiration of creating truly personalised formulations of peptide vaccines, such as APVAC2, is understandable, however the lengthy and tedious manufacturing process could become incredibly costly.
Another regimen that could boost a patient’s anti-tumour response is an autologous tumour-lysate pulsed DC vaccine that showed a good safety profile. Of note, this vaccine can be potentially produced within a maximum of several weeks after the collection of patient’s tumour sample.211 Priming of patient’s own DCs with their own tumour lysate should ensure that the therapy could target a wide array of TAAs and TSAs. Moreover, DC vaccinations could benefit from the prior immune checkpoint blockade in the same patient.
Unfortunately, none of the above-mentioned therapies involve direct tumour-killing actions, which could be executed by CAR-T and CAR-NK cell therapies or by the oncolytic viruses. Their highly potent, inherently polyvalent oncolytic activity, coupled with intracavitary, local administration and a high safety profile, make them good candidates for the creation of safe combinatorial treatment regimens. Moreover, the new NK cell lines with higher expansion properties and with a full repertoire of stimulating receptors, such as CD16, could be used as “off-the-shelf” therapeutics. The direct tumour-killing action evoked by these cells could prepare the tumour resection margins for the host’s anti-tumour response primed by the subsequent DC vaccination. Many of these therapeutic options amplify each other’s potential, however the combinatorial approaches should not be considered without scepticism. Clinicians should ensure that the patient’s safety remains paramount at any time, as adding to the current SoC additional immunomodulatory molecules and cellular compounds could lead to unacceptable and fatal AEs.
