")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Gephyrin and CYP2C9 Genetic Polymorphisms in Patients with Pharmacoresistant Epilepsy
Authors El-Tallawy HN, Abuhamdah S , Nassar AY, Farghaly WMA , Saleem TH , Atta SA, Sayed AA, Tohamy AM, Hassan MH
Received 4 July 2021
Accepted for publication 25 August 2021
Published 9 September 2021 Volume 2021:14 Pages 1133—1140
DOI https://doi.org/10.2147/PGPM.S327808
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Hamdy N El-Tallawy,1 Sawsan Abuhamdah,2,3 Ahmed Y Nassar,4 Wafaa MA Farghaly,1 Tahia H Saleem,4 Sara A Atta,4 Ayat A Sayed,4 Amal M Tohamy,1 Mohammed H Hassan5
1Department of Neurology and Psychiatry, Faculty of Medicine, Assiut University, Assiut, Egypt; 2Department of Pharmaceutical Sciences, College of Pharmacy, Al Ain University, Abu Dhabi, United Arab Emirates; 3Department of Biopharmaceutics and Clinical Pharmacy, Faculty of Pharmacy, University of Jordan, Amman, Jordan; 4Department of Medical Biochemistry, Faculty of Medicine, Assiut University, Assiut, Egypt; 5Department of Medical Biochemistry, Faculty of Medicine, South Valley University, Qena, 83523, Egypt
Correspondence: Mohammed H Hassan Email [email protected]
Purpose: Gephyrin (GPHN) is an essential protein in the regulation of inhibitory postsynaptic density and polymorphism in the corresponding gene may have a role in the development of pharmacoresistant epilepsy (PRE). For the first time, we aimed to evaluate the association of rs928553T/C variants with PRE susceptibility. Moreover, we have analyzed the genetic polymorphism affecting CYP2C9 “rs12782374G/A” in the same population to detect the effect of SNP on the drug-metabolizing ability of patients with PRE.
Patients and Methods: This case-control study enrolled 100 patients (group A) and 100 healthy, age and sex-matched controls, unrelated to patients (group B). TaqMan™ assays using real-time PCR were run for genotyping of rs928553T/C and rs12782374G/A in all participants.
Results: GPHN T>C polymorphism revealed significant risk association with occurrence of PRE using dominant, recessive and codominant models as follows: TT vs (TC+CC): OR 0.23, 95%CI: 0.13– 0.43, P< 0.001. In addition, (TT+TC vs CC): OR 0.38, 95%CI: 0.18– 0.77, P< 0.001. Also, T vs C (OR 0.34, 95%CI: 0.22– 0.51, P=< 0.001). Similarly, CYP2C9 G>A polymorphism showed a significant increased risk of PRE (GG vs (GA+AA): OR 0.11, 95%CI: 0.05– 0.23, P< 0.001). Furthermore, (GG+GA vs AA): OR 0.18, 95%CI: 0.084– 0.39, P< 0.001. Also, G vs A (OR 0.24, 95%CI: 0.15– 0.366, P=< 0.001).
Conclusion: Mutation of both GPHN (rs928553) and CYP2C9 (rs1278237) genes may be implicated as a genetic mediators of resistance in patients with PRE.
