")
Back to Journals » International Medical Case Reports Journal » Volume 8
Giant malignant peripheral nerve sheath tumor of thigh in an adolescent with neurofibromatosis type 1: a case report
Authors Tosun HB , Serbest S, Turk BA, Gumustas SA, Uludag A
Received 8 July 2015
Accepted for publication 15 September 2015
Published 28 October 2015 Volume 2015:8 Pages 267—271
DOI https://doi.org/10.2147/IMCRJ.S92015
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Haci Bayram Tosun,1 Sancar Serbest,2 Bilge Aydin Turk,3 Seyit Ali Gumustas,1 Abuzer Uludag1
1Department of Orthopaedics and Traumatology, Faculty of Medicine, Adiyaman University, Adiyaman, 2Department of Orthopaedics and Traumatology, Faculty of Medicine, Kirikkale University, Kirikkale, 3Department of Pathology, Faculty of Medicine, Adiyaman University, Adiyaman, Turkey
Abstract: Malignant peripheral nerve sheath tumors (MPNSTs) are rare sarcomas of children and adolescents, and they are aggressive tumors with a high rate of local recurrence. We present a 15-year-old boy with neurofibromatosis type 1 (NF1), who had a giant MPNST on the right thigh taking into account the available literature. Diagnosis of MPNST may be delayed in NF1 patients due to confusion with a neurofibroma and/or a plexiform neurofibroma. Malignancy should be considered, especially in cases with big masses, with heterogeneous involvement, or in the presence of cysts or necrotic nodules. The aim of surgical treatment is complete surgical excision.
Keywords: nerve sheath neoplasm, sarcoma, adolescent, neurofibromatosis, lower extremity
Introduction
Malignant peripheral nerve sheath tumors (MPNSTs) are very rare high-grade sarcomas with a poor prognosis at childhood and adolescence, and only 10%–20% of MPNSTs are seen in these periods.1–4 MPNSTs often present with pain associated with mass lesions or neuronal involvement in the head–neck, abdomen, chest wall, and other anatomic regions, but extremity involvement is uncommon, especially in children or adolescents.3,5,6 The most important risk factor for MPNST development is the presence of neurofibromatosis type 1 (NF1), and half of the MPNST cases occur in patients with NF1 history.2,7,8 MPNSTs may be confused with neurofibromas and/or plexiform neurofibromas in patients with NF1.7,9,10 We present a case of giant MPNST located at the right thigh of a patient with NF1 taking into account the available literature.
In our study, electronic searches for reports of similar cases and other rare manifestations using the keywords “neurofibroma”, “neural sheath tumors”, “soft tissue sarcomas in children with neurofibromatosis”, “neurofibroma and adolescents”, and “neurofibromatosis and malignancy” were conducted in Google, PubMed, SpringerLink, EMBASE, Elsevier, and Ovid databases for all English articles with abstracts. Only 30 reports focusing on neurofibroma and MPNSTs in the patient – with or without neurofibromatosis – or those that provided important theoretical contributions are referenced in the current work.
Case report
A 15-year-old male presented to our clinic with a swelling and light pain at the right thigh for a year. On physical examination, a mass with indeterminate borders was palpated at the anteromedial area of the right thigh. There were typical NF1 skin findings such as café au lait spots and peripheral neurofibromas all over the body (Figure 1). Magnetic resonance imaging (MRI) revealed a 13×9 cm mass at the anteromedial thigh with posterior extension, which was isointense in T1 weighted (T1W) images, heterogeneously iso- to hyperintense in T2 weighted (T2W) images, and diffusely contrast enhancing except the central cystic areas in the postcontrast images (Figure 2). With a suspicion of malignancy, fine-needle-aspiration biopsy was performed. Histopathological evaluation led to an initial diagnosis of benign neurofibroma, and total surgical excision was planned. Ethical approval for this study was not taken, but we informed both the patient and the patient’s parents about this study and obtained written informed consent. The mass was excised under general anesthesia. During surgery, the mass was observed to have a hard, fixed, and large capsule. Histopathological evaluation revealed a smooth-surfaced, grayish-brown 13×9×9 cm solid mass weighing 610 g, which appears nodular and heterogeneous (Figure 3). A malign mesenchymal tumor involving diffuse necrotic areas was observed. It was generally hypercellular, but in some areas, it was hypocellular. There were hyperchromatic spindle cells at myxoid areas with a storiform pattern. There were occasional atypical mesenchymal cells with clear cytoplasms and vesicular nuclei, as well as diffuse pleomorphic multinucleated giant cells. Diffuse atypical mitosis (42 mitoses per 10 HPF [high- power field]) was striking. Immunohistochemical analysis revealed strong (+) staining with S-100, Vimentin, CD-56, CD-99, and P-53, as well as 80% positivity for Ki-67 staining (Figure 4). Considering these data, the lesion was diagnosed as MPNST (Grade-3; Federation Nationale des Centres de Lutte Contre le Cancer).
