")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Ginsenoside Rd Promotes Cardiac Repair After Myocardial Infarction by Modulating Monocytes/Macrophages Subsets Conversion
Authors Zhao T, Wang X , Liu Q, Yang T, Qu H, Zhou H
Received 7 June 2022
Accepted for publication 12 August 2022
Published 22 August 2022 Volume 2022:16 Pages 2767—2782
DOI https://doi.org/10.2147/DDDT.S377624
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Tingyao Zhao,1,* Xinting Wang,2,* Qian Liu,2 Tianshu Yang,1 Huiyan Qu,1 Hua Zhou2
1Department of Cardiovascular Disease, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 2Institute of Cardiovascular Disease of Integrated Traditional Chinese and Western Medicine, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hua Zhou; Huiyan Qu, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, No. 528, Zhangheng Road, Pudong New Area, Shanghai, 201203, People’s Republic of China, Email [email protected]; [email protected]
Abstract:
Purpose: This study aimed to elucidate the potential molecular mechanisms by which GSRd improves cardiac inflammation and immune environment after MI.
Materials and Methods: The potential target genes of GSRd were predicted using the STITCH database. In vivo, MI mice models were established by left anterior descending ligation and were divided into the sham group, MI + Vehicle group, and MI + GSRd group. DMSO, DMSO, and GSRd 50 μL/day were intraperitoneally injected, respectively. After 28 days, echocardiography, Masson staining, immunofluorescence staining, flow cytometry, RT-PCR, and Western blot were performed. Mice peritoneal macrophages were extracted in vitro, and Western blot was performed after GSRd and/or Akt inhibitor MK2206 intervention.
Results: GSRd significantly improved mouse myocardial function, attenuated cardiac fibrosis, and inhibited inflammation and apoptosis in myocardial tissues after myocardial infarction. Meanwhile, GSRd increased non-classical Ly6Clow Mos/Mps while reduced of classical Ly6Chigh Mos/Mps at the same time in myocardial tissues. In addition, GSRd significantly reversed the activity of p-Akt and p-mTOR in the heart Mos/Mps after MI. In vitro studies showed that the activity of p-Akt and p-mTOR in peritoneal macrophages were significantly increased in a dose-dependent manner after GSRd treatment. Furthermore, the AKT inhibitor MK2206 was found to block the enhanced activity of p-Akt and p-mTOR induced by GSRd in peritoneal macrophages.
Conclusion: GSRd can enhance the transformation of Ly6Chigh Mos/Mps to Ly6Clow Mos/Mps in mice after MI by activating the Akt/mTOR signaling pathway, inhibiting cardiac dysfunction and promoting cardiac repair.
Keywords: myocardial infarction, ginsenoside rd, monocytes/macrophages, Akt/mTOR
Introduction
Acute myocardial infarction (MI) is one of the major causes of death in coronary artery disease, and its incidence showed an upgraded trend in recent years.1,2 In adult mammals, the limited regenerative capacity of the myocardium after myocardial infarction cannot compensate for a large number of necrotic cardiomyocytes while the lost cardiomyocytes are replaced by proliferating fibroblasts and secreting dysregulated extracellular matrix, leading to ventricular remodeling, arrhythmias, heart failure, and life-threatening events.3 Therefore, how to inhibit ventricular remodeling after myocardial infarction, reduce the area of myocardial infarction and delay the occurrence of heart failure is an effective means to improve the clinical treatment effect and the living quality of patients.
After MI, monocytes/macrophages (Mos/Mps) are recruited into the heart to play key roles as inflammatory actors and central regulators of ventricular remodeling.4,5 Two different Mos/Mps subpopulations, including Ly6Chigh and Ly6Clow, were participated in ventricular remodeling after myocardial infarction at different periods.6,7 During 1–3 days after myocardial infarction Early reactive Mos/Mps (Ly6Chigh) exert phagocytic and proinflammatory effects by massively secreting interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), myeloperoxidase, and cathepsin in myocardial tissue.8 And during 4–7 days after myocardial infarction, late responding Mos/Mps (Ly6Clow) plays an anti-inflammatory role and promotes cardiac repair by secreting repair cytokines including transforming growth factor-β1 (TGF-β1), interleukin-10 (IL-10), and vascular endothelial growth factor (VEGF).7 Furthermore, Ly6Chigh Mos can be converted to Ly6Clow Mos in circulation9,10 and inflamed tissues including infarcted hearts.8,11 Delayed conversions of Ly6Chigh to Ly6Clow Mos/Mps may attenuate wound repair after myocardial infarction, as in animals with atherosclerosis, diabetes, or myocardial infarction.7,12,13 Therefore, the enhanced transformation of Ly6Chigh Mos/Mps to Ly6Clow Mos/Mps may help to slow down ventricular remodeling after MI and delay the development of heart failure.
Traditional Chinese Medicine (TCM) has a long history and remarkable efficacy in preventing and treating MI. The pathogenesis of MI is heart-Qi deficiency and failure to stimulate blood flow, then blood stasis, resulting in myocardial ischemic necrosis caused by transient or persistent occlusion of the distal coronary artery. Ginseng is one of the most valuable natural herbal medicines in China and worldwide and is a crucial herb for treating MI.14 Ginsenosides have pharmacological and therapeutic effects on the immune,15 cardiovascular,16,17 endocrine,18 and nervous systems.19 Ginsenoside Rd (GSRd) is identified as one of the most important active ingredients in Ginseng and has anti-inflammatory effects on natural immune components.20 GSRd has a wide range of applications in ischemic disease research. GSRd activated the Akt/GSK-3beta signaling pathway, increased the Bcl-2/Bax ratio, inhibited the mitochondria-dependent apoptotic pathway to attenuate myocardial ischemia-reperfusion injury in rats, and reduced infarct size.21 GSRd enhances antioxidant enzyme activity, stabilizes mitochondrial membrane potential, increases intracellular ATP levels, and activates the PI3K/Akt signaling pathway to promote neuronal survival, and ameliorates ischemic brain injury.22 GSRd protects against renal ischemia/reperfusion injury by inhibiting M1 macrophage polarization23 and attenuates experimental autoimmune neuritis in mice by regulating monocyte subset conversion.24 However, the role of GSRd in Mos/Mop phenotypic transformation after myocardial infarction and its molecular mechanism remain unclear.
In this study, we established the mouse model of myocardial infarction to evaluate the therapeutic effect of GSRd on fibrosis and cardiac function after acute myocardial infarction and explore its potential mechanism, providing a new choice and theoretical basis for the prevention and treatment of ventricular remodeling after myocardial infarction.
Materials and Methods
Reagents
Ginsenoside Rd (Selleck, Houston, USA) standards (purity > 95%) were dissolved in dimethyl sulfoxide (DMSO) to obtain a stock solution for further investigation.
Animals
Eight-week-old male C57BL/6J mice (22–25 g) were purchased from SLAC Laboratory Animal Co., Ltd. (Shanghai, China) and housed in an environment with controlled temperature (22°C ± 1°C) and relative humidity (50% ± 5%) on a 12-h:12-h light/dark cycle, with free access to sterile food and water. Mice were randomly divided into three groups: (a) sham group, (b) MI + Vehicle group, and (c) MI + GSRd group. Mice in the sham and MI groups were intraperitoneally injected with vehicle DMSO, 50 μL/day. Mice in the GSRd group received an intraperitoneal injection of GSRd, 50 μL/day (GSRd dissolved in DMSO, 50 μg/kg/day). All procedures in this study were approved by the Experimental Animal Welfare and Ethics Committee of Shanghai University of Traditional Chinese Medicine (NO. PZSHUTCM210226013). All animal experiments were conducted following the animal experimental guidelines set by the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
MI Mouse Model
Male mice aged 6–8 weeks underwent permanent ligation of the left anterior descending (LAD) branch coronary artery.25 Briefly, Mice were anesthetized with 1–1.5% isoflurane gas and mechanically ventilated with rodent respirators. Left thoracotomy was performed to open the chest cavity and expose the heart so that the LAD could be seen and permanently ligated with 6–0 silk thread outside the left atrium. The presence of myocardial branches in the perfusion bed confirmed complete vascular occlusion. Mice that died within 24 hours of surgery were excluded from the experiment. The sham animals underwent the same procedure, but without coronary ligation.
