")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 15
Genome Editing and Human Pluripotent Stem Cell Technologies for in vitro Monogenic Diabetes Modeling
Authors Dabi YT , Degechisa ST
Received 25 March 2022
Accepted for publication 8 June 2022
Published 11 June 2022 Volume 2022:15 Pages 1785—1797
DOI https://doi.org/10.2147/DMSO.S366967
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ming-Hui Zou
Yosef Tsegaye Dabi,1,2 Sisay Teka Degechisa1,3
1Department of Medical Biochemistry, School of Medicine, College of Health Sciences, Addis Ababa University, Addis Ababa, Ethiopia; 2Department of Medical Laboratory Science, Wollega University, Nekemte, Ethiopia; 3Department of Medical Laboratory Sciences, College of Medicine and Health Sciences, Arba Minch University, Arba Minch, Ethiopia
Correspondence: Yosef Tsegaye Dabi, Email [email protected]
Abstract: Diabetes is a metabolic disease characterized by chronic hyperglycemia. Polygenic diabetes, which encompasses type-1 and type-2 diabetes, is the most prevalent kind of diabetes and is caused by a combination of different genetic and environmental factors, whereas rare phenotype monogenic diabetes is caused by a single gene mutation. Monogenic diabetes includes Neonatal diabetes mellitus and Maturity-onset diabetes of the young. The majority of our current knowledge about the pathogenesis of diabetes stems from studies done on animal models. However, the genetic difference between these creatures and humans makes it difficult to mimic human clinical pathophysiology, limiting their value in modeling key aspects of human disease. Human pluripotent stem cell technologies combined with genome editing techniques have been shown to be better alternatives for creating in vitro models that can provide crucial knowledge about disease etiology. This review paper addresses genome editing and human pluripotent stem cell technologies for in vitro monogenic diabetes modeling.
Keywords: monogenic diabetes, MODY, NDM, genome editing, pluripotent stem cell
Introduction
Diabetes mellitus (DM) is a metabolic disorder characterized by pancreatic β cell loss and hyperglycemia due to altered insulin production and/or action. Type 1 diabetes mellitus (T1DM) is an autoimmune disorder characterized by destruction of pancreatic β cells,1 whereas the pathogenesis that defines Type 2 diabetes mellitus (T2DM) is insulin resistance in the insulin-target tissues and β cell dysfunction.2 Polygenic diabetes, which encompasses T1DM and T2DM, is the most prevalent kind of diabetes and involves alteration in different genes, whereas rare phenotype monogenic diabetes (MD) is caused by a single gene mutation linked to pancreatic development and β cell activity.3
Increased glucose across the GLUT 2 transporter is processed by the enzyme glucokinase in the normal pancreatic beta-cell, resulting in increased ATP generation. This results in the closing of the KATP channel, which depolarizes the cell membrane and activates calcium influx through voltage-gated calcium channels, allowing insulin granule exocytosis.4 The inner subunit (Kir6.2) of the KATP channel is encoded by KCNJ11, while the outer subunit (ABCC8) is encoded by ABCC8 (SUR1).5 Even in the presence of hyperglycemia, mutations in either gene cause the KATP channels to remain abnormally “stuck open”. The cell membrane cannot properly depolarize without channel closure, and hence insulin cannot be released from the beta-cell.5,6
For many years, animal studies have been employed extensively in biomedical research to acquire insights into the normal and aberrant biological processes that occur in humans. However, the variation in disease conditions between animal models and humans limits their utility in mimicking crucial aspects of human disease.7,8 Human pluripotent stem cells (hPSCs), which include human embryonic stem cells (ESCs) and human induced pluripotent stem cells (iPSCs), can be used as an alternative for disease modeling because of their ability to differentiate into all cell types.9,10
ESCs and iPSCs have provided a critical opportunity for biological research, allowing us to address problems that were previously unanswerable due to a lack of a human model and replicating the development process with differentiation techniques to generate cell populations of therapeutic significance.11,12 Furthermore, the recent discovery of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) gene manipulation enables the correction or introduction of specific pathogenic mutations, the generation of isogenic cell lines, and the investigation of the same genetic variant in multiple genetic backgrounds.13,14 The purpose of this review paper is to describe recent achievements in modeling monogenic diabetes with genome editing and pluripotent stem cell technology.
