")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Ginsenoside Rb1 Alleviates Bleomycin-Induced Pulmonary Inflammation and Fibrosis by Suppressing Central Nucleotide-Binding Oligomerization-, Leucine-Rich Repeat-, and Pyrin Domains-Containing Protein Three Inflammasome Activation and the NF-κB Pathway
Authors Liu J , Fan G , Tao N, Feng F, Meng C, Sun T
Received 14 February 2022
Accepted for publication 8 June 2022
Published 13 June 2022 Volume 2022:16 Pages 1793—1809
DOI https://doi.org/10.2147/DDDT.S361748
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Jingjing Liu,1– 3 Guoqing Fan,1– 3 Ningning Tao,4 Feifei Feng,5 Chao Meng,1– 3 Tieying Sun1,2
1Department of Respiratory Medicine and Critical Care, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 2Graduate School of Peking Union Medical College, Beijing, People’s Republic of China; 3The MOH Key Laboratory of Geriatrics, Beijing Hospital, National Center of Gerontology, Beijing, People’s Republic of China; 4Department of Respiratory & Critical Care Medicine, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, People’s Republic of China; 5Department of Respiratory & Critical Care Medicine, The Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, People’s Republic of China
Correspondence: Tieying Sun, Department of Respiratory Medicine and Critical Care, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China, Tel +15153169108, Email [email protected]
Purpose: Idiopathic pulmonary fibrosis is a chronic and irreversible fibrotic interstitial pneumonia of unknown etiology and therapeutic strategies are limited. Emerging evidence suggests that the continuous activation of the central nucleotide-binding oligomerization-, leucine-rich repeat-, and pyrin domain-containing protein 3 (NLRP3) inflammasome is involved in the pathogenesis of pulmonary fibrosis. Ginsenoside Rb1 (G-Rb1) is the most abundant component in the traditional Chinese herb ginseng and has anti-inflammatory and anti-fibrotic activities. The purpose of this study was to explore whether G-Rb1 exerts anti-inflammatory and anti-fibrotic activities in vivo and in vitro by suppressing the activation of the NLRP3 inflammasome and NF-κB pathway.
Methods: Forty-eight male C57BL/6 mice were randomly divided into four groups (n=12/group) as follows: control, bleomycin (BLM), BLM/G-Rb1, and G-Rb1. A pulmonary fibrosis model was developed via an intratracheal injection of BLM. Six mice from each group were euthanized on days 3 and 21. The degree of pulmonary fibrosis was examined by histological evaluation and assessing α-smooth muscle actin levels. THP-1 cells were differentiated into macrophages, and stimulated by lipopolysaccharide and adenosine triphosphate. Activation of the NLRP3 inflammasome and NF-κB pathway was determined by Western blotting. Interleukin-1 beta and interleukin-18 levels were measured by ELISA. MRC-5 cells were cultured in the conditioned medium of the treated macrophages, after which markers of myofibroblasts were determined by Western blotting.
Results: G-Rb1 ameliorated BLM-induced pulmonary inflammation and fibrosis in mice, and suppressed NLRP3 inflammasome activation and the NF-κB pathway in lung tissues. Moreover, interleukin-1 beta secreted after NLRP3 inflammasome activation in macrophages promoted fibroblast differentiation. G-Rb1 inhibited lipopolysaccharide- and adenosine triphosphate-induced NLRP3 inflammasome activation in macrophages and disturbed the crosstalk between macrophages and fibroblasts.
Conclusion: G-Rb1 ameliorates BLM-induced pulmonary inflammation and fibrosis by suppressing NLRP3 inflammasome activation and the NF-κB pathway. Hence, G-Rb1 is a potential novel therapeutic drug for idiopathic pulmonary fibrosis.
Keywords: G-Rb1, pulmonary fibrosis, NLRP3 inflammasome, macrophages
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive and irreversible fibrotic disease characterized by alveolar epithelial cell injury, chronic inflammation, an unusual activation of myofibroblasts, and the excessive deposition of collagen.1–3 Patients with IPF have an overall poor prognosis and high mortality.4 Until now, the only effective curative therapy for IPF has been lung transplantation; however, the high cost of this procedure and lack of donors limit its clinical application.2 Additionally, although nintedanib and pirfenidone, which reduce the progression of IPF, have been authorized, neither can reverse the outcome of IPF.2 Therefore, new research elucidating the pathogenesis of pulmonary fibrosis to discover novel therapeutic drugs is urgently needed.
The inflammasome is an intracellular multi-protein complex, consisting of a sensor protein, an adaptor protein, and the inflammatory caspase-1 family protease.5 More specifically, the central nucleotide-binding oligomerization-, leucine-rich repeat-, and pyrin domain-containing protein 3 (NLRP3) inflammasome is the best characterized and extensively studied inflammasome. The NLRP3 inflammasome is triggered by microorganisms (eg bacteria or viruses), endogenous damage mediators (eg adenosine triphosphate [ATP] and reactive oxygen species), and foreign particulate matter to serve as a platform for caspase-1 activation.6–8 Then, bioactive caspase-1 cleaves the interleukin-1 beta (IL-1β) and interleukin-18 (IL-18) precursor into mature forms.6 The NF-κB pathway is the upstream initiator of NLRP3 inflammasome activation, which upregulates the transcription of NLRP3.9 The NLRP3 inflammasome is expressed in various innate immune cells, including dendritic cells, macrophages, and T cells.9 Alveolar macrophages, as resident immune cells in the lung, release a large amount of profibrotic soluble mediators, chemokines, extracellular matrix proteins, and metalloproteases, which provide an extracellular environment for fibroblasts to differentiate into myofibroblasts.10–12 Emerging evidence revealed that the NLRP3 inflammasome is involved in the pathogenesis of lung fibrosis in experimental models and patients.5,13–15 In addition, NLRP3 inflammasome activation in aged mice increases their susceptibility to pulmonary fibrosis, whereas, in NLRP3-/- mice, IL-1β production and progression of fibrosis are reduced.13 IL-1β participates in the development of pulmonary fibrosis, which promotes the production of TGF-β1.16 Furthermore, IL-1β drives the transformation and activation of myofibroblasts and stimulates collagen expression in vitro.17,18 Thus, suppressing NLRP3 inflammasome activation in alveolar macrophages has the potential to become a feasible treatment strategy for patients with IPF.
Ginsenosides, a class of triterpene saponins, are the major active compounds in the traditional Chinese herb ginseng, and ginsenoside Rb1 (G-Rb1) is the most abundant active compound among the bioactive ginsenosides.19 Previous studies have demonstrated that G-Rb1 exerts anti-fibrotic effects on the liver and kidneys20,21 and suppresses collagen I level.22 In addition, G-Rb1 attenuates lipopolysaccharide (LPS)-induced acute lung injury through the NF-κB pathway.23 However, although a protective effect of ginsenoside Rg1, a protopanaxatriol-type ginsenoside, against bleomycin (BLM)-induced fibrosis has been demonstrated in rats,24 the efficacy of the protopanaxadiol-type ginsenoside G-Rb1 in BLM-induced pulmonary fibrosis has not been reported. Therefore, in this study, we investigated whether G-Rb1 can alleviate pulmonary fibrosis by suppressing NLRP3 inflammasome activation.