Echocardiography
Transthoracic echocardiography was performed to assess mouse cardiac function after surgery using an echocardiograph (Vevo2100, Visual Sonics, USA) with M-mode analysis. All mice (8–10 weeks old) were anesthetized by isoflurane inhalation (1%–2%). As previously described,26 the mitral valve lobe was visualized in a parasternal long-axis view to assess cardiac function.
Immunofluorescence
Cryosections of heart (7μm) were prepared and stained with CD68 monoclonal antibody (FA-11) (1:200, eBioscience, USA) overnight at 4 °C. Follow, the sections were incubated with Alexa Fluor 488-conjected secondary antibodies for 1 h in the dark. Nuclei were stained with DAPI (1:2000, Thermo Fisher, USA). All slides were mounted with ProLongTM Gold (Thermo Fisher, USA). Confocal microscope from Zeiss were used to take image. Image J software were applied to analysis the obtained images.
Quantitative Reverse Transcription Polymerase Chain Reaction (RT-PCR) Analysis
RNA was isolated from cells using TRIzol (Invitrogen, USA) and 1μg of RNA was used as a template for cDNA synthesis using reverse transcription kits (Takara, Dalian, China), then cDNA was subjected to real-time PCR analysis using TB green mixture (Takara, Dalian, China) on an ABI 7900 Q-PCR system from Life Technologies (Carlsbad, CA, USA) according to the manufacturer’s protocol. All mRNA expression levels were normalized by comparing them to the housekeeping gene GAPDH. Sequences of primers for real-time PCR analysis of mice samples were verified against the National Center for Biotechnology Information database to confirm specificity and are listed in Table 1.
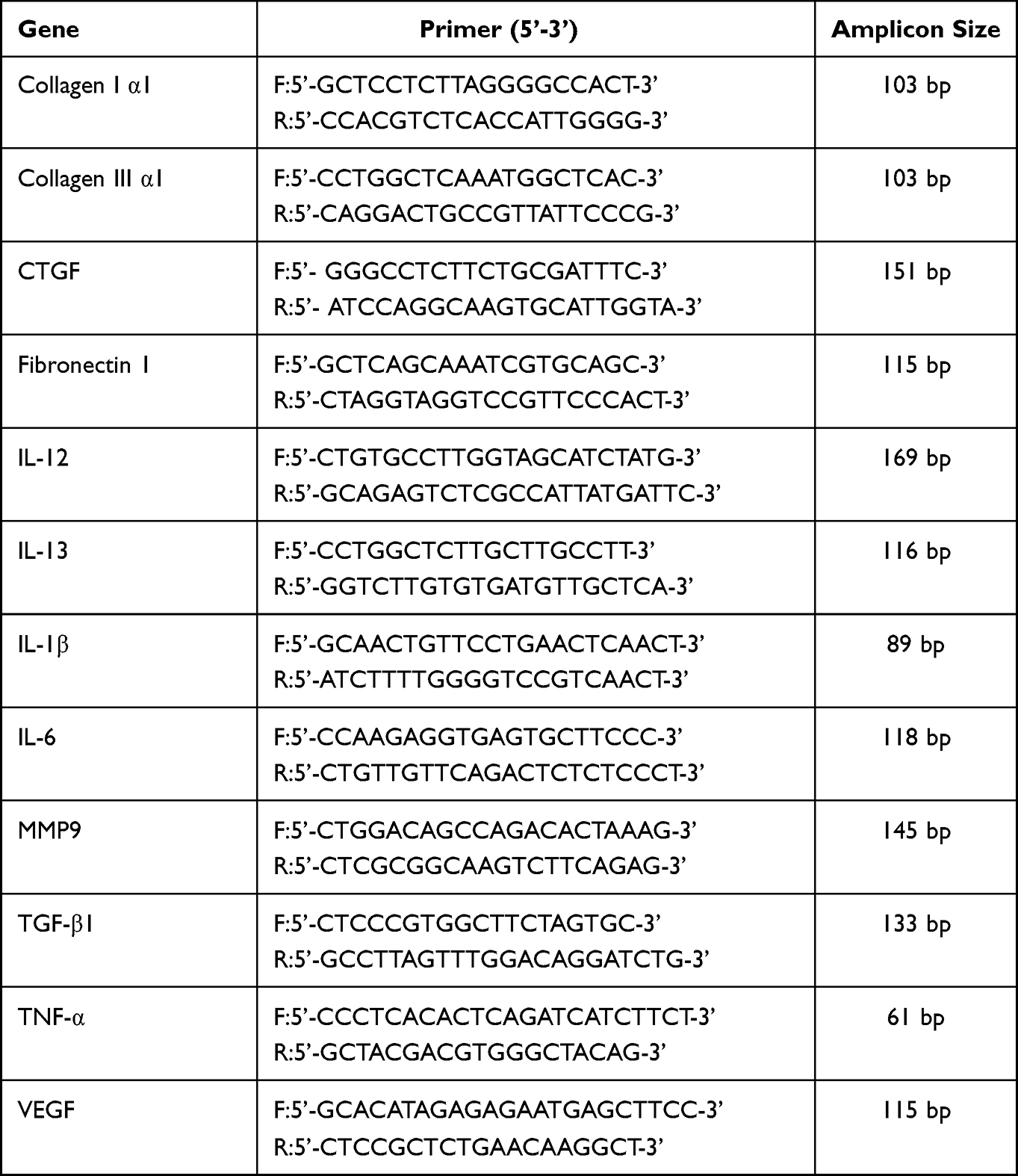 |
Table 1 Sequence for RT-PCR Primer |
Heart Masson’s Trichrome Staining
Heart samples were harvested and fixed in 10% phosphate-buffered formalin for 24 h, subsequently embedded in paraffin and cut into 5 μm slices. Histologic sections of tissues were stained with Masson’s trichrome (Solaribio, Beijing, China). The mean cross-sectional area and fibrosis of the left ventricular apex up to the aortic valve leaflet attachment point were assessed by Image-Pro Plus software.
Cell Extract and Culture
Peritoneal macrophages were obtained from abdominal cavity and cultured in medium 1640 containing 10% fetal bovine serum. Single-cell suspensions of infarcted hearts were prepared. Briefly, the heart was dissected, minced with delicate scissors, and enzymatically digested with a digested with a collagenase mix (including 125 U/ml collagenase IX, 450 U/ml collagenase I, 60 U/ml DNase I, and 60/U ml hyaluronidase I-S (Sigma -Aldrich, St Louis, MO, USA) for 1 h at 37°C with gentle agitation. After digestion, the tissues were triturated, passed through 70-mm cell strainers, washed, lysed red blood cells, and stained with antibodies for fluorescence activated cell sorting (FACS) analysis. For monocyte/macrophage sorting, cells were then stained with APC-Cy7-CD11b (M1/70, 1:50, BD, USA), - CD115 (CSF-1R, 1:50, BioLegend, USA), FITC-Ly6C (HK1.4, 1:50, BioLegend), and Brilliant Violet 421-Ly6G (1A8, 1:50, BD, USA). BD FACS Aria flow cytometry system (BD Biosciences, San Jose, CA, USA) was used to perform flow cytometry, and FlowJo software (Tree Star, Ashland, OR, USA) were used to analysis the flow cytometry data. For sorted monocyte/macrophages, RT-PCR was performed.
Flow Cytometry
Cells were collected from mice and stained with fluorescent-labeled antibodies according to the manufacturer’s protocol. All fluorochrome-conjugated antibodies as follow: APC-Cy7-CD11b (M1/70, 1:50, BD, USA), - CD115 (CSF-1R, 1:50, BioLegend, USA), FITC-Ly6C (HK1.4, 1:50, BioLegend), and Brilliant Violet 421-Ly6G (1A8, 1:50, BD, USA). BD FACS Aria flow cytometry system (BD Biosciences, San Jose, CA, USA) was used to perform flow cytometry, and FlowJo software (Tree Star, Ashland, OR, USA) were used to analysis the flow cytometry data.
