")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Genomic insights into the etiology of Alzheimer's disease: a review
Authors Reitz C
Received 4 March 2014
Accepted for publication 7 April 2014
Published 29 May 2014 Volume 2014:4 Pages 59—66
DOI https://doi.org/10.2147/AGG.S51293
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Christiane Reitz1–3
1Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, 2Gertrude H Sergievsky Center, 3Department of Neurology, Columbia University, New York, NY, USA
Abstract: Over the past decade, studies capitalizing on high-throughput genome technologies have significantly advanced the knowledge on the genetic underpinnings of Alzheimer's disease (AD) by identifying a wide set of pathophysiological mechanisms implicated in the disease in addition to amyloid precursor protein (APP) metabolism. These include: innate immune response and inflammation, lipid metabolism, endocytosis, cell migration, tau pathology, hippocampal synaptic function and axonal transport, regulation of gene expression and posttranslational modification of proteins, and microglial and myeloid cell function. The cumulative population attributable fraction associated with known genetic variants amounts now to ~80%. High-throughput sequencing studies have started to map specific causative variants in these genes and have provided invaluable evidence for an involvement of rare variants in AD, overturning the “common disease–common variant” hypothesis. The ongoing and future large-scale translational studies and next generation whole genome or whole exome sequencing efforts hold the promise of mapping the specific causative variants in these genes; of identifying additional risk variants, including rare and structural variants; and of identifying novel targets for genetic testing, prevention, and treatment.
Keywords: genetics, gene, variation, polymorphism, genome-wide association study, sequencing
Introduction
Late-onset Alzheimer’s disease (LOAD) is the most frequent form of dementia in Western societies leading to a considerable health care burden. It is estimated that 17 million people worldwide have Alzheimer’s disease (AD).1 In the US alone, five million people have been affected, leading to a direct estimated health care cost of US$157–$215 billion dollars per year.1,2 The annual incidence rate of AD increases from 1% among people aged 65 years to approximately 8% for people aged 85 years and older. The duration of illness can be as long as 20 years, but the average is between 4 and 8 years.3 The aging of those born in the post-World War II era suggests that these numbers may triple by the year 2050, resulting in an increase of nearly 80% in total societal costs per adult.2
AD typically begins with the onset of symptoms after age 60 and evolves slowly from mildly impaired memory function to severe cognitive loss, finally terminating inevitably in complete incapacity and death. Although in recent years there have been significant advances in biomarkers for the disease (plasma amyloid beta [Abeta], cerebrospinal fluid [CSF] Abeta and tau, and amyloid imaging)4–6 and prediction of cognitive decline, to date, there are no definitive diagnostic tests or biological markers of the disease, and the diagnosis during life is based on a clinical examination. At death, the pathological manifestations in brain include deposits of extracellular β-amyloid protein (Aβ) in diffuse plaques and plaques containing elements of degenerating neurons (“neuritic plaques”). Intracellular changes include deposits of abnormally hyperphosphorylated tau protein, a microtubule assembly protein, in the form of neurofibrillary tangles. Activation of microglia and loss of neurons and synapses is also widespread.
Efforts to limit the impact of AD are being hindered by the lack of success of experimental drugs, which is attributable to incomplete characterization of the basic pathologic mechanisms. It is clear that there is a significant genetic component, with a heritability of 58% to 79%.7–9 Consequently, determining which genes and gene networks contribute to AD risk is expected to reveal major basic pathogenic mechanisms, highlighting key proteins and pathways for drug development (“druggable targets”); and to inform the development of genetic testing methods for identifying those at greatest risk for AD when preventive measures become available.
In recent years, the genetic analysis of LOAD has focused on the identification of common variants through genome-wide association studies (GWASs) and has identified several novel susceptibility genes, implicating specific pathways in the disease. This review article summarizes the current genomic insight into AD and provides suggestions for future research.
Data source and study selection
The primary sources of the studies addressed in this review article were abstracts and full-text articles published in English in the PubMed database between January 2008 and 2014. The key words used for searching PubMed were “Alzheimer’s disease”, “dementia”, “gene”, “genetics”, “genomics”, “epigenetics”, “endophenotype”, “genome-wide association study”, “rare variants”, “common variants”, and “sequencing”. The retrieved abstracts were read to identify studies addressing the topics included in this review. We also performed a manual search of references cited in published articles. The studies were read in their entirety to assess their appropriateness for inclusion in this article.
Genetic epidemiology of Alzheimer’s disease
Consistent with the role of a significant genetic contribution, the heritability of AD is 58% to 79%, and a positive family history is the strongest risk factor.10,11 Families that are multiply affected by AD are at increased risk for dementia, but the distribution of secondary cases is not consistent with Mendelian inheritance. AD is more frequent among monozygotic than dizygotic twins,9,10,12 and first-degree relatives of patients with AD have approximately twice the expected lifetime risk of developing the disease.