Materials and Methods
Reagents
Bleomycin (BLM) hydrochloride was purchased from Nippon Kayaku Co. Ltd. (Tokyo, Japan). Ginsenoside Rb1 was purchased from Solarbio (Beijing, China). MCC950 was purchased from APExBIO (Houston, TX, USA). LPS (Escherichia coli 0111:B4), ATP, and phorbol-12-myristate-13-acetate (PMA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against NLRP3 (ab263899), IL-1β (ab234437), pro-caspase-1 (ab179515), and phospho-NF-κBS536 p65 (ab76302) were purchased from Abcam (Waltham, MA, USA). Antibodies against apoptosis-associated speck-like (ASC) (#67824), phospho-IκBαSer32 (#2859), and β-actin (#4970) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against NF-κB p65 (10745–1), IκBα (10268–1), α-smooth muscle actin (α-SMA; 80008–1), collagen type I (Col I) (66761–1), and CD11c (17342–1), as well as secondary antibodies were purchased from Proteintech (Rosemont, IL, USA). Recombinant human IL-1β was purchased from Proteintech. Lipofectamine™ 3000 Transfection Reagent was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Thiazolyl blue tetrazolium bromide, hematoxylin-eosin solution (H&E), and Masson’s trichrome staining kit, and Giemsa Stain kit were purchased from Solarbio. IL-1β and IL-18 ELISA kits were purchased from R&D Systems (Wiesbaden-Nordenstadt, Germany). Hydroxyproline assay kit were purchased from NanJing JianCheng Taihao Biotechnology (NanJing, China).
Animals and Treatments
Eight-week-old male C57BL/6 mice, weighing approximately 22 g were obtained from SPF Biotechnology (Beijing, China). All mice were maintained under standard humidity, temperature, and light conditions (55±5% humidity, 22±2 ℃, 12 h light-dark cycle, respectively) and had free access to food and water. After a week of adaptation, 48 mice were randomly divided into four groups (12 mice per group) as follows: Control group (control); BLM group (BLM); BLM/G-Rb1 group (BLM+G-Rb1); and G-Rb1 group (G-Rb1). Pulmonary fibrosis was initiated via an intratracheal injection of BLM (5 mg/kg, 50 μL), whereas control and G-Rb1 groups were administered the same dose of saline intratracheally. Twenty-four hours after BLM provocation, the G-Rb1 mice (20 mg/kg, once per day) were injected intraperitoneally every day until they were euthanized. Six mice from each group were euthanized on days 3 and 21. Lung tissues, serum, and broncho-alveolar lavage fluid (BALF) were collected for further experiments. The procedures involving experimental animals were authorized by the Medical Ethics Committee of the Second Hospital of Shandong University (KYLL-2019(KJ)A-0150) and conducted according to the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and the Institutional Animal Care and Use Committee (IACUC) guidelines.
Cell Culture
THP-1 cells and MRC-5 cells were obtained from Peking Union Medical College. THP-1 cells were cultured in RPMI 1640 media (Gibco, Amarillo, TX, USA) with 10% fetal bovine serum (Gibco) and 1% penicillin and streptomycin. MRC-5 cells were maintained in Minimum Essential Medium with Earle’s Balanced Salts (Gibco), 10% fetal bovine serum, and 1% penicillin and streptomycin. All cells were grown at 37 ℃ with 5% CO2. THP-1 cells were differentiated into macrophages by adding PMA (100 ng/mL) for 24 h in a serum-free medium, and were then used in the subsequent experiments. The macrophages were pre-treated with or without G-Rb1 (20 μM) for 24 h or MCC950 (1 μM) for 1 h, then stimulated by LPS (100 ng/mL) for 5.5 h and ATP (5 mM) for 30 min or with the vehicle control. Protein extraction was performed for Western blot analyses.
The conditioned medium of the treated macrophages was replaced with a fresh culture medium for 24 h. Conditioned medium obtained from the cell supernatants was gathered, centrifuged, and mixed with a fresh culture medium. MRC-5 cells were cultured in the conditioned media; the cells were collected after 48 h for further experiments.
Small Interfering RNA (siRNA)-Mediated Knockdown of NLRP3 in THP-1 Cells
siRNA (GenePharma, Shanghai, China) was used to knockdown NLRP3 expression. The sequence of the NLRP3 siRNA was 5’ CAACAGGAGAGACCUUUAUTT AUAAAGGUCUCUCCUGUUGTT 3’. The macrophages were transfected with control siRNA or NLRP3 siRNA using Lipofectamine 3000. NLRP3 siRNA-transfected cells were cultured for 48 h before LPS and ATP stimulation. NLRP3-knockdown was confirmed via Western blot analysis.
Histological Evaluation
Lung tissue samples were cut into 5 -μm sections and stained with H&E and the Masson’s Trichrome stain kit. The staining results were observed by light microscopy (Olympus, Tokyo, Japan).
BALF Analysis
The total number of cells in BALF was counted by hemocytometer. The number of neutrophils and macrophages were counted by Giemsa Stain kit.
Hydroxyproline Assay
Briefly, the lung tissue collected on day 21 was homogenized and evaluated using a hydroxyproline assay kit (NanJing Jiancheng Taihao Biotechnology).
Immunofluorescence Staining
Macrophages were treated as described above. Subsequently, the cells were fixed and blocked. The cells were incubated with rabbit anti-NF-κB p65 antibody overnight at 4 ℃, followed by subsequent incubation with fluorescently labeled secondary antibodies at 37 ℃ for 30 min. Finally, the nuclei were stained with 4’, 6-diamidino-2-phenylindole and observed using fluorescence microscopy (Olympus).
MRC-5 cells were cultured in the conditioned medium for 48 h. Then, they were subjected to immunofluorescence staining as mentioned above. The rabbit anti-α-SMA primary antibody was used for immunofluorescence staining.
The lung sections were deparaffinized, and the antigen was repaired with sodium citrate and sealed with 5% bovine serum albumin. The sections were incubated with rabbit anti-α-SMA antibody overnight at 4 ℃ and then with fluorescently labeled secondary antibodies at 37 ℃ for 30 min. The nuclei were stained with 4’, 6-diamidino-2-phenylindole and imaged by fluorescence microscopy (Olympus).
The primary antibodies used for the immunofluorescence co-localization of lung tissue sections were rabbit anti-CD11c and mouse anti-NLRP3. The images were captured via fluorescence microscopy (Olympus).
ELISA
The IL-1β and IL-18 levels of mouse BALF and cell supernatants were determined using ELISA kits (R&D Systems).
Cell Viability Assay
THP-1 cells (1×104 cells/well) were seeded onto a 96-well plate. The cells were treated with 0.1% dimethyl sulfoxide (vehicle control) and with G-Rb1 at different doses (0, 5, 10, 20, 40, 80, 160, and 320 μM) for 24 h. The cells were analyzed using a thiazolyl blue tetrazolium bromide assay kit to determine cell viability. The optical density was measured at 490 nm using a microplate reader (Thermo Fisher Scientific).
Western Blot Analysis
Lung tissues or cells were lysed with a RIPA lysis buffer supplemented with phosphatase and protease inhibitors and quantified using a BCA Protein Assay Kit (Thermo Fisher Scientific). The total protein in the cell supernatant was extracted with acetone. Equal amounts of proteins were separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes. The membranes were blocked with 5% skim milk and then incubated with the primary antibodies against β-actin, α-SMA, Col I, NLRP3, ASC, pro-caspase-1, IL-1β, phospho-NF-κBS536 p65, NF-κB p65, phospho-IκBαSer32, and IκBα. The membranes were then incubated with HRP-conjugated goat anti-rabbit or goat anti-mouse antibodies. The results were evaluated using ImageJ (National Institute of Mental Health, Bethesda, MD, USA).