Keywords: pharmacoresistant epilepsy, rs928553T/C, rs12782374G/A, real time-PCR
Introduction
The debilitating health issue of pharmacoresistant epilepsy (PRE) is associated with elevated morbidity and mortality, including neuropsychological symptoms, and a group of adverse effects associated with antiepileptic drugs (AEDs).1 Epilepsy affects over 70 million people worldwide. One-third of those patients exhibit resistance to pharmacotherapy with the highest percentage in patients with focal epilepsies, whereup to 60% of them manifest resistance to AEDs.2,3 Pharmacoresistant epilepsy defined (according to the International League Against Epilepsy, ILAE, 2010) as the failure of responding to adequate trials of two tolerated and appropriately chosen AEDs (monotherapy or in combination).4,5 Another definition of pharmacoresistant epilepsy by the ILAE in 2011, as the presence of seizures in the previous six months, even under proper therapeutic regimens (either monotherapy or in combination).5 The prevalence of definite intractable epilepsy in Al-Kharga District, New Valley (Egypt) was 0.7/1000.6
The gene variation hypothesis, predictably, proposes that abnormalities (mutations or polymorphisms) in genes producing enzymes, receptors, ion channels, and other critical components may have an essential role in determining the phenotype of epilepsy.7 CYP2C9 is an isoform of phase I drug-metabolizing cytochrome P450 (CYP450) enzyme, which plays a major role in the oxidation of both xenobiotics and endogenous compounds.8 It is an essential source of variability in drug-response and plays a role not only in phase I-dependent metabolism of xenobiotics, but also in the metabolism of endogenous compounds such as steroids, vitamins, fatty acids, and over 90% of drugs with a narrow therapeutic index (eg warfarin, phenytoin, and valproic acid).9,10 The gene coding for the CYP2C9 enzyme is clustered in a 500 kb region on chromosome 10q24 and characterized by being highly polymorphic.11,12 The CYP2C9 polymorphism alleles in epileptic patients range from 4.5 to 13.6%, being less frequent in African Americans and Asians.13 Genetic variants in CYP2C9 lead to changes in metabolic activity and accordingly low enzyme activity that play a major role in the pathogenesis of adverse drug reactions and significant differences in AED serum concentrations.11,14 For example, because CYP2C9 is responsible for 90% of phenytoin metabolism, a variation in this gene can reduce phenytoin metabolism, increasing the risk of concentration-dependent toxicity and other negative side effects.15,16
Gephyrin (GPHN) is an essential core scaffolding protein responsible for organizing the inhibitory glycine and γ-aminobutyric acid type A (GABAA) receptors at postsynaptic density.17,18 They ensure the accurate accumulation of those receptors in precise apposition to presynaptic release sites, also interact with cytoskeletal anchoring elements to provide a physical platform for maintaining receptors at synapses and regulate downstream signaling pathways.18 Besides its synaptic function, gephyrin has also a role in the biosynthesis of the molybdenum cofactor (MoCo),19 which is required for the activity of four molybdo enzymes that catalyze redox reactions. Mutations in GPHN can cause MoCo deficiency that is characterized by untreatable neonatal seizures starting at birth.17 So it is not surprising that gephyrin dysfunction and deletion can result in the expression of truncated gephyrin variants in patients with epilepsy and many other neurological disorders such as autism, schizophrenia, and seizures.20 The aim of the current study aimed is to identify the role of polymorphisms in GPHN as a risk factor for development of PRE and genetic variants of CYP2C9 enzyme that may play a major role in pathogenesis of therapeutic failure to anti-epileptic drugs in patients with PRE.
Patients and Methods
Study Design and Participants
This case-control study has been conducted on 200 subjects categorized into two groups: 100 patients with PRE (group A) who were selected from the outpatients’ epilepsy clinics and inpatients departments of Neurology and Psychiatry, Assiut University Hospital, in addition to 100 healthy, age and sex-matched controls unrelated to patients (group B) who were selected randomly from the same population as the patients, with no known personal history or family history of epilepsy or any neurological or psychiatric diseases. The study was performed in collaboration with the Medical Biochemistry department of Faculties of Medicine, Assiut and South Valley Universities, Egypt. The recruitment of patients and controls in the present study was done in accordance with the guidelines laid down in the Declaration of Helsinki. The study was conducted in the period from March 2016 to July 2019 and the protocol was approved by the Ethics Committee of the Faculty of Medicine, Assiut University, Egypt (code number: IRB00008718). All participants gave written informed consent after the purpose and method of the study were briefly discussed. Sample size calculation was carried out using G*Power 3 software. A calculated sample of 98 respondents (cases or controls) was needed, with an error probability of 0.05 and 80% power on one-tailed test (type I error).
Estimation of serum level of AEDs was done for those with probable medically uncontrolled seizures to ensure adequate dose compliance at therapeutic drug monitoring unit, Assiut University Hospitals, Egypt. Epileptic patients with other types of neurological diseases, chronic medical diseases (cardiac, hepatic or renal or malignancies, or those currently pregnant or breast-feeding) all were excluded from the study. None of the controls had a history of the cerebrovascular, nervous system, or any chronic disease excluded by medical history and classical EEG.