Western Blot
Heart tissues and cells were extracted in lysis buffer containing protease inhibitors. Pierce BCA Protein Assay Kit (Pierce, Rockford, IL, USA) was used to assess protein concentration. Equal quantities of protein were denatured, dissolved in 10% SDS-PAGE gel, transferred to a nitrocellulose membrane, incubated with 5% skim milk for 1–1.5 h, and then incubated with primary antibody at 4° C overnight. Primary antibodies were diluted as follows: chemoattractant protein-1 (Mcp-1) (1:1000, Proteintech, USA), C-C motif Chemokine receptor 2 (CCR2) (1:500, Abcam, UK), p-AKT (1:1000, CST, USA), AKT (1:1000, CST, USA), p-mTOR (1:1000, CST, USA), mTOR (1:1000, CST, USA), Bax (1:1000, ABclonal, USA), Bcl-1 (1:1000, ABclonal, USA), and GAPDH (1:2000, CST, USA), used as the load control. The membranes were then incubated at room temperature in a blocking buffer containing an HRP-labeled secondary antibody for 2 h at room temperature. The enhanced chemiluminescence reagent (Thermo Fisher Scientific, USA) were used to develop western blots.
Statistics Analysis
Data analysis were achieved by the GraphPad Prism version 5.0 (GraphPad Software, Inc.). Results are shown as mean ± standard error of the mean (SEM). Comparison between groups was performed using two-tailed Student’s t-test and one- or two-way analysis of variance with Bonferroni post-hoc analyses. p < 0.05 was considered statistically significant. According to the literature,27 sufficient statistical sample size was designed in the experiment. Randomization and blinding strategies were used whenever possible.
Results
GSRd Improves Mouse Cardiac Function After Myocardial Infarction
To explore the effect of GSRd (Figure 1A) on cardiac contractile function in vivo, WT mice were injected with vehicle DMSO or GSRd (50 μg/kg body weight) and underwent sham or LAD artery ligation surgery for 4 weeks (Figure 1B). The survival of mice was observed after 4 weeks (Figure 1C). No mice in the sham group died, 55% of mice in the MI + vehicle group died, and 30% of MI mice treated with GSRd died. Survival was significantly decreased in the MI + vehicle group compared with the sham group; GSRd significantly improved survival compared with the MI + vehicle group. GSRd intervention significantly improved the survival of MI mice.
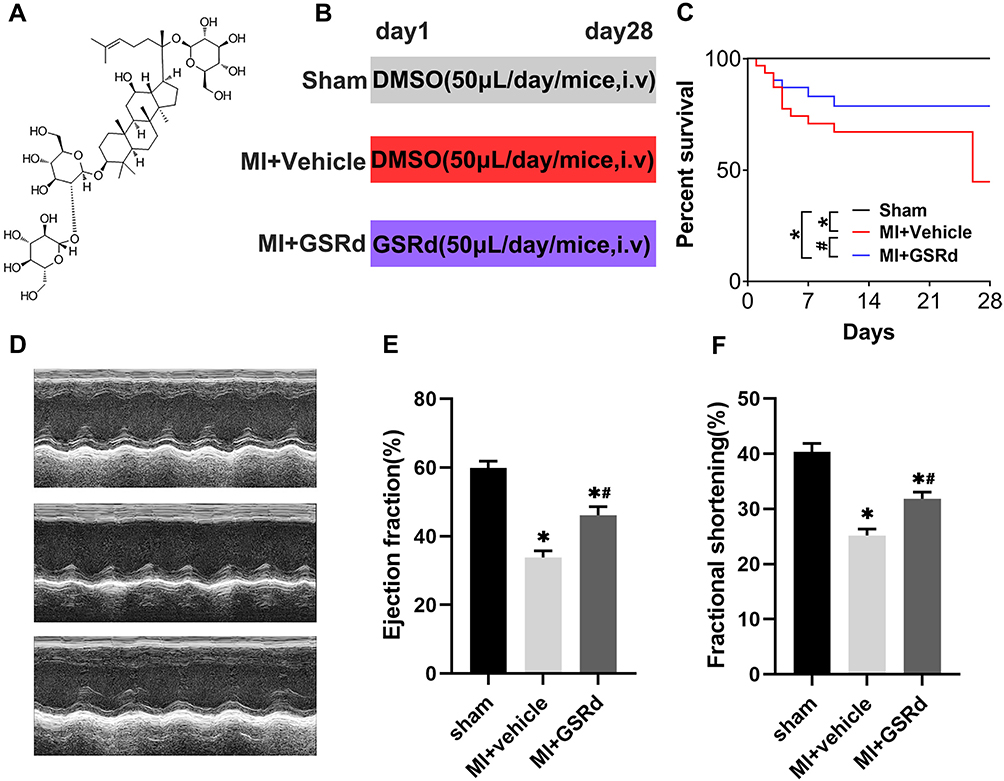 |
Figure 1 Effects of GSRd on cardiac dysfunction following MI. (A) Structure formula information of GSRd. n = 20 per group. (B) Experimental protocols for MI and GSRd treatment. (C) Changes in the survival rates of mice following MI (n = 20). (D) Effects of GSRd on echocardiographic images. (E) cardiac ejection fraction (n = 6), (F) fraction shortening (n = 6). *P < 0.05 versus sham group; #P < 0.05 versus MI + vehicle group. |
Furthermore, a high-resolution echocardiography imaging system with M-model analysis revealed that ejection fraction and fractional shortening were significantly decreased in MI + vehicle group mice compared with the sham group, and GSRd treated mice exhibited a significant increase in ejection fraction and fractional shortening after MI compared with the MI + vehicle group (Figure 1D-F). These results indicated that the MI model mice modeling was successful, and GSRd could improve the cardiac function of mice after MI.
GSRd Attenuates Cardiac Fibrosis After Myocardial Infarction
To explore whether GSRd affects cardiac fibrosis following MI, Masson’s trichrome staining (Figure 2A) was performed. As shown in Figure 2B and C, the infarct zone and border zone fibrosis area of MI + vehicle group mice increased significantly compared with the sham group. Compared with untreated MI mice, GSRd-treated MI mice significantly reduced the infarct zone and border zone fibrosis area. The mRNA levels of myocardial fibrosis markers (connective tissue growth factor (CTGF), collagen Iα1, collagen IIIα1, and fibronectin (Fn)) in the heart tissues with the infarct zone were measured in each group, as shown in Figure 2D. Compared with the sham group, the cardiac infarct zone mRNA levels of CTGF, collagen Iα1, collagen IIIα1, and Fn were significantly increased in MI + vehicle group mice. GSRd-treated MI mice significantly reduced cardiac infarct zone CTGF, collagen Iα1, collagen IIIα1, and Fn mRNA levels compared with untreated MI mice. These data suggest that GSRd therapy inhibits myocardial fibrosis after MI and alleviates cardiac remodeling.