Yet another area that is noted to have abrogating effects on immunotherapeutics are immunosuppressive drugs such as corticosteroids. These remain currently as the first-line of anti-oedematous drugs. Clinical practice demands creation of an equilibrium between oedema treatment and potentially life-saving immune therapies. The paradigm of systemic DEX administration might change in favour of other anti-oedematous treatments such as antiangiogenic drugs (i.e., bevacizumab).
Our review has discussed multiple clinical trials at different phases. The select trials discussed above are gathered in Table 1 which should make the differences in their designs more evident. As most of these trials remain unblinded or even unrandomized and involve small groups of patients, the post hoc meta-analyses remain crucial in finding positive treatment effects in specific patients. Currently, it is very challenging as the clinical trials often recruit patients with tumours of different molecular features (e.g., different status of IDH or MGMT promoter mutation). These tumours collectively classified as high-grade gliomas, actually form different diseases which require distinct therapeutic regimens. The differences in clinical trial designs appear also in SoC treatments which are different for newly diagnosed and recurrent GBM.
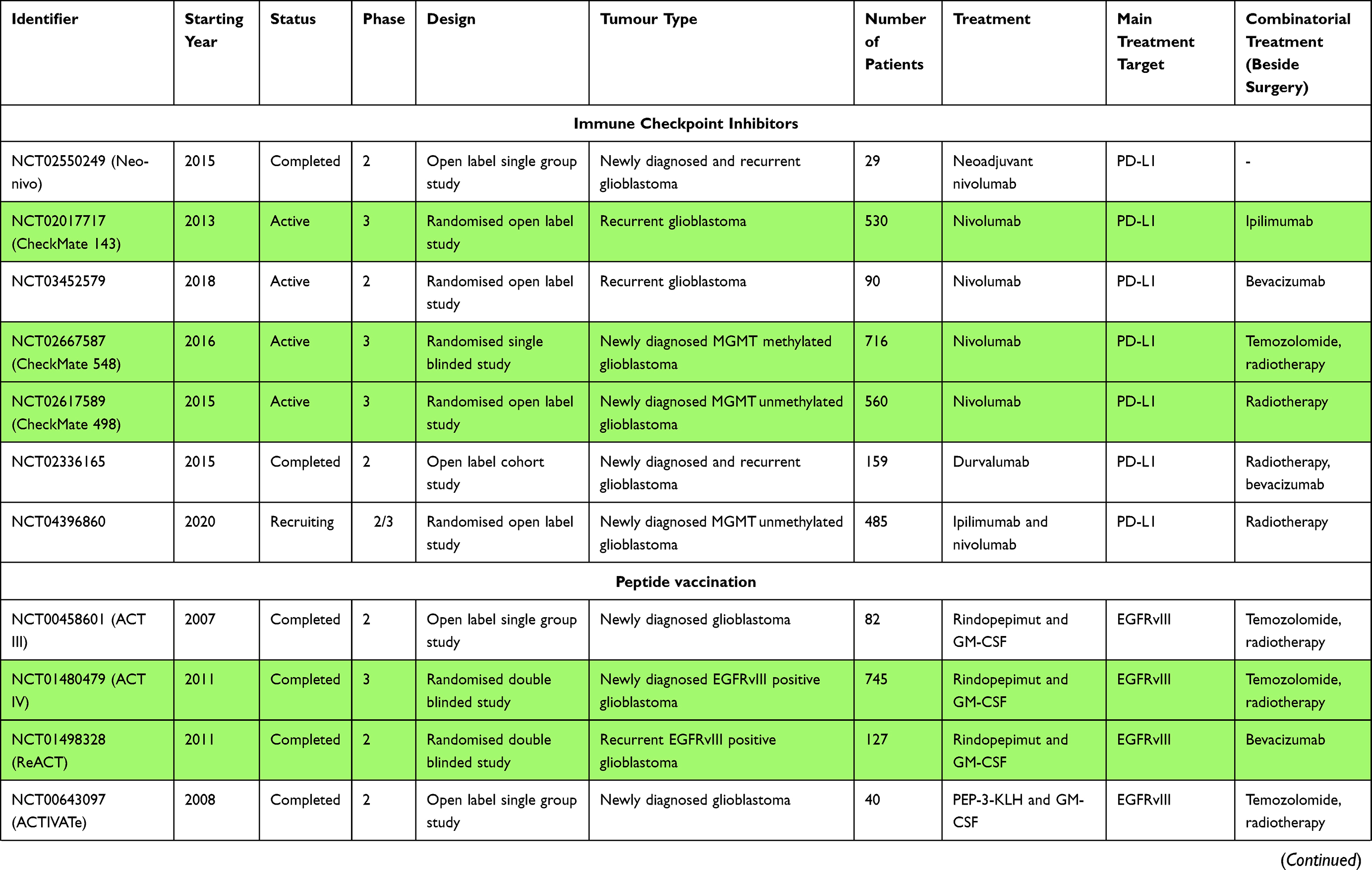 | 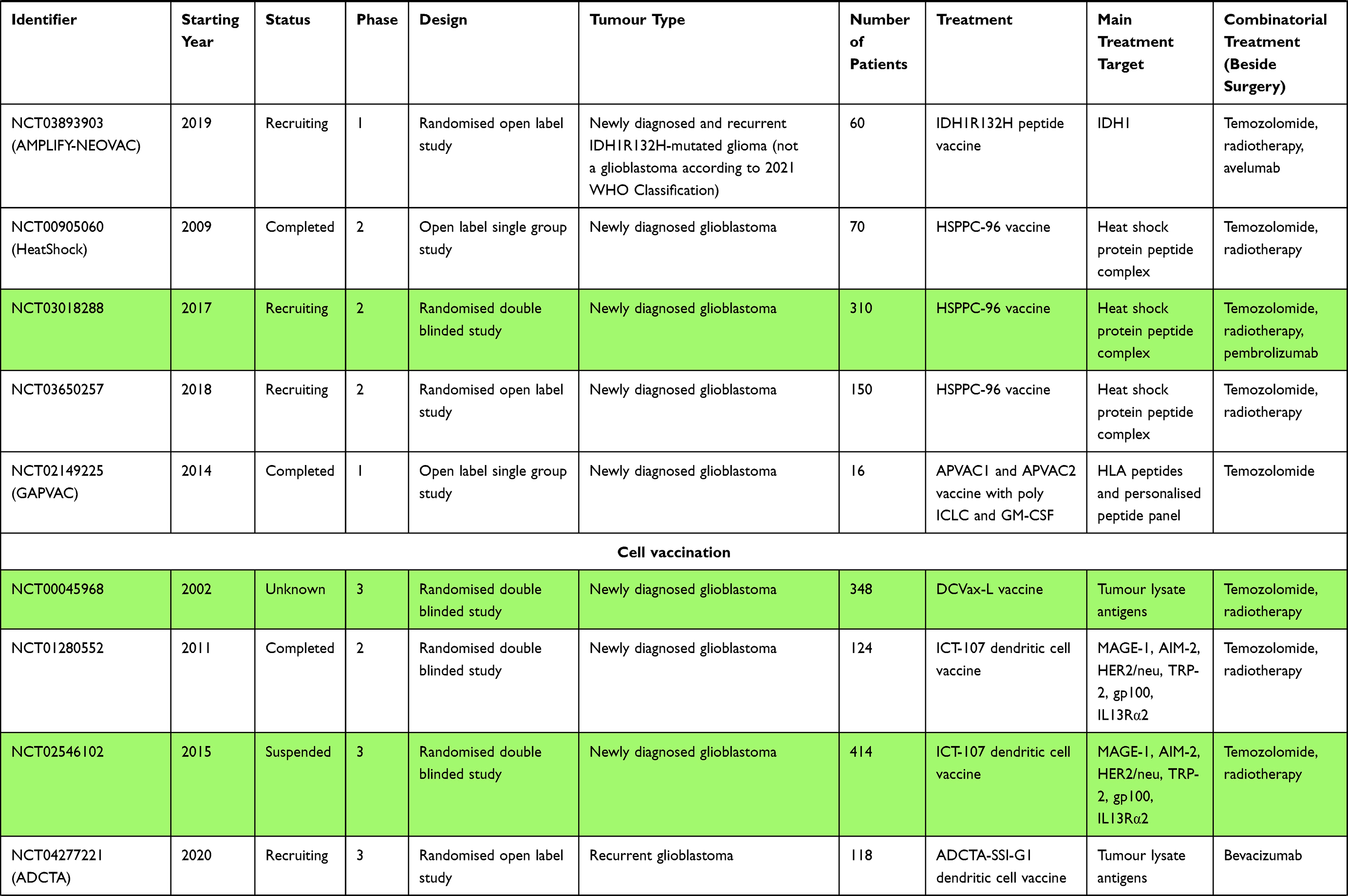 |  | 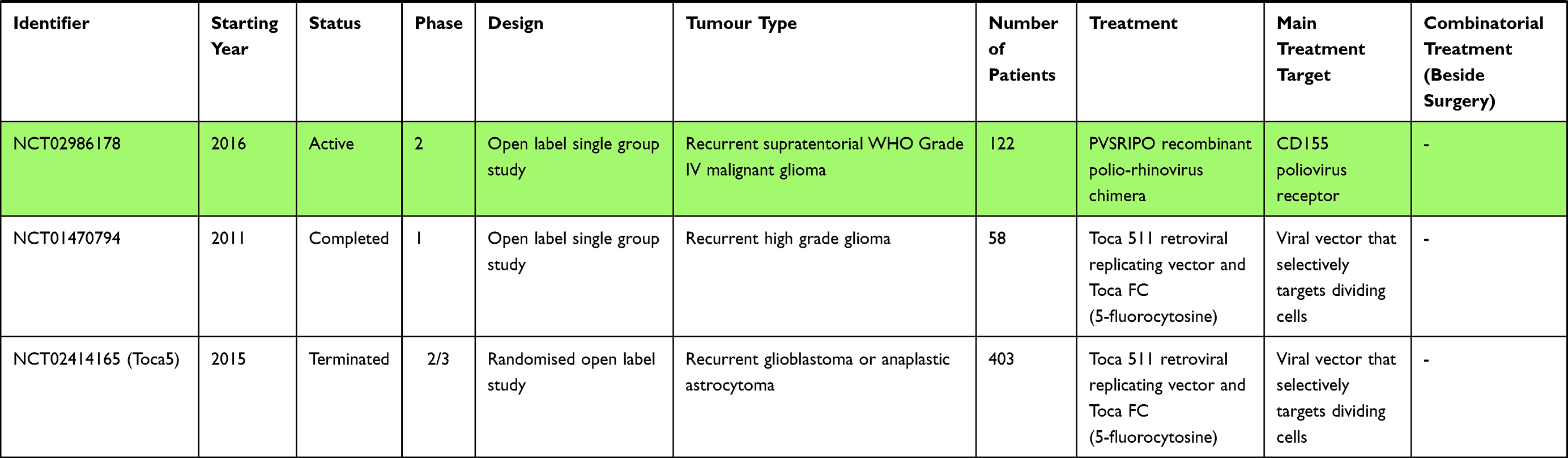 |
Table 1 Notable GBM Immunotherapy Clinical Trials. Green Background Indicates the Most Prominent and Influential Trials |
Last but not least, the recent changes made to the 2021 WHO Classification of Tumors of the Central Nervous System will hamper the comparison of previous studies with the newest clinical trials. To ensure that the post hoc analyses will remain possible, researchers should meticulously report the results of their trials and the worldwide scientific society should be encouraged to design large-scale, randomised, blinded clinical trials.