Statistical Analysis
All data from three to six independent experiments were analyzed using GraphPad Prism 8 software (GraphPad Software, San Diego, CA, USA) and are presented as the mean ± standard error of the mean. Student’s t-test with Welch’s correction was applied to compare two distinct groups. The differences in multiple comparisons were analyzed using one-way analysis of variance (ANOVA) with Tukey’s post-hoc test correction for repeated measurements. P values <0.05 were defined as statistically significant.
Results
G-Rb1 Alleviated BLM-Induced Pulmonary Inflammation
The therapeutic impact of G-Rb1 was examined using a BLM-induced pulmonary inflammation and fibrosis mouse model (Figure 1A). Mice were injected with BLM through their airways to induce pulmonary inflammation over 72 h. According to the H&E staining results, the BLM group showed severe alveolitis, pulmonary edema, and serious inflammatory cell infiltration compared with the control group. The administration of G-Rb1 markedly reduced the intensity of alveolitis, edema, and inflammatory cell accumulation (Figure 1B). The Szapiel score was used to assess the degree of alveolitis in the H&E-stained samples. The results showed that the BLM group showed more severe alveolitis than the control group, and that the treatment with G-Rb1 alleviated the severe alveolitis (Figure 1C). To assess the anti-inflammatory effect of G-Rb1, we examined the inflammatory cytokine levels in the BALF. ELISA demonstrated that the levels of cytokines, including those of IL-1β and IL-18, in the BALF were higher in the BLM group than in the control group. G-Rb1 treatment decreased the inflammatory mediator levels induced by BLM (Figure 1D). Furthermore, we detected the total cell count and cell classifications in the BALF. G-Rb1 treatment also reduced the total cell count, and the numbers of macrophages and neutrophils in the BALF (Figure 1E).
 |
Figure 1 Ginsenoside Rb1 attenuates BLM-induced pulmonary inflammation in mice on day 3. (A) Forty-eight mice were randomly divided into four groups (n=12): Control group (control); BLM group (BLM); BLM/ginsenoside Rb1 group (BLM+G-Rb1); ginsenoside Rb1 group (G-Rb1). Mice were intratracheally injected with either saline or bleomycin (BLM, 5 mg/kg, 50 μL) on day 0. Twenty-four hours after the establishment of the model, the mice were treated with G-Rb1 (20 mg/kg) every day until they were euthanized. Six mice in each group were euthanized on days 3 and 21. (B) Hematoxylin-eosin (H&E) staining of lung tissues. Scale bar: 100 μm. (C) Szapiel score of lung tissue in mice (n=6). (D) IL-1β and IL-18 levels in mouse BALF were measured using ELISA (n=6). (E) Total numbers of cells, macrophages, and neutrophils in BALF. (n=6, **P< 0.01, ***P < 0.001, ****P < 0.0001). |
G-Rb1 Inhibited NLRP3 Inflammasome Activation in BLM-Induced Pulmonary Inflammation
We further explored whether G-Rb1 can inhibit NLRP3 inflammasome activation. The expression levels of NLRP3, ASC, caspase-1 p10, and pro-IL-1β increased significantly in the lungs on day 3 of BLM exposure compared with those in the control group (Figure 2A). Compared with the BLM group, the levels of NLRP3, ASC, caspase-1 p10, and pro-IL-1β were markedly inhibited in the G-Rb1 group (Figure 2A, and B).
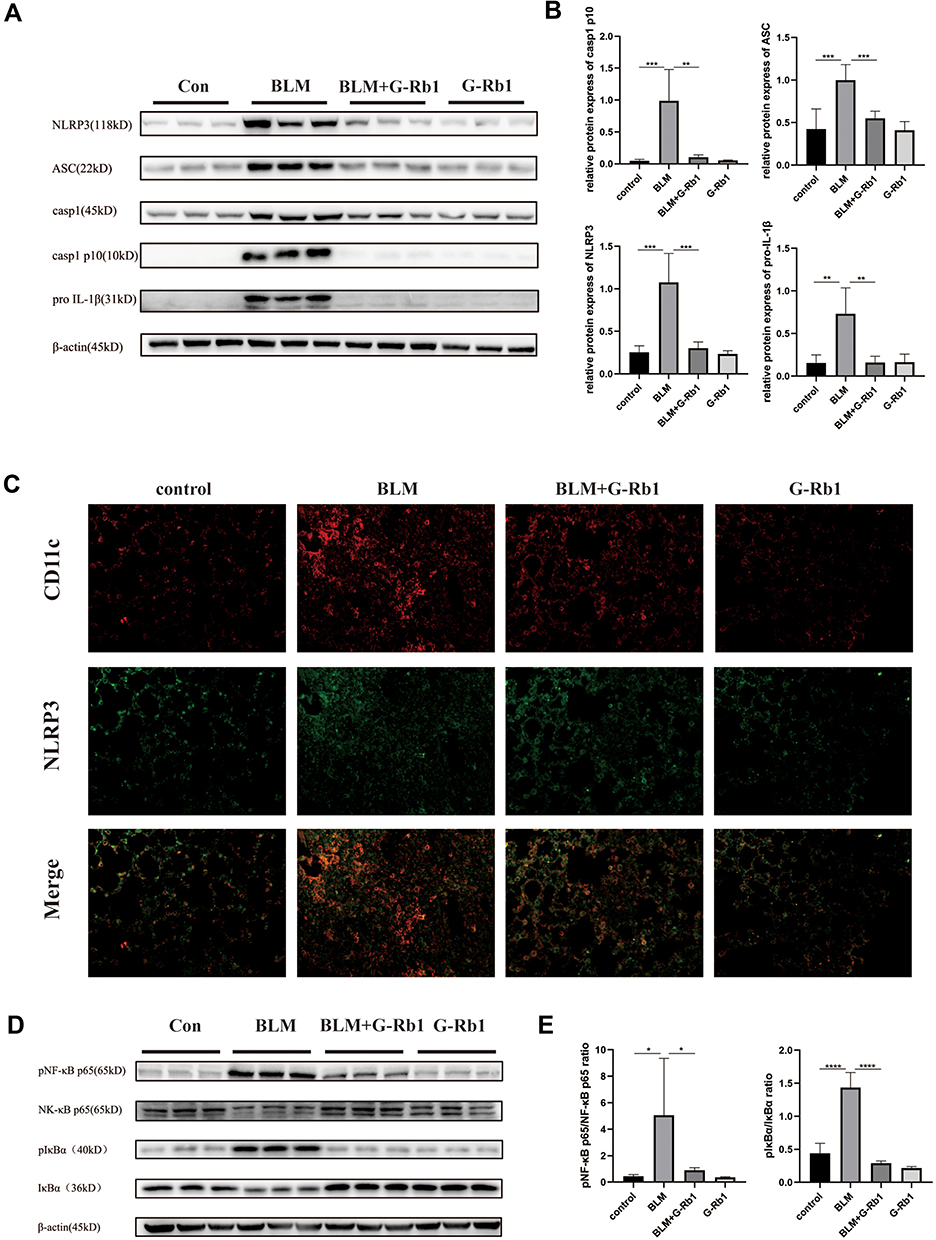 |
Figure 2 Ginsenoside Rb1 inhibits NLRP3 inflammasome activation and the NF-κB pathway in BLM-induced pulmonary inflammation in mice on day 3. (A) The expression of NLRP3, ASC, caspase-1, caspase-1 p10, and pro IL-1β in lung tissues, measured using Western blotting (n=6). (C) Immunofluorescence co-localization of NLRP3 and CD11c. (D) The expression of phosphorylated IκBα and NF-κB p65 in lung tissues, measured using Western blotting (n=6). (B) (E) Quantitative analysis of the Western blots shown in (A) and (D). (n=6, *P < 0.05, **P< 0.01, ***P < 0.001, ****P < 0.0001). |
To further elucidate the cellular source of the NLRP3 inflammasome in the BLM-induced fibrotic lungs, we examined the co-localization of NLRP3 with the alveolar macrophage marker CD11c by immunofluorescence. NLRP3 staining was highly colocalized with CD11c+ alveolar macrophages in the lungs of BLM mice (Figure 2C). The results confirmed that NLRP3 was mainly located in alveolar macrophages in BLM-induced pulmonary fibrosis.