Studies of numerous large pedigrees with early-onset AD (onset age: 30–50 years) led to the discovery of autosomal dominant mutations in the amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes.13–15 These studies suggested a common pathogenic mechanism involving enhanced generation and aggregation of Aβ. According to this “amyloid hypothesis”, β-secretase cleaves APP near the N terminus of the Aβ peptide; then, the membrane-bound C-terminal APP fragment is cleaved by γ-secretase, leading to accumulation of Aβ40 and Aβ42.
Role of rare and common variants in Alzheimer’s disease
For several decades, the main hypothesis assumed to underlie genetically complex diseases including AD was the “common disease–common variant” hypothesis. According to this hypothesis, the genetic factors underlying common diseases will be alleles that are themselves quite common in the population at large.16 In line with this notion, over the past several years, the most common strategy for finding novel AD gene candidates has been the GWAS. In GWASs, as many as several million genetic markers (single nucleotide polymorphisms [SNPs]) are tested for genetic association with disease risk and/or phenotypic endophenotypes, such as age of onset, biomarkers, imaging results, and neuropathological end points. These studies, however, have identified loci accounting for only part of the heritability of most complex diseases. Although some of this “missing heritability” may be ascribed to a large number of SNPs with weak effect, there is increasing evidence that there is a substantial contribution from rare variants with large effect that are not readily identifiable by SNP-based methods17,18 unless very large sample sizes (for example, >250,000 unselected individuals) are assessed.19 Thus, in recent work, resequencing of known risk loci and whole genome and whole exome sequencing has been pursued to reveal rare point mutations that may have an appreciable impact on disease risk or severity and explain part of this missing heritability.
Apolipoprotein (APO)E region
For more than a decade, only one genetic risk factor, the APOE-ε4 allele, located on chromosome 19q13, was an unequivocally established “susceptibility” gene in non-Hispanic Whites of European ancestry. APOE is a lipid-binding protein and is expressed in humans as three common isoforms coded for by three alleles, APOE-ε2, -ε3, and -ε4. A single APOE-ε4 allele is associated with a two- to threefold increased risk, and having two copies is associated with a fivefold or more increase.20 In addition, each inherited APOE-ε4 allele lowers the age at onset by 6–7 years.21–28 APOE-ε4 is also associated with lower cognitive performance, in particular the memory domain, and is associated with mild cognitive impairment29–32 and with progression from mild cognitive impairment to dementia.29–39 While the population attributable risk for APOE-ε4 is estimated at 20%–50%,40 the presence of -ε4 is neither necessary nor sufficient for developing the disease.41 In ethnic groups other than non-Hispanic Whites, the association between APOE and LOAD is largely inconsistent across studies.42,43
Findings from candidate gene and genome-wide association studies
Table 1 lists the major GWAS studies conducted. Due to a paucity of data in other ethnic groups, most genetic association studies have restricted their efforts to non-Hispanic White populations. In addition, there are differences in linkage disequilibrium and allele frequencies between ethnic groups, which can lead to genetic background noise and the likelihood of false-positive findings due to confounding in combined analyses. Consequently, the first two sets of large-scale GWASs were performed in individuals of European ancestry and identified CLU, PICALM, CR1, BIN1, MS4A4A, ABCA7, CD2AP, CD33, and EPHA1 as AD susceptibility loci.44–47 CLU, also known as APOJ, is a lipoprotein highly expressed in both the periphery and the brain.48 Like APOE, it is involved in lipid transport.49 CLU is also hypothesized to act as an extracellular chaperone that influences Aβ aggregation and receptor-mediated Aβ clearance by endocytosis.48 Unlike APOE, there are no known coding variants that account for the observed genetic association to CLU, suggesting that genetic variation in expression levels may be responsible for the altered risk for LOAD.50 BIN1 (amphiphysin II) is a member of the Bin1/amphiphysin/RVS167 (BAR) family of genes, which are involved in diverse cellular processes, including actin dynamics, membrane trafficking, and clathrin-mediated endocytosis,51 affecting APP-processing and Aβ-production or Aβ-clearance from brain. PICALM is also involved in clathrin-mediated endocytosis and recruits clathrin and adaptor protein complex 2 to sites of vesicle assembly.52 Complement receptor 1 (CR1) is a cell surface receptor that is part of the complement system. It has binding sites for the complement factors C3b and C4b and is involved in clearing immune complexes containing these two proteins. Since Aβ oligomers can bind C3b, CR1 may participate in the clearance of Aβ. CR1 may also play a role in neuroinflammation, which is a prominent feature in AD.53 Interestingly, CLU may play a role in this process as an inhibitor.54 In summary, this first set of GWASs identified loci mainly clustering in four pathways, namely, immune response, APP-processing, lipid metabolism, and endocytosis/intracellular trafficking.