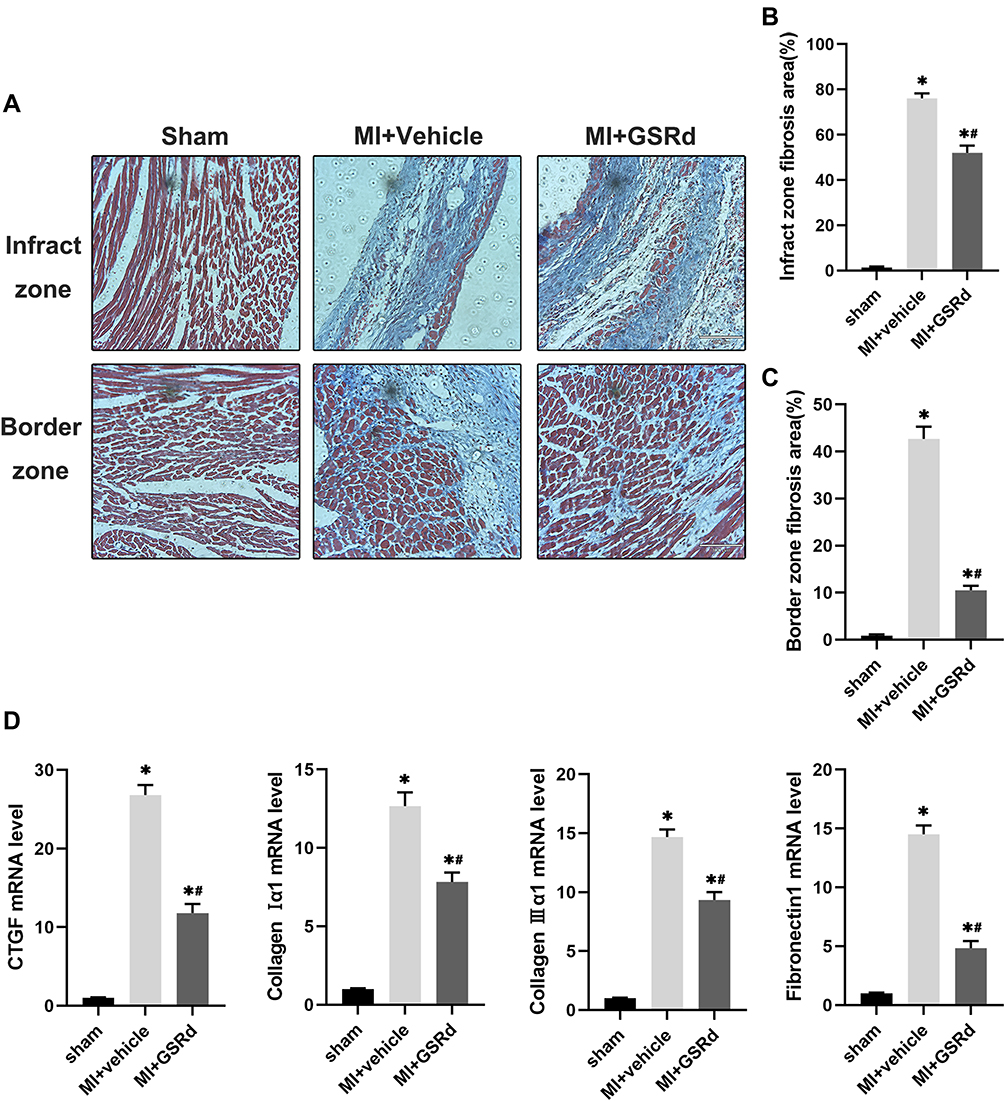 |
Figure 2 Effects of GSRd on cardiac fibrosis following MI. (A) Masson’s trichrome staining of histological sections at 4 weeks after MI. Scale bar = 100μm. (B) Experimental protocols for MI and GSRd treatment. (C) Percent survival. (D) Reverse transcription‑quantitative polymerase chain reaction analyses of CTGF, Collagen I α1, Collagen III α1 and fibronectin 1 (n=6). *P < 0.05 versus sham group; #P < 0.05 versus MI + vehicle group. |
GSRd Inhibits Inflammation and Apoptosis in Myocardial Tissues
As shown in Figure 3A and B, macrophages in the myocardial tissue were marked by CD68 immunohistochemical staining. Compared with the sham group, the cardiac macrophage infiltration in the MI + vehicle group was significantly increased. GSRd significantly reduced cardiac macrophage infiltration after MI compared with the MI + vehicle group.
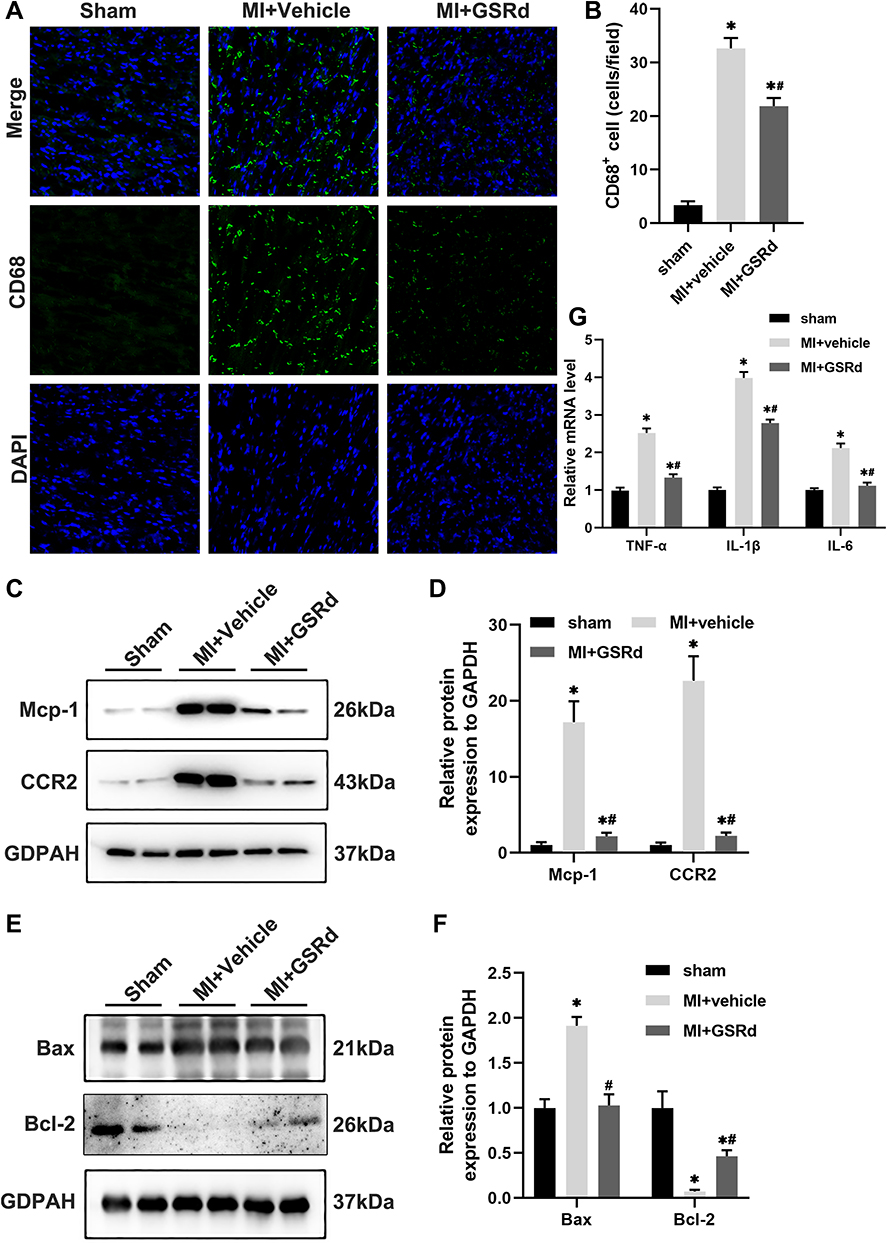 |
Figure 3 Effects of GSRd on cardiac macrophage infiltration and inflammation tissues following MI. (A) Representative immunofluorescent images of CD68 in the heart tissues. Scale bar, 50μm. (B) Quantitation of CD68+ area per field in (A), n=3-5 per group. (C) Mcp-1 and CCR2 protein expression in heart tissue of each group mice detected by Western blot. (D) Mcp-1 and CCR2 relative protein expression to GAPDH. (E) Bax and Bcl-2 protein expression in heart tissue of each group mice detected by Western blot. (F) Bax and Bcl-2 relative protein expression to GAPDH. (G) Reverse transcription‑quantitative polymerase chain reaction analyses of TNFα, IL-1β and IL-6 (n=6) in heart tissue of each group mice. *P < 0.05 versus sham group; #P < 0.05 versus MI + vehicle group. |
Pro-inflammatory Ly6Chigh monocytes rely on Mcp-1/CCR2 to reach the inflammatory site of MI. As shown in Figure 3C and D, Western blot analysis was used to detect the effect of GSRd on Mcp-1/CCR2 protein expression in MI heart tissue. Compared with the sham group, Mcp-1/CCR2 protein expression was significantly up-regulated in the MI + vehicle group. Mcp-1/CCR2 protein expression was significantly down-regulated after GSRd treatment compared with the MI + vehicle group.
As shown in Figure 3E and F, Western blot analysis was used to detect the effect of GSRd on apoptosis-related protein Bcl-2/Bax of MI mice cardiomyocytes. Bcl-2 was an anti-apoptotic protein, and Bax was a pro-apoptotic protein. Compared with the sham group, Bcl-2 protein expression was significantly down-regulated, and Bax protein expression was significantly up-regulated in the MI + vehicle group. Compared with the MI + vehicle group, Bcl-2 protein expression was significantly up-regulated, and Bax protein expression was significantly down-regulated after GSRd treatment.
As shown in Figure 3G, mRNA expression levels of pro-inflammatory mediators TNF-α, IL-1β, and IL-6 in MI heart tissue were detected by RT-PCR. Compared with the sham group, the mRNA expression levels of pro-inflammatory mediators TNF-α, IL-1β, and IL-6 in the MI + vehicle group were significantly increased. Compared with the MI + vehicle group, the mRNA expression levels of TNF-α, IL-1β and IL-6 decreased considerably after GSRd treatment.
