")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Ginkgolide B ameliorates myocardial ischemia reperfusion injury in rats via inhibiting endoplasmic reticulum stress
Authors Guo C, Zhang J, Zhang P, Si A, Zhang Z, Zhao L, Lv F, Zhao G
Received 17 July 2018
Accepted for publication 15 January 2019
Published 26 February 2019 Volume 2019:13 Pages 767—774
DOI https://doi.org/10.2147/DDDT.S179101
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Jianbo Sun
This paper has been retracted.
Changlei Guo, Junbiao Zhang, Peiyong Zhang, Aoyang Si, Zhenling Zhang, Liangping Zhao, Fenghua Lv, Guoan Zhao
Department of Cardiology, The First Affiliated Hospital Xinxiang Medical University, Weihui 452100, Henan, China
Purpose: Ginkgolide B (GB) is a terpene lactone component found in Ginkgo biloba, which has a protective role on ischemia reperfusion (I/R) injury. This study was aimed at exploring the protective mechanism of GB on the myocardial I/R.
Patients and methods: Myocardial I/R model was established on Sprague Dawley rats. The levels of cardiac troponin I, cardiac troponin T, lactic dehydrogenase, and myoglobin were determined by a 200FR NEO automatic biochemical analyzer. Histological examination was performed through HE and terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling staining. The expression levels of p-PERK, p-IRE1α, ATF6, p-AKT, and mTOR were detected by Western blot.
Results: The results exhibited that GB treatment suppressed the high levels of cardiac troponin I, cardiac troponin T, lactic dehydrogenase, and myoglobin and ameliorated the damaged and irregularly arranged myocardial cells induced by I/R injury significantly, indicating that GB could ameliorate myocardial I/R injury. Moreover, the high expression levels of endoplasmic reticulum (ER) stress key proteins caused by I/R injury were suppressed significantly by GB treatment, including p-PERK, p-IRE1α, and ATF6. GB treatment also decreased the number of apoptotic cells compared with I/R group. In addition, activation of ER stress by Tunicamycin treatment could counteract the protective effects of GB on I/R injury, suggesting that GB ameliorated myocardial I/R injury through inhibition of ER stress-induced apoptosis. Finally, the decreased p-AKT and p-mTOR expressions caused by I/R injury were upregulated by GB and inhibition of PI3K/AKT/mTOR pathway by LY294002 abolished the protective effects of GB on I/R injury, indicating that GB activated PI3K/AKT/mTOR pathway during I/R injury.
Conclusion: GB protected against myocardial I/R injury through inhibiting ER stress-induced apoptosis via PI3K/AKT/mTOR signaling pathway.
Keywords: myocardial injury, Ginkgolide B, endoplasmic reticulum stress, apoptosis PI3K/AKT/mTOR pathway
Introduction
Atherosclerotic disease and stroke are the leading causes of death and disability around the world, and myocardial infarction contributes to most of the morbidity and mortality.1,2 Currently, reperfusion, including percutaneous coronary intervention and thrombolysis, is considered to be the most effective therapy to protect ischemic damage during myocardial infarction. However, reperfusion after myocardial infarction may cause myocardial ischemia reperfusion (I/R) injury, which becomes a major cause of death in acute myocardial infarction patients after reperfusion.3
Previous studies reported that I/R might cause injury through induction of excessive myocardial endoplasmic reticulum (ER) stress. ER is a vital organelle for protein folding and maturation in eukaryotic cells.4,5 Moderate ER stress can upregulate the release of chaperone proteins including glucose-regulating protein 78 and calreticulin, which is protective and plays an important role in recognition and degradation of misfolded proteins.6 However, prolonged or severe ER stress, which can be triggered by I/R injury, will cause cell apoptosis through increasing proapoptotic proteins (Bax) while decreasing antiapoptotic proteins (Bcl-2), and activation of caspases.7 Hence, suppressing ER stress-induced apoptosis has become an effective therapy for the treatment of myocardial I/R injury.8
Ginkgolide B (GB) is one of the major terpene lactone components found in Ginkgo biloba extracts. Previous researches indicated that GB (also named BN 52021) was a specific and potent antagonist of platelet-activating factor (PAF) receptor, and it could inhibit PAF-induced cascade effect in inflammatory reactions.9–12 Several studies suggested that GB showed remarkable neuroprotective effects in animal models of focal cerebral I/R injury.13–15 Studies have examined the protective role of GB against myocardial infarction since the late 1980s.16–18 In addition, a study showed that GB could improve the contractile function in isolated cardiomyocytes from I/R rats, which indicate GB can partly prevent IR injury in rat heart.19 Recently, GB was proved to be a promising candidate against IR-induced myocardial dysfunction via reducing degradation of the membrane phospholipids.20 In particular, GB exerted cardioprotective properties against doxorubicin-induced cardiotoxicity by reducing ROS via regulating Akt and calcium signaling pathways.21 Thus, the underlying cardioprotective mechanism of GB in myocardial I/R requires further investigations.