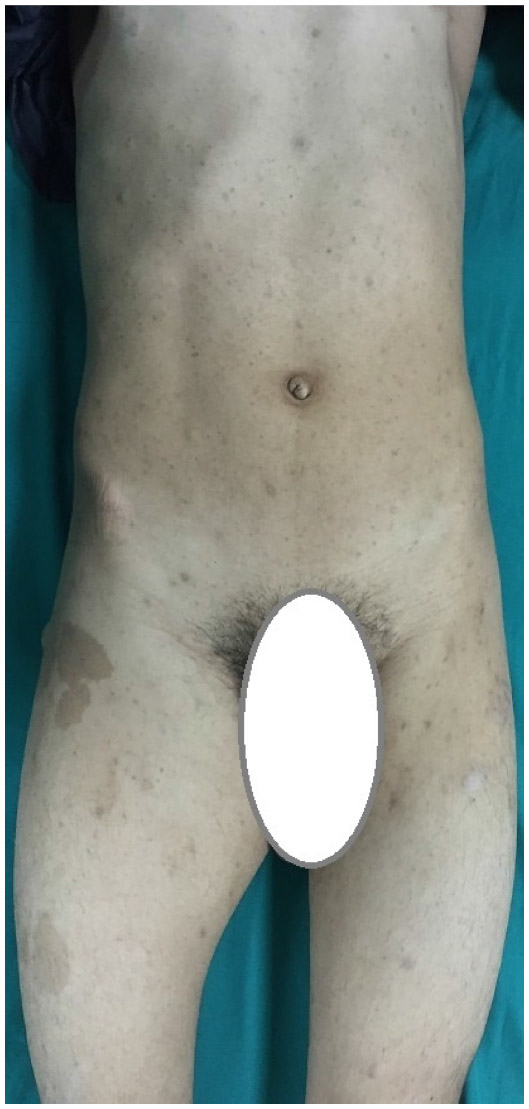 | Figure 1 External appearance of a 15-year-old boy. |
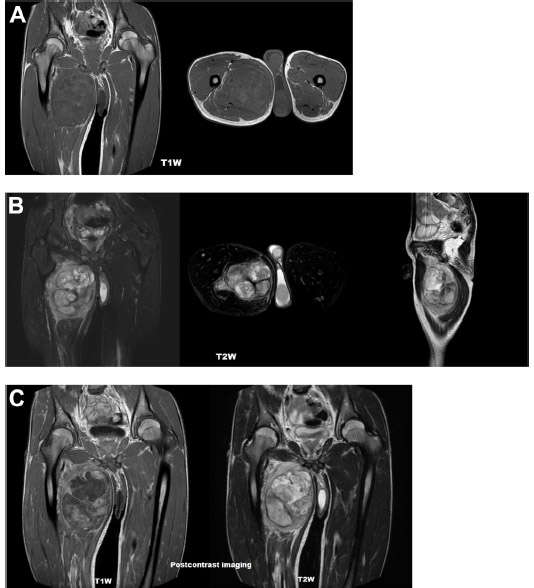 | Figure 2 MRI appearance of a 13×9 cm mass at the anteromedial thigh. |
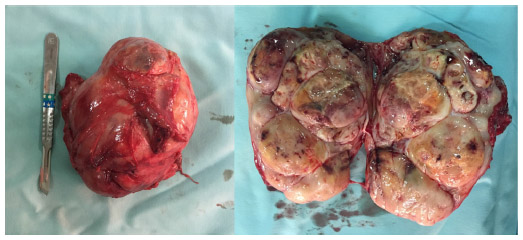 | Figure 3 Macroscopic appearance of the mass after surgical excision. |
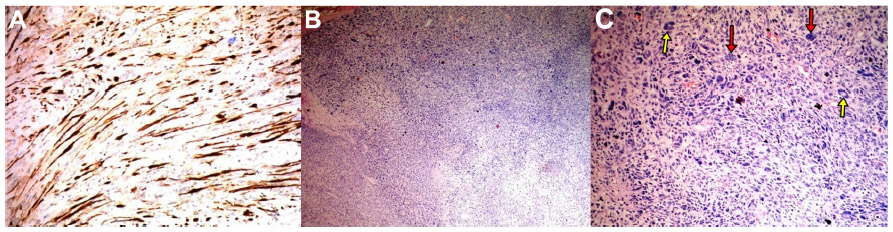 | Figure 4 Appearance after histopathological and immunochemical staining. |
Discussion
MPNSTs are rare malignant tumors originating from peripheral nerve sheath cells. They may behave aggressively and make up 5%–10% of all soft tissue sarcomas.2,4,7,11–13 MPNSTs may arise from all peripheral nerves and frequently reported localizations are the extremities, trunk, head, and neck.1,2,5,9,11,13,14 In patients with neurofibromatosis, MPNST most frequently occurs at ages of 30–40 years. When compared with benign tendon sheath tumors, MPNSTs more frequently occur at extremities.15 MPNSTs are rare in childhood and have an poor prognosis.5,10 Although MPNST can be found in any part of the body, extremity involvement is observed less frequently in pediatric cases.3,5,6,16 Patients with MPNST frequently are admitted with complaints of a palpable mass, pain, and/or other neurological symptoms due to neuronal involvement.11 Solitary and/or plexiform neurofibromas in teenagers or children can be confused with MPNSTs, but signs of atypia or significant mitotic activity are not present.17–21 Malignant transformation and recurrence of the solitary neurofibromas without neurofibromatosis are unusual.20 Other cases of solitary and/or plexiform neurofibroma in pediatric patients without neurofibromatosis have been reported in body regions with slightly less frequency. These include the mons pubis,17 eyelid dermis,18 intrascrotal and extratesticular sites,19 abdominal wall,20 and parotid region.17,21
MPNSTs can resemble benign tumors, both histologically and radiologically.4 Demir et al5 suggested that the diagnosis is easily made in patients without NF1 who present with a palpable mass and pain, but diagnosis is frequently delayed in patients with NF1 due to confusion of these lesions as neurofibroma and/or plexiform neurofibroma. To