Clinical Assessments and Data Collection
Diagnosis of PRE is dependent on the presence of uncontrolled seizures with an average frequency of one seizure per month for two years with the usage of at least three different tolerated and appropriately chosen AEDs (singly or in combination),21 in association with video EEG by specialized neurologist done at the epidemiological center for neurological disease, Faculty of Medicine, Assiut University. The medical history and diagnosis of each patient were revised by the two qualified neurologists participating in the study. Data collected before genotyping including: patient age and gender, family history of epilepsy, and age at the onset of epilepsy.
Genomic DNA Extraction and TaqMan™ SNPs Genotyping Assays
Five millimeters of venous blood collected in EDTA tubes was obtained from all patients and controls. DNA was extracted from whole blood using Qiagen (QIAamp DNA blood mini kits for genomic DNA purification, catalogue no. 51104 supplied by Qiagen, Hilden, Germany), according to the guidelines of the manufacturer. DNA samples were genotyped by Rotor-Gene Q, Applied Biosystems (Foster City, CA, USA) using an automated method on purified genomic DNA by a TaqMan™ assay specifically designed to distinguish the variants of both gephyrin T/C (rs928553) and CYP2C9 G/A (rs12782374) polymorphisms using fluorescent-labeled probes (VIC/FAM) GCGCTTTCAGTGGGGCGGCCGGCCA[C/T]TCCCGAGGGTTGCAAGAGGAGGCCC and GGCTACATACTGTATGATTCCAACC[A/G]TATTACATTTTGGAAAAGGCAAAAC respectively with their catalogue no. 4351379 and 4351379 respectively, supplied by Thermo Fisher Scientific, Waltham, MA, USA. DNA was genotyped according to the manufacturer’s protocol. Briefly, the reaction components for each genotyping reaction were as follows: 10 ng of DNA, 5 µL of TaqMan™ Genotyping Master Mix, 0.5 µL of assay mix, and water to a total volume of 10 µL. The thermocycler conditions were as follows: 50°C for 2 min, 95°C for 10 min, and 50 cycles of 92°C for 15 seconds and 60°C for 90 seconds. Randomized sample duplications between plates were set to check the accuracy of the genotyping. “No-template negative controls” (NTCs) were applied in each PCR run to confirm free reaction contamination. The allele distribution and amplification plots of rs928553 and rs12782374 were demonstrated in (Figure 1A–C).
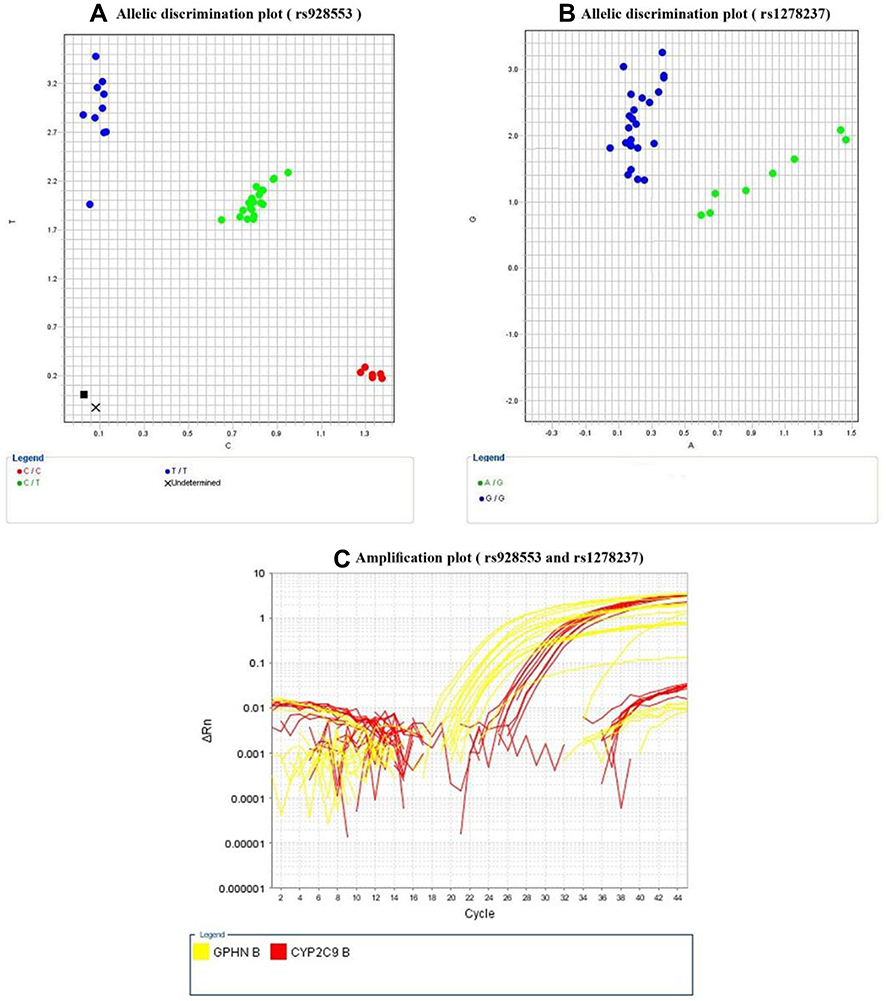 |