In our present study, animal models of myocardial I/R injury were constructed to investigate the effect of GB treatment on myocardial I/R injury and the underlying mechanism. We observed that GB treatment ameliorated myocardial I/R injury through inhibiting ER stress-induced apoptosis. This study may shed light on therapeutic strategy for myocardial I/R injury.
Materials and methods
Construction of animal model and grouping
The animal study followed the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication 85-23, revised 1996). The research was approved by the Medical Ethics Committee of the First Affiliated Hospital Xinxiang Medical University. Pathogen-free, healthy male Sprague Dawley rats (280±20 g) were purchased from Experimental Animal Center of the First Affiliated Hospital Xinxiang Medical University.
Coronary artery ligation method was used to establish the I/R injury model.22 Rats received endotracheal intubation and artificial ventilation after anesthetizing with sodium pentobarbital intraperitoneal injection (30 mg/kg). Thread was passed through the left coronary artery after exposure of heart. Another two threads were drawn from the knot to loosen the ligation. Left coronary artery was ligated to produce ischemia, and local myocardium appeared cyanosis after ligation. Then, after 1 hour of ischemia, the ligation was loosened to restore blood flow, leading to reperfusion which lasted for 1 hour in this study.
Rats were randomly divided into six groups as follows (n=8 per group): 1) Sham group was operated with no ischemia. 2) Sham+GB group was operated with no ischemia and given GB at 15 mg/kg. 3) I/R model group was operated with I/R. 4) I/R+GB group was operated with I/R and given GB 15 mg/kg.20 5) IR+tunicamycin group was operated with I/R and given tunicamycin (10 mg/mL).23 6) I/R+GB+tunicamycin group was operated with I/R and given GB (15 mg/kg) with tunicamycin (10 mg/mL).7 I/R+LY294002 group was operated with I/R and given LY294002 (10 mg/kg).8 I/R+GB+LY294002 group was operated with I/R and given GB (15 mg/kg) with LY294002 (10 mg/kg).9 I/R+AG490 group was operated with I/R and given AG490 (10 mg/kg).10 I/R+GB+AG490 group was operated with I/R and given GB (15 mg/kg) with AG490 (1 mg/kg).24 GB was dissolved in 1 M NaOH, diluted with isotonic saline, and adjusted to a pH of 7.0–8.0 with HCl. Rats were administered GB through intravenous infusion 10 minutes prior to the left coronary artery the left occlusion.20 After 25 minutes reperfusion, LY294002/tunicamycin/AG490 was present for comparison of effects upon followed IR. At the end of the study, blood of each rat was collected for detection. Rats were sacrificed and their heart tissues were collected for the following experiments.
Detection of myocardial enzyme levels in serum
Rats were sacrificed and blood was collected; then, serum was separated to measure the heart muscle damage, and some indicators, including cardiac troponin I (cTnI), cardiac troponin T (cTnT), lactic dehydrogenase (LDH), and myoglobin (Mb), were evaluated in the serum. The levels of enzyme were determined using a 200FR NEO automatic biochemical analyzer (Toshiba, Tokyo, Japan).
HE staining
At the end of the study, myocardial samples of left ventricle were collected and fixed in a 4% buffered paraformaldehyde solution for 24 hours. Then, tissues were dehydrated, embedded in paraffin, and cut into 4-μm-thick sections for HE staining and then examined under light microscopy in six randomly selected areas.25
Western blot assay
Proteins were extracted using RIPA lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer’s instructions. The same amount (20 μg) of proteins was separated by 10% SDS-PAGE gel and then transferred onto polyvinylidene fluoride (PVDF) membranes. Following blocking, the membrane was incubated with the following primary antibodies (Cell Signaling, San Jose, CA, USA) at 4°C overnight: p-PERK, p-IRE1α, ATF6, p-AKT, mTOR, and GAPDH. After a secondary antibody (Cell Signaling) was added for incubation of 1 hour, the membranes were detected using a ChemiDoc XRS imaging system.
TUNEL staining
The 4-μm-thick sections of myocardial tissues were used for TUNEL staining to label the nuclei of apoptotic cells. Sections were incubated in 3% H2O2 and then in the TUNEL reaction mixture. The sections were rinsed and visualized using DAB. Hematoxylin was used for counterstaining. The numbers of TUNEL-positive cells of five random fields were counted under light microscopy. The apoptosis index was calculated as the percent of TUNEL-positive cells relative to the total cells.
Statistical analysis
The data were expressed as the mean±SD of independent experiments. Statistical analysis was performed using Student’s t-test or one-way ANOVA. The difference was considered statistically significant at P<0.05. All statistical analyses were performed with SPSS 19.0.
Results
GB ameliorates myocardial I/R injury
To evaluate the protective effect of GB against myocardial I/R injury, we detected the levels of cTnI, cTnT, LDH, and Mb in the serum of rats from different groups. Significantly increased levels of cTnI, cTnT, LDH, and Mb were observed in I/R group compared to that in Sham group. However, GB treatment suppressed the increased cTnI, cTnT, LDH, and Mb levels induced by I/R remarkably (Figure 1A–D, P<0.05). In addition, as shown in Figure 1E, GB treatment also ameliorated the damaged and irregularly arranged myocardial cells induced by I/R injury (Figure 1E). These results indicated that GB could ameliorate myocardial I/R injury.