G-Rb1 Inhibited the NF-κB Pathway in BLM-Induced Pulmonary Inflammation
To further characterize the molecular mechanism by which G-Rb1 inhibits inflammation and NLRP3 inflammasome activation, we investigated the IκBα/NF-κB pathway in BLM mice. After BLM stimulation, the phosphorylation of IκBα and NF-κB p65 increased obviously, and the G-Rb1 treatment inhibited phosphorylation compared with that following the BLM treatment (Figure 2D, and E).
G-Rb1 Attenuated BLM-Induced Pulmonary Fibrosis
More vascular hemorrhage, excessive collagen accumulation, and chaotic alveolar structure were observed in the BLM group compared with the control group, whereas the G-Rb1 treatment resulted in a major reduction in inflammation and fibrosis (Figure 3A). Furthermore, evident collagen accumulation in lung tissues after BLM stimulation was detected using Masson’s trichrome staining, whereas the administration of G-Rb1 attenuated the destruction of alveolar structures and collagen accumulation (Figure 3B). The Ashcroft score was used to assess the degree of pulmonary inflammation and fibrosis in the H&E-stained and Masson’s trichrome-stained samples. The Ashcroft semi-quantitative score results showed that the degree of pulmonary fibrosis in the BLM group was significantly higher than that in the control group, whereas the administration of G-Rb1 effectively alleviated pulmonary fibrosis (Figure 3C). α-SMA, as a typical fibrosis marker, was detected using Western blotting and immunofluorescence staining. The α-SMA level increased markedly in the lungs of mice in the BLM group compared with that in the control group, which was blocked by G-Rb1 (Figure 3D, and E). The hydroxyproline content in lung tissue, which can reflect the degree of pulmonary fibrosis, was significantly higher in BLM group mice than in control group animals, which was blocked by G-Rb1 (Figure 3F). The expression of IL-1β and IL-18 in the BALF from the BLM group on day 21 was significantly higher than that from the control group, whereas G-Rb1 treatment reduced the cytokine levels (Figure 3G).
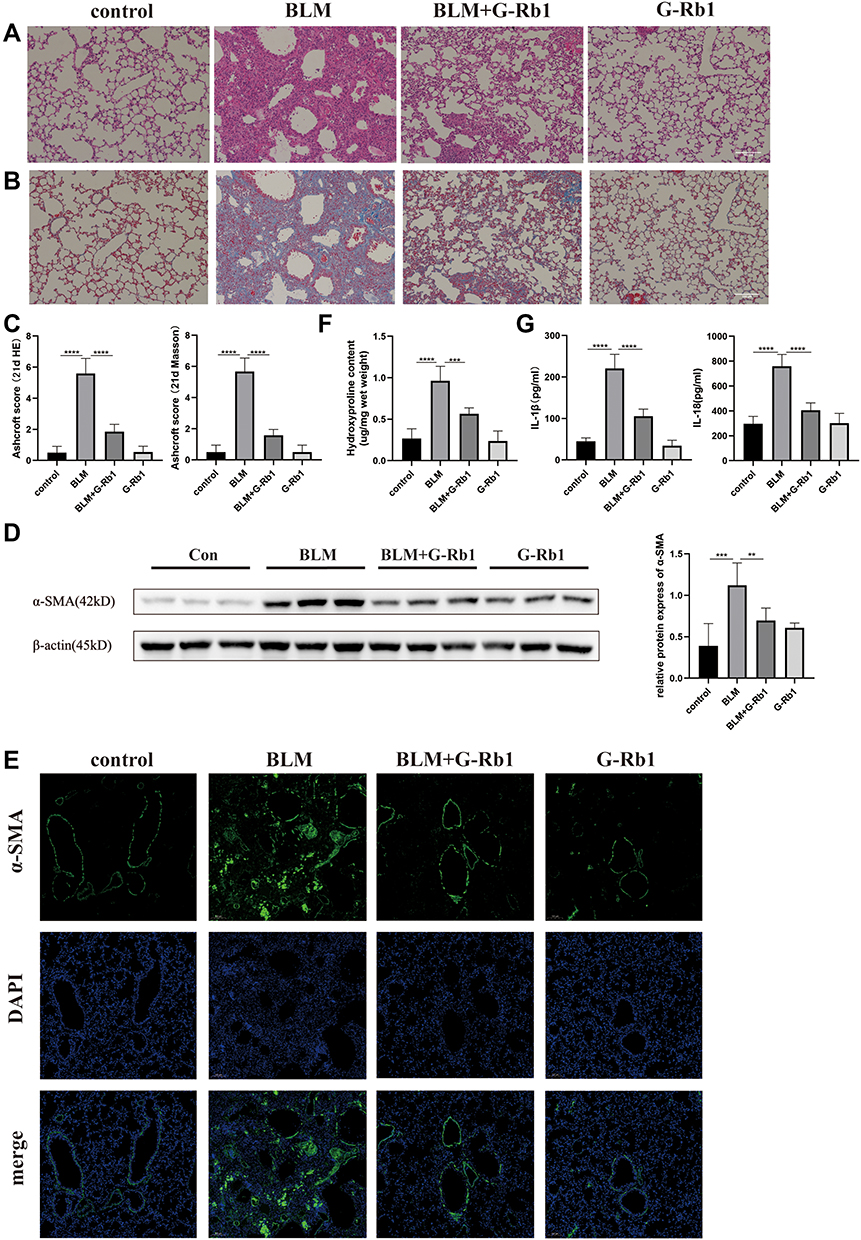 |
Figure 3 Ginsenoside Rb1 attenuates BLM-induced pulmonary fibrosis. Mice were euthanized on day 21. (A) H&E staining of lung tissues. Scale bar: 100 μm. (B) Masson’s trichrome staining of lung tissues. Scale bar: 100 μm. (C) Semi-quantitative assessment of pulmonary fibrosis by Ashcroft score (n=6). (D) (E) The expression of α-SMA in the lung tissues was evaluated using Western blotting and immunofluorescence (n=6). (F) The hydroxyproline content in lung tissues (n=6). (G) The IL-1β and IL-18 levels of BALF were evaluated using ELISA. (n=6, **P< 0.01, ***P < 0.001, ****P < 0.0001). |
G-Rb1 Inhibited BLM-Induced NLRP3 Inflammasome Activation and the NF-κB Pathway in the Lungs
The expression levels of NLRP3, ASC, caspase-1 p10, and pro-IL-1β were significantly increased in the lungs of mice 21 days after BLM administration in comparison with those in the control group (Figure 4A, B). Additionally, NLRP3 inflammasome activation was remarkably blocked upon G-Rb1 administration in lung tissues. Consistent with this, the phosphorylation of IκBα and NF-κB p65 increased significantly, whereas G-Rb1 treatment reversed this effect (Figure 4C, and D). Therefore, it was postulated that G-Rb1 could suppress BLM-induced fibrosis by suppressing NLRP3 inflammasome activation and the NF-κB signaling pathway.