These results indicate that GSRd inhibits inflammation and apoptosis in the myocardial tissues.
GSRd Modulating Mos/Mps Subsets Conversion Following MI
Circulating Mos/Mps have the important feature of being recruited to inflammatory sites, and subsets of different phenotypes show different functions. For instance, macrophages differentiated from monocytes can replenish the tissue macrophage pool and modulate the local inflammatory environment resulting in cardiac injury. In order to study the effect of GSRd on the in-situ transformation of Ly6Chigh - Ly6Clow Mos/Mps, the mouse heart tissue was collected immediately for flow cytometry analysis and quantitative RT-PCR analysis after sacrifice.
As shown in Figure 4A-D, the total Mos/Mps and Ly6Chigh Mos/Mps cardiac infiltration significantly increased in the MI + vehicle group compared with the sham group. In contrast, the Ly6Clow Mos/Mps cardiac infiltration significantly decreased. Compared with the MI + vehicle group, GSRd significantly reduced the total Mos/Mps and Ly6Chigh Mos/Mps cardiac infiltration after MI and significantly increased the cardiac infiltration of Ly6Clow Mos/Mps.
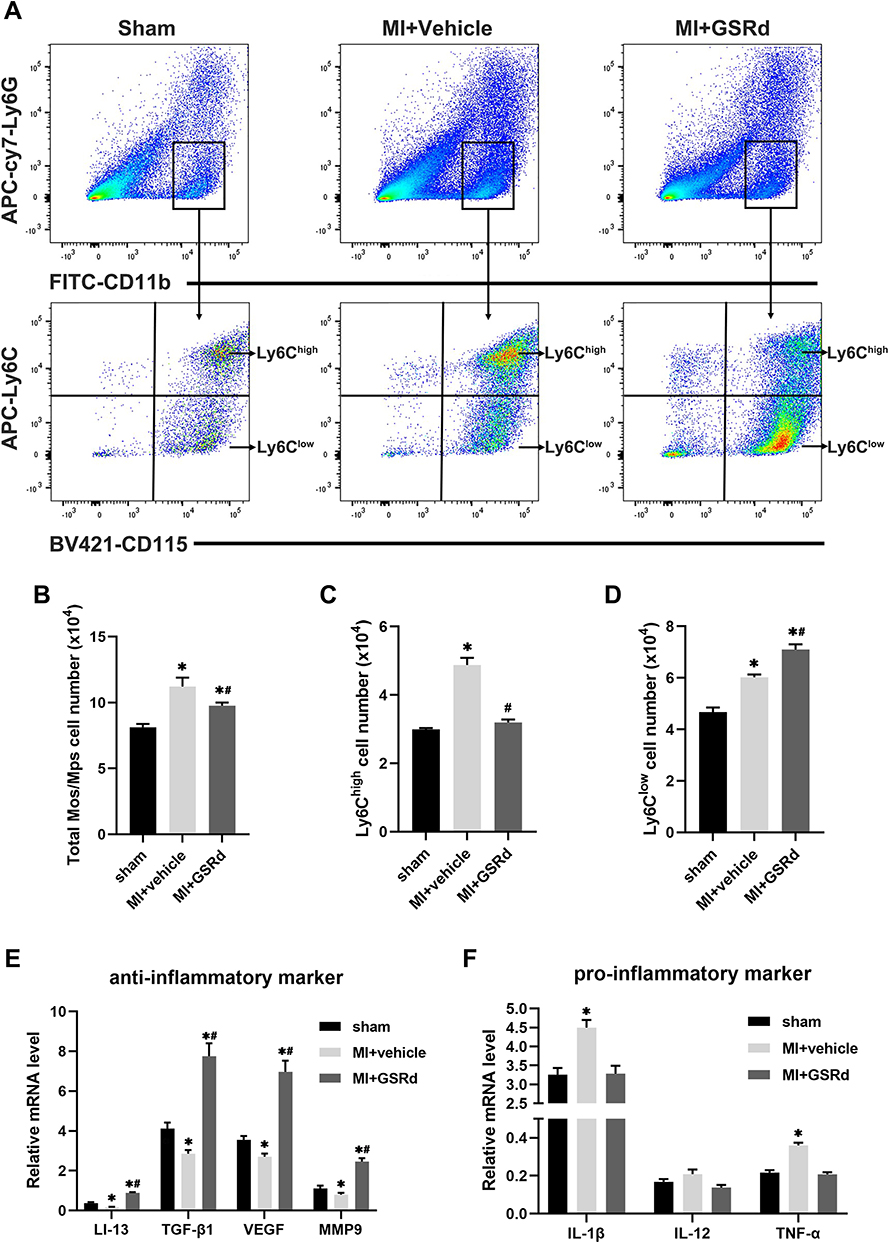 |
Figure 4 Effects of GSRd on modulating Mos/Mps subsets conversion following MI. (A) Gating strategy for CD11b+CD115+Ly6G- Ly6Chigh and CD11b+CD115+Ly6G-Ly6Clow Mos/Mps in hearts of each group mice. (B-D) Total Mos/Mps (B), Ly6Chigh (C), and Ly6Clow (D) Mos/Mps in the hearts of each group of mice. (E and F) Reverse transcription‑quantitative polymerase chain reaction analyses of anti-inflammatory markers (E) and pro-inflammatory markers (F) in the infiltrating Mos/Mps (n=6). *P < 0.05 versus sham group; #P < 0.05 versus MI + vehicle group. |
As shown in Figure 4E and F, the mRNA expressions of repairing and pro-angiogenic related cytokines (IL-13, TGFβ1, VEGF, and MMP9) and pro-inflammatory related cytokines (IL-1β, IL-12, and TNFα) in the infiltrating Mos/Mps were detected by RT-PCR. Compared with the sham group, the mRNA expressions of repair and pro-angiogenic related cytokines (IL-13, TGFβ1, VEGF, and MMP9) were significantly down-regulated in the MI + vehicle group, and the mRNA expressions of pro-inflammatory related cytokines (IL-1β and TNFα) were significantly up-regulated. Compared with the MI + vehicle group, the mRNA expressions of repairable and pro-angiogenic related cytokines (IL-13, TGFβ1, VEGF, and MMP9) were significantly up-regulated after GSRd treatment, while the mRNA expressions of pro-inflammatory related cytokines were not significantly affected.
Taken together, these results suggest that GSRd promotes the recruitment of Ly6Clow Mos/Mps in the heart following MI.