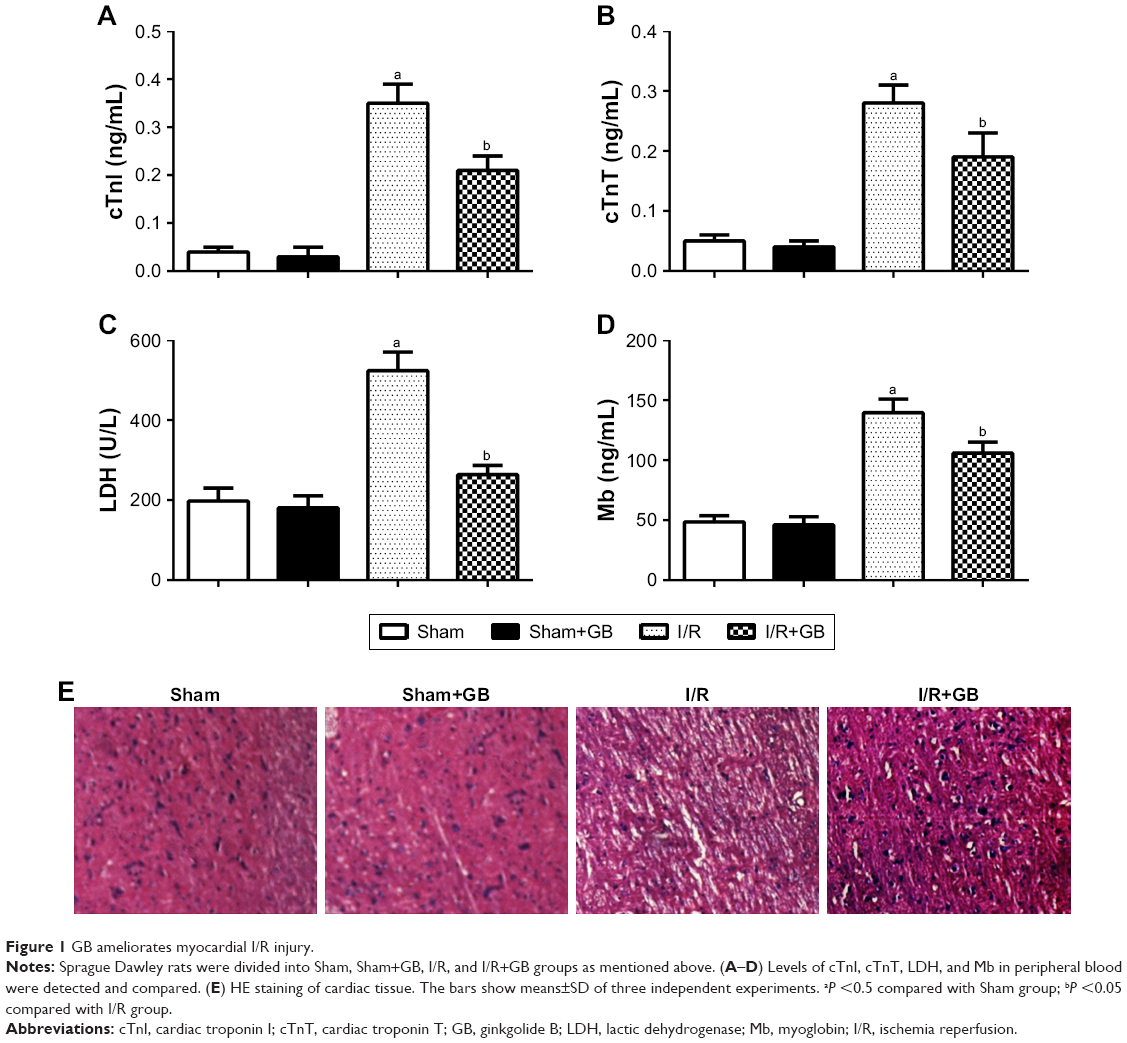 | Figure 1 GB ameliorates myocardial I/R injury. |
GB inhibits ER stress induced by I/R
During ER stress, the subsequent unfolded protein response (UPR), which consists of three highly coordinated signaling pathways (PERK pathway, IRE1α pathway, and ATF6 pathway), was activated.15 Thus, we determined the phosphorylation levels of PERK and IRE1α and the relative expression of ATF6, Bax, and Bcl-2 by Western blot. Our data showed that elevated levels of p-PERK, p-IRE1α, Bax, and ATF6 caused by I/R injury was suppressed significantly by GB treatment (Figure 2A and B, P<0.05). Moreover, results from TUNEL assay also showed that GB treatment decreased the number of apoptosis cells compared to I/R group (Figure 2C and D, P<0.05). Our data suggested that GB inhibited ER stress induced by I/R.