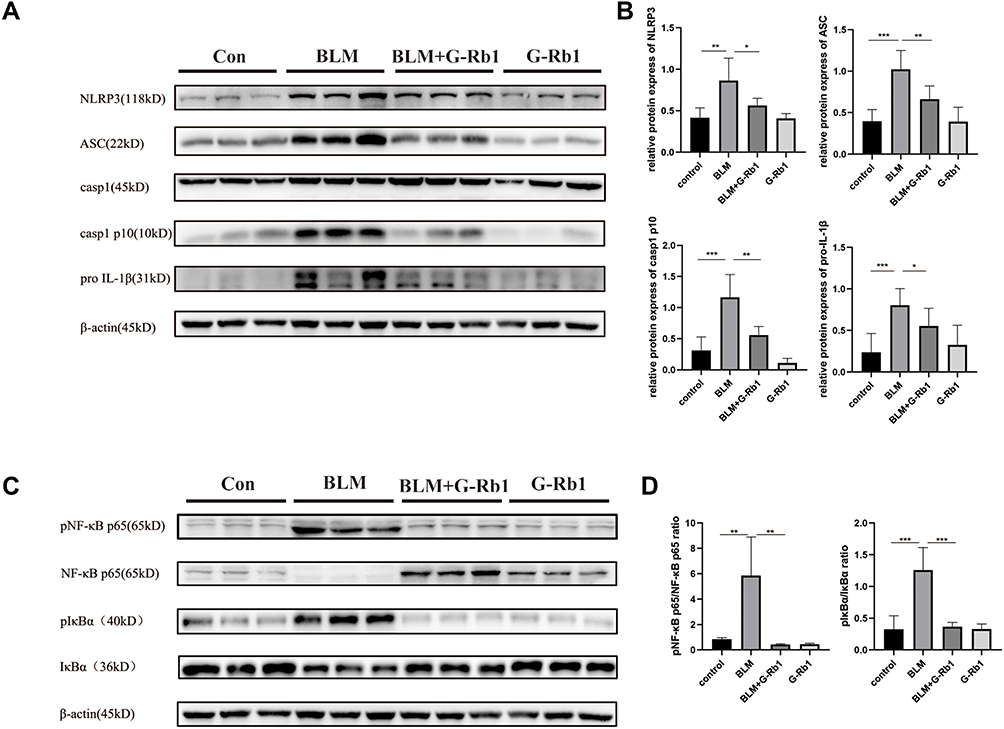 |
Figure 4 G-Rb1 inhibits BLM-induced NLRP3 inflammasome activation and the NF-κB pathway in lungs on day 21. (A) Representative Western blots of NLRP3, ASC, caspase-1, caspase-1 p10, and pro IL-1β expression in lung tissues (n=6). (C) Representative Western blots of IκBα and NF-κB p65 phosphorylation in lung tissues (n=6). (B) (D) Quantitative analysis of the Western blots shown in A and C. (n=6, *P < 0.05, **P < 0.01, ***P < 0.001). |
G-Rb1 Suppressed NLRP3 Inflammasome Activation and IL-1β Maturation in Macrophages in vitro
To further verify the mechanism of how G-Rb1 affects NLRP3 inflammasome activation in vitro, the effect of different concentrations of G-Rb1 on THP-1 cell viability was assessed using a thiazolyl blue tetrazolium bromide assay (Figure 5A). The results showed that G-Rb1 was not cytotoxic to THP-1 cells at doses below 20 μM but was slightly toxic at doses above 40 μM. Therefore, the G-Rb1 concentration of 20 μM was selected for further experiments. The macrophages were exposed to LPS and ATP to activate the NLRP3 inflammasome. As expected, upon LPS and ATP stimulation, the NLRP3 level in cells increased and abundant cleaved caspase-1 was detected in the cell supernatants, which was blocked by G-Rb1 (Figure 5B, and C). Correspondingly, more mature cleaved IL-1β was present in cell supernatants exposed to LPS/ATP, whereas G-Rb1 pretreatment inhibited the release of cleaved IL-1β (Figure 5D). Therefore, our results suggested that G-Rb1 pretreatment inhibited NLRP3 inflammasome activation and IL-1β secretion.
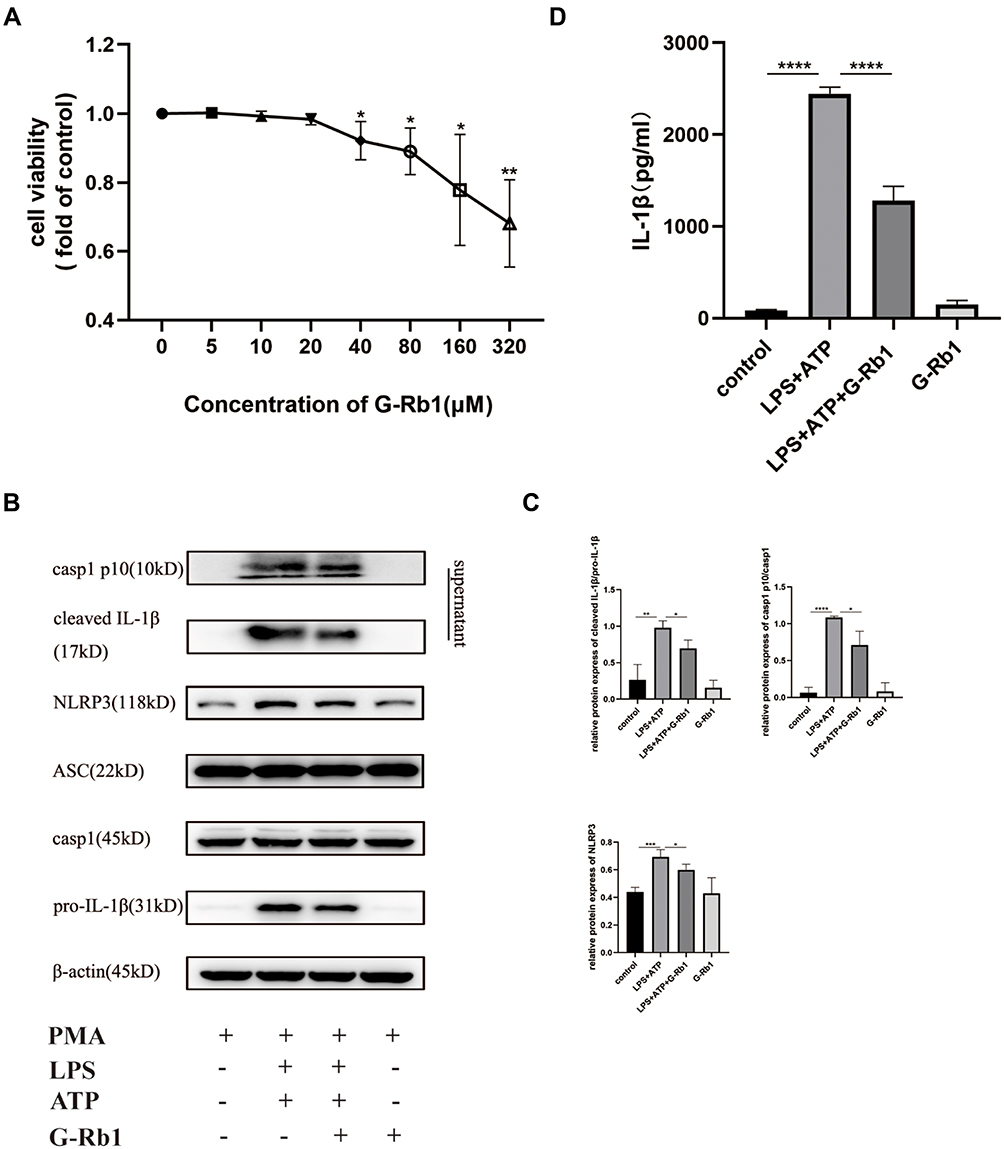 |
Figure 5 G-Rb1 inhibits NLRP3 inflammasome activation in macrophages. (A) Effect of G-Rb1 at various doses on the viability of THP-1 cells. (B) Representative Western blots of NLRP3, ASC, pro-caspase-1, caspase-1 p10, pro-IL-1β, and cleaved IL-1β. (C) Quantitative analysis of the Western blots shown in B. (D) IL-1β levels in the cell supernatants, detected using ELISA. (n≥3, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). |
Effect of G-Rb1 on the NF-κB Pathway in Macrophages
To further demonstrate the effect of G-Rb1 on the NF-κB pathway in vitro, we investigated the expression of IκBα and NF-κB p65 in THP-1 cells upon LPS/ATP stimulation. Compared with the control group, the protein expression levels of phosphorylated IκBα and NF-κB p65 increased markedly in THP-1 cells stimulated by LPS/ATP, and this effect was suppressed by G-Rb1 (Figure 6A, and B). To further clarify the molecular mechanism by which G-Rb1 inhibited the NF-κB pathway, the nuclear translocation of NF-κB p65, which is a marker of NF-κB activation, was investigated using immunofluorescence. Immunofluorescence images showed that the nuclear translocation of NF-κB p65 increased upon LPS/ATP stimulation, whereas G-Rb1 pretreatment significantly reduced the LPS/ATP-induced nuclear translocation of NF-κB p65 from the cytoplasm (Figure 6C). We also compared the effects of G-Rb1 and MCC950 on the NF-κB pathway. MCC950 pretreatment had no inhibitory effect on IκBα and NF-κB p65 phosphorylation (Figure 6D, and E).
 |
Figure 6 G-Rb1 suppresses the NF-κB pathway in macrophages whereas MCC950 has no inhibitory effect on the NF-κB pathway. (A) (D) Representative Western blots of pIκBα/IκBα and pNF-κB p65/NF-κB p65. (B) (E) Quantitative analysis of the Western blots shown in (A) and (D). (C) Nuclear translocation of NF-κB p65 in macrophages examined using immunofluorescence. (n≥3, *P < 0.05, **P < 0.01, ***P < 0.001). |
The Activation of the NLRP3 Inflammasome in Macrophages Promoted Fibroblast to Myofibroblast Differentiation, and G-Rb1 Inhibited the Crosstalk Between Macrophages and Fibroblasts