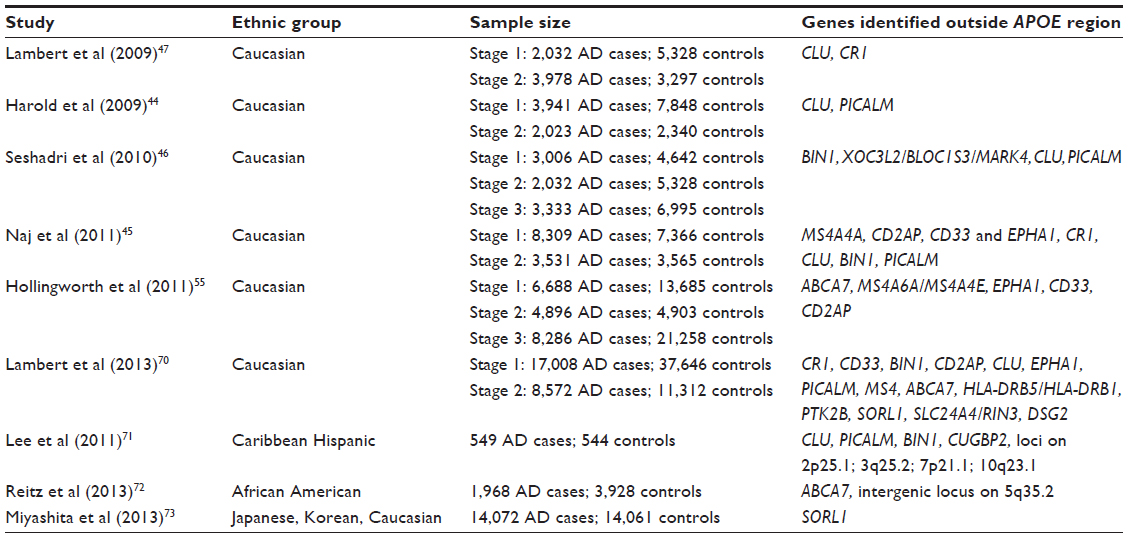 | Table 1 Major LOAD GWASs conducted |
The second set of large GWASs identified additional susceptibility genes (CD33, MS4A4A/MS4A4E/MS4A6E cluster, ABCA7, CD2AP, and EPHA1).45,55 In line with the pathways identified by the first set of GWASs, all of these five loci are likely involved in the immune system, while ABCA7 is in addition involved in lipid metabolism and APP-processing (Table 2). The CD33 gene encodes a protein that is a member of a family of cell surface immune receptors that bind extracellular sialylated glycans and signal via a cytoplasmic domain called the immunoreceptor tyrosine inhibitory motif.56,57 CD33 has primarily been studied in the peripheral immune system, where it is expressed on myeloid progenitors and monocytes and also in the brain. In the periphery, CD33 appears to inhibit proliferation of myeloid cells.58 The MS4A4A/MS4A4E/MS4A6E locus is part of a cluster of 15 MS4A genes on chromosome 11 and encodes proteins with multiple membrane-spanning domains that were initially identified by their homology to CD20, a B-lymphocyte cell surface molecule. Little is known about the function of MS4A4A gene products; however, like CD33, MS4A4A is expressed on myeloid cells and monocytes and likely has an immune-related function. EPHA1 encodes a member of the ephrin family of cell surface receptors, which interact with ephrin ligands on adjacent cells to modulate cell adhesion, migration, and axon guidance and synapse formation and plasticity. While there is a substantial body of research on the function of ephrin receptors in general, little is known about the EPHA1 gene product. Like other ephrin receptors, it regulates cell morphology and motility,59 and early work implicated this receptor in regulating vascular morphogenesis and angiogenesis.60 Knocking out the EPHA1 gene in mice results in abnormal tail and reproductive tract development61 but no effects on the brain. Consistent with this notion, in mice, expression is restricted to epithelial tissue. In humans, EPHA1 is expressed by CD4-positive T lymphocytes,62 monocytes,63 intestinal epithelium, including colon epithelium. Combined with the lack of evidence for brain expression, this may suggest that, like CD33, CR1, and MS4A4/MS4A6E, the role of the EPHA1 gene product in AD may be mediated through the immune system. The CD2-associated protein gene (CD2AP) encodes a scaffolding protein that binds directly to actin,64 nephrin, and other proteins involved in cytoskeletal organization. In the immune system, CD2AP is required for synapse formation65 in a process that involves clathrin-dependent actin polymerization. ABCA7 is an integral transmembrane adenosine triphosphate (ATP)-binding cassette transporter belonging to the ABC family proteins that mediate the biogenesis of high-density lipoprotein with cellular lipid and helical apolipoproteins.66 It binds APOA-1 and functions in APO-mediated phospholipid and cholesterol efflux from cells.67 In addition, ABCA7 affects the transport of other important proteins (including Aβ protein)67 through the cell membrane and is involved in host defense through effects on phagocytosis of apoptotic cells by macrophages.66
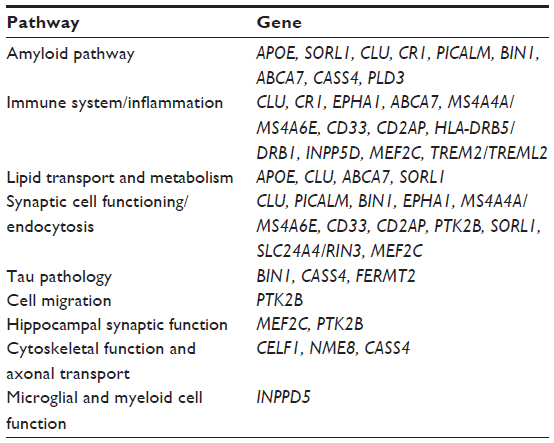 | Table 2 Major pathways identified by genomic studies |