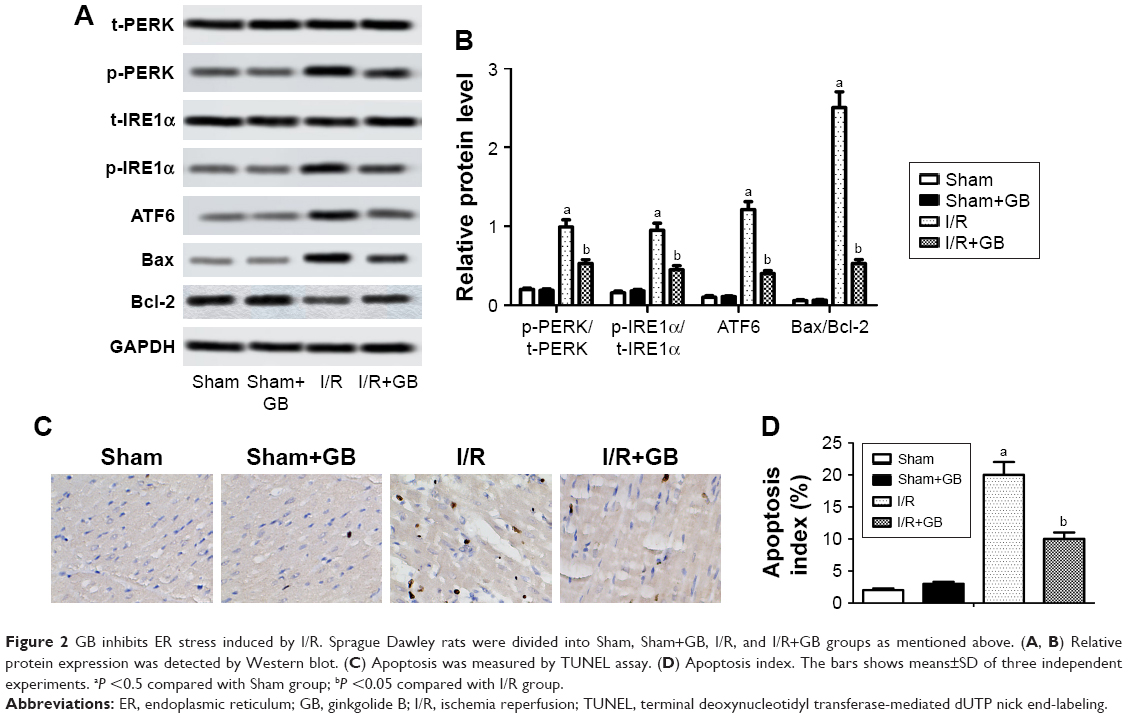 | Figure 2 GB inhibits ER stress induced by I/R. Sprague Dawley rats were divided into Sham, Sham+GB, I/R, and I/R+GB groups as mentioned above. (A, B) Relative protein expression was detected by Western blot. (C) Apoptosis was measured by TUNEL assay. (D) Apoptosis index. The bars shows means±SD of three independent experiments. aP <0.5 compared with Sham group; bP <0.05 compared with I/R group. |
ER stress activator tunicamycin counteracts the effects of GB on I/R injury
To demonstrate that GB could ameliorate ER stress during I/R injury, ER stress tunicamycin was used to treat rats in the experiments. We observed that tunicamycin increased ATF6, cleaved Caspase-12, and p-IRE1α expressions in I/R group and I/R+tunicamycin group (Figure 3A and B, P<0.05). In addition, tunicamycin counteracted the protective effects of GB on myocardial damage and also increased apoptosis index compared to I/R+GB group (Figure 3C–E, P<0.05). Besides that, tunicamycin treatment increased cTnI and cTnT levels compared to I/R+GB group remarkably (Figure 3F and G, P<0.05). In summary, these results demonstrated that activation of ER stress could counteract the protective effects of GB on I/R injury.