Monogenic Diabetes
MD is linked to mutations in a variety of genes involved in pancreatic development and ß cell activity.15 MD can be divided into two categories: Neonatal diabetes mellitus (NDM) and Maturity-onset diabetes of the young (MODY).16 Depending on the time of diagnosis NDM is diagnosed within the first six months of life, whereas MODY is diagnosed before the age of 25.17
MODY
MODY is the most frequent kind of monogenic diabetes, accounting for 1–5% of all diabetes mellitus cases.18 It is a clinically and genetically heterogeneous collection of endocrine diseases caused by mutations in a single gene implicated in pancreatic beta cell function.19 These abnormalities are mostly caused by reduced glucose phosphorylation, decreased activity of pancreatic beta cell expressed transcription factors or altered insulin biosynthesis.20 MODY is non-insulin-dependent diabetes that is often diagnosed before the age of 25 and is autosomal dominant. Multiple kinds of monogenic diabetes, each with a different clinical presentation, have been linked to mutations in more than ten different genes.20,21
MODY is thought to be caused by a variety of genetic mutations, including missense, frameshift, splice site, nonsense, indel, deletion, and insertion.22–24 According to their underlying molecular etiology, MODY subtypes may be divided into five groups: transcriptional regulatory disorders, enzyme disorders, protein misfolding disorders, ion channel disorders, and signal transduction disorders.25 Table 1 shows the several types of mutations linked to MODY, as well as the affected chromosomes, subtypes, and molecular etiology.
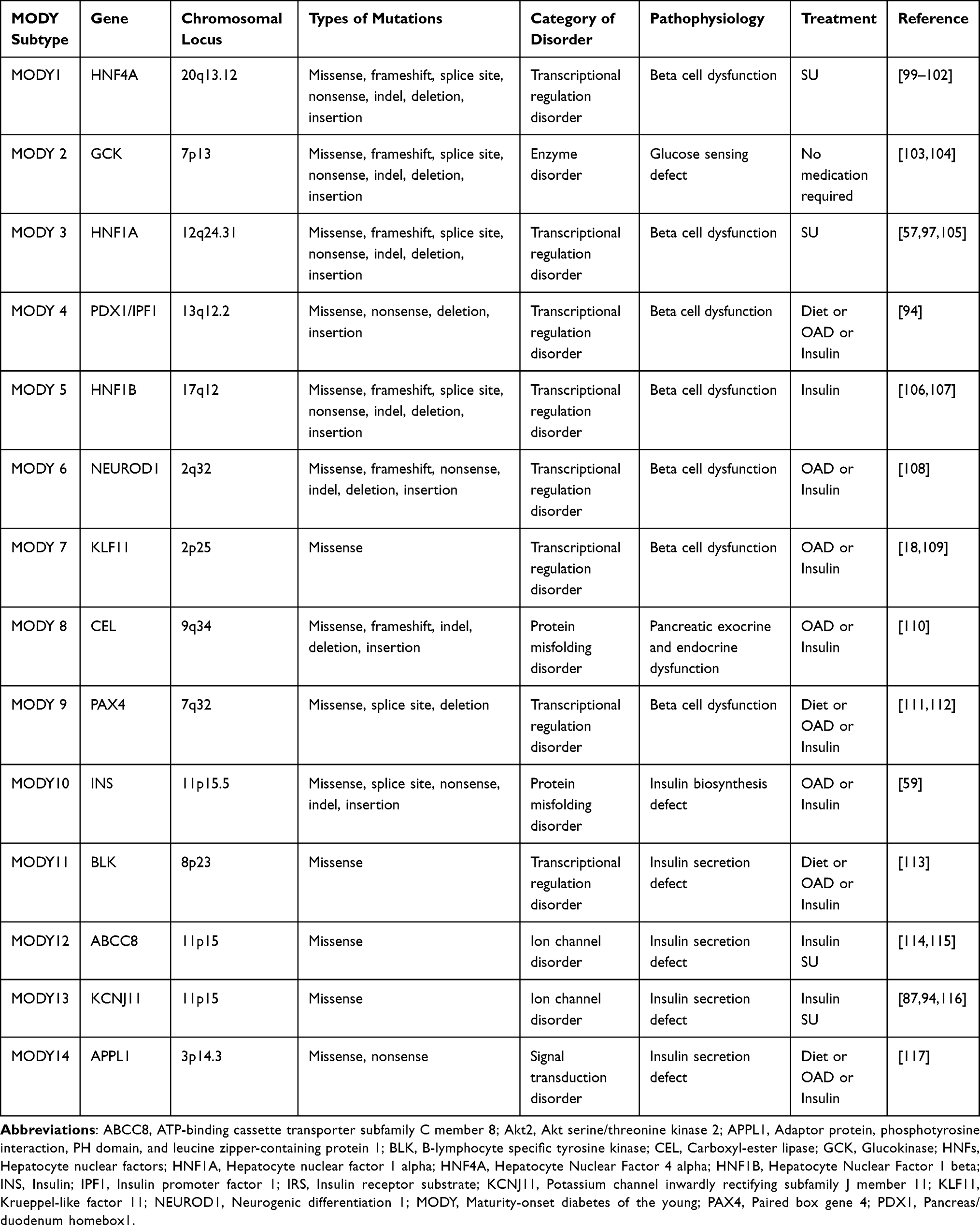 |
Table 1 Molecular Pathogenesis of Different Types of MODY |
NDM
NDM is a rare type of monogenic diabetes compared to MODY and it is diagnosed within the first six months of life, includes a variety of clinically and genetically diverse disorders.26,27 NDM can be permanent (PNDM) and require lifetime therapy, or transient (TNDM), with insulin reliance for the first few months and spontaneous remission of diabetes by the age of 18 months.26,28–30