Figure 1 Allelic discrimination plots for rs928553 (A) where blue dots represent wild homozygous genotype (TT), and green dots represent mutant heterozygous genotype (TC) and the red dots represent mutant homozygous genotype (CC), and rs1278237 (B) where blue dots represent wild homozygous genotype (GG), and green dots represent mutant heterozygous genotype (GA); amplification plots for both rs928553 and rs1278237 (C), using real time-PCR. |
Statistical Analysis
Date entry and data analysis were done using SPSS version 22 (Statistical Package for Social Science). Data were presented as number, percentage, mean, standard deviation. Chi-squared test and Fisher's Exact test were used to compare qualitative variables. Odds ratio (OR) with 95% confidence intervals (CI) was calculated. Pearson's correlation was done to measure correlation between quantitative variables. The following models were used to test multiple logistic regression analysis and Fisher’s exact test: dominant (major allele homozygotes vs heterozygotes+minor allele homozygotes), recessive (major allele homozygotes+heterozygotes vs minor allele homozygotes), and codominant (major allele homozygotes vs heterozygote and minor allele homozygotes vs major allele homozygotes). P-value considered statistically significant when P<0.05. The studied SNPs were followed the Hardy–Weinberg (HW) equation.22,23
Results
Demographic and Clinical Data of the Study Groups
One hundred patients with PRE, and an equal number of healthy unrelated controls were included in the study. The mean age of PRE patients and controls were (22.56±9.45, and 22.09±6.78 years respectively). There was male predominance among the included PRE patients with male to female ratio 1.8:1. Positive family history of epilepsy was present in PRE patients with negative family history among the controls.
Regarding to the clinical characteristics of the included patients, the age of onset of epilepsy in studied patients was highly frequent in adolescence (32%). The majority of PRE patients have daily (56%) or weekly (42%) fits. Twenty percent have status epilepticus with positive consanguinity in 58% and positive family history of epilepsy was present in 40% of patients, (Table 1). The included PRE patients received AEDs (Depakine®, Trileptal®, oxaleptal, apetryl, oxycarbazine) in different combinations. The clinical types of epileptic seizures of the included patients were also presented in (Table 1).