GSRd Modulating Mos/Mps Subsets Conversion via Akt/mTOR Activity After MI
The potential target genes of GSRd were predicted using STITCH (http://stitch.embl.de/),28 which identified AKT1, CYCS, and TRPM7 directly interacting with GSRd (Figure 5A). It is noteworthy that mTOR, as a downstream molecule of PI3K/Akt activated by Toll-like receptors, has been previously reported to be critical in innate immune coordination of macrophages and monocytes.29,30
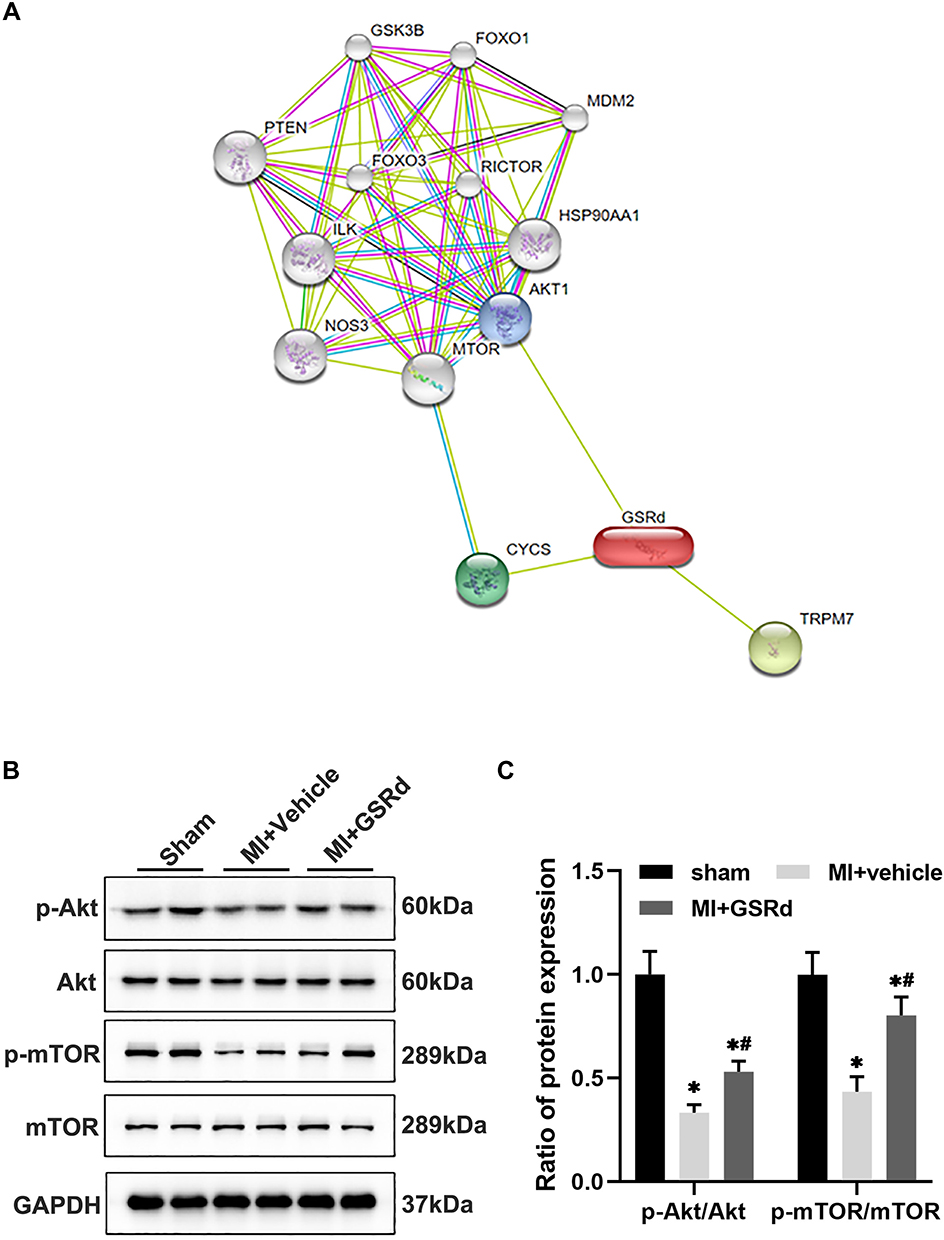 |
Figure 5 GSRd modulates Mos/Mps subsets conversion following MI via AKT/mTOR pathway. (A) The potential targets gene of GSRd were predicted by the STITCH. (B) total-Akt, p-Akt, total-mTOR, and p-mTOR protein expression in Mos/Mps collected from heart tissue of each group mice detected by Western blot. (C) total-Akt, p-Akt, total-mTOR, and p-mTOR protein expression in Mos/Mps relative protein expression to GAPDH. *P < 0.05 versus sham group; #P < 0.05 versus MI + vehicle group. |
As shown in Figure 5B and C, we examined the Akt/mTOR pathway activity in Mos/Mps in the heart of each group via western blot analysis. Compared with the sham group, the p-Akt, and p-mTOR activity were significantly decreased in the MI + vehicle group. Compared with the MI + vehicle group, p-Akt, and p-mTOR activity increased after GSRd treatment.
Gsrd regulates cardiac Mos/Mps subsets conversion after MI, and the possible mechanism is to activate Akt/mTOR pathway activity.
GSRd Regulates Akt/mTOR Activity of Peritoneal Macrophages
To further elucidate the effect of GSRd on the Akt/mTOR pathway activity in macrophages, mice peritoneal macrophages were extracted and treated with AKT activation inhibitor MK2206.
As shown in Figure 6A and B, compared with the blank control group, GSRd at 20μM and 50μM concentrations significantly increased the Akt/mTOR pathway activity in peritoneal macrophages, especially at 50μM concentrations. This dose-effect experiment suggested that the activity of the Akt/mTOR pathway in peritoneal macrophages depended on the concentration of GSRd.
 |
Figure 6 GSRd regulates Akt/mTOR activity of peritoneal macrophages. (A) total-Akt, p-Akt, total-mTOR and p-mTOR protein expression in peritoneal macrophage with GSRd treated detected by Western blot. (B) total-Akt, p-Akt, total-mTOR and p-mTOR protein expression in peritoneal macrophage relative protein expression to GAPDH. (C) total-Akt, p-Akt, total-mTOR and p-mTOR protein expression in peritoneal macrophage with MK2206 and/or GSRd treated detected by Western blot. Ginsenoside Rd; MI, myocardial infarction; MK2206, an AKT inhibitor. (D) total-Akt, p-Akt, total-mTOR and p-mTOR protein expression in peritoneal macrophage with MK2206 and/or GSRd relative protein expression to GAPDH. *P < 0.05 versus sham group; #P < 0.05 versus MI + vehicle group. |
As shown in Figure 6C and D, the Akt activation inhibitor MK2206 was used to further verify the Akt/mTOR pathway activity of GSRd peritoneal macrophages. Compared with the blank control group, GSRd(50μM) significantly increased the activity of the Akt/mTOR pathway, MK2206(5.5nM) significantly decreased the Akt/mTOR pathway activity, GSRd(50μM) combined with MK2206(5.5nM) significantly decreased the activity of Akt/mTOR pathway in peritoneal macrophages.
The Akt/mTOR pathway activity was increased in peritoneal macrophages dependent on GSRd concentration, while the Akt/mTOR pathway activity was inhibited by MK2206 (AKT inhibitor) treatment. Thus, the evidence supports the contention that GSRd converts the Mos/Mps subsets phenotype via Akt/mTOR activity following MI.
Discussion
This study found that GSRd treatment improved mouse myocardial function, attenuated cardiac fibrosis, and inhibited inflammation and apoptosis in myocardial tissues after MI. After the treatment of GSRd, increased non-classical Ly6Clow Mos/Mps and reduced classical Ly6Chigh Mos/Mps in myocardial tissue after MI, and the phenotype of tissue Mos/Mps was transformed into a regression phase. Furthermore, in vitro studies confirmed that GSRd has a similar regulatory effect on monocyte/macrophage differentiation, which may be related to the AKT/mTOR pathway. As shown in Figure 7A, pathological manifestations of MI; As shown in Figure 7B, the pharmacological mechanism of GSRd action on macrophages is related to Akt/mTOR pathway.
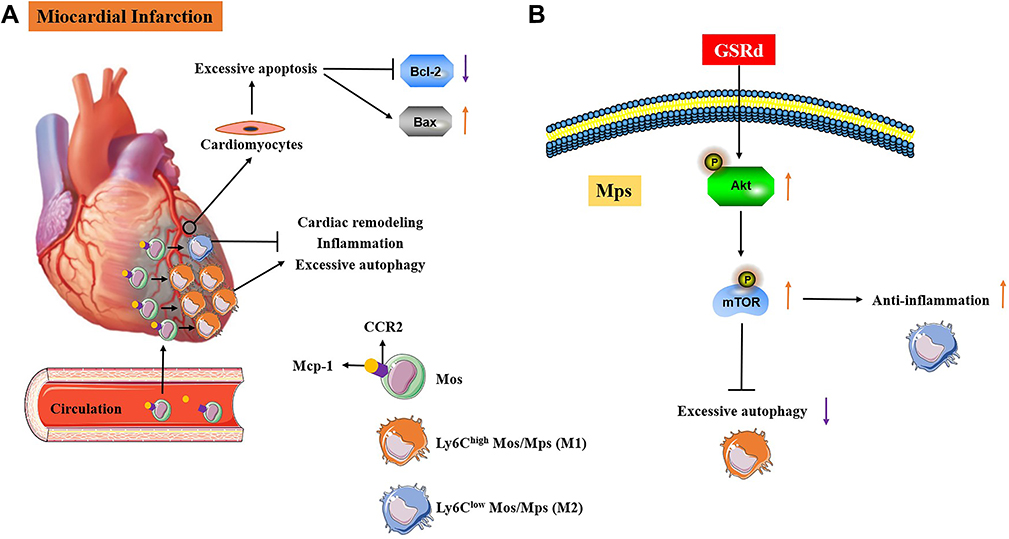 |
Figure 7 MI pathological manifestations and possible intervention mechanisms of GSRd. (A) Pathological manifestations of MI. (B) The pharmacological mechanism of GSRd action on macrophages is related to Akt/mTOR pathway. |
Ginsenoside is the most bioactive pharmacological compound extracted from ginseng rhizomes and is used to treat several clinical diseases.23 It is worth noting that the anti-oxidative stress, anti-apoptotic and anti-inflammatory effects of GSRd have attracted the attention of many researchers in recent years.20,24,31 In particular, recent studies have shown that GSRd can mitigate experimental autoimmune nerve injury by regulating monocyte conversion.24 GSRd has been widely used in the study of ischemic diseases and inflammatory diseases. MI is a typical ischemic disease and aseptic inflammatory disease. In this study, GSRd was selected as the research object to explore the possible mechanism of its treatment of MI.