The largest GWAS in persons of European ancestry to date was recently performed by the International Genomics of Alzheimer’s Project. This study, by Lambert et al,68 consisted of a large, two-stage meta-analysis of the aforementioned major GWASs of individuals of European ancestry and included in total 74,046 subjects. In addition to the APOE locus, 19 loci (CR1, BIN1, CD2AP, EPHA1, CLU, MS4A6A, PICALM, ABCA7, HLA-DRB5/HLA-DRB1, PTK2B, SORL1, SLC24A4/RIN3, INPP5D, MEF2C, NME8, ZCWPW1, CELF1, FERMT2, CASS4) reached genome-wide significance, defined as P<5×10−8, in the combined stage 1 and stage 2 analysis. Out of these, eleven (HLA-DRB5/HLA-DRB1, PTK2B, SORL1, SLC24A4/RIN3, INPP5D, MEF2C, NME8, ZCWPW1, CELF1, FERMT2, CASS4) were novel. Out of the 12 novel loci reaching genome-wide significance, several cluster in the specific pathways previously identified, ie, immune response (HLA-DRB5/DRB1, INPP5D, MEF2C), APP-processing (SORL1, CASS4), tau pathology (CASS4, FERMT2), cell migration (PTK2B), and lipid transport and endocytosis (SORL1). The SORL1 (sortilin-related receptor, L[DLR class] 1) gene has previously been demonstrated to modulate trafficking and processing of APP, in a candidate gene approach.69,70 The results of this study68 further suggested the existence of additional pathways, such as hippocampal synaptic function (MEF2C, PTK2B), cytoskeletal function and axonal transport (CELF1, NME8, CASS4), regulation of gene expression and posttranslational modification of proteins, and microglial and myeloid cell function (INPP5D).
Consistent with the notion of a genetically complex disorder, the odds ratios (ORs) of all disease-associated loci other than the APOE-ε4 allele range from 1.08 to 1.29. Examining the genetic effect attributable to all the associated loci, the most strongly associated SNPs at each locus other than APOE show population attributable fractions (PAFs) or preventive fractions between 1.0%–8.0%.68 The cumulative PAF has been estimated at 89.4%.68 Finally, the results from the combined stage 1 and stage 2 data sets also identified 13 suggestive loci (an intergenic locus at chr1q31.2, HS3ST1, SQSTM1, TREML2, NDUFAF6, ECHDC3, AP2A2, ADAMTS20, IGH, SPPL2A, TRIP4, SCIMP, ACE) with association P-values <10−6.
Few studies have been performed in minority groups. In the largest GWAS performed to date in Caribbean Hispanics,71 associations in CLU, PICALM, and BIN1 were replicated, and several additional loci on 2p25.1, 3q25.2, 7p21.1 and 10q23.1 – which could be replicated in an independent cohort of non-Hispanic Whites of European ancestry from the National Institute on Aging Late-Onset Alzheimer’s Disease Family Study – were observed. Finally, in the largest GWAS of African Americans performed, Reitz et al72 identified ABCA7 as a major susceptibility locus in this ethnic group. Interestingly, in contrast to all GWAS loci identified in Caucasians, the ABCA7 locus had, in African Americans, an effect size as strong as that of APOE-ε4 (ie, a 70%–80% increase in risk compared with a 10%–20% increase in risk through the GWAS loci observed in Whites). This finding may represent a winner’s curse (ie, inflation of the estimated effect in a discovery set, in relation to follow-up studies) and needs to be confirmed by independent studies in African Americans and functional methods; however, this finding may have major implications for developing targets for genetic testing, prevention, and treatment in this ethnic group if proven true. In addition, this study confirmed APOE as a susceptibility gene in this ethnic group, which had been, prior to this study, inconsistently seen across studies. This study also replicated CR1, BIN1, EPHA1, and CD33. A large-scale GWAS study in Asian (Japanese and Korean) populations identified SORL1 as a susceptibility gene,73 and candidate gene studies performed in Asian populations identified MS4A6A, CD33, PICALM, CR1, CLU.74–79 The effect of the APOE-ε4 allele was universally observed in all GWASs performed across ethnic groups. (These findings suggest that there is, at least to some degree, overlap in causative genetic loci across different ethnic groups, although the causative variants within the disease-associated genes may differ.)