make an early diagnosis of MPNST, MRI evaluation and biopsy should be performed immediately when malignancy is suspected.22,23 However, both plexiform neurofibroma and MPNSTs have similar MRI findings, which demonstrates areas of low-to-intermediate signal intensity on T1W sequence and high signal intensity on T2W sequence with heterogeneous enhancement.21 Wasa et al24 reported that patients with MPNST generally have giant masses, and presence of peripheral enhancement pattern, perilesional edema-like zone, and intratumoral cystic lesions are observed more frequently. Moreover, they reported that heterogeneity in T1-weighted images support MPNST in patients with NF1. In addition, presence of markedly increased nodularity also suggests malignancy.25 Recent studies suggest that positron emission tomography–computed tomography (CT) scanning is especially helpful to differentiate MPNST from neurofibroma and/or plexiform neurofibroma.16,22–25 Hemorrhagic or necrotic heterogeneity in MRI or CT may suggest malignancy, but this finding may also be observed in benign peripheral nerve sheath tumors.10,21 Garg et al17 reported that radiological investigations and fine-needle-aspiration cytology may not be very helpful for diagnosis. The rarity of positive cytology may be secondary to the adhesive nature of cells in such tumors. Microscopy reveals proliferation of all elements of nerves, including axons, Schwann cells, and fibroblasts. Immunoreactivity for S-100 protein, NSE, and VIM is characteristic.17 Our case had findings of NF1. After MRI, adequate sample was obtained with fine-needle-aspiration biopsy due to suspicion of malignancy. Histopathological evaluation was consistent with benign neurofibroma, and therefore total surgical excision was planned. But histopathological evaluation of the excised material was interpreted as MPNST with malignant cells at the surgical border. So revision surgery was planned and oncological consultation was obtained. We suggest that malignancy should be suspected in NF1 cases that show masses with heterogeneous contrast involvement and occasional cystic and necrotic places. Radical surgical excision should be considered in such cases.