NDM is genetically diverse, with over 25 identified genetic causes that are linked to a variety of pancreatic and non-pancreatic characteristics.31,32 Mutations in KCNJ11/Kir6.2 and ABCC8/SUR1, which encode the two subunits that make up the hetero-octameric ATP-dependent K+ (KATP) channel, in INS and ZFP57 genes, or chromosomal 6q24 abnormalities are the most common NDM genetic subtypes.31,33
One of the main causes of TNDM is the overexpression of genes on chromosome 6q24. On chromosome 6q24, TNDM is associated with the overexpression of at least two imprinted genes, Pleomorphic adenoma gene-like 1 (PLAGL1) and Hydatidiform Mole Associated and Imprinted (HYMAI).34,35 Paternal uniparental chromosome 6 (UPD6), paternal allele 6q24 duplication, or a loss of methylation mutation (LOM) within the differentially methylated region (DMR) could all be genetic mechanisms causing 6q24-related neonatal diabetes.35
Another key gene linked to NDM is the INS gene. It is the second most prevalent cause of newborn diabetes that is permanent.36 In most cases, mutations in the insulin gene appear to result in insulin protein misfolding.36 These proteins appear to aggregate in numerous subcellular compartments, causing endoplasmic reticulum (ER) stress and beta-cell death.37 Table 2 shows monogenic causes of neonatal diabetes with associated clinical features.
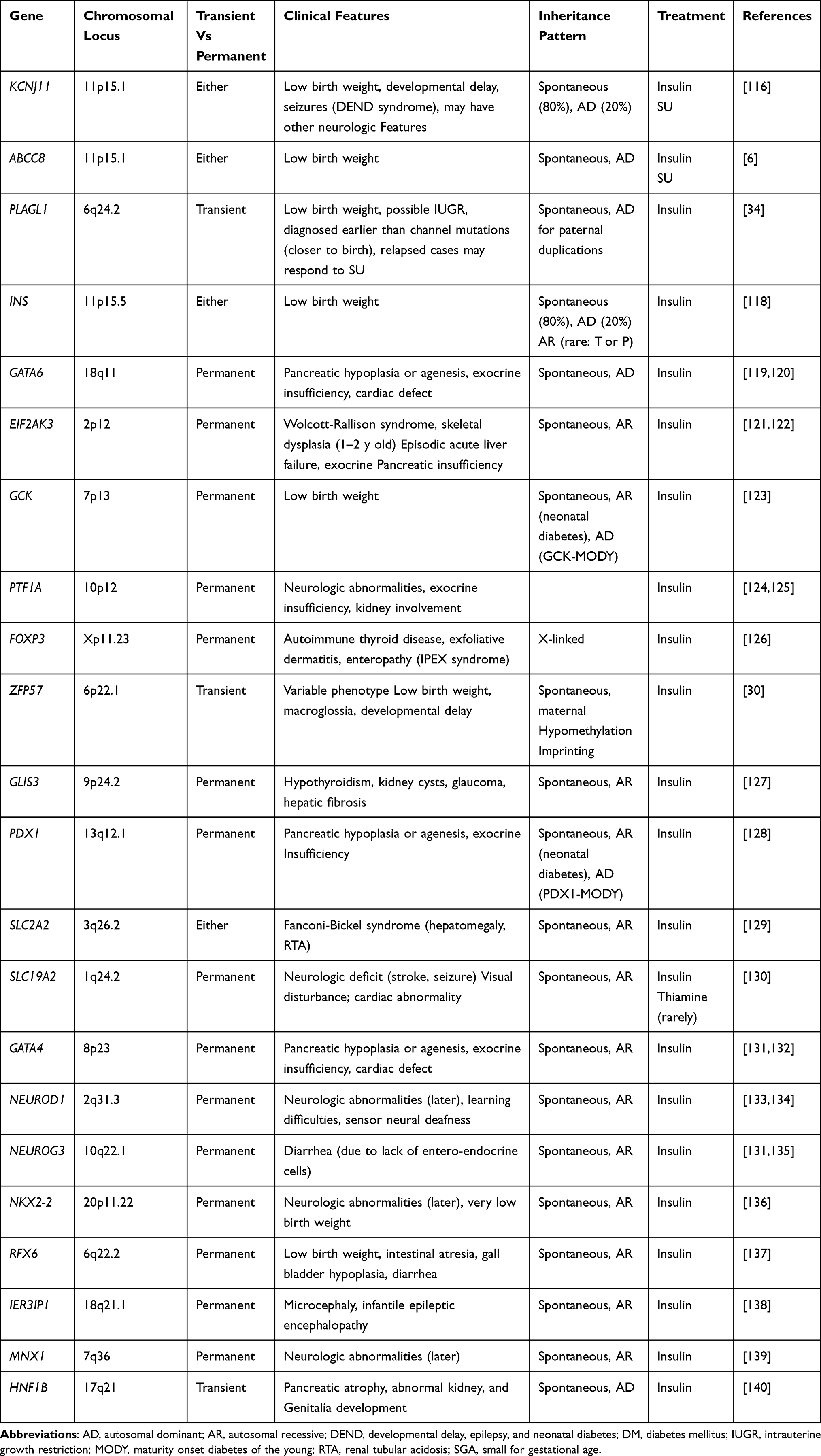 |
Table 2 Monogenic Causes of Neonatal Diabetes Mellitus |
In-Vitro Modeling of MD Using hPSCs
Overview of hPSCs
Stem cells are distinguished by their key property to self-renew and differentiate into various cell types based on their potency. Based on their ability to differentiate into a wide range of cell lineages they are classified as pluripotent, multipotent, or unipotent.38 Human ESCs and iPSCs are examples of hPSCs. They can, in principle, grow into any somatic cell present in the human body and might be used as a disease model to examine various molecular mechanisms involved in disease causation.39,40