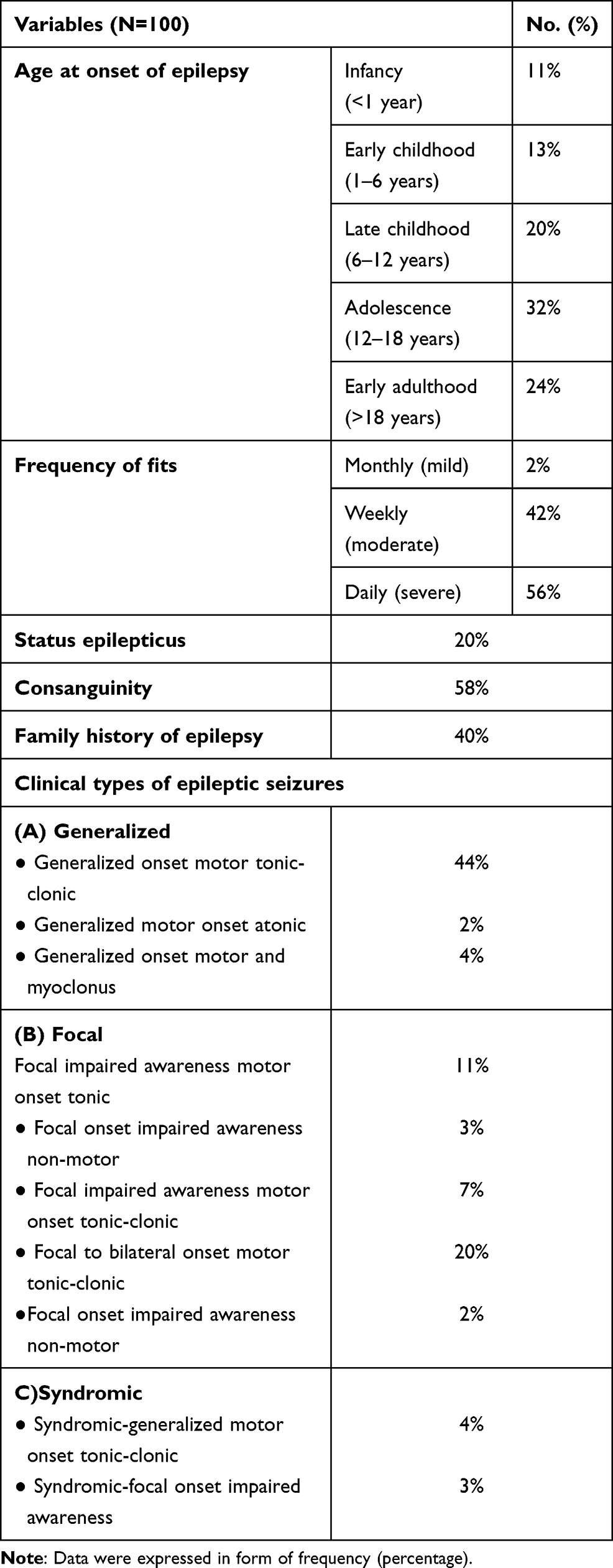 |
Table 1 Clinical Characteristics of PRE Patients |
Allelic and Genotype Frequencies of GPHN T>C in the Study Groups
The genotypic distribution of GPHN T>C (rs 928553) polymorphism and allelic frequency of T and C alleles in patients and unrelated healthy controls have been described in (Table 2). The frequencies of GPHN T>C genotypes determined from PRE were 30.2% for TT, 62.9% for TC and 68.2% for CC, while in unrelated healthy controls, the frequency of TT, TC and CC were present in a percentage of 69.8%, 37.1%, and 31.8%, respectively (P<0.001), with significantly higher allelic frequency for (C) among patients vs unrelated healthy controls (52% and 27%, respectively) and significantly higher frequency for T allele among the unrelated healthy controls vs the patients group (73% and 48% respectively). There was significantly higher (TT+TC) frequency among unrelated healthy controls compared to the patients with PRE (55.1% and 44.9%, respectively, P=0.006) with significantly higher frequency of (TC+CC) genotypes among PRE patients vs the unrelated healthy controls (64.9% and 35.1%, respectively, P<0.001). This indicated that GPHN T>C polymorphism is associated with an increased risk of PRE (TT vs (TC+CC): OR 0.23, 95%CI: 0.13–0.43, P<0.001). In addition, (TT+TC vs CC): OR 0.38, 95%CI: 0.18–0.77, P<0.001. Also, T vs C (OR 0.34, 95%CI: 0.22–0.51, P=<0.001).