The conditioned medium was applied to assess the influence of macrophages on the differentiation of fibroblasts into myofibroblasts (Figure 7A). Differentiation of MRC-5 cells to myofibroblasts was observed after they were stimulated using the conditioned medium. The result was supported by the increased levels of α-SMA and collagen I (Figure 7B and C). Furthermore, G-Rb1 pretreatment restrained the macrophage-fibroblast crosstalk. These results showed that the NLRP3 inflammasome in macrophages promoted fibroblast differentiation, and G-Rb1 inhibited the crosstalk between macrophages and fibroblasts.
 |
Figure 7 G-Rb1 attenuates macrophage-induced myofibroblast differentiation of fibroblasts. (A) Illustration of the conditioned medium. THP-1 cells were differentiated into macrophages. Cells were pretreated with or without G-Rb1 and then stimulated with LPS and ATP or the vehicle control. The conditioned medium was then collected and diluted for culture of the MRC-5 cells. (B) The expression of α-SMA and collagen I in MRC-5 cells exposed to the conditioned medium, detected using Western blotting. (C) The expression of α-SMA in MRC-5 cells exposed to the conditioned medium, detected using immunofluorescence. (n≥3, *P < 0.05, **P < 0.01, ***P < 0.001). |
NLRP3 siRNA was used to downregulate the expression of NLRP3 (Figure 8A), ELISA results demonstrated that upon NLRP3 knockdown, IL-1β levels in cell supernatants exposed to LPS/ATP decreased markedly (Figure 8B). These results confirmed that IL-1β production by macrophages in response to LPS/ATP was NLRP3-dependent. Conditioned medium was then obtained from the macrophages that were transfected with control siRNA or NLRP3 siRNA, and the differentiation of MRC-5 cells to myofibroblasts was observed following stimulation using the conditioned medium. The results suggested that the differentiation of MRC-5 cells to myofibroblasts was inhibited upon NLRP3 knockdown in macrophages (Figure 8C). Moreover, the results following the addition of rhIL-1β (10 ng/mL) to MRC-5 cell culture confirmed the role of the NLRP3-IL-1β axis in the crosstalk between macrophages and fibroblasts (Figure 8D).
 |
Figure 8 The role of the NLRP3-IL-1β axis in the crosstalk between macrophages and fibroblasts. (A) The expression of NLRP3 after NLRP3 siRNA transfection. (B) IL-1β levels in cell supernatants after NLRP3 siRNA transfection. (C) The expression of α-SMA in MRC-5 cells exposed to the conditioned medium, detected using immunofluorescence. (D) The expression of α-SMA and collagen I in MRC-5 cells stimulated by IL-1β. (n≥3, *P < 0.05, **P < 0.01, ***P < 0.001). |
Discussion
IPF is an interstitial lung disease with high mortality and poor prognosis.1,4 To date, the treatment of IPF remains a major challenge worldwide. Emerging evidence has demonstrated that NLRP3 inflammasome activation is involved in the development of multiple pulmonary fibrosis, including silicosis-, nanoparticle-, LPS-, or BLM-induced pulmonary fibrosis.13,18,25–27 Previous research has demonstrated that G-Rb1 can delay the progression of inflammation and fibrosis.20–23 In this work, to our knowledge, the pharmacological mechanism of the effect of G-Rb1 on BLM-induced pulmonary fibrosis and inflammation was evaluated for the first time. In vivo, G-Rb1 inhibited extracellular matrix deposition and inflammation by suppressing the NLRP3 inflammasome and the NF-κB pathway. In addition, G-Rb1 interrupted the macrophage-fibroblast communication by suppressing NLRP3 inflammasome activation in vitro.