Rare variants in late-onset Alzheimer’s disease
Several loci containing rare, causative sequencing variants have been identified, some of which overlap with the aforementioned common variant loci identified by GWASs. In large multiplex pedigrees with LOAD, targeted exome sequencing of APP, PSEN1, PSEN2, APOE, GRN, and MAPT has identified 33 missense, nonsense, and splice site variants in 60 families (13.7%), 18 of which were novel.80 A recent study of whole-genome sequence data from 1,795 Icelanders identified a rare, coding mutation (A673T) in the APP gene protecting against LOAD and cognitive decline in the elderly without cognitive impairment.81 A673T is adjacent to the aspartyl protease β-site in APP and results in an approximately 40% reduction in the formation of Aβ in vitro. This strong protective effect of the A673T substitution against LOAD provides proof of principle for the hypothesis that reducing the β-cleavage of APP may protect against the disease. The TREM2/TREML2 locus associated with LOAD in a case-control GWAS had previously been reported by two independent multistage studies that combined sequencing analyses followed by a meta-analysis of independent imputed data sets (see below), with additional direct genotyping in other independent samples and functional analyses. These studies identified a rare exonic variant (R47H) associated with a three- to fourfold increased risk of developing AD.82,83 An exon sequencing study of PICALM in 48 AD cases and 48 controls identified an exonic splicing enhancer variation in exon 5 in linkage disequilibrium with GWAS SNP rs3851179.84 Other genes with rare variants for LOAD include SORL1,85 ADAM10,86 and PLD3.87 PLD3 is a poorly characterized member of the PLD superfamily of phospholipases but seems to affect APP-processing.80
Discussion and significance
In conclusion, over the past decade, studies capitalizing on high-throughput genome technologies have significantly advanced the knowledge on the genetic underpinnings of AD, implicating a wide set of pathophysiological mechanisms in addition to APP metabolism, including: innate immune response and inflammation, lipid metabolism, endocytosis, cell migration, tau pathology, hippocampal synaptic function and axonal transport, regulation of gene expression and posttranslational modification of proteins, and microglial and myeloid cell function. As described above, the cumulative PAF of the common GWAS loci has been estimated at 89.4%.68 However, it remains possible that this estimation is partly explained by the winner’s curse (ie, an overestimation of genetic effect size in initial studies). Consistent with this notion are 1) the fact that high-throughput sequencing studies have started to map additional causative rare variants in these genes, providing invaluable evidence for an involvement of rare variants in this complex disease and clearly overturning the common disease–common variant hypothesis; and 2) the increasing evidence for a significant role of noncoding ribonucleic acid (RNA) in complex disease.88 While the human genome contains about 20,000 protein-coding genes, 98.8% consists of non-protein-coding DNA sequence.88 The majority of these sequences are dynamically transcribed, mainly into non-protein-coding RNAs, with tens if not hundreds of thousands that show specific expression patterns and subcellular locations, as well as many classes of small regulatory RNAs. The emerging evidence indicates that these RNAs control the epigenetic states that underpin development and that many are dysregulated in cancer and other complex diseases, including AD.88 In addition, there is evidence that animals, particularly primates, have evolved plasticity in these RNA regulatory systems, especially in the brain. Thus, it appears that what was dismissed as “junk”, because it was not understood, may hold a significant part of the key to understanding human evolution, development, and cognition.
Overall, the specification of the pathways involved in AD etiology bears a strong potential for the development of therapeutic targets. The current basis for diagnostic biomarkers and therapies in clinical trials are the identified variants in APP, PSEN1, PSEN2, and APOE in AD. However, before this novel information can be used in clinical settings and safely considered for pharmaceutical intervention, ongoing and future large-scale next-generation sequencing approaches (both hypothesis-driven and hypothesis-free) as well as extensive functional studies are needed, to identify the specific causative variants and the specific factors with which they interact. In addition, a significant part of the missing heritability of AD, which is expected to be explained by several additional loci with small effect sizes each, needs to be identified. Such studies, including the Alzheimer’s Disease Sequencing Project, a major collaborative whole exome and whole genome sequencing effort by the National Institutes of Health (NIH), are now underway.