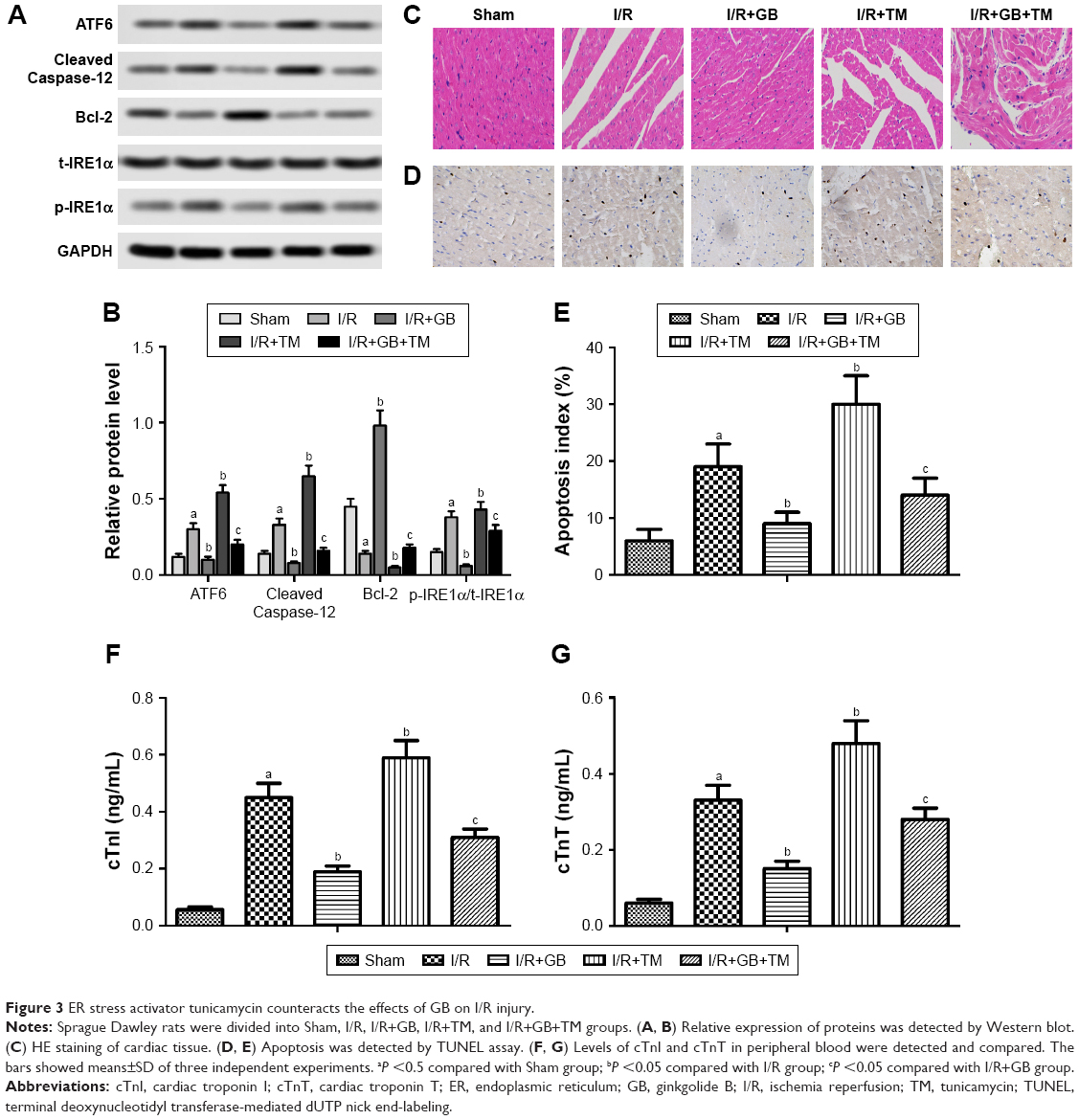 | Figure 3 ER stress activator tunicamycin counteracts the effects of GB on I/R injury. |
GB exerts protective effects against I/R injury through activation of PI3K/AKT/mTOR pathway
PI3K/AKT/mTOR and SAFE signaling pathways are reported to be involved in ER stress-triggered apoptosis.16,24 Results from Western blot showed that relative expressions of p-AKT, p-mTOR, and p-STAT3 were downregulated in I/R group while upregulated in I/R+GB group, suggesting that GB activated PI3K/AKT/mTOR and SAFE signaling pathways in I/R (Figure 4A and B, P<0.05). Inhibition of PI3K/AKT/mTOR pathway by LY294002 increased cTnI and cTnT levels as well as apoptosis index compared to I/R+GB group (Figure 4C and D, P<0.05). Inhibition of the SAFE pathway by AG490 showed the same results. These results indicated that GB exerted protective effects against I/R injury via activation of PI3K/AKT/mTOR and SAFE signaling pathways.