Despite the fact that ESCs have a high potential for use in biological research to better understand fundamental biological processes, they have certain limitations in future therapeutic settings because of the same rejection issues that current organ transplants encounter. Individual members of society are also concerned about using human embryos for research objectives.41,42 On the other hand iPSC technology enables the generation of pluripotent cells from nearly any individual, and protocols are being designed to differentiate those cell lines into a wide range of cell types present in the body. IPSCs are being utilized to investigate the impact of genetic variations in disease pathogenesis in a variety of derived cell types, such as pancreatic beta like cells and various kinds of neurons.43,44
hPSCs Models of MD
Because pancreatic beta cells are the primary cells affected in all kinds of diabetes, a robust strategy for generating functional cells in vitro is required for a better understanding of the pathophysiology associated with different types of diabetes. Small molecules and growth factors are used in in-vitro differentiation techniques to enhance or inhibit certain pathways at each stage of the differentiation process.10,45 Pancreatic beta cells are known to be derived from pancreatic progenitors that express PDX1 and NKX6.1.46,47 In-vitro, hPSCs are differentiated into several stages of pancreatic β cells. The expression of essential transcription factors (TFs) that regulate β cell development and functionality distinguishes each stage.15
While ESCs are derived from cells isolated from the blastocyst’s inner cell mass,48 iPSCs allow for the production of pluripotent cells by transcriptional reprogramming of terminal cells from adult patients.49 The capacity to produce patient-specific human tissue in vitro has enabled genetic disease models which can mimic the disease phenotype in humans.