Despite these challenges, efforts should still be made to recruit GBM patients and offer them the avant-garde therapies that could potentially improve their poor prognosis.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. Lancet. 2018;392(10145):432–446. doi:10.1016/S0140-6736(18)30990-5
2. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi:10.1056/NEJMoa043330
3. Feldheim J, Kessler AF, Monoranu CM, Ernestus RI, Löhr M, Hagemann C. Changes of O(6)-methylguanine DNA methyltransferase (MGMT) promoter methylation in glioblastoma relapse-a meta-analysis type literature review. Cancers. 2019;11(12):1837. doi:10.3390/cancers11121837
4. Li Y, Ali S, Clarke J, Cha S. Bevacizumab in recurrent glioma: patterns of treatment failure and implications. Brain Tumor Res Treat. 2017;5(1):1–9. doi:10.14791/btrt.2017.5.1.1
5. Stupp R, Taillibert S, Kanner A, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA. 2017;318(23):2306–2316. doi:10.1001/jama.2017.18718
6. Ballman KV, Buckner JC, Brown PD, et al. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro Oncol. 2007;9(1):29–38. doi:10.1215/15228517-2006-025
7. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol. 2017;19(suppl_5):v1–v88. doi:10.1093/neuonc/nox158
8. Montemurro N, Fanelli GN, Scatena C, et al. Surgical outcome and molecular pattern characterization of recurrent glioblastoma multiforme: a single-center retrospective series. Clin Neurol Neurosurg. 2021;207:106735. doi:10.1016/j.clineuro.2021.106735
9. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231–1251. doi:10.1093/neuonc/noab106
10. Uhm J. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi:10.1038/nature07385
11. Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi:10.1016/j.ccr.2009.12.020
12. Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. doi:10.1016/j.ccr.2006.02.019
13. Wen PY, Weller M, Lee EQ, et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020;22(8):1073–1113. doi:10.1093/neuonc/noaa106
14. Sidaway P. CNS cancer: glioblastoma subtypes revisited. Nat Rev Clin Oncol. 2017;14(10):587. doi:10.1038/nrclinonc.2017.122
15. Wang Q, Hu B, Hu X, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32(1):42–56.e46. doi:10.1016/j.ccell.2017.06.003
16. Gabrusiewicz K, Rodriguez B, Wei J, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016;1(2). doi:10.1172/jci.insight.85841
17. Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2012;60(3):502–514. doi:10.1002/glia.21264
18. Buonfiglioli A, Hambardzumyan D. Macrophages and microglia: the cerberus of glioblastoma. Acta Neuropathol Commun. 2021;9(1):54. doi:10.1186/s40478-021-01156-z
19. Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front Immunol. 2018;9:1004. doi:10.3389/fimmu.2018.01004
20. Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213(1):48–65. doi:10.1111/j.1600-065X.2006.00441.x
21. Huang J, Liu F, Liu Z, et al. Immune checkpoint in glioblastoma: promising and challenging. Front Pharmacol. 2017;8. doi:10.3389/fphar.2017.00242
22. Korn T, Kallies A. T cell responses in the central nervous system. Nat Rev Immunol. 2017;17(3):179–194. doi:10.1038/nri.2016.144
23. Zhang ET, Inman CB, Weller RO. Interrelationships of the pia mater and the perivascular (Virchow-Robin) spaces in the human cerebrum. J Anat. 1990;170:111–123.
24. Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–341. doi:10.1038/nature14432
25. Johnston M, Nagra G, Koh L, Zakharov A, Armstrong D. Cerebrospinal fluid transport across the cribriform plate into extracranial lymphatics in rats: development and quantification. Cerebrospinal Fluid Res. 2006;3(S1). doi:10.1186/1743-8454-3-S1-S9
26. Aspelund A, Antila S, Proulx ST, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212(7):991–999. doi:10.1084/jem.20142290
27. Matias D, Balca-Silva J, da Graca GC, et al. Microglia/astrocytes-glioblastoma crosstalk: crucial molecular mechanisms and microenvironmental factors. Front Cell Neurosci. 2018;12:235. doi:10.3389/fncel.2018.00235
28. Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood–brain barriers. Trends Immunol. 2012;33(12):579–589. doi:10.1016/j.it.2012.07.004
29. Desai K, Hubben A, Ahluwalia M. The role of checkpoint inhibitors in glioblastoma. Target Oncol. 2019;14(4):375–394. doi:10.1007/s11523-019-00655-3
30. Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Investig. 2017;97(5):498–518. doi:10.1038/labinvest.2017.19
31. Fanelli GN, Grassini D, Ortenzi V, et al. Decipher the glioblastoma microenvironment: the first milestone for new groundbreaking therapeutic strategies. Genes. 2021;12(3):445. doi:10.3390/genes12030445
32. Kuang D-M, Zhao Q, Peng C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206(6):1327–1337. doi:10.1084/jem.20082173
33. Paolino M, Penninger J. The role of TAM family receptors in immune cell function: implications for cancer therapy. Cancers. 2016;8(10):97. doi:10.3390/cancers8100097
34. Ruffell B, Chang-Strachan D, Chan V, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623–637. doi:10.1016/j.ccell.2014.09.006
35. Silver DJ, Sinyuk M, Vogelbaum MA, Ahluwalia MS, Lathia JD. The intersection of cancer, cancer stem cells, and the immune system: therapeutic opportunities. Neuro-Oncology. 2016;18(2):153–159. doi:10.1093/neuonc/nov157
36. Urbantat RM, Vajkoczy P, Brandenburg S. Advances in chemokine signaling pathways as therapeutic targets in glioblastoma. Cancers. 2021;13(12):2983. doi:10.3390/cancers13122983
37. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–27. doi:10.1038/nn.4185
38. Annovazzi L, Mellai M, Bovio E, Mazzetti S, Pollo B, Schiffer D. Microglia immunophenotyping in gliomas. Oncol Lett. 2018;15(1):998–1006. doi:10.3892/ol.2017.7386
39. Leblond MM, Peres EA, Helaine C, et al. M2 macrophages are more resistant than M1 macrophages following radiation therapy in the context of glioblastoma. Oncotarget. 2017;8(42):72597–72612. doi:10.18632/oncotarget.19994
40. Franzin R, Netti GS, Spadaccino F, et al. The use of immune checkpoint inhibitors in oncology and the occurrence of AKI: where do we stand? Front Immunol. 2020;11:574271. doi:10.3389/fimmu.2020.574271
41. Maxwell R, Jackson CM, Lim M. Clinical trials investigating immune checkpoint blockade in glioblastoma. Curr Treat Options Oncol. 2017;18(8):51. doi:10.1007/s11864-017-0492-y
42. Cheng X, Veverka V, Radhakrishnan A, et al. Structure and interactions of the human programmed cell death 1 receptor. J Biol Chem. 2013;288(17):11771–11785. doi:10.1074/jbc.M112.448126
43. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5(12):1365–1369. doi:10.1038/70932
44. Francisco LM, Salinas VH, Brown KE, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015–3029. doi:10.1084/jem.20090847
45. Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-pd-1 therapy. Clin Cancer Res. 2014;20(19):5064–5074. doi:10.1158/1078-0432.CCR-13-3271
46. Berghoff AS, Kiesel B, Widhalm G, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncology. 2015;17(8):1064–1075. doi:10.1093/neuonc/nou307
47. Nduom EK, Wei J, Yaghi NK, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology. 2016;18(2):195–205. doi:10.1093/neuonc/nov172
48. Hao C, Chen G, Zhao H, et al. PD-L1 expression in glioblastoma, the clinical and prognostic significance: a systematic literature review and meta-analysis. Front Oncol. 2020;10:1015. doi:10.3389/fonc.2020.01015
49. Chen RQ, Liu F, Qiu XY, Chen XQ. The prognostic and therapeutic value of PD-L1 in glioma. Front Pharmacol. 2018;9:1503. doi:10.3389/fphar.2018.01503
50. Raedler LA. Opdivo (Nivolumab): second PD-1 inhibitor receives FDA approval for unresectable or metastatic melanoma. Am Health Drug Benefits. 2015;8(Spec Feature):180–183.
51. Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol. 2015;23:32–38. doi:10.1016/j.coph.2015.05.011
52. Huang BY, Zhan YP, Zong WJ, et al. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS One. 2015;10(8):e0134715. doi:10.1371/journal.pone.0134715
53. Reardon DA, Gokhale PC, Klein SR, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016;4(2):124–135. doi:10.1158/2326-6066.CIR-15-0151
54. Zeng J, See AP, Phallen J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Physics. 2013;86(2):343–349. doi:10.1016/j.ijrobp.2012.12.025
55. Chamberlain MC, Kim BT. Nivolumab for patients with recurrent glioblastoma progressing on bevacizumab: a retrospective case series. J Neurooncol. 2017;133(3):561–569. doi:10.1007/s11060-017-2466-0
56. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25(3):470–476. doi:10.1038/s41591-018-0339-5
57. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477–486. doi:10.1038/s41591-018-0337-7
58. de Groot J, Penas-Prado M, Alfaro-Munoz K, et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. 2020;22(4):539–549. doi:10.1093/neuonc/noz185
59. Reardon DA, Brandes AA, Omuro A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma. JAMA Oncol. 2020;6(7):1003. doi:10.1001/jamaoncol.2020.1024
60. Reardon DA, Omuro A, Brandes AA, et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro-Oncology. 2017;19(suppl_3):iii21. doi:10.1093/neuonc/nox036.071
61. Omuro A, Vlahovic G, Baehring J, et al. OS07.3 nivolumab in combination with radiotherapy with or without temozolomide in patients with newly diagnosed glioblastoma: updated results from CheckMate 143. Neuro-Oncology. 2017;19(suppl_3):iii13. doi:10.1093/neuonc/nox036.044
62. Ahluwalia MS, Rauf Y, Li H, Wen PY, Peereboom DM, Reardon DA. Randomized Phase 2 study of nivolumab (nivo) plus either standard or reduced dose bevacizumab (bev) in recurrent glioblastoma (rGBM). J Clin Oncol. 2021;39(15_suppl):2015. doi:10.1200/JCO.2021.39.15_suppl.2015
63. Chokshi CR, Brakel BA, Tatari N, et al. Advances in immunotherapy for adult glioblastoma. Cancers. 2021;13(14):3400. doi:10.3390/cancers13143400
64. Sampson JH, Omuro AMP, Preusser M, et al. A randomized, phase 3, open-label study of nivolumab versus temozolomide (TMZ) in combination with radiotherapy (RT) in adult patients (pts) with newly diagnosed, O-6-methylguanine DNA methyltransferase (MGMT)-unmethylated glioblastoma (GBM): CheckMate-498. J Clin Oncol. 2016;34(15_suppl):TPS2079. doi:10.1200/JCO.2016.34.15_suppl.TPS2079
65. Bristol Myers Squibb announces update on phase 3 CheckMate −548 trial evaluating patients with newly diagnosed MGMT-methylated glioblastoma multiforme; 2021. Available from: https://news.bms.com/news/details/2020/Bristol-Myers-Squibb-Announces-Update-on-Phase-3-CheckMate–548-Trial-Evaluating-Patients-with-Newly-Diagnosed-MGMT-Methylated-Glioblastoma-Multiforme/default.aspx.
66. Reardon DA, Kim TM, Frenel JS, et al. Treatment with pembrolizumab in programmed death ligand 1-positive recurrent glioblastoma: results from the multicohort Phase 1 KEYNOTE-028 trial. Cancer. 2021;127(10):1620–1629. doi:10.1002/cncr.33378
67. Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. JNCI. 2000;92(3):205–216. doi:10.1093/jnci/92.3.205
68. Reardon DA, Kim T-M, Frenel J-S, et al. ATIM-35 results of the phase Ib KEYNOTE-028 multi-cohort trial of pembrolizumab monotherapy in patients with recurrent PD-L1-positive glioblastoma multiforme (GBM). Neuro-Oncology. 2016;18(suppl_6):vi25–vi26. doi:10.1093/neuonc/now212.100
69. Reardon DA, Nayak L, Peters KB, et al. Phase II study of pembrolizumab or pembrolizumab plus bevacizumab for recurrent glioblastoma (rGBM) patients. J Clin Oncol. 2018;36(15_suppl):2006. doi:10.1200/JCO.2018.36.15_suppl.2006
70. Sahebjam S, Forsyth P, Arrington J, et al. Atim-18. A phase I trial of hypofractionated stereotactic irradiation (Hfsrt) with pembrolizumab and bevacizumab in patients with recurrent high grade glioma (Nct02313272). Neuro-Oncology. 2017;19(suppl_6):vi30. doi:10.1093/neuonc/nox168.113
71. Reardon DA, Kaley TJ, Dietrich J, et al. Phase II study to evaluate safety and efficacy of MEDI4736 (durvalumab) + radiotherapy in patients with newly diagnosed unmethylated MGMT glioblastoma (new unmeth GBM). J Clin Oncol. 2019;37(15_suppl):2032. doi:10.1200/JCO.2019.37.15_suppl.2032
72. Reardon DA, Kaley TJ, Dietrich J, et al. Phase 2 study to evaluate safety and efficacy of MEDI4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: an update. J Clin Oncol. 2017;35(15_suppl):2042. doi:10.1200/JCO.2017.35.15_suppl.2042
73. Reardon D, Kaley T, Dietrich J, et al. Atim-38. Phase 2 study to evaluate the clinical efficacy and safety of Medi4736 (Durvalumab, Durva) + bevacizumab (Bev) in Bev-naïve patients with recurrent glioblastoma (Gbm). Neuro-Oncology. 2018;20(suppl_6):vi10. doi:10.1093/neuonc/noy148.033
74. Reardon D, Kaley T, Dietrich J, et al. Atim-12. Phase 2 study to evaluate the clinical efficacy and safety of Medi4736 (Durvalumab [Dur]) in patients with bevacizumab (Bev)-refractory recurrent glioblastoma (Gbm). Neuro-Oncology. 2017;19(suppl_6):vi28.