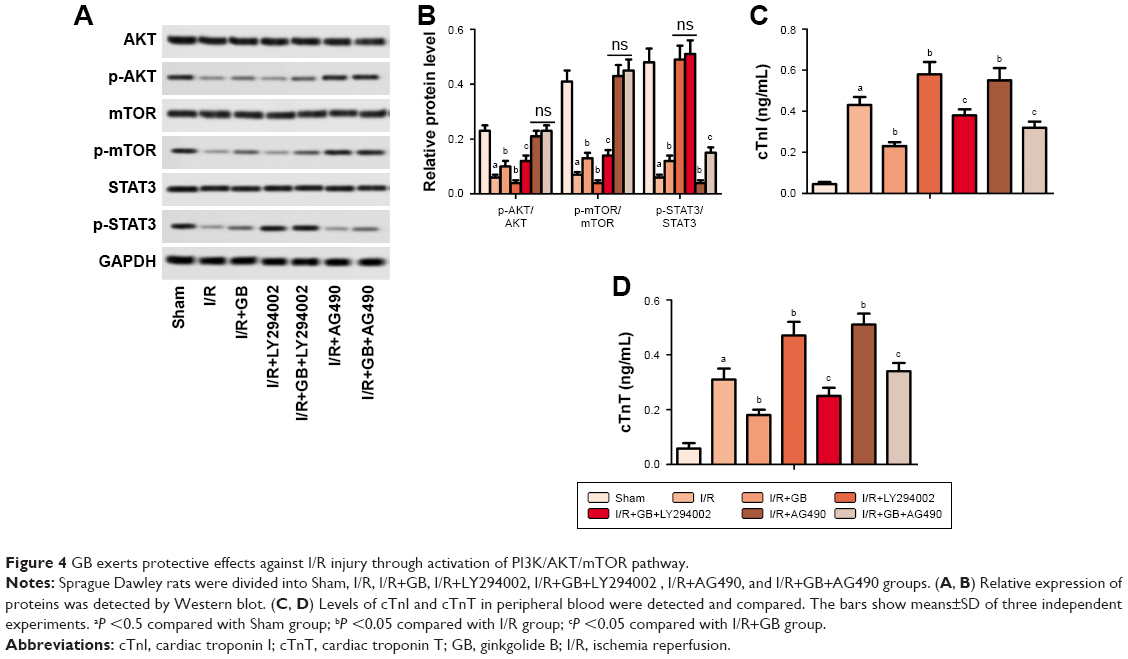 | Figure 4 GB exerts protective effects against I/R injury through activation of PI3K/AKT/mTOR pathway. |
Discussion
Myocardial I/R injury has caused high morbidity and mortality worldwide, making it a considerable challenge to solve this problem. In our present study, we showed that GB ameliorated I/R injury in myocardial I/R animal model through inhibiting ER stress-induced apoptosis via activation of PI3K/AKT/mTOR signaling pathway. The myocardial protective effects of GB may make it an effective agent for the treatment of I/R injury.
GB is a lactone component isolated from G. biloba. It has been reported that GB has neuroprotective effects against ischemia-induced brain injury both in vitro and in vivo.26–28 GB is reported to reduce neuronal damage in rats following transient forebrain ischemia, and the beneficial effects of GB were due to inhibition of PAF, which was a potent mediator of many inflammatory processes.9 Besides, several studies elucidated that GB acted as a PAF antagonist and had a protective effect against myocardial I/R injury.16–21 Here, we explored another mechanism of how GB exerted protective effects against myocardial I/R injury.
During myocardial ischemia, the membranes of cardiac muscle cells were damaged, leading to the release of myocardial enzymes and proteins including cTnI, cTnT, LDH, and Mb into peripheral blood. Therefore, detecting the levels of cTnI, cTnT, LDH, and Mb in peripheral blood can reflect the degree of myocardial ischemia necrosis.29 In our study, we observed that GB treatment downregulated the high levels of cTnI, cTnT, LDH, and Mb and also improved the damaged myocardial cells induced by I/R injury significantly, indicating that GB mitigated myocardial injury in myocardial I/R model rats. Accumulating evidences indicated that excessive, prolonged ER stress played an important role in inducing cell death, especially apoptosis.30,31 Recent studies demonstrated that overproduction of ROS could activate ER stress-induced myocardial apoptosis. Thus, inhibition of ER stress could be cardioprotective in the setting of myocardial I/R injury.30,32 In response of ER stress, the UPR which can minimize the accumulation and aggregation of misfolded proteins is activated. UPR consists of three highly coordinated signaling pathways including PERK, IRE1α, and ATF6 pathways. These three key proteins are also responsible for the proapoptotic pathways of UPR during ER stress-mediated apoptosis caused by prolonged stress.33 In this present study, we detected the phosphorylation levels of PERK, IRE1α, and the relative expression of ATF6 by Western blot, and as a result, these three proteins were all significantly increased after I/R injury. I/R injury also increased apoptosis index compared to control group. However, GB treatment suppressed the expressions of p-PERK, p-IRE1α, and ATF6 and also reduced apoptosis index compared to I/R group, indicating that GB exerted protective effects against myocardial I/R injury through suppressing ER stress-induced apoptosis. To verify our conclusion, ER stress activator tunicamycin was used to treat rats in combination with GB. Our results showed that activation of ER stress by tunicamycin counteracted the inhibitory effects of GB on cTnI and cTnT levels and apoptosis index, verifying our conclusion that GB ameliorated myocardial I/R injury via suppressing ER stress-induced apoptosis. As for the clinical relevance, the nanoparticle delivery of mitoprotective agents has applied to ischemic heart disease.34 In this study, GB, as a mitoprotective agent, may be applied to the treatment in I/R by nanoparticle delivery.