Various methodologies might be used to generate relevant cell types from hPSCs for modeling MD. The most popular strategy is to develop iPSCs from affected patients’ somatic cells containing a specific mutation/variant (patient-derived iPSCs) and compare them to iPSCs generated from healthy controls (Ctr-iPSCs). Another option is to employ genome-editing methods to induce MD-relevant mutations into existing hPSC lines (ESCs and iPSCs) in order to explore the function of the genes linked with MD.50
Patient-Derived iPSCs for Modeling MD
Takahashi K. and Yamanaka S. reported the generation of the first induced pluripotent stem cells (iPSCs) in mouse fetal fibroblasts by transduction of four transcription factors (Oct3/4, Sox2, c-Myc, and Klf4), revealing that somatic cells can be converted to a pluripotent stage iPSCs. The iPSCs formed have the characteristics of ESCs and may be utilized to develop into different cell types.49 The generation of iPSCs from patients and their differentiation into disease-relevant cell types has enormous potential for in vitro disease modeling.51 Keratinocytes, dermal fibroblasts, adipocytes, and peripheral blood cells are among the human somatic cells that may be reprogrammed. Retroviruses, lentiviruses, plasmids, adenoviruses, Sendai viruses, transposons, protein, modified RNA, and miRNA are being used in reprogramming.52
Rezania et al developed a seven-stage protocol for generating reprogrammed hiPSC lines into insulin-producing cells. During static in vitro incubations, stage (S) 7 cells exhibited critical markers of mature pancreatic beta cells and secreted glucose-stimulated insulin identical to human islets. This can be generalized to in-vitro models of monogenic diabetes.53
Diabetes stem cell models should allow for the investigation of specific processes leading to human β-cell failure as well as the testing of techniques to retain or restore β-cell function. Teo et al were the first to report the successful generation of hiPSCs from patients with five different MODY conditions (MODY1, MODY2, MODY3, MODY5, and MODY8) as a valuable resource for studying the role of these MODY genes/transcription factors in the development of human pancreas and beta cells, as well as the regulation of beta-cell secretory function.54
Shang et al used induced pluripotent stem cells derived from skin fibroblasts of Wolfram syndrome patients and discovered that these WFS1 mutant cells have insulin processing and secretion in response to various secretagogues comparable to healthy controls, but with a lower insulin content and increased activity of UPR pathways.55 Mutations in the WFS1 gene (wolframin), which is abundantly expressed in human islets as well as the heart, brain, placenta, and lung, can cause childhood-onset insulin-dependent diabetes.56
Other researchers investigated the influence of MODY1/HNF4A mutation on the development of the foregut lineage in humans using a hiPSCs-based disease modeling method. Their findings indicate that in MODY1, liver and pancreas development is disrupted early on, contributing to patients’ altered hepatic proteins and β-cell abnormalities.57 Conditional ablation of Hnf4a in mouse pancreatic β-cells, on the other hand, did not result in a diabetic phenotype, despite reduced glucose-stimulated insulin secretion (GSIS),58 showing that rodent models do not adequately reproduce the MODY1 phenotype in humans.