75. Lukas RV, Rodon J, Becker K, et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J Neurooncol. 2018;140(2):317–328. doi:10.1007/s11060-018-2955-9
76. Weathers S-PS, Kamiya-Matsuoka C, Harrison RA, et al. Phase I/II study to evaluate the safety and clinical efficacy of atezolizumab (atezo; aPDL1) in combination with temozolomide (TMZ) and radiation in patients with newly diagnosed glioblastoma (GBM). J Clin Oncol. 2020;38(15_suppl):2511. doi:10.1200/JCO.2020.38.15_suppl.2511
77. Kurz S, Silverman JS, Hochman T, et al. Atim-37. Phase II, open-label, single arm, multicenter study of avelumab with hypofractionated radiation (Hfrt) for adult patients with secondarily transformed Idh-mutant glioblastoma (Gbm). Neuro-Oncology. 2019;21(Supplement_6):vi9–vi10. doi:10.1093/neuonc/noz175.036
78. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229(1):12–26. doi:10.1111/j.1600-065X.2009.00770.x
79. Qureshi OS, Zheng Y, Nakamura K, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–603. doi:10.1126/science.1202947
80. Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by Cd25+Cd4+regulatory T cells constitutively expressing cytotoxic T lymphocyte–associated antigen 4. J Exp Med. 2000;192(2):303–310. doi:10.1084/jem.192.2.303
81. Fecci PE, Ochiai H, Mitchell DA, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13(7):2158–2167. doi:10.1158/1078-0432.CCR-06-2070
82. Prieto PA, Yang JC, Sherry RM, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012;18(7):2039–2047. doi:10.1158/1078-0432.CCR-11-1823
83. Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459–465. doi:10.1016/S1470-2045(12)70090-6
84. Queirolo P, Spagnolo F, Ascierto PA, et al. Efficacy and safety of ipilimumab in patients with advanced melanoma and brain metastases. J Neurooncol. 2014;118(1):109–116. doi:10.1007/s11060-014-1400-y
85. Omuro A, Vlahovic G, Lim M, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro-Oncology. 2018;20(5):674–686. doi:10.1093/neuonc/nox208
86. Schwarze JK, Duerinck J, Dufait I, et al. A phase I clinical trial on intratumoral and intracavitary administration of ipilimumab and nivolumab in patients with recurrent glioblastoma. J Clin Oncol. 2020;38(15_suppl):2534. doi:10.1200/JCO.2020.38.15_suppl.2534
87. Sloan AE, Gilbert MR, Zhang P, et al. NRG BN002: phase I study of checkpoint inhibitors anti-CTLA-4, anti-PD-1, the combination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2018;36(15_suppl):2053. doi:10.1200/JCO.2018.36.15_suppl.2053
88. Lee JB, Ha SJ, Kim HR. Clinical insights into novel immune checkpoint inhibitors. Front Pharmacol. 2021;12:681320. doi:10.3389/fphar.2021.681320
89. Zhai L, Lauing KL, Chang AL, et al. The role of IDO in brain tumor immunotherapy. J Neurooncol. 2015;123(3):395–403. doi:10.1007/s11060-014-1687-8
90. Mitsuka K, Kawataki T, Satoh E, Asahara T, Horikoshi T, Kinouchi H. Expression of indoleamine 2,3-dioxygenase and correlation with pathological malignancy in gliomas. Neurosurgery. 2013;72(6):1031–1039. doi:10.1227/NEU.0b013e31828cf945
91. Wainwright DA, Balyasnikova IV, Chang AL, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18(22):6110–6121. doi:10.1158/1078-0432.CCR-12-2130
92. Goldberg MV, Drake CG. LAG-3 in cancer immunotherapy. NIH Public Access. 2010;344:269–278.
93. Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3 — potential mechanisms of action. Nat Rev Immunol. 2015;15(1):45–56. doi:10.1038/nri3790
94. Harris-Bookman S, Mathios D, Martin AM, et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int J Cancer. 2018;143(12):3201–3208. doi:10.1002/ijc.31661
95. Lim M, Ye X, Piotrowski AF, et al. Updated safety phase I trial of anti-LAG-3 alone and in combination with anti-PD-1 in patients with recurrent GBM. J Clin Oncol. 2020;38(15_suppl):2512. doi:10.1200/JCO.2020.38.15_suppl.2512
96. Lynes J, Jackson S, Sanchez V, et al. Cytokine microdialysis for real-time immune monitoring in glioblastoma patients undergoing checkpoint blockade. Neurosurgery. 2019;84(4):945–953. doi:10.1093/neuros/nyy392
97. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017;276(1):97–111. doi:10.1111/imr.12520
98. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi:10.1038/nri3862
99. Li G, Wang Z, Zhang C, et al. Molecular and clinical characterization of TIM-3 in glioma through 1024 samples. OncoImmunology. 2017;6(8):e1328339. doi:10.1080/2162402X.2017.1328339
100. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–2194. doi:10.1084/jem.20100643
101. Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7(1):10501. doi:10.1038/ncomms10501
102. Kim JE, Patel MA, Mangraviti A, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23(1):124–136. doi:10.1158/1078-0432.CCR-15-1535
103. Pelloski CE, Ballman KV, Furth AF, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. 2007;25(16):2288–2294. doi:10.1200/JCO.2006.08.0705
104. Swartz AM, Li QJ, Sampson JH. Rindopepimut: a promising immunotherapeutic for the treatment of glioblastoma multiforme. Future Med. 2014;6:679–690.
105. Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722–4729. doi:10.1200/JCO.2010.28.6963
106. Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-Oncology. 2011;13(3):324–333. doi:10.1093/neuonc/noq157
107. Schuster J, Lai RK, Recht LD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro-Oncology. 2015;17(6):854–861. doi:10.1093/neuonc/nou348
108. Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18(10):1373–1385. doi:10.1016/S1470-2045(17)30517-X
109. Celldex. Data safety and monitoring board recommends celldex’s phase 3 study of RINTEGA® (rindopepimut) in newly diagnosed glioblastoma be discontinued as it is unlikely to meet primary overall survival endpoint in patients with minimal residual disease. Press Release Web site; 2016. Available from: https://ir.celldex.com/news-releases/news-release-details/data-safety-and-monitoring-board-recommends-celldexs-phase-3.