Histopathological evaluation of the needle biopsy material may yield a false diagnosis, but larger samples that are obtained after surgery show marked nuclear polymorphism, mitosis, vascular invasion, necrosis, hypercellularity, a high proliferative index (immunopositivity to Ki67), and many more atypical spindle cells.3,12,25,26 The minimum amount of histological examination to establish diagnosis includes immunohistochemical stains for DES, MYOG, VIM, S-100, proliferative activity marker MIB-1, potential expression of p53, cerbB2, p27, p16 oncogenes, or tumor suppressor genes. Expression of p53 plays an important role in the evolution of MPNST.24,27,28 Our case had histopathological and immunochemical findings similar to the earlier findings, and diagnosis of MPNST (Grade-3; FNCLCC) was made.
Despite the fact that MPNSTs have high local recurrence rates and are generally associated with poor prognosis, little is known about prognostic factors or effective clinical management for this tumor type.4,10,13,16 The most significant factors affecting the prognosis of MPNST in the pediatric age group are surgical resectability, local invasiveness, tumor size (>5 cm), extent of surgery, Intergroup Rhabdomyosarcoma Study grade, tumor location (central vs peripheral), presence of NF1, and adjuvant treatment.2,5,7,9,15,22 Recurrence is seen in 75% of the cases, and 5-year survival ranges between 16% and 53%. Poor prognosis is associated with a tumor size >5 cm, subtotal resection of tumor, and preexisting NF1.7,29,30
Because MPNST is an aggressive tumor, complete surgical resection is necessary for a successful treatment. However, complete surgical resection is rarely possible, and the most important reason of treatment failure is the difficulty in achieving local control. Information about clinical management of this tumor is sparse because there are very few reports in the literature, and this tumor is rarely seen in the pediatric population.2 Although response to radiotherapy and chemotherapy is weak, adjuvant radiotherapy and chemotherapy are frequently required for MPNST patients.2,4,14,22 Carli et al2 reported that postoperative radiotherapy may help to achieve local control in patients with minimal residual tumors. In addition, they reported that chemotherapy may be effective in tumors thought to be unresectable.2,12 Because MPNST has greater chance for recurrence than other sarcomas, current treatment is radical surgical resection. Recurrence may occur years after the surgery; therefore, long period of follow-up is warranted.2,3,7,25
Conclusion
In conclusion, MPNST is a rare, soft tissue sarcoma of childhood and adolescence, which may be misdiagnosed as neurofibroma or plexiform neurofibroma in patients with NF1. Especially, a giant mass and MRI features of heterogeneous involvement, along with a cystic and nodular mass, should suggest malignancy. The main target of surgical treatment is complete surgical excision. In young patients with MPNST, the most important risk factor is the presence of NF, and in such cases, total excision of the mass is very difficult.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
deCou JM, Rao BN, Parham DM, et al. Malignant peripheral nerve sheath tumors: the St Jude Children’s Research Hospital experience. Ann Surg Oncol. 1995;2(6):524–529. | |
Carli M, Ferrari A, Mattke A, et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol. 2005;23(33):8422–8430. | |
Pourtsidis A, Doganis D, Baka M, et al. Malignant peripheral nerve sheath tumors in children with neurofibromatosis type 1. Case Rep Oncol Med. 2014;2014:843749. | |
Amirian ES, Goodman JC, New P, Scheurer ME. Pediatric and adultmalignant peripheral nerve sheath tumors: an analysis of data from the surveillance, epidemiology, and end results program. J Neurooncol. 2014;116(3):609–616. | |
Demir HA, Varan A, Yalçn B, Akyüz C, Kutluk T, Büyükpamukçu M. Malignant peripheral nerve sheath tumors in childhood: 13 cases from a single center. J Pediatr Hematol Oncol. 2012;34(3):204–207. | |