Balboa et al developed an in-vitro model using hiPSCs from patients with INS mutations and showed that the INS mutations cause proinsulin misfolding, higher symptoms of ER stress, and decreased proliferation in INS mutant beta-like cells compared to corrected controls.59 Other researchers generated hiPSCs from fibroblasts of a patient with PNDM and undetectable insulin at birth owing to a homozygous mutation in the insulin gene’s translation start site in another recent study. Their findings reveal that INS mutant cells differentiated into hormone-negative hiPSCs.60
Application of Genome Editing Technologies for Monogenic Diabetes Modeling Using hPSCs
Methods in Genome Editing
Zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the clustered regularly interspaced short palindromic repeats-associated Cas protein system (CRISPR/Cas) are three cutting-edge gene technologies that use enzyme “scissors” to cut DNA at specified gene loci.61 The mode of action is common for the three methods, in which these enzymes bind to a sequence of interest in the genomic DNA and induce targeted double-strand break (DSB) followed by DNA repair mechanisms, either homologous recombination (HR) or non-homologous end-joining (NHEJ).62
ZFNs
ZFNs are artificial structures created by combining restriction endonucleases with zinc-finger-binding domain proteins designed to target user-specified sequences within the genome.62 ZFNs are made up of N-terminal DNA binding domain that has been custom-engineered linked together via a linker peptide to a C-terminal endonuclease.63 The endonuclease is a non-specific cleavage domain from the bacterial FokI endonuclease that dimerizes to cleave DNA, whereas the DNA binding domain is an array of several zinc-finger domains, each of which recognizes a distinct three base pair sequence of DNA.64
When there are DSBs in the genome, an intracellular signaling cascade known as the DNA damage response is activated to repair the double-strand breaks caused by ZFNs.65 The mechanisms include the following:1 ZFNs containing FokI endonucleases and protein-binding domains are introduced into the cell,2 FokI and protein-binding domains are released to enter the nucleus,3 protein-binding domains attach with DNA fragment to be removed,4 FokI cuts out the identified DNA segment by creating double-stranded DNA break, and5 the desirable DNA segment is inserted and integrated into the DNA sequence.61
TALENs
TALENs, like ZFNs, have a modular structure with N-terminal transcription activator-like effector (TALE) DNA-binding domain fused to a C-terminal FokI endonuclease domain.66,67 TALENs are somewhat more site-specific than ZFNs, with fewer off-target effects because TALE repeats can target single base pair (bp), but Zinc fingers recognize DNA triplets.68
TALE DNA binding domain is composed of 34-residue repeat domains.69 At positions 12 and 13, the repeat variable residues (RVDs) make contact with DNA.69 For constructing synthetic TALE arrays, the most often employed RVDs are NI for adenine, HD for cytosine, NG for thymine, and NN or HN for guanine or adenine.70,71 TALEs are commonly built to recognize 12- to 20-bps of DNA, with more bases resulting in greater genome-editing specificity.72 TALE DNA-binding domains may be built in a variety of ways, the easiest being Golden Gate assembly.66
Recent TALEN assembly improvements have focused on the development of strategies to improve their performance, such as specificity profiling to uncover nonconventional RVDs that increase TALEN activity.73,74 The FokI cleavage domain, which cuts on the inside of a 12- to 19-bp spacer region that separates each TALE binding site, mediates dimerization of TALEN proteins.67 Targeted nucleases cause DSBs, which are repaired by NHEJ or, in the presence of a donor template, HDR.75
CRISPR/Cas9
Bacteria have a distinct adaptive immune system that destroys foreign DNA using an RNA-guided DNA endonuclease.76 The three processes of CRISPR immunity are spacer acquisition, CRISPR RNA (crRNA) synthesis, and interference.77 CRISPR/Cas evolved from prokaryotes’ adaptive resistance to bacteriophages, invading plasmids, and viruses. A–T-rich leader sequences are found right adjacent to 27–42 bp palindromic repeats in the bacterial genome. The palindromes are divided by “interspaced” DNA known as protospacers, which is a template of previously identified bacteriophage DNA and acts as a marker of earlier infection.78,79
Past foreign DNA fragments are used as spacers in the CRISPR genomic locus. CRISPR RNAs (crRNAs) are transcribed from CRISPR loci and contain a unique sequence complementary to target DNA (called protospacer) as well as a repeat region that hybridizes to a short RNA termed transactivated crRNA (tracrRNA). The crRNA then assembles with Cas proteins to form the effector complex; Cas proteins are guided to break target DNA by the crRNA and tracrRNA combination.79 Furthermore, crRNA and tracrRNA may be combined into a single-guide RNA (sgRNA) that can target and activate Cas9 endonuclease activity.80