Acknowledgments
Dr Reitz was supported by a Paul B Beeson Career Development Award (number K23AG034550).
Disclosure
The author reports no conflicts of interest in this work.
References
Hurd MD, Martorell P, Delavande A, Mullen KJ, Langa KM. Monetary costs of dementia in the United States. N Engl J Med. 2013;368(14):1326–1334. | |
2010 Alzheimer’s disease facts and figures. Alzheimers Dement. 2010;6(2):158–194. | |
Mayeux R. Epidemiology of neurodegeneration. Annu Rev Neurosci. 2003;26:81–104. | |
Roe CM, Fagan AM, Grant EA, Holtzman DM, Morris JC. CSF biomarkers of Alzheimer disease: “noncognitive” outcomes. Neurology. 2013;81(23):2028–2031. | |
Rowe CC, Bourgeat P, Ellis KA, et al. Predicting Alzheimer disease with β-amyloid imaging: results from the Australian imaging, biomarkers, and lifestyle study of ageing. Ann Neurol. 2013;74(6):905–913. | |
Laske C. Blood-based biomarkers in Alzheimer disease: where are we now and where have we to go? JAMA Neurol. 2013;70(1):133. | |
Alsop DC, Detre JA. Reduced transit-time sensitivity in noninvasive magnetic resonance imaging of human cerebral blood flow. J Cereb Blood Flow Metab. 1996;16(6):1236–1249. | |
Gatz M, Pedersen NL, Berg S, et al. Heritability for Alzheimer’s disease: the study of dementia in Swedish twins. J Gerontol A Biol Sci Med Sci. 1997;52(2):M117–M125. | |
Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168–174. | |
Lautenschlager NT, Cupples LA, Rao VS, et al. Risk of dementia among relatives of Alzheimer’s disease patients in the MIRAGE study: What is in store for the oldest old? Neurology. 1996;46(3):641–650. | |
Mayeux R, Sano M, Chen J, Tatemichi T, Stern Y. Risk of dementia in first-degree relatives of patients with Alzheimer’s disease and related disorders. Arch Neurol. 1991;48(3):269–273. | |
Bergem AL, Lannfelt L. Apolipoprotein E type epsilon4 allele, heritability and age at onset in twins with Alzheimer disease and vascular dementia. Clin Genet. 1997;52(5):408–413. | |
Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–706. | |
Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977. | |
Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760. | |
Lander ES. The new genomics: global views of biology. Science. 1996;274(5287):536–539. | |
Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet. 2001;69(1):124–137. | |
Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. | |
Agarwala V, Flannick J, Sunyaev S, Altshuler D; GoT2D Consortium. Evaluating empirical bounds on complex disease genetic architecture. Nat Genet. 2013;45(12):1418–1427. | |
Kuusisto J, Koivisto K, Kervinen K, et al. Association of apolipoprotein E phenotypes with late onset Alzheimer’s disease: population based study. BMJ. 1994;309(6955):636–638. | |
Breitner JC, Wyse BW, Anthony JC, et al. APOE-epsilon4 count predicts age when prevalence of AD increases, then declines: the Cache County Study. Neurology. 1999;53(2):321–331. | |
Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. | |
Gomez-Isla T, West HL, Rebeck GW, et al. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer’s disease. Ann Neurol. 1996;39(1):62–70. | |
Holmes C, Levy R, McLoughlin DM, Powell JF, Lovestone S. Apolipoprotein E: non-cognitive symptoms and cognitive decline in late onset Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1996;61(6):580–583. | |
Kurz A, Altland K, Lautenschlager N, et al. Apolipoprotein E type 4 allele and Alzheimer’s disease: effect on age at onset and relative risk in different age groups. J Neurol. 1996;243(6):452–456. | |
Murman DL, Foster NL, Kilgore SP, McDonagh CA, Fink JK. Apolipoprotein E and Alzheimer’s disease: strength of association is related to age at onset. Dementia. 1996;7(5):251–255. | |
Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342(8873):697–699. | |
Roses AD. Alzheimer’s disease: the genetics of risk. Hosp Pract (Minneap). 1997;32(7):51–55, 58–63, 67–69. | |
Barabash A, Marcos A, Ancín I, et al. APOE, ACT and CHRNA7 genes in the conversion from amnestic mild cognitive impairment to Alzheimer’s disease. Neurobiol Aging. 2009;30(8):1254–1264. | |
Petersen RC, Smith GE, Ivnik RJ, et al. Apolipoprotein E status as a predictor of the development of Alzheimer’s disease in memory-impaired individuals. JAMA. 1995;273(16):1274–1278. | |
Sasaki M, Kodama C, Hidaka S, et al. Prevalence of four subtypes of mild cognitive impairment and APOE in a Japanese community. Int J Geriatr Psychiatry. 2009;24(10):1119–1126. | |