Although the application of percutaneous coronary intervention has significantly reduced the mortality rate of AMI,32 malignant events caused by ventricular remodeling after myocardial infarction remain prominent. Many factors are involved in ventricular remodeling, including cardiomyocyte hypertrophy, apoptosis, and myocardial fibrosis.33 In particular, the myocardial fibrosis caused by excessive deposition of ECM protein in the myocardium distorts the myocardial structure and leads to the development of arrhythmia and cardiac dysfunction, thus affecting the clinical course and outcome of patients with heart failure.34 Therefore, inhibition of myocardial fibrosis can be an effective means for the prevention and treatment of myocardial infarction. In this study, GSRd significantly inhibited MI myocardial fibrosis and its related markers (CTGF, Collagen I α1, Collagen III α1, Fn) mRNA expression. CTGF is one of the extracellular matrix proteins secreted by the CCN family, also known as CCN2. CTGF expression is elevated in human fibrotic diseases.35 Treatment of MI mice with CTGF monoclonal antibody can improve survival rate, preserve left ventricular systolic function, reduce inflammation, inhibit myocardial fibrosis, and promote cardiac repair.36 Collagen is the main component of the extracellular matrix, of which Collagen I and III account for 85% and 11% respectively.37 Fn polymerization is a necessary condition for collagen matrix deposition. From 12 h to 14 days after MI, Fn was significantly increased in the infarct area and marginal area of MI patients.38 Inhibition of Fn expression in ischemia-reperfusion model mice significantly reduced cardiac injury fibroblast markers and neutrophil infiltration after 1 week. Cardiac dysfunction, pathological cardiac remodeling, and myocardial fibrosis were significantly improved after 4 weeks.39
MI is accompanied by excessive apoptosis of cardiomyocytes, which plays a role in regulating cell mass and structure in many tissues.40 The Bcl-2 proto-oncogene family is essential for regulating apoptosis, and Bcl-2 exerts anti-apoptotic effects by binding to the Bax domain, which has pro-apoptotic effects and inhibiting Bax activation.41 Clinical studies have shown overexpression of Bax in the infarcted area of MI patients and accelerated cardiomyocyte apoptosis; In the acute phase of MI, Bcl-2 is induced to express processes involved in anti-cardiomyocyte apoptosis.42 The relative abundance of the pro-apoptotic protein Bax and the anti-apoptotic protein Bcl-2 determines the susceptibility of cells to death.43 This study showed that GSRd ameliorated the excessive apoptosis of cardiomyocytes in MI by significantly upregulating anti-apoptotic Bcl-2 protein expression and downregulating pro-apoptotic Bax protein expression.
Mcp-1, also known as Chemokine ligand 2 (CCL2), regulates monocyte recruitment in inflammatory diseases. Inflammatory Ly6Chigh monocytes rely on Mcp-1/CCR2 to reach the inflammatory zone.44 Mcp-1 or CCR2 gene deletion can reduce the inflammatory response in MI models.44,45 Targeted silencing of CCR2 increased cardiac ejection fraction from 29% to 35% in mice 21 days after MI, reduced recruitment of Ly6Chigh monocytes to injured myocardium, alleviated inflammatory response after MI, and inhibited left ventricular remodeling.46 Clinical studies have shown that intensive atorvastatin therapy down-regulates the expression of Mcp-1/CCR2 in patients with unstable angina pectoris and alleviates the inflammatory response of CD14+ monocytes recruitment.47 In this study, GSRd significantly reduced cardiac Mcp-1/CCR2 protein expression and infiltration of cardio-inflammatory Ly6Chigh Mos/Mps in MI mice.
Monocyte and macrophage subsets are critical for cardiac fibrosis48 and MI recovery.49 Previous studies have shown that Mos/Mps could be divided into two subsets (pro-inflammatory Ly6Chigh and less inflammatory and patrolling Ly6Clow) according to Ly6C expression level in mice.7 Meanwhile, CD14+ CD16− Mos resembles mouse Ly6Chigh cells, while CD14Dim Mos resembles mouse Ly6Clow Mos in humans.50 In myocardial infarction model mice, two types of monocytes/macrophages play an important role in cardiac healing. In the early stage (1–3 days after MI), Ly6Chigh (CD16-negative) pro-inflammatory monocytes lead to digestion of damaged tissue through phagocytosis and proteolysis, and in the late stage (3–5 days after MI), Ly6Clow (CD16-positive) anti-inflammatory monocytes promote healing through myofibroblast aggregation, angiogenesis, and collagen deposition.7 Consequently, regulating the monocyte /macrophage transformation from Ly6Chigh to Ly6Clow to inhibit myocardial fibrosis is beneficial to the repair of the heart after myocardial infarction. In this study, after the treatment of GSRd, increased non-classical Ly6Clow Mos/Mps and reduced classical Ly6Chigh Mos/Mps in myocardial tissue after MI, and the phenotype of tissue Mos/Mps was transformed into a regression phase.
Furthermore, in vivo and in vitro studies have confirmed that GSRd improves the cardiac immune environment after MI by enhancing cardiac Ly6Clow Mos/Mps recruitment. The possible mechanism may be related to the activation of the Akt/mTOR pathway to regulate the transformation of macrophages from excessive autophagy M1 type to anti-inflammatory protective M2 type polarization, namely, Ly6Chigh to Ly6Clow. The Akt/mTOR pathway is a major signal transduction pathway that regulates cell proliferation, survival, autophagy, and apoptosis. mTOR, a downstream molecule of Akt, is a crucial regulator of cellular protein synthesis, growth, expansion, autophagy, lysosomal function, and cellular metabolism.51 Myocardial cell death after MI largely depends on inhibiting the Akt/mTOR signaling pathway.52 Akt1−/− macrophages reduce mTOR activity and induce M1-type polarization.53 This study confirmed that GSRd regulates the Akt/mTOR pathway activity in peritoneal macrophages.
Conclusions
GSRd improves cardiac function, inhibits cardiac fibrosis and cardiomyocyte apoptosis in MI mice, suppresses cardiac inflammation by regulating the polarization of Mos/Mps subpopulations, enhances cardiac Ly6Clow Mos/Mps recruitment to alleviate the immune environment in post-MI hearts, and promotes cardiac repair. The possible mechanism is related to activating the Akt/mTOR pathway to regulate the macrophage polarization from excessive autophagic M1 type to anti-inflammatory protective M2 type, that is, the conversion of Ly6Chigh to Ly6Clow.
Acknowledgments
The work was supported by the Three-year Action Plan of Shanghai Shenkang Medical Development Center (Grant/Award Number: SHDC2020CR1053B); the National Natural Science Foundation of China (Grant/Award Number: 81973656, 81803881). Tingyao Zhao and Xinting Wang are the co-first authors. Huiyan Qu and Hua Zhou are the co-corresponding authors.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Roth GA, Johnson C, Abajobir A, et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1–25.
2. Benjamin EJ, Muntner P, Alonso A, et al. Heart Disease and Stroke Statistics-2019 Update: a Report From the American Heart Association. Circulation. 2019;139(10):e56–e528.
3. Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: from Inflammation to Fibrosis. Circ Res. 2016;119(1):91–112.
4. Hulsmans M, Sam F, Nahrendorf M. Monocyte and macrophage contributions to cardiac remodeling. J Mol Cell Cardiol. 2016;93:149–155.
5. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res. 2020;116(6):1101–1112.
6. Lee WW, Marinelli B, van der Laan AM, et al. PET/MRI of inflammation in myocardial infarction. J Am Coll Cardiol. 2012;59(2):153–163.
7. Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204(12):3037–3047.