The CRISPR/Cas9 genome editing system may be broken down into three steps: recognition, cleavage, and repair.81 Through its 5’ crRNA complementary base pair component, the designed sgRNA drives Cas9 and detects the target sequence in the gene of interest. In the absence of sgRNA, the Cas9 protein stays inactive. Cas9 nuclease causes DSBs three base pairs upstream of Protospacer Adjacent Motif (PAM).82 The PAM sequence is a short (2–5 bp) conserved DNA sequence downstream of the cut site that changes in length depending on the bacterial species. Cas9 protein, the most extensively used nuclease in genome editing tools, detects the PAM sequence at 5-NGG-3 (N can be any nucleotide base). The Cas9 protein is then activated for DNA cleavage.83,84 The HNH domain cleaves the complementary strand of target DNA, whereas the RuvC domain cleaves the non-complementary strand, resulting in primarily blunt-ended DSBs. Finally, the host cellular machinery repairs the DSB.85
CRISPR/Cas-9 Based Genetic Manipulation of hPSCs for Modeling MD
The use of hPSCs and in vitro differentiation protocols that mimic in vivo pancreatic development is well suited to investigate pancreatic monogenic diseases,86,87 as are genome editing tools such as CRISPR/Cas9 to modify hPSCs for human in vitro disease modeling to understand the role of MD relevant mutations in pancreatic development and beta-cell function.88,89
Several pancreatic differentiation protocols have been employed for modeling MD using hPSCs.47,90,91 hPSCs can be successfully differentiated into definitive endoderm, which can then be shaped into a posterior-foregut-like population capable of upregulating PDX1 expression in response to retinoic acid and fibroblast growth factor 10 (FGF10), as well as inhibitors of the bone morphogenetic protein (BMP) and hedgehog signaling pathways.47
The genome editing of a particular cell line that differentiates rapidly to beta-cells may be utilized to develop knock-out models of pancreatic development genes and used to understand their function.92 The CRISPR-Cas9 nuclease system may be used to introduce various genomic changes. The following steps are taken to alter hPSCs for modeling MD using the CRISPR-Cas9 nuclease system: (a) sgRNA design, (b) sgRNA synthesis, (c) single or multiplex sgRNA transfection in hPSCs, (d) assessment of Indel frequency, and (e) clonal propagation of knockout lines.93
Huangfu et al use TALEN and CRISPR-Cas-mediated gene editing in combination with hPSC-directed differentiation to model MD in vitro. This study examines the involvement of PDX1 and seven other pancreatic transcription factors (RFX6, PTF1A, GLIS3, MNX1, NGN3, HES1, and ARX) in pancreatic cell commitment.94 This phenotype correlates with the observation that patients with heterozygous PDX1 mutations develop diabetes at a young age, and it also validates that low levels of PDX1 can cause beta-cell dysfunction, a decrease in beta-cell mass during fetal development, and/or the maintenance of beta-cell mass in adults.95,96
Cardenas-Diaz et al employed the CRISPR-Cas9 technology to genetically alter ESCs to ablate one or two alleles of HNF1A and differentiate these stem cell lines into pancreatic beta-like cells in MODY3, one of the most frequent forms of MDs caused by mutations in the transcription factor HNF1A. Their findings imply that HNF1A plays an important role in endocrine cell development since deletion of HNF1A results in increased production of alpha cell markers such as glucagon and reduced expression of PAX4, a transcription factor that regulates beta cell development.97
Shi et al employed CRISPR/Cas9 to generate hPSCs with frameshift mutations in GATA6, either alone or in conjunction with mutations in GATA4. GATA6± haploinsufficiency changes pancreatic progenitor cell development, resulting in a lower proportion of glucose-responsive beta-like cells, according to their findings.88 Chia et al studied the role of GATA6 using both gene-edited and patient-derived hPSCs and discovered that GATA6 heterozygous hPSCs had a slight decrease in endoderm development, but GATA6-null hPSCs can only form mesoderm-like cells. According to their findings, GATA6 appears to be upstream of the endoderm program in humans.98
Conclusion
Monogenic diabetes is an interesting disease category to model in vitro using hPSCs since it is caused by a single mutation. Over the last decade, great progress in the field of hPSCs has been made in terms of developing hPSC-based models and differentiation processes for studying diabetes pathogenesis. By modifying the genotype of the initial hPSCs or producing hiPSCs from patients with monogenic diabetes, researchers are seeking to understand the mechanisms behind the development of various types of monogenic diabetes. Detailed understanding of the molecular underpinnings behind pancreatic beta-cell development and function, as well as using reliable models that can precisely mimic the effects in humans, can considerably improve the chances of developing effective diabetes treatments.
Disclosure
The authors report no conflicts of interest in relation to this work.