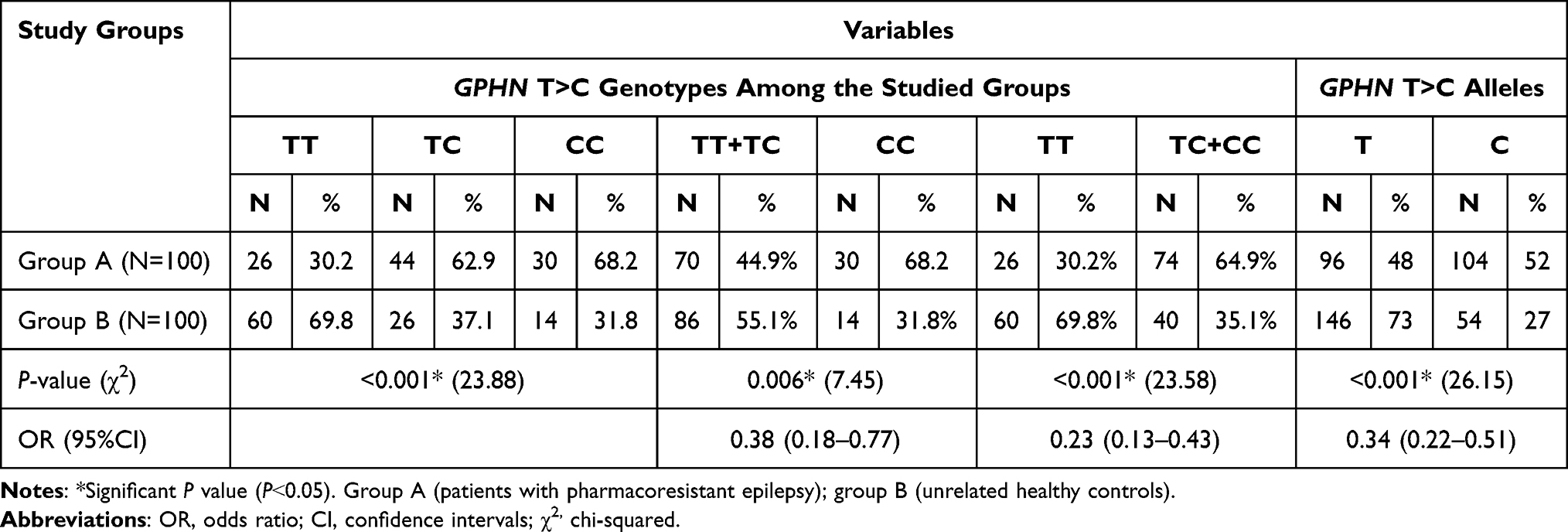 |
Table 2 Genotypes and Alleles Frequencies of GPHN T>C in the Study Groups |
Genotypes and Alleles Frequencies of CYP2C9 G>A in the Study Groups
The genotypic distribution of CYP2C9 G>A (rs2782374) polymorphism and allelic frequency of (G) and (A) alleles in patients and unrelated healthy controls have been described in (Table 3). The frequencies of CYP2C9 G>A genotypes determined from PRE were 16.7% for GG, 56.5% for GA and 79.2% for AA, while in unrelated healthy controls, the frequency of GG, GA and AA were present in a percentage of 83.3%, 43.5%, and 20.8%, respectively (P<0.001), with significantly higher allelic frequency for (A) among patients vs unrelated healthy controls (64% and 30%, respectively) and significantly higher frequency for G allele among the unrelated healthy controls vs the patients group (70% and 36%, respectively). There was significantly higher (GG+GA) frequency among unrelated healthy controls compared to the patients with PRE (59.2% and 40.8%, respectively, P<0.001) with significantly higher frequency of (GA+AA) genotypes among PRE patients vs the unrelated healthy controls (64.3% and 35.7%, respectively, P<0.001). This indicated that CYP2C9 G>A polymorphism is associated with an increased risk of PRE (GG vs (GA+AA): OR 0.11, 95%CI: 0.05–0.23, P<0.001). In addition, (GG+GA vs AA): OR 0.18, 95%CI: 0.084–0.39, P<0.001. Also, G vs A (OR 0.24, 95%CI: 0.15–0.366, P=<0.001).