Tyas SL, Salazar JC, Snowdon DA, et al. Transitions to mild cognitive impairments, dementia, and death: findings from the Nun Study. Am J Epidemiol. 2007;165(11):1231–1238. | |
Blom ES, Giedraitis V, Zetterberg H, et al. Rapid progression from mild cognitive impairment to Alzheimer’s disease in subjects with elevated levels of tau in cerebrospinal fluid and the APOE epsilon4/epsilon4 genotype. Dement Geriatr Cogn Disord. 2009;27(5):458–464. | |
Devanand DP, Pelton GH, Zamora D, et al. Predictive utility of apolipoprotein E genotype for Alzheimer disease in outpatients with mild cognitive impairment. Arch Neurol. 2005;62(6):975–980. | |
Hämäläinen A, Grau-Olivares M, Tervo S, et al. Apolipoprotein E epsilon 4 allele is associated with increased atrophy in progressive mild cognitive impairment: a voxel-based morphometric study. Neurodegener Dis. 2008;5(3–4):186–189. | |
Hsiung GY, Sadovnick AD, Feldman H. Apolipoprotein E epsilon4 genotype as a risk factor for cognitive decline and dementia: data from the Canadian Study of Health and Aging. CMAJ. 2004;171(8):863–867. | |
Jack CR, Petersen RC, Xu YC, et al. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology. 1999;52(7):1397–1403. | |
Ramakers IH, Visser PJ, Aalten P, et al. The association between APOE genotype and memory dysfunction in subjects with mild cognitive impairment is related to age and Alzheimer pathology. Dement Geriatr Cogn Disord. 2008;26(2):101–108. | |
Tierney MC, Szalai JP, Snow WG, et al. A prospective study of the clinical utility of ApoE genotype in the prediction of outcome in patients with memory impairment. Neurology. 1996;46(1):149–154. | |
Slooter AJ, Cruts M, Kalmijn S, et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Arch Neurol. 1998;55(7):964–968. | |
Myers RH, Schaefer EJ, Wilson PW, et al. Apolipoprotein E epsilon4 association with dementia in a population-based study: The Framingham study. Neurology. 1996;46(3):673–677. | |
Tang MX, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, et al. The APOE-epsilon4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. JAMA. March 11, 1998;279(10):751–755. | |
Tang MX1, Maestre G, Tsai WY, Liu XH, Feng L, Chung WY, et al. Effect of age, ethnicity, and head injury on the association between APOE genotypes and Alzheimer’s disease. Ann N Y Acad Sci. December 16, 1996;802:6–15. | |
Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–1093. | |
Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43(5):436–441. | |
Seshadri S, Fitzpatrick AL, Ikram MA, et al; CHARGE Consortium; GERAD1 Consortium; EADI1 Consortium. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303(18):1832–1840. | |
Lambert JC, Heath S, Even G, et al; European Alzheimer’s Disease Initiative Investigators. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–1099. | |
Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Clusterin: a forgotten player in Alzheimer’s disease. Brain Res Rev. 2009;61(2):89–104. | |
Wollmer MA, Sleegers K, Ingelsson M, et al. Association study of cholesterol-related genes in Alzheimer’s disease. Neurogenetics. 2007;8(3):179–188. | |
DeMattos RB, Cirrito JR, Parsadanian M, et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41(2):193–202. | |
Pant S, Sharma M, Patel K, Caplan S, Carr CM, Grant BD. AMPH-1/Amphiphysin/Bin1 functions with RME-1/Ehd1 in endocytic recycling. Nat Cell Biol. 2009;11(12):1399–1410. | |
Tebar F, Bohlander SK, Sorkin A. Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol Biol Cell. 1999;10(8):2687–2702. | |
Crehan H, Holton P, Wray S, Pocock J, Guerreiro R, Hardy J. Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology. 2012;217(2):244–250. | |
McGeer PL, Rogers J. Anti-inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology. 1992;42(2):447–449. | |
Hollingworth P, Harold D, Sims R, et al; Alzheimer’s Disease Neuroimaging Initiative; CHARGE consortium; EADI1 consortium. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43(5):429–435. | |
Cao H, Crocker PR. Evolution of CD33-related siglecs: regulating host immune functions and escaping pathogen exploitation? Immunology. 2011;132(1):18–26. | |
von Gunten S, Bochner BS. Basic and clinical immunology of Siglecs. Ann N Y Acad Sci. 2008;1143:61–82. | |
Vitale C, Romagnani C, Falco M, et al. Engagement of p75/AIRM1 or CD33 inhibits the proliferation of normal or leukemic myeloid cells. Proc Natl Acad Sci U S A. 1999;96(26):15091–15096. | |