Wu L, Deng X, Yang C, Xu Y. Spinal intradural malignant peripheral nerve sheath tumor in a child with neurofibromatosis type 2: the first reported case and literature review. Turk Neurosurg. 2014;24(1):135–139. | |
Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006–2021. | |
Okada K, Hasegawa T, Tajino T, et al. Clinical relevance of pathological grades of malignant peripheral nerve sheath tumor: a multi-institution TMTS study of 56 cases in Northern Japan. Ann Surg Oncol. 2007; 14(12):597–604. | |
Neville H, Corpron C, Blakely ML, Andrassy R. Pediatric neurofibrosarcoma. J Pediatr Surg. 2003;38(3):343–346. | |
Gupta G, Maniker A. Malignant peripheral nerve sheath tumors. Neurosurg Focus. 2007;22(6):E12. | |
D’Augustino AN, Soule EH, Miller RH. Sarcomas of the peripheral nerves and somatic soft tissues associated with multiple neurofibromatosis (von Recklinghausen’s disease). Cancer. 1963;16:1015–1027. | |
Gahlot GP, Mridha AR, Nath D, Khan SA, Gamanagatti S. Intraosseous primary malignant peripheral nerve sheath tumor of the calcaneus: an unusual case and review of literature. Indian J Pathol Microbiol. 2015;58(2):220–222. | |
Ferrari A, Bisogno G, Macaluso A, et al. Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer. 2007;109(7):1406–1412. | |
Kim JG, Sung WJ, Kim DH, Kim YH, Sohn SK, Lee KB. Malignant peripheral nerve sheath tumor in neurofibromatosis type I: unusualpresentation of intraabdominal or intrathoracicmass. Korean J Intern Med. 2005;20(1):100–104. | |
Casanova M, Ferrari A, Spreafico F, et al. Malignant peripheral nerve sheath tumors in children: a single-institution twenty-year experience. J Pediatr Hematol Oncol. 1999;21(6):509–513. | |
Yoon JH, Lee HS, Chun JI, Park SY, Park HJ, Park BK. Huge intrathoracic malignant peripheral nerve sheath tumor in an adolescent with neurofibromatosis type 1. Case Rep Pediatr. 2014;2014: 951252. | |
Garg C, Agrawal A, Agrawal R, Kumar P. Neurofibroma at unusual locations: report of two cases in teenage girls. J Clin Diagn Res. 2015;9(4):ED03–ED4. | |
Stagner AM, Jakobiec FA. Peripheral nerve sheath tumors of the eyelid dermis: a clinicopathologic and immunohistochemical analysis. Ophthal Plast Reconstr Surg. 2015. Epub 2015 Mar 18. | |
Moradi A, Kazemzadehazad B. A childhood case of solitary intrascrotal and extratesticular neurofibroma. Urol J. 2015;12(1):2042–2043. | |
Barajas-Gamboa JS, Flórez-Salamanca L. Solitary neurofibroma in the abdominal wall of a patient without neurofibromatosis: case report. Biomedica. 2009;29(4):501–505. | |
Khalid M, Khalid S, Zaheer S, Bhatnagar S, Ahmad M. Solitary parotid plexiform neurofibroma – diagnostic difficulty in a clinically unsuspected case. Indian J Cancer. 2014;51(3):373–374. | |
Gnanalingham K, Bhattacharjee S, O’Neill K. Intraosseousmalignant peripheral nerve sheath tumor (MPNST) of the thoracicspine: a rarecause of spinal cord compression. Spine (Phila Pa 1976). 2004;29(18):E402–E405. | |
Moharir M, London K, Howman-Giles R, North K. Utility of positron emittion tomography for tumor surveillance in children with neurofibromatosis type 1. Eur J Nucl Med Mol Imaging. 2010;37(7):1309–1317. | |
Wasa J, Nishida Y, Tsukushi S, et al. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010;194(6):1568–1574. | |
Hyodo T, Sugawara Y, Sakayama K, Kito K. Early-stage malignant peripheral nerve sheath tumour arising from a solitary neurofibroma. Br J Radiol. 2012;85(1010):e26–e30. | |
Wick MR, Swanson PE, Scheithauer BW, Manivel JC. Malignant peripheral nerve sheath tumour: an immunohistochemical study of 62 cases. Am J Clin Pathol. 1987;87(4):425–433. | |
Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62(5):1573–1577. | |
Kosmas C, Tsakonas G, Evgenidi K, et al. Malignant peripheral nerve sheath tumor in neurofibromatosis type-1: twocasereports. Cases J. 2009;2:7612. | |
Sordillo PP, Helson L, Hajdu SI, et al. Malignant schwannoma – clinical characteristics, survival, a response to therapy. Cancer. 1981; 47(10):2503–2509. | |
Wong WW, Hirose T, Scheithauer BW, Schild SE, Gunderson LL. Malignant peripheral nerve sheath tumour: analysis of treatment outcome. Int J Radiat Oncol Biol Phys. 1998;42(2):351–360. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.