 |
Table 3 Genotypes and Alleles Frequencies of CYP2C9 G>A in the Study Groups |
Discussion
Although monogenic polymorphism can explain part of the mechanism of pharmacoresistance, most genes are multifactorial-controlled including gene regulatory sequences. So the current study included two gene polymorphisms acting on two different proteins gephyrin and cytochrome P2C9. Many studies tried to find out the genetic and nongenetic causes of PRE due to the continued presence of disease despite the usage of many drugs at maximum tolerated dose.5 Although any structural lesion can lead to pharmacoresistance, its occurrence in the absence of a structural lesion could be due to genetic cause. Even in presence of structural causes, the course of disease and response to treatment is difficult to predict. Therefore, mutation of genes encoding proteins responsible for the health of synaptic transmission as ion channels or polymorphism in neurotransmitter receptors is the major cause of pharmacoresistance.24
There are many factors affecting neurotransmitter level and their impact on neuronal transduction including genes responsible for synthesis of neurotransmitter or their receptors. Gephyrin gene is one of scaffolding proteins responsible for clustering of those amino acids and neurotransmitters, that analyzed in our study and found that the mutant alleles T>C and C>C were significantly frequent in patients compared to unrelated healthy controls. A study by Dejanovic et al20 showed that stress-induced irregular splicing of GPHN results in the expression of abnormal gephyrin variants with defective function as identified in epilepsy, autism, and schizophrenia that go with the results of our study.
It was found that CYP2C9 genetic polymorphism can lead to a significant change in metabolic activity and so differences in metabolism of AEDs especially, valproic acid.25 These findings are consistent with our results where there is a significant increase in mutated genotypes G>C and C>C in patients compared to controls. Change in phenotype within and between different individuals may be due to change in genotype both in regulatory and exonic sequences. Furthermore, accelerated metabolism of valproic acid and other AEDs may occur with concurrent administration of other drugs that leads to displacement of the first from protein binding and decrease blood concentration.26
Conclusion
This study revealed that the genetic variation of GPHN “rs928553T/C” and CYP2C9 “rs12782374G/A” could be molecular determinants for increased risk of PRE patients. Further larger scale studies are required to confirm the significant association of the studied variants with PRE susceptibility and to clarify the role of these polymorphisms as potential biomarkers of responsiveness to antiepileptic drugs and their molecular mechanisms, and thus may help in early expectation and proper management strategies of such patients.
Study Limitations
Lack of comparison with other experimental group including patients with nondrug-resistant epilepsy was the main study limitation that could be included in future studies. Another study limitation was the small sample size, which impeded further examination of the studied gene polymorphisms in relation to the clinical characteristics of the patients.
Data Sharing Statement
The datasets used and/or analyzed in this study are available from the corresponding author on reasonable request.
Author Contributions
Study concept and design: HNE-T, SA, AYN, THS, WMAF, SAA, AAS, AMT and MHH; clinical assessments and data collection: HNE-T, WMAF, and AMT; blood sampling and genetic assays: AAS, SAA and MHH; statistical analysis: AAS and MHH; literature research: HNE-T, SA, AYN, THS, WMAF, SAA, AAS, AMT and MHH; first manuscript drafting: MHH. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The current research has been funded by Faculties of Medcine, Assiut and South Valley Universities, Egypt.
Disclosure
The authors report no competing interests in this work.
References
1. Alexopoulos AV. Pharmacoresistant epilepsy: definition and explanation. Epileptology. 2013;1(1):38–42. doi:10.1016/j.epilep.2013.01.001
2. Fountain NB, Van Ness PC, Swain-Eng R, Tonn S, Bever CT