8. Hilgendorf I, Gerhardt LM, Tan TC, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114(10):1611–1622.
9. Sunderkotter C, Nikolic T, Dillon MJ, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172(7):4410–4417.
10. Yona S, Kim KW, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91.
11. Varga T, Mounier R, Gogolak P, Poliska S, Chazaud B, Nagy L. Tissue LyC6- macrophages are generated in the absence of circulating LyC6- monocytes and Nur77 in a model of muscle regeneration. J Immunol. 2013;191(11):5695–5701.
12. Singla DK, Singla RD, Abdelli LS, Glass C. Fibroblast growth factor-9 enhances M2 macrophage differentiation and attenuates adverse cardiac remodeling in the infarcted diabetic heart. PLoS One. 2015;10(3):e120739.
13. Tang J, Shen Y, Chen G, et al. Activation of E-prostanoid 3 receptor in macrophages facilitates cardiac healing after myocardial infarction. Nat Commun. 2017;8:14656.
14. Ma J, Yin C, Ma S, et al. Shensong Yangxin capsule reduces atrial fibrillation susceptibility by inhibiting atrial fibrosis in rats with post-myocardial infarction heart failure. Drug Des Devel Ther. 2018;12:3407–3418.
15. Kim HJ, Kim P, Shin CY. A comprehensive review of the therapeutic and pharmacological effects of ginseng and ginsenosides in central nervous system. J Ginseng Res. 2013;37(1):8–29.
16. Peng L, Sun S, Xie LH, Wicks SM, Xie JT. Ginsenoside Re: pharmacological effects on cardiovascular system. Cardiovasc Ther. 2012;30(4):e183–e188.
17. Li X, Xiang N, Wang Z. Ginsenoside Rg2 attenuates myocardial fibrosis and improves cardiac function after myocardial infarction via AKT signaling pathway. Biosci Biotechnol Biochem. 2020;84(11):2199–2206.
18. Yokozawa T, Liu ZW, Dong E. A study of ginsenoside-Rd in a renal ischemia-reperfusion model. Nephron. 1998;78(2):201–206.
19. Wang Y, Kan H, Yin Y, et al. Protective effects of ginsenoside Rg1 on chronic restraint stress induced learning and memory impairments in male mice. Pharmacol Biochem Behav. 2014;120:73–81.
20. Kim DH, Chung JH, Yoon JS, et al. Ginsenoside Rd inhibits the expressions of iNOS and COX-2 by suppressing NF-kappaB in LPS-stimulated RAW264.7 cells and mouse liver. J Ginseng Res. 2013;37(1):54–63.
21. Wang Y, Li X, Wang X, et al. Ginsenoside Rd attenuates myocardial ischemia/reperfusion injury via Akt/GSK-3beta signaling and inhibition of the mitochondria-dependent apoptotic pathway. PLoS One. 2013;8(8):e70956.
22. Liu Y, Zhang RY, Zhao J, et al. Ginsenoside Rd Protects SH-SY5Y Cells against 1-Methyl-4-phenylpyridinium Induced Injury. Int J Mol Sci. 2015;16(7):14395–14408.
23. Ren K, Jin C, Ma P, Ren Q, Jia Z, Zhu D. Ginsenoside Rd alleviates mouse acute renal ischemia/reperfusion injury by modulating macrophage phenotype. J Ginseng Res. 2016;40(2):196–202.
24. Ren K, Li S, Ding J, et al. Ginsenoside Rd attenuates mouse experimental autoimmune neuritis by modulating monocyte subsets conversion. Biomed Pharmacother. 2021;138:111489.
25. Szklarczyk D, Santos A, von Mering C, Jensen LJ, Bork P, Kuhn M. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016;44(D1):D380–D384.
26. Sun H, Zhu X, Cai W, Qiu L. Hypaphorine Attenuates Lipopolysaccharide-Induced Endothelial Inflammation via Regulation of TLR4 and PPAR-gamma Dependent on PI3K/Akt/mTOR Signal Pathway. Int J Mol Sci. 2017;18:4.
27. Yang HY, Ahn MJ, Jeong MH, et al. Predictors of In-Hospital Mortality in Korean Patients with Acute Myocardial Infarction. Chonnam Med J. 2019;55(1):40–46.
28. Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, et al. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. 2018;186:73–87.
29. Li L, Zhao Q, Kong W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018;68-69:490–506.
30. Glezeva N, Horgan S, Baugh JA. Monocyte and macrophage subsets along the continuum to heart failure: misguided heroes or targetable villains?. J Mol Cell Cardiol. 2015;89(Pt B):136–145.
31. Bonnet S, Provencher S, Guignabert C, et al. Translating Research into Improved Patient Care in Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2017;195(5):583–595.
32. Ben-Mordechai T, Palevski D, Glucksam-Galnoy Y, Elron-Gross I, Margalit R, Leor J. Targeting macrophage subsets for infarct repair. J Cardiovasc Pharmacol Ther. 2015;20(1):36–51.
33. Ingersoll MA, Spanbroek R, Lottaz C, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. 2010;115(3):e10–e19.
34. Zhang N, An X, Lang P, Wang F, Xie Y. Ginsenoside Rd contributes the attenuation of cardiac hypertrophy in vivo and in vitro. Biomed Pharmacother. 2019;109:1016–1023.
35. Jun JI, Lau LF. Taking AIM at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov. 2011;10(12):945–963.
36. Vainio LE, Szabo Z, Lin R, et al. Connective Tissue Growth Factor Inhibition Enhances Cardiac Repair and Limits Fibrosis After Myocardial Infarction. JACC Basic Transl Sci. 2019;4(1):83–94.
37. Eijgenraam TR, Sillje H, de Boer RA. Current understanding of fibrosis in genetic cardiomyopathies. Trends Cardiovasc Med. 2020;30(6):353–361.
38. van Dijk A, Niessen HW, Ursem W, Twisk JW, Visser FC, van Milligen FJ. Accumulation of fibronectin in the heart after myocardial infarction: a putative stimulator of adhesion and proliferation of adipose-derived stem cells. Cell Tissue Res. 2008;332(2):289–298.
39. Valiente-Alandi I, Potter SJ, Salvador AM, et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation. 2018;138(12):1236–1252.
40. Wyllie AH. Apoptosis: cell death in tissue regulation. J Pathol. 1987;153(4):313–316.
41. Lindqvist LM, Vaux DL. BCL2 and related prosurvival proteins require BAK1 and BAX to affect autophagy. Autophagy. 2014;10(8):1474–1475.
42. Misao J, Hayakawa Y, Ohno M, Kato S, Fujiwara T, Fujiwara H. Expression of bcl-2 protein, an inhibitor of apoptosis, and Bax, an accelerator of apoptosis, in ventricular myocytes of human hearts with myocardial infarction. Circulation. 1996;94(7):1506–1512.
43. Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3(6):614–620.
44. Dewald O, Zymek P, Winkelmann K, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96(8):881–889.
45. Abdi R, Means TK, Ito T, et al. Differential role of CCR2 in islet and heart allograft rejection: tissue specificity of chemokine/chemokine receptor function in vivo. J Immunol. 2004;172(2):767–775.
46. Majmudar MD, Keliher EJ, Heidt T, et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation. 2013;127(20):2038–2046.
47. Yang J, Liu C, Zhang L, et al. Intensive Atorvastatin Therapy Attenuates the Inflammatory Responses in Monocytes of Patients with Unstable Angina Undergoing Percutaneous Coronary Intervention via Peroxisome Proliferator-Activated Receptor gamma Activation. Inflammation. 2015;38(4):1415–1423.
48. Kumar D, Hacker TA, Buck J, et al. Distinct mouse coronary anatomy and myocardial infarction consequent to ligation. Coron Artery Dis. 2005;16(1):41–44.
49. Stapel B, Kohlhaas M, Ricke-Hoch M, et al. Low STAT3 expression sensitizes to toxic effects of beta-adrenergic receptor stimulation in peripartum cardiomyopathy. Eur Heart J. 2017;38(5):349–361.
50. Yan X, Anzai A, Katsumata Y, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35.
51. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;169(2):361–371.
52. He S, Shen J, Li L, et al. A Novel Molecular Mechanism of IKKepsilon-Mediated Akt/mTOR Inhibition in the Cardiomyocyte Autophagy after Myocardial Infarction. Oxid Med Cell Longev. 2020;2020:7046923.
53. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J Immunol. 2017;198(3):1006–1014.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.