110. Reardon DA, Desjardins A, Vredenburgh JJ, et al. Rindopepimut with bevacizumab for patients with relapsed EGFRvIII-expressing glioblastoma (REACT): results of a double-blind randomized phase II trial. Clin Cancer Res. 2020;26(7):1586–1594. doi:10.1158/1078-0432.CCR-18-1140
111. Kaminska B, Czapski B, Guzik R, Król SK, Gielniewski B. Consequences of IDH1/2 mutations in gliomas and an assessment of inhibitors targeting mutated IDH proteins. Molecules. 2019;24:MDPI AG. doi:10.3390/molecules24050968
112. Szopa W, Burley TA, Kramer-Marek G, Kaspera W. Diagnostic and therapeutic biomarkers in glioblastoma: current status and future perspectives. BioMed Res Int. 2017;2017. doi:10.1155/2017/8013575
113. Platten M, Schilling D, Bunse L, et al. A mutation-specific peptide vaccine targeting IDH1R132H in patients with newly diagnosed malignant astrocytomas: a first-in-man multicenter phase I clinical trial of the German Neurooncology Working Group (NOA-16). J Clin Oncol. 2018;36(15_suppl):2001. doi:10.1200/JCO.2018.36.15_suppl.2001
114. Platten M, Bunse L, Wick A, et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature. 2021;592(7854):463–468. doi:10.1038/s41586-021-03363-z
115. Migliorini D, Dutoit V, Allard M, et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro-Oncology. 2019;21(7):923–933. doi:10.1093/neuonc/noz040
116. Boydell E, Marinari E, Migliorini D, Dietrich PY, Patrikidou A, Dutoit V. Exploratory study of the effect of IMA950/poly-ICLC vaccination on response to bevacizumab in relapsing high-grade glioma patients. Cancers. 2019;11(4):464. doi:10.3390/cancers11040464
117. Rampling R, Peoples S, Mulholland PJ, et al. A cancer research UK first time in human phase I trial of IMA950 (novel multipeptide therapeutic vaccine) in patients with newly diagnosed glioblastoma. Clin Cancer Res. 2016;22(19):4776–4785. doi:10.1158/1078-0432.CCR-16-0506
118. ClinicalTrials.gov. A study of varlilumab and IMA950 vaccine plus poly-ICLC in patients with WHO grade II low-grade glioma (LGG); 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT02924038.
119. Graner MW, Cumming RI, Bigner DD. The heat shock response and chaperones/heat shock proteins in brain tumors: surface expression, release, and possible immune consequences. J Neurosci. 2007;27(42):11214–11227. doi:10.1523/JNEUROSCI.3588-07.2007
120. Hermisson M, Strik H, Rieger J, Dichgans J, Meyermann R, Weller M. Expression and functional activity of heat shock proteins in human glioblastoma multiforme. Neurology. 2000;54(6):1357–1365. doi:10.1212/WNL.54.6.1357
121. Crane CA, Han SJ, Ahn B, et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res. 2013;19(1):205–214. doi:10.1158/1078-0432.CCR-11-3358
122. Bloch O, Crane CA, Fuks Y, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol. 2014;16(2):274–279. doi:10.1093/neuonc/not203
123. GP96 heat shock protein-peptide complex vaccine in treating patients with recurrent or progressive glioma. Available from: https://clinicaltrials.gov/ct2/show/NCT00293423.
124. Bloch O, Lim M, Sughrue ME, et al. Autologous heat shock protein peptide vaccination for newly diagnosed glioblastoma: impact of peripheral PD-L1 expression on response to therapy. Clin Cancer Res. 2017;23(14):3575–3584. doi:10.1158/1078-0432.CCR-16-1369
125. Vaccine therapy with bevacizumab versus bevacizumab alone in treating patients with recurrent glioblastoma multiforme that can be removed by surgery. Available from: https://clinicaltrials.gov/ct2/show/NCT01814813.
126. Bloch O, Shi Q, Anderson SK, et al. Atim-14. Alliance A071101: a phase II randomized trial comparing the efficacy of heat shock protein peptide complex-96 (Hsppc-96) vaccine given with bevacizumab versus bevacizumab alone in the treatment of surgically resectable recurrent glioblastoma. Neuro-Oncology. 2017;19(suppl_6):vi29–vi29. doi:10.1093/neuonc/nox168.110
127. A large-scale research for immunotherapy of glioblastoma with autologous heat shock protein gp96. Available from: https://clinicaltrials.gov/ct2/show/NCT03650257.
128. Hilf N, Kuttruff-Coqui S, Frenzel K, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565(7738):240–245. doi:10.1038/s41586-018-0810-y
129. GAPVAC phase I trial in newly diagnosed glioblastoma patients. Available from: https://clinicaltrials.gov/ct2/show/NCT02149225.
130. Personalized NeoAntigen cancer vaccine with RT plus pembrolizumab for patients with MGMT unmethylated, newly diagnosed GBM. Available from: https://clinicaltrials.gov/ct2/show/NCT02287428.
131. Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–239. doi:10.1038/s41586-018-0792-9
132. Kong Z, Wang Y, Ma W. Vaccination in the immunotherapy of glioblastoma. Hum Vaccin Immunother. 2018;14(2):255–268. doi:10.1080/21645515.2017.1388481
133. Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity. 2014;40:642–656. doi:10.1016/j.immuni.2014.04.016
134. FDA approves provenge - a cellular immunotherapy for men with advanced prostate cancer. @drugscom; 2022. Available from: https://www.drugs.com/newdrugs/fda-approves-provenge-cellular-immunotherapy-men-advanced-prostate-cancer-2130.html.
135. Wang QT, Nie Y, Sun SN, et al. Tumor-associated antigen-based personalized dendritic cell vaccine in solid tumor patients. Cancer Immunol Immunother. 2020;69(7):1375–1387. doi:10.1007/s00262-020-02496-w
136. Liau LM, Ashkan K, Tran DD, et al. First results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16(1):1. doi:10.1186/s12967-017-1374-6
137. Study of a drug [DCVax®-L] to treat newly diagnosed GBM brain cancer. Available from: https://clinicaltrials.gov/ct2/show/NCT00045968.
138. Wick W, van den Bent MJ. First results on the DCVax phase III trial: raising more questions than providing answers. Neuro-Oncology. 2018;20(10):1283–1284. doi:10.1093/neuonc/noy125
139. Expanded access protocol for GBM patients with already manufactured DCVax®-L who have screen-failed protocol 020221. Available from: https://clinicaltrials.gov/ct2/show/NCT02146066.
140. Phuphanich S, Wheeler CJ, Rudnick JD, et al. Phase i trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013;62(1):125–135. doi:10.1007/s00262-012-1319-0
141. A study of ICT-107 immunotherapy in glioblastoma multiforme (GBM). Available from: https://clinicaltrials.gov/ct2/show/NCT01280552.
142. Wen PY, Reardon DA, Armstrong TS, et al. A randomized double-blind placebo-controlled phase II trial of dendritic cell vaccine ICT-107 in newly diagnosed patients with glioblastoma. Clin Cancer Res. 2019;25(19):5799–5807. doi:10.1158/1078-0432.CCR-19-0261
143. Phase 3 randomized, double-blind, controlled study of ICT-107 in glioblastoma. Available from: https://clinicaltrials.gov/ct2/show/NCT02546102.