Yamazaki T, Masuda J, Omori T, Usui R, Akiyama H, Maru Y. EphA1 interacts with integrin-linked kinase and regulates cell morphology and motility. J Cell Sci. 2009;122(Pt 2):243–255. | |
Adams RH, Klein R. Eph receptors and ephrin ligands. Essential mediators of vascular development. Trends Cardiovasc Med. 2000;10(5):183–188. | |
Duffy SL, Coulthard MG, Spanevello MD, et al. Generation and characterization of EphA1 receptor tyrosine kinase reporter knockout mice. Genesis. 2008;46(10):553–561. | |
Holen HL, Nustad K, Aasheim HC. Activation of EphA receptors on CD4+CD45RO+ memory cells stimulates migration. J Leukoc Biol. 2010;87(6):1059–1068. | |
Sakamoto A, Sugamoto Y, Tokunaga Y, et al. Expression profiling of the ephrin (EFN) and Eph receptor (EPH) family of genes in atherosclerosis-related human cells. J Int Med Res. 2011;39(2):522–527. | |
Lehtonen S, Zhao F, Lehtonen E. CD2-associated protein directly interacts with the actin cytoskeleton. Am J Physiol Renal Physiol. 2002;283(4):F734–F743. | |
Dustin ML, Olszowy MW, Holdorf AD, et al. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell. 1998;94(5):667–677. | |
Tanaka N, Abe-Dohmae S, Iwamoto N, Yokoyama S. Roles of ATP-binding cassette transporter A7 in cholesterol homeostasis and host defense system. J Atheroscler Thromb. 2011;18(4):274–281. | |
Chan SL, Kim WS, Kwok JB, et al. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J Neurochem. 2008;106(2):793–804. | |
Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–1458. | |
Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39(2):168–177. | |
Reitz C, Cheng R, Rogaeva E, et al; Genetic and Environmental Risk in Alzheimer Disease 1 Consortium. Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch Neurol. 2011;68(1):99–106. | |
Lee JH, Cheng R, Barral S, et al. Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch Neurol. 2011;68(3):320–328. | |
Reitz C, Jun G, Naj A, et al. Alzheimer Disease Genetics Consortium. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ε4, and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013;309(14):1483–1492. | |
Miyashita A, Koike A, Jun G, et al. Alzheimer Disease Genetics Consortium. SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS One. 2013;8(4):e58618. | |
Chen LH, Kao PY, Fan YH, et al. Polymorphisms of CR1, CLU and PICALM confer susceptibility of Alzheimer’s disease in a southern Chinese population. Neurobiol Aging. 2012;33(1):210. e1–e7. | |
Chung SJ, Lee JH, Kim SY, et al. Association of GWAS top hits with late-onset Alzheimer disease in Korean population. Alzheimer Dis Assoc Disord. 2013;27(3):250–257. | |
Tan L, Yu JT, Zhang W, et al. Association of GWAS-linked loci with late-onset Alzheimer’s disease in a northern Han Chinese population. Alzheimers Dement. 2013;9(5):546–553. | |
Yuan Q, Chu C, Jia J. Association studies of 19 candidate SNPs with sporadic Alzheimer’s disease in the North Chinese Han population. Neurol Sci. 2012;33(5):1021–1028. | |
Li HL, Shi SS, Guo QH, et al. PICALM and CR1 variants are not associated with sporadic Alzheimer’s disease in Chinese patients. J Alzheimers Dis. 2011;25(1):111–117. | |
Yu JT, Song JH, Ma T, et al. Genetic association of PICALM polymorphisms with Alzheimer’s disease in Han Chinese. J Neurol Sci. 2011;300(1–2):78–80. | |
Cruchaga C, Haller G, Chakraverty S, et al; NIA-LOAD/NCRAD Family Study Consortium. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS One. 2012;7(2):e31039. | |
Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488(7409):96–99. | |
Guerreiro R, Wojtas A, Bras J, et al; Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. | |
Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–116. | |
Schnetz-Boutaud NC, Hoffman J, Coe JE, Murdock DG, Pericak- Vance MA, Haines JL. Identification and confirmation of an exonic splicing enhancer variation in exon 5 of the Alzheimer disease associated PICALM gene. Ann Hum Genet. 2012;76(6):448–453. | |
Pottier C, Hannequin D, Coutant S, et al; PHRC GMAJ Collaborators. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry. 2012;17(9):875–879. | |
Kim M, Suh J, Romano D, et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum Mol Genet. 2009;18(20):3987–3996. | |
Cruchaga C, Karch CM, Jin SC, et al; UK Brain Expression Consortium; Alzheimer’s Research UK Consortium. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014;505(7484):550–554. | |
Mattick JS. The central role of RNA in human development and cognition. FEBS Lett. 2011;585(11):1600–1616. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.