3. Tang F, Hartz AMS, Bauer B. Drug-resistant epilepsy: multiple hypotheses, few answers. Front Neurol. 2017;8:301. doi:10.3389/fneur.2017.00301
4. Berg AT, Kelly MM. Defining intractability: comparisons among published definitions. Epilepsia. 2006;47(2):431–436. doi:10.1111/j.1528-1167.2006.00440.x
5. Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia. 2010;51(6):1069–1077. doi:10.1111/j.1528-1167.2009.02397.x
6. Farghaly WM, El-Tallawy HN, Rageh TA, et al. Epidemiology of uncontrolled epilepsy in the Al-Kharga District, New Valley, Egypt. Seizure. 2013;22(8):611–616. doi:10.1016/j.seizure.2013.04.010
7. Balestrini S, Sisodiya SM. Pharmacogenomics in epilepsy. Neurosci Lett. 2018;667:27–39. doi:10.1016/j.neulet.2017.01.014
8. Rettie AE, Jones JP. Clinical and toxicological relevance of CYP2C9: drug-drug interactions and pharmacogenetics. Annu Rev Pharmacol Toxicol. 2005;45(1):477–494. doi:10.1146/annurev.pharmtox.45.120403.095821
9. Lee CR, Goldstein JA, Pieper JA. Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics. 2002;12(3):251–263. doi:10.1097/00008571-200204000-00010
10. Arici M, Özhan G. CYP2C9, CYPC19 and CYP2D6 gene profiles and gene susceptibility to drug response and toxicity in Turkish population. Saudi Pharm J. 2017;25(3):376–380. doi:10.1016/j.jsps.2016.09.003
11. Pirmohamed M, Park BK. Cytochrome P450 enzyme polymorphisms and adverse drug reactions. Toxicology. 2003;192(1):23–32. doi:10.1016/s0300-483x(03)00247-6
12. Naraharisetti SB, Lin YS, Rieder MJ, et al. Human liver expression of CYP2C8: gender, age, and genotype effects. Drug Metab Dispos. 2010;38(6):889–893. doi:10.1124/dmd.109.031542
13. Silvado CE, Terra VC, Twardowschy CA. CYP2C9 polymorphisms in epilepsy: influence on phenytoin treatment. Pharmgenomics Pers Med. 2018;11:51–58. doi:10.2147/PGPM.S108113
14. Lopez-Garcia MA, Feria-Romero IA, Fernando-Serrano H, Escalante-Santiago D, Grijalva I, Orozco-Suarez S. Genetic polymorphisms associated with antiepileptic metabolism. Front Biosci (Elite Ed). 2014;6(2):377–386. doi:10.2741/713
15. Depondt C, Godard P, Espel RS, Da Cruz AL, Lienard P, Pandolfo M. A candidate gene study of antiepileptic drug tolerability and efficacy identifies an association of CYP2C9 variants with phenytoin toxicity. Eur J Neurol. 2011;18(9):1159–1164. doi:10.1111/j.1468-1331.2011.03361.x
16. Chung WH, Chang WC, Lee YS, et al. Genetic variants associated with phenytoin-related severe cutaneous adverse reactions. JAMA. 2014;312(5):525–534. doi:10.1001/jama.2014.7859
17. Petrini EM, Ravasenga T, Hausrat TJ, et al. Synaptic recruitment of gephyrin regulates surface GABAA receptor dynamics for the expression of inhibitory LTP. Nat Commun. 2014;5(1):3921. doi:10.1038/ncomms4921
18. Choii G, Ko J. Gephyrin: a central GABAergic synapse organizer. Exp Mol Med. 2015;47(4):e158. doi:10.1038/emm.2015.5
19. Kamdar KP, Shelton ME, Finnerty V. The Drosophila molybdenum cofactor gene cinnamon is homologous to three Escherichia coli cofactor proteins and to the rat protein gephyrin. Genetics. 1994;137(3):791–801. doi:10.1093/genetics/137.3.791
20. Dejanovic B, Djémié T, Grünewald N, et al. Simultaneous impairment of neuronal and metabolic function of mutated gephyrin in a patient with epileptic encephalopathy. EMBO Mol Med. 2015;7(12):1580–1594. doi:10.15252/emmm.201505323
21. Giussani G, Canelli V, Bianchi E, et al. A population-based study of active and drug-resistant epilepsies in Northern Italy. Epilepsy Behav. 2016;55:30–37. doi:10.1016/j.yebeh.2015.11.021
22. Hardy GH. Mendelian proportions in a mixed population. Science. 1908;28(706):49–50. doi:10.1126/science.28.706.49
23. Stark AE. A clarification of the Hardy-Weinberg law. Genetics. 2006;174:1695–1697.
24. Kobow K, Blümcke I. Epigenetics in epilepsy. Neurosci Lett. 2018;667:40–46. doi:10.1016/j.neulet.2017.01.012
25. Monostory K, Nagy A, Tóth K, et al. Relevance of CYP2C9 function in valproate therapy. Curr Neuropharmacol. 2019;17(1):99–106. doi:10.2174/1570159X15666171109143654
26. Morath B, Green K, Zaradzki M, Heid J, Karck M, Hoppe-Tichy T. Subtherapeutic valproic acid plasma concentrations under concomitant dipyrone therapy in an epilepsy patient-a case report. Eur J Clin Pharmacol. 2018;74(5):675–677. doi:10.1007/s00228-018-2417-5
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.