The PI3K/AKT/mTOR signaling pathway is one of the most important signaling pathways in the regulation of multiple cellular processes including cell proliferation, growth, and apoptosis.35,36 UPR initiates programmed cell death via the transcriptional regulation of genes involved in cell death, including TRB3 which can inhibit activation of AKT and preventing the phosphorylation of AKT.37 Therefore, there exists a cross-talk between the UPR and Akt signaling pathways. Zhang’s study reported that increased ROS production might account for inhibition of the PI3K/Akt pathway and led to ER stress and mitochondrial dysfunction, which consequently induced apoptosis in L-02 hepatocytes.38 Hence, we hypothesized that PI3K/AKT/mTOR signaling pathway might play a vital role in the ER stress-induced apoptosis. In this study, we found that GB increased the levels of p-AKT, p-mTOR, and p-STAT3 and counteracted with LY294002/AG490, suggesting that GB might suppress ER stress through activation of PI3K/AKT/mTOR and SAFE signaling pathways.
Limitations
Due to the limitation of equipment and fund, we did not evaluate left ventricular function or triphenyltetrazolium chloride staining, either with electrocardiograph. We evaluated the myocardial damage through the determination of cTnI, cTnT, LDH, and Mb levels as well as HE staining, which can also reflect the level of myocardial damage. Meanwhile, further studies need to be done on whether GB transmits signals from outside the cell or penetrates into the cell and induces a signal in the cytoplasm.
Conclusion
Taken together, GB protected against myocardial I/R injury through inhibiting ER stress-induced apoptosis via PI3K/AKT/mTOR signaling pathway. This finding suggests that GB may be a potential therapeutic approach in the treatment of I/R injury.
Acknowledgments
We sincerely appreciate the technical support from The First Affiliated Hospital Xinxiang Medical University.
Disclosure
The author reports no conflicts of interest in this work.
References
Santos-Gallego CG, Bayón J, Badimón JJ. Thrombi of different pathologies: implications for diagnosis and treatment. Curr Treat Options Cardiovasc Med. 2010;12(3):274–291. | ||
Santos-Gallego CG, Picatoste B, Badimón JJ. Pathophysiology of acute coronary syndrome. Curr Atheroscler Rep. 2014;16(4):401. | ||
Heusch G. Critical issues for the translation of cardioprotection. Circ Res. 2017;120(9):1477–1486. | ||
Lin YD, Chen S, Yue P, et al. CAAT/enhancer binding protein homologous protein-dependent death receptor 5 induction is a major component of SHetA2-induced apoptosis in lung cancer cells. Cancer Res. 2008;68(13):5335–5344. | ||
Ron D, Hubbard SR. How IRE1 reacts to ER stress. Cell. 2008;132(1):24–26. | ||
Xia JG, Xu FF, Qu Y, Song DG, Shen H, Liu XH. Atorvastatin post-conditioning attenuates myocardial ischemia reperfusion injury via inhibiting endoplasmic reticulum stress-related apoptosis. Shock. 2014;42(4):365–371. | ||
Mcguckin MA, Eri RD, das I, Lourie R, Florin TH. ER stress and the unfolded protein response in intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2010;298(6):G820–G832. | ||
Kim DS, Kwon DY, Kim MS, et al. The involvement of endoplasmic reticulum stress in flavonoid-induced protection on cardiac cell death caused by ischaemia/reperfusion. J Pharm Pharmacol. 2010;62(2):197–204. | ||
Braquet P. Proofs of involvement of PAF-acether in various immune disorders using BN 52021 (ginkgolide B): a powerful PAF-acether antagonist isolated from Ginkgo biloba L. Adv Prostaglandin Thromboxane Leukot Res. 1986;16:179–198. | ||
Yang Y, Nemoto EM, Harvey SA, Subbotin VM, Gandhi CR. Increased hepatic platelet activating factor (PAF) and PAF receptors in carbon tetrachloride induced liver cirrhosis. Gut. 2004;53(6):877–883. | ||
Xia SH, Hu CX, Fang JM, Di Y, Zhao ZL, Liu LR. G[alpha]i2 and G[alpha]q expression change in pancreatic tissues and BN52021 effects in rats with severe acute pancreatitis. Pancreas. 2008;37(2):170–175. | ||
Mahmoud F, Abul H, Onadeko B, Khadadah M, Haines D, Morgan G. In vitro effects of Ginkgolide B on lymphocyte activation in atopic asthma: comparison with cyclosporin A. Jpn J Pharmacol. 2000;83(3):241–245. | ||
Qin XF, Lu XJ, Ge JB, Xu HZ, Qin HD, Xu F. Ginkgolide B prevents cathepsin-mediated cell death following cerebral ischemia/reperfusion injury. Neuroreport. 2014;25(4):267–273. | ||
Gu JH, Ge JB, Li M, Wu F, Zhang W, Qin ZH. Inhibition of NF-κB activation is associated with anti-inflammatory and anti-apoptotic effects of Ginkgolide B in a mouse model of cerebral ischemia/reperfusion injury. Eur J Pharm Sci. 2012;47(4):652–660. | ||