144. Dendritic cell immunotherapy against cancer stem cells in glioblastoma patients receiving standard therapy. Available from: https://clinicaltrials.gov/ct2/show/NCT03548571.
145. Jan CI, Tsai WC, Harn HJ, et al. Predictors of response to autologous dendritic cell therapy in Glioblastoma multiforme. Front Immunol. 2018;9:727. doi:10.3389/fimmu.2018.00727
146. ADCTA for adjuvant immunotherapy in standard treatment of recurrent glioblastoma multiforme (GBM). Available from: https://clinicaltrials.gov/ct2/show/NCT04277221.
147. DC migration study for newly-diagnosed GBM (ELEVATE). 2015. Available from: https://clinicaltrials.gov/ct2/show/NCT02366728.
148. ClinicalTrials.gov. Vaccine therapy in treating patients with newly diagnosed glioblastoma multiforme; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT00639639.
149. Batich KA, Mitchell DA, Healy P, Herndon JE, Sampson JH. Once, twice, three times a finding: reproducibility of dendritic cell vaccine trials targeting cytomegalovirus in glioblastoma. Clin Cancer Res. 2020;26(20):5297–5303. doi:10.1158/1078-0432.CCR-20-1082
150. Batich KA, Reap EA, Archer GE, et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res. 2017;23(8):1898–1909. doi:10.1158/1078-0432.CCR-16-2057
151. NCT02465268 vaccine therapy for the treatment of newly diagnosed glioblastoma multiforme (ATTAC-II); 2015. Available from: https://clinicaltrials.gov/ct2/show/NCT02465268.
152. ITI announces publication of results from 3 ATTAC studies of CMV-specific dendritic cell vaccines for the treatment of GBM; 2020. Available from: https://www.immunomix.com/immunomic-therapeutics-announces-publication-of-results-from-3-attac-studies-of-cmv-specific-dendritic-cell-vaccines-for-The-treatment-of-gbm/.
153. Fecci PE, Sampson JH. The current state of immunotherapy for gliomas: an eye toward the future JNSPG 75th anniversary invited review article. J Neurosurg. 2019;131(3):657–666. doi:10.3171/2019.5.JNS181762
154. Choi BD, Maus MV, June CH, Sampson JH. Immunotherapy for glioblastoma: adoptive T-cell Strategies. Clin Cancer Res. 2019;25(7):2042–2048. doi:10.1158/1078-0432.CCR-18-1625
155. Morgan RA, Johnson LA, Davis JL, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23(10):1043–1053. doi:10.1089/hum.2012.041
156. US Food and Drug Administration. Oncology Nursing Society. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome; 2017. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tisagenlecleucel-b-cell-all-and-tocilizumab-cytokine-release-syndrome. Accessed April 14, 2022.
157. Brown CE, Badie B, Barish ME, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–4072. doi:10.1158/1078-0432.CCR-15-0428
158. Cellular adoptive immunotherapy using genetically modified T-lymphocytes in treating patients with recurrent or refractory high-grade malignant glioma. Available from: https://clinicaltrials.gov/ct2/show/NCT00730613.
159. Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of interleukin-13 receptor α2 in glioblastoma multiforme: implications for targeted therapies. Cancer Res. 2007;67(17):7983–7986. doi:10.1158/0008-5472.CAN-07-1493
160. Genetically modified T-cells in treating patients with recurrent or refractory malignant glioma. Available from: https://clinicaltrials.gov/ct2/show/NCT02208362.
161. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi:10.1056/NEJMoa1610497
162. Brown CE, Starr R, Weng L, et al. 247. Phase I study of second generation chimeric antigen receptor-engineered t cells targeting IL13Rα2 for the treatment of glioblastoma. Mol Ther. 2016;24:S97. doi:10.1016/S1525-0016(16)33056-8
163. CAR T cell receptor immunotherapy targeting EGFRvIII for patients with malignant gliomas expressing EGFRvIII; 2020. Available from: ClinicalTrials.gov.
164. Goff SL, Morgan RA, Yang JC, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. 2019;42(4):126–135. doi:10.1097/CJI.0000000000000260
165. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399). doi:10.1126/scitranslmed.aaa0984
166. Ahmed N, Brawley V, Hegde M, et al. HER2-specific chimeric antigen receptor–modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094–1101. doi:10.1001/jamaoncol.2017.0184
167. CMV-specific cytotoxic T lymphocytes expressing CAR targeting HER2 in patients with GBM. Available from: https://clinicaltrials.gov/ct2/show/NCT01109095.
168. Memory-enriched T cells in treating patients with recurrent or refractory grade III-IV glioma. Available from: https://clinicaltrials.gov/ct2/show/NCT03389230.
169. Lin Q, Ba T, Ho J, et al. First-in-human trial of EphA2-redirected CAR T-cells in patients with recurrent glioblastoma: a preliminary report of three cases at the starting dose. Front Oncol. 2021;11:694941. doi:10.3389/fonc.2021.694941
170. Phase I EGFR BATs in newly diagnosed glioblastoma. Available from: https://clinicaltrials.gov/ct2/show/NCT03344250.
171. Bielamowicz K, Fousek K, Byrd TT, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology. 2018;20(4):506–518. doi:10.1093/neuonc/nox182
172. Wing A, Fajardo CA, Posey AD, et al. Improving CART-cell therapy of solid tumors with oncolytic virus–driven production of a bispecific T-cell engager. Cancer Immunol Res. 2018;6(5):605–616. doi:10.1158/2326-6066.CIR-17-0314
173. Choi BD, Yu X, Castano AP, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. 2019;37(9):1049–1058. doi:10.1038/s41587-019-0192-1
174. Choe JH, Watchmaker PB, Simic MS, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13(591). doi:10.1126/scitranslmed.abe7378
175. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep. 2017;7(1):1–14. doi:10.1038/s41598-017-10940-8
176. Kuhn NF, Purdon TJ, Van leeuwen DG, et al. CD40 ligand-modified chimeric antigen receptor T cells enhance antitumor function by eliciting an endogenous antitumor response. Cancer Cell. 2019;35(3):473–488.e476. doi:10.1016/j.ccell.2019.02.006
177. Burger MC, Zhang C, Harter PN, et al. CAR-engineered NK cells for the treatment of glioblastoma: turning innate effectors into precision tools for cancer immunotherapy. Front Immunol. 2019;10:2683. doi:10.3389/fimmu.2019.02683
178. Pan C, Zhai Y, Li G, Jiang T, Zhang W. NK cell-based immunotherapy and therapeutic perspective in gliomas. Front Oncol. 2021;11:751183. doi:10.3389/fonc.2021.751183
179. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23(2):181–192 e185. doi:10.1016/j.stem.2018.06.002
180. Albinger N, Hartmann J, Ullrich E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021;28(9):513–527. doi:10.1038/s41434-021-00246-w
181. Strassheimer F, Strecker MI, Alekseeva T, et al. OS12.6.A combination therapy of CAR-NK-cells and anti-PD-1 results in high efficacy against advanced-stage glioblastoma in a syngeneic mouse model and induces protective anti-tumor immunity in vivo. Neuro-Oncology. 2021;23(Supplement_2):ii15–ii15. doi:10.1093/neuonc/noab180.049
182. Strassheimer F, Strecker M, Alekseeva T, et al. P06.12 Combination therapy of CAR-NK-cells and anti-PD-1 antibody results in high efficacy against advanced-stage glioblastoma in a syngeneic mouse model and induces protective anti-tumor immunity in vivo. J Immunother Cancer. 2020;8(Suppl2):
183. ClinicalTrials.gov. Engineered NK cells containing deleted TGF-BetaR2 and NR3C1 for the treatment of recurrent glioblastoma; 2022. https://clinicaltrials.gov/ct2/show/NCT04991870.