Our results confirm that G-Rb1 alleviated BLM-induced pulmonary inflammation. According to the results of H&E staining, G-Rb ameliorated the severe alveolitis, pulmonary edema, abundant inflammatory cell infiltration, and thickening of alveolar septa induced by the exposure to BLM. Furthermore, BLM stimulation triggered the NLRP3 inflammasome in the lung tissues whereas treatment with G-Rb1 suppressed the activation of the NLRP3 inflammasome. Our results are in agreement with those of other studies, which have reported that BLM induces acute lung injury and activates the NLRP3 inflammasome.28–30 The NLRP3 inflammasome is expressed in various cells. To locate the main source of the NLRP3 inflammasome in the lung tissue, double immunofluorescence staining of NLRP3 and the alveolar macrophage marker CD11c was performed. The results indicated that the NLRP3 inflammasome was mainly expressed in alveolar macrophages during lung inflammation, which was consistent with previous reports.13 Additionally, the phosphorylation of IκBα and NF-κB p65 increased notably in the BLM group, which was subsequently blocked by G-Rb1. Our results were consistent with those of previous studies, which suggested that the NF-κB signaling pathway is activated in BLM-induced acute lung injury.31,32 A previous study by Yuan et al confirmed that G-Rb1 attenuates acute lung injury by suppressing the NF-κB pathway.23 Therefore, our results demonstrated that G-Rb1 alleviated lung inflammation triggered by BLM in mice by suppressing the NLRP3 inflammasome and NF-κB pathway.
We demonstrated that G-Rb1 ameliorated pulmonary fibrosis in mice. The pathological results suggested that vascular hemorrhage, excessive collagen accumulation, and alveolar structure destruction were detected in the BLM group, whereas G-Rb1 treatment yielded a significant reduction in fibrosis. Previous studies have demonstrated that G-Rb1 delayed the progression of carbon tetrachloride-induced liver fibrosis and unilateral ureter obstruction-induced renal fibrosis.20,21 However, the therapeutic effect of G-Rb1 in pulmonary fibrosis has not been evaluated. Moreover, the NLRP3 inflammasome was activated and the phosphorylation of IκBα and NF-κB p65 in the NF-κB pathway was elevated in lung tissues exposed to BLM, whereas these effects were blocked by G-Rb1. Previous studies have indicated that the NLRP3 inflammasome is involved in the process of BLM-induced pulmonary fibrosis.5,13,14 In addition, the NF-κB signaling pathway is activated in BLM-induced pulmonary fibrosis, leading to the upregulated transcription of NLRP3 and pro-IL-1β and upregulated expression of pro-fibrotic factors such as CXCL12 and MCP-1.9,31,33 Our results demonstrated, for the first time, that G-Rb1 alleviated pulmonary fibrosis induced by BLM in mice by blocking the NLRP3 inflammasome and the NF-κB pathway.
In addition, we verified the mechanism of G-Rb1 in vitro. Macrophages were exposed to LPS/ATP to trigger NLRP3 inflammasome activation. G-Rb1 pretreatment suppressed NLRP3 inflammasome activation. Correspondingly, G-Rb1 administration effectively inhibited the phosphorylation of IκBα and NF-κB p65, as well as the nuclear translocation of the latter. Research shows that the NF-κB pathway regulates the production of IL-1β due to the upregulation of pro-IL-1β.34 Moreover, the NLRP3 inflammasome promotes the maturation of IL-1β through bioactive caspase-1 to cleave pro-IL-1β. Therefore, G-Rb1 can block the production and maturation of IL-1β. Emerging evidence suggests that MCC950, a specific small-molecule inhibitor of NLRP3 inflammasome, alleviates BLM-induced pulmonary fibrosis in mice.35 To compare the effect of G-Rb1 and MCC950 on the NF-κB pathway, MCC950 was evaluated for its role in the NF-κB pathway. However, the results confirmed that MCC950 has no impact on the repression of the NF-κB pathway.
Myofibroblasts are the primary effector cells in the progression of IPF and they contribute to the synthesis of extracellular matrix and collagen deposition.36 Myofibroblast sources include epithelial-mesenchymal transition, bone marrow-derived progenitors, and lung fibroblast differentiation; the differentiated lung fibroblasts are key contributors to myofibroblasts.37 Macrophages secrete abundant cytokines to promote the proliferation and differentiation of fibroblasts. Therefore, we used conditioned medium to confirm the influence of cytokines secreted by macrophages on fibroblast differentiation. Our results showed that cytokines secreted by macrophages promoted fibroblast differentiation, whereas G-Rb1 inhibited the interaction between macrophages and fibroblasts. IL-1β, the main cytokine produced by macrophages, plays an indispensable role in fibroblast differentiation and the deposition of extracellular matrix.15,16,18 To confirm the role of IL-1β in the differentiation of myofibroblasts, exogenous IL-1β was added to the MRC-5 cell culture. Our results demonstrated that the α-SMA and collagen I levels increased more in the rhIL-1β group than that in the control group. Furthermore, NLRP3 inflammasome activation is the upstream regulatory platform for the production and secretion of IL-1β. In our study, NLRP3 expression was knocked down in macrophages by NLRP3 siRNA, and IL-1β levels in cell supernatants were detected by ELISA after LPS/ATP stimulation. The results demonstrated that IL-1β expression decreased significantly after NLRP3 siRNA treatment. The results confirmed that IL-1β production in macrophages was NLRP3-specific and that the NLRP3 inflammasome-IL-1β axis participates in the progression of macrophage-initiated fibroblast differentiation, which may underlie the early phase fibrotic response to lung injury. Furthermore, G-Rb1 inhibited the crosstalk between macrophages and fibroblasts by suppressing NLRP3 inflammasome activation in macrophages.
Our study has some limitations. First of all, it is controversial whether a BLM-induced pulmonary fibrosis mouse model can completely reproduce the pathogenesis of IPF in humans. Second, the pharmacological mechanism of G-Rb1 and its direct molecular target were not elucidated adequately. Finally, no positive control drug was used in the animal experiments.
Conclusion
In brief, the results of this study showed that G-Rb1 effectively ameliorated BLM-induced pulmonary inflammation and fibrosis by inhibiting NLRP3 inflammasome activation and the NF-κB pathway in alveolar macrophages. Therefore, ginsenosides may be a candidate drug for IPF treatment in the future.
Abbreviations
ASC, adaptor protein apoptosis-associated speck-like; ATP, adenosine triphosphate; BALF, broncho-alveolar lavage fluid; BLM, bleomycin; G-Rb1, ginsenoside Rb1; H&E, hematoxylin-eosin; IL-1β, interleukin 1 beta; IL-18, interleukin 18; IPF, idiopathic pulmonary fibrosis; LPS, lipopolysaccharide; NLRP3, central nucleotide-binding oligomerization, leucine-rich repeat, and pyrin domain-containing protein 3 inflammasome; PMA, phorbol-12-myristate-13-acetate; α-SMA, smooth muscle actin.