Fang W, Deng Y, Li Y, et al. Blood brain barrier permeability and therapeutic time window of Ginkgolide B in ischemia-reperfusion injury. Eur J Pharm Sci. 2010;39(1–3):8–14. | ||
Chopra K, Singh M, Gupta S, Ganguly NK. Involvement of oxygen free radicals in the action of BN 52021 (PAF antagonist) to limit myocardial infarct size. Methods Find Exp Clin Pharmacol. 1993;15(7):437–445. | ||
Chakrabarty S, Thomas P, Sheridan DJ. Contribution of platelets and platelet-activating factor (PAF) to the arrhythmogenic, haemodynamic and necrotic effects of acute myocardial ischaemia. Eur Heart J. 1991;12(5):583–589. | ||
Baranes J, Hellegouarch A, Le Hegarat M, et al. The effects of PAF-acether on the cardiovascular system and their inhibition by a new highly specific PAF-acether receptor antagonist BN 52021. Pharmacol Res Commun. 1986;18(8):717–737. | ||
Hao Y, Sun Y, Xu C, et al. Improvement of contractile function in isolated cardiomyocytes from ischemia-reperfusion rats by ginkgolide B pretreatment. J Cardiovasc Pharmacol. 2009;54(1):3–9. | ||
Pei HX, Hua R, Guan CX, Fang X. Ginkgolide B reduces the degradation of membrane phospholipids to prevent ischemia/reperfusion myocardial injury in rats. Pharmacology. 2015;96(5–6):233–239. | ||
Gao J, Chen T, Zhao D, Zheng J, Liu Z. Ginkgolide B exerts cardioprotective properties against doxorubicin-induced cardiotoxicity by regulating reactive oxygen species, Akt and calcium signaling pathways in vitro and in vivo. PLoS One. 2016;11(12):e0168219. | ||
Xu Z, Alloush J, Beck E, Weisleder N. A murine model of myocardial ischemia-reperfusion injury through ligation of the left anterior descending artery. J Vis Exp. 2014;(86). | ||
Teng X, Song J, Zhang G, et al. Inhibition of endoplasmic reticulum stress by intermedin (1-53) protects against myocardial injury through a PI3 kinase-Akt signaling pathway. J Mol Med. 2011;89(12):1195–1205. | ||
Santos-Gallego CG, Vahl TP, Goliasch G, et al.Sphingosine-1-phosphate receptor agonist fingolimod increases myocardial salvage and decreases adverse postinfarction left ventricular remodeling in a porcine model of ischemia/reperfusion. Circulation. 2016;133(10):954–966. | ||
Chen C, Nong Z, Meng M, et al. Toxicological evaluation of Yulangsan polysaccharide in Wistar rats: A 26-week oral gavage study. Environ Toxicol Pharmacol. 2016;41:1–7. | ||
Oberpichler H, Sauer D, Rossberg C, Mennel HD, Krieglstein J. PAF antagonist ginkgolide B reduces postischemic neuronal damage in rat brain hippocampus. J Cereb Blood Flow Metab. 1990;10(1):133–135. | ||
Lv P, Fang W, Geng X, Yang Q, Li Y, Sha L. Therapeutic neuroprotective effects of Ginkgolide B on cortex and basal ganglia in a rat model of transient focal ischemia. Eur J Pharm Sci. 2011;44(3):235–240. | ||
Yang ZZ, Li J, Li SX, Feng W, Wang H. Effect of Ginkgolide B on striatal extracellular amino acids in middle cerebral artery occluded rats. J Ethnopharmacol. 2011;136(1):117–122. | ||
Venge P, van Lippen L, Blaschke S, et al. Equal clinical performance of a novel point-of-care cardiac troponin I (cTnI) assay with a commonly used high-sensitivity cTnI assay. Clinica Chimica Acta. 2017;469:119–125. | ||
Miyazaki Y, Kaikita K, Endo M, et al. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):1124–1132. | ||
Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115(10):2656–2664. | ||
Jian L, Lu Y, Lu S, Lu C. Chemical chaperone 4-phenylbutyric acid protects H9c2 cardiomyocytes from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis. Mol Med Rep. 2016;13(5):4386–4392. | ||
Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22(1):487–508. | ||
Ong SB, Lu S, Katwadi K, Ismail NI, Kwek XY, Hausenloy DJ. Nanoparticle delivery of mitoprotective agents to target ischemic heart disease. Future Cardiol. 2017;13(3):195–198. | ||
Huang KF, Zhang GD, Huang YQ, Diao Y. Wogonin induces apoptosis and down-regulates survivin in human breast cancer MCF-7 cells by modulating PI3K-Akt pathway. Int Immunopharmacol. 2012;12(2):334–341. | ||
Sui T, Ma L, Bai X, Li Q, Xu X. Resveratrol inhibits the phosphatidylinositide 3-kinase/protein kinase B/mammalian target of rapamycin signaling pathway in the human chronic myeloid leukemia K562 cell line. Oncol Lett. 2014;7(6):2093–2098. | ||
Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279(44):45495–45502. | ||
Zhang Y, Xiao F, Liu X, Liu K, Zhou X, Zhong C. Cr(VI) induces cytotoxicity in vitro through activation of ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction via the PI3K/Akt signaling pathway. Toxicol In Vitro. 2017;41:232–244. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.