184. Han J, Chu J, Keung Chan W, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. 2015;5(1):11483. doi:10.1038/srep11483
185. Shaim H, Shanley M, Basar R, et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J Clin Invest. 2021;131(14). doi:10.1172/JCI142116
186. Wang J, Toregrosa-Allen S, Elzey BD, et al. Multispecific targeting of glioblastoma with tumor microenvironment-responsive multifunctional engineered NK cells. Proc Natl Acad Sci U S A. 2021;118(45):e2107507118.
187. Genßler S, Burger MC, Zhang C, et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. OncoImmunology. 2016;5(4):e1119354. doi:10.1080/2162402X.2015.1119354
188. Giotta Lucifero A, Luzzi S. Against the resilience of high-grade gliomas: gene therapies (Part II). Brain Sci. 2021;11(8):976. doi:10.3390/brainsci11080976
189. Caffery B, Lee JS, Alexander-Bryant AA. Vectors for glioblastoma gene therapy: viral & non-viral delivery strategies. Nanomaterials. 2019;9(1). doi:10.3390/nano9010105
190. Banerjee K, Nunez FJ, Haase S, et al. Current approaches for glioma gene therapy and virotherapy. Front Mol Neurosci. 2021;14:621831. doi:10.3389/fnmol.2021.621831
191. Wu W, Klockow JL, Zhang M, et al. Glioblastoma multiforme (GBM): an overview of current therapies and mechanisms of resistance. Pharmacol Res. 2021;171:105780. doi:10.1016/j.phrs.2021.105780
192. Kim SS, Rait A, Kim E, Pirollo KF, Chang EH. A tumor-targeting p53 nanodelivery system limits chemoresistance to temozolomide prolonging survival in a mouse model of glioblastoma multiforme. Nanomedicine. 2015;11(2):301–311. doi:10.1016/j.nano.2014.09.005
193. Kumthekar P, Ko CH, Paunesku T, et al. A first-in-human phase 0 clinical study of RNA interference-based spherical nucleic acids in patients with recurrent glioblastoma. Sci Transl Med. 2021;13(584). doi:10.1126/scitranslmed.abb3945
194. DNX-2401 (formerly known as delta-24-RGD-4C) for recurrent malignant gliomas. Available from: https://clinicaltrials.gov/ct2/show/NCT00805376.
195. Lang FF, Conrad C, Gomez-Manzano C, et al. Phase I study of DNX-2401 (delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. 2018;36(14):1419–1427. doi:10.1200/JCO.2017.75.8219
196. Combination adenovirus + pembrolizumab to trigger immune virus effects. Available from: https://clinicaltrials.gov/ct2/show/NCT02798406.
197. Aiken R, Chen C, Cloughesy T, et al. Atim-33. Interim results of a phase II multi-center study of oncolytic adenovirus Dnx-2401 with pembrolizumab for recurrent glioblastoma; captive study (Keynote-192). Neuro-Oncology. 2019;21(Supplement_6):vi8–vi9. doi:10.1093/neuonc/noz175.032
198. Desjardins A, Gromeier M, Herndon JE, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018;379(2):150–161. doi:10.1056/NEJMoa1716435
199. CD147-CART cells in patients with recurrent malignant glioma. Available from: https://clinicaltrials.gov/ct2/show/NCT04045847.
200. ClinicalTrials.gov. Combination of PVSRIPO and atezolizumab for adults with recurrent malignant glioma; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT03973879.
201. A study of a retroviral replicating vector combined with a prodrug administered to patients with recurrent malignant glioma. Available from: https://clinicaltrials.gov/ct2/show/NCT01156584.
202. Study of a retroviral replicating vector to treat patients undergoing surgery for a recurrent malignant brain tumor; 2012. Available from: https://clinicaltrials.gov/ct2/show/NCT01470794.
203. Cloughesy TF, Landolfi J, Hogan DJ, et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med. 2016;8(341):341ra375–341ra375. doi:10.1126/scitranslmed.aad9784
204. Cloughesy TF, Landolfi J, Vogelbaum MA, et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro-Oncology. 2018;20(10):1383–1392. doi:10.1093/neuonc/noy075
205. The Toca 5 trial: Toca 511 & Toca FC versus standard of care in patients with recurrent high grade glioma. Available from: https://clinicaltrials.gov/ct2/show/NCT02414165.
206. Tocagen. Toca 5 phase 3 trial results presented at the society for neuro-oncology annual meeting. Available from: https://ir.tocagen.com/news-releases/news-release-details/toca-5-phase-3-trial-results-presented-society-neuro-oncology.
207. Tocagen. Tocagen reports results of Toca 5 phase 3 trial in recurrent brain cancer. Available from: https://ir.tocagen.com/news-releases/news-release-details/tocagen-reports-results-toca-5-phase-3-trial-recurrent-brain.
208. Park AK, Fong Y, Kim SI, et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci Transl Med. 2020;12(559). doi:10.1126/scitranslmed.aaz1863
209. Woroniecka K, Fecci PE. Immuno-synergy? Neoantigen vaccines and checkpoint blockade in glioblastoma. Neuro Oncol. 2020;22(9):1233–1234. doi:10.1093/neuonc/noaa170
210. Woroniecka K, Chongsathidkiet P, Rhodin K, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. 2018;24(17):4175–4186. doi:10.1158/1078-0432.CCR-17-1846
211. Nava S, Dossena M, Pogliani S, et al. An optimized method for manufacturing a clinical scale dendritic cell-based vaccine for the treatment of glioblastoma. PLoS One. 2012;7(12):e52301. doi:10.1371/journal.pone.0052301
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.