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Author Contributions
TYS and JJL designed the entire study. JJL, GQF, NNT, FFF and CM completed the animal and cell experiments. TYS and JJL wrote the article. All authors approved the final manuscript. All authors made a significant contribution to the work reported, whether in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The research was funded by the National Key Research and Development Program of China, key special projects of major chronic non-communicable disease prevention and control (2016YFC1304600), the Shandong Provincial Natural Science Foundation Youth Project (ZR2020QH289), and the National Natural Science Foundation of China youth project (82103803).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Du Bois RM. Strategies for treating idiopathic pulmonary fibrosis. Nat Rev Drug Discov. 2010;9(2):129–140. doi:10.1038/nrd2958
2. Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3–e19. doi:10.1164/rccm.201506-1063ST
3. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208(7):1339–1350. doi:10.1084/jem.20110551
4. Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old. Eur Respir J. 2016;48(1):179–186. doi:10.1183/13993003.01653-2015
5. Brusselle GG, Provoost S, Bracke KR, Kuchmiy A, Lamkanfi M. Inflammasomes in respiratory disease: from bench to bedside. Chest. 2014;145(5):1121–1133. doi:10.1378/chest.13-1885
6. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi:10.1016/s1097-2765(02)00599-3.7
7. Gasse P, Riteau N, Charron S, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med. 2009;179(10):903–913. doi:10.1164/rccm.200808-1274OC
8. De Nardo D, De Nardo CM, Latz E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am J Pathol. 2014;184(1):42–54. doi:10.1016/j.ajpath.2013.09.007
9. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. doi:10.1038/nri3452
10. Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol Med. 2016;22(4):303–316. doi:10.1016/j.molmed.2016.02.004
11. Desai O, Winkler J, Minasyan M, Herzog EL. The role of immune and inflammatory cells in idiopathic pulmonary fibrosis. Front Med. 2018;5:43. doi:10.3389/fmed.2018.00043
12. Liu G, Zhai H, Zhang T, et al. New therapeutic strategies for IPF: based on the “phagocytosis-secretion-immunization” network regulation mechanism of pulmonary macrophages. Biomed Pharmacother. 2019;118:109230. doi:10.1016/j.biopha.2019.109230
13. Stout-Delgado HW, Cho SJ, Chu SG, et al. Age-dependent susceptibility to pulmonary fibrosis is associated with NLRP3 inflammasome activation. Am J Respir Cell Mol Biol. 2016;55(2):252–263. doi:10.1165/rcmb.2015-0222OC
14. Liang Q, Cai W, Zhao Y, et al. Lycorine ameliorates bleomycin-induced pulmonary fibrosis via inhibiting NLRP3 inflammasome activation and pyroptosis. Pharmacol Res. 2020;158:104884. doi:10.1016/j.phrs.2020.104884
15. Jäger B, Seeliger B, Terwolbeck O, et al. The NLRP3-Inflammasome-Caspase-1 pathway is upregulated in idiopathic pulmonary fibrosis and acute exacerbations and is inducible by apoptotic A549 cells. Front Immunol. 2021;12:642855. doi:10.3389/fimmu.2021.642855
16. Liu RM. Oxidative stress, plasminogen activator inhibitor 1, and lung fibrosis. Antioxid Redox Signal. 2008;10(2):303–319. doi:10.1089/ars.2007.1903
17. Postlethwaite AE, Raghow R, Stricklin GP, Poppleton H, Seyer JM, Kang AH. Modulation of fibroblast functions by interleukin 1: increased steady-state accumulation of type I procollagen messenger RNAs and stimulation of other functions but not chemotaxis by human recombinant interleukin 1 alpha and beta. J Cell Biol. 1988;106(2):311–318. doi:10.1083/jcb.106.2.311
18. Hindman B, Ma Q. Carbon nanotubes and crystalline silica stimulate robust ROS production, inflammasome activation, and IL-1β secretion in macrophages to induce myofibroblast transformation. Arch Toxicol. 2019;93(4):887–907. doi:10.1007/s00204-019-02411-y
19. Washida D, Kitanaka S. Determination of polyacetylenes and ginsenosides in Panax species using high performance liquid chromatography. Chem Pharm Bull. 2003;51(11):1314–1317. doi:10.1248/cpb.51.1314
20. Hou YL, Tsai YH, Lin YH, Chao JC. Ginseng extract and ginsenoside Rb1 attenuate carbon tetrachloride-induced liver fibrosis in rats. BMC Complement Altern Med. 2014;14:415. doi:10.1186/1472-6882-14-415
21. Liu X, Chen J, Sun N, et al. Ginsenoside Rb1 ameliorates autophagy via the AMPK/mTOR pathway in renal tubular epithelial cells in vitro and in vivo. Int J Biol Macromol. 2020;163:996–1009. doi:10.1016/j.ijbiomac.2020.07.060
22. Tark KC, Lee DW, Lew DH, Kang EH, Roh H, Lee MC. Effects of ginsenoside Rb1 on hypertrophic scar remodeling in rabbit model. Eur J Pharmacol. 2015;750:151–159. doi:10.1016/j.ejphar.2015.01.011
23. Yuan Q, Jiang YW, Ma TT, Fang QH, Pan L. Attenuating effect of ginsenoside Rb1 on LPS-induced lung injury in rats. J Inflamm. 2014;11(1):40. doi:10.1186/s12950-014-0040-5
24. Zhan H, Huang F, Ma W, Zhao Z, Zhang H, Zhang C. Protective effect of ginsenoside Rg1 on bleomycin-induced pulmonary fibrosis in rats: involvement of Caveolin-1 and TGF-β1 signal pathway. Biol Pharm Bull. 2016;39(8):1284–1292. doi:10.1248/bpb.b16-00046
25. Peeters PM, Perkins TN, Wouters EF, Mossman BT, Reynaert NL. Silica induces NLRP3 inflammasome activation in human lung epithelial cells. Part Fibre Toxicol. 2013;10:3. doi:10.1186/1743-8977-10-3
26. Li X, Yan X, Wang Y, et al. NLRP3 inflammasome inhibition attenuates silica-induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells. Exp Cell Res. 2018;362(2):489–497. doi:10.1016/j.yexcr.2017.12.013
27. Xu WJ, Wang XX, Jin JJ, et al. Inhibition of GGPPS1 attenuated LPS-induced acute lung injury and was associated with NLRP3 inflammasome suppression. Am J Physiol Lung Cell Mol Physiol. 2019;316(3):L567–L577. doi:10.1152/ajplung.00190.2018
28. Smith RE, Strieter RM, Phan SH, Kunkel SL. C-C chemokines: novel mediators of the profibrotic inflammatory response to bleomycin challenge. Am J Respir Cell Mol Biol. 1996;15(6):693–702. doi:10.1165/ajrcmb.15.6.8969262
29. Brar SS, Meyer JN, Bortner CD, Van Houten B, Martin WJ
30. Xu JF, Washko GR, Nakahira K, et al. Statins and pulmonary fibrosis: the potential role of NLRP3 inflammasome activation. Am J Respir Crit Care Med. 2012;185(5):547–556. doi:10.1164/rccm.201108-1574OC
31. Dong J, Ma Q. In vivo activation and pro-fibrotic function of NF-κB in fibroblastic cells during pulmonary inflammation and fibrosis induced by carbon nanotubes. Front Pharmacol. 2019;10:1140. doi:10.3389/fphar.2019.01140
32. Wang Z, Li X, Chen H, et al. Resveratrol alleviates bleomycin-induced pulmonary fibrosis via suppressing HIF-1α and NF-κB expression. Aging. 2021;13(3):4605–4616. doi:10.18632/aging.202420
33. Krug LT, Torres-González E, Qin Q, et al. Inhibition of NF-kappaB signaling reduces virus load and gammaherpesvirus-induced pulmonary fibrosis. Am J Pathol. 2010;177(2):608–621. doi:10.2353/ajpath.2010.091122
34. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. 2016;165(4):792–800. doi:10.1016/j.cell.2016.03.046
35. Ji J, Hou J, Xia Y, Xiang Z, Han X. NLRP3 inflammasome activation in alveolar epithelial cells promotes myofibroblast differentiation of lung-resident mesenchymal stem cells during pulmonary fibrogenesis. Biochim Biophys Acta Mol Basis Dis. 2021;1867(5):166077. doi:10.1016/j.bbadis.2021.166077
36. Pardo A, Selman M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 2016;13(suppl 5):S417–S421. doi:10.1513/AnnalsATS.201605-341AW
37. Tanjore H, Xu XC, Polosukhin VV, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180(7):657–665. doi:10.1164/rccm.200903-0322OC
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.