References
1. Atkinson MA, von Herrath M, Powers AC, Clare-Salzler M. Current concepts on the pathogenesis of type 1 diabetes—considerations for attempts to prevent and reverse the disease. Diabetes Care. 2015;38(6):979–988. doi:10.2337/dc15-0144
2. Tangvarasittichai S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J Diabetes. 2015;6(3):456. doi:10.4239/wjd.v6.i3.456
3. Tallapragada DSP, Bhaskar S, Chandak GR. New insights from monogenic diabetes for “common” type 2 diabetes. Front Genet. 2015;6:251. doi:10.3389/fgene.2015.00251
4. James DE, Stöckli J, Birnbaum MJ. The aetiology and molecular landscape of insulin resistance. Nat Rev Mol Cell Biol. 2021;22(11):751–771. doi:10.1038/s41580-021-00390-6
5. Pipatpolkai T, Usher S, Stansfeld PJ, Ashcroft FM. New insights into K ATP channel gene mutations and neonatal diabetes mellitus. Nat Rev Endocrinol. 2020;16(7):378–393. doi:10.1038/s41574-020-0351-y
6. Lin Y-W, Akrouh A, Hsu Y, Hughes N, Nichols CG, De León DD. Compound heterozygous mutations in the SUR1 (ABCC 8) subunit of pancreatic KATP channels cause neonatal diabetes by perturbing the coupling between Kir6. 2 and SUR1 subunits. Channels. 2012;6(2):133–138. doi:10.4161/chan.19980
7. Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol. 2016;17(3):170–182. doi:10.1038/nrm.2015.27
8. Merkle FT, Eggan K. Modeling human disease with pluripotent stem cells: from genome association to function. Cell Stem Cell. 2013;12(6):656–668. doi:10.1016/j.stem.2013.05.016
9. Abdelalim EM, Bonnefond A, Bennaceur-Griscelli A, Froguel P. Pluripotent stem cells as a potential tool for disease modelling and cell therapy in diabetes. Stem Cell Rev Rep. 2014;10(3):327–337. doi:10.1007/s12015-014-9503-6
10. Al‐Khawaga S, Memon B, Butler AE, Taheri S, Abou‐Samra AB, Abdelalim EM. Pathways governing development of stem cell‐derived pancreatic β cells: lessons from embryogenesis. Biol Rev. 2018;93(1):364–389. doi:10.1111/brv.12349
11. Lim WF, Inoue-Yokoo T, Tan KS, Lai MI, Sugiyama D. Hematopoietic cell differentiation from embryonic and induced pluripotent stem cells. Stem Cell Res Ther. 2013;4(3):1–11. doi:10.1186/scrt222
12. Sauer V, Roy-Chowdhury N, Guha C, Roy-Chowdhury J. Induced pluripotent stem cells as a source of hepatocytes. Curr Pathobiol Rep. 2014;2(1):11–20. doi:10.1007/s40139-013-0039-2
13. Bassett AR. Editing the genome of hiPSC with CRISPR/Cas9: disease models. Mamm Genome. 2017;28(7):348–364. doi:10.1007/s00335-017-9684-9
14. Cong L, Zhang F. Genome engineering using CRISPR-Cas9 system. In: Chromosomal Mutagenesis. Springer; 2015:197–217.
15. Abdelalim EM. Modeling different types of diabetes using human pluripotent stem cells. Cell Mol Life Sci. 2021;78(6):2459–2483. doi:10.1007/s00018-020-03710-9
16. Misra S, Owen KR. Genetics of monogenic diabetes: present clinical challenges. Curr Diab Rep. 2018;18(12):1–11. doi:10.1007/s11892-018-1111-4
17. Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic β-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4(4):200–213.
18. Anık A, Çatlı G, Abacı A, Böber E. Maturity-onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab. 2015;28(3–4):251–263. doi:10.1515/jpem-2014-0384
19. Sanyoura M, Philipson LH, Naylor R. Monogenic diabetes in children and adolescents: recognition and treatment options. Curr Diab Rep. 2018;18(8):1–13. doi:10.1007/s11892-018-1024-2
20. Vaxillaire M, Bonnefond A, Froguel P. The lessons of early-onset monogenic diabetes for the understanding of diabetes pathogenesis. Best Pract Res Clin Endocrinol Metab. 2012;26(2):171–187. doi:10.1016/j.beem.2011.12.001
21. Hattersley AT, Patel KA. Precision diabetes: learning from monogenic diabetes. Diabetologia. 2017;60(5):769–777. doi:10.1007/s00125-017-4226-2
22. Inoue I, Nakaoka H. Genetics of diabetes: are they thrifty genotype? In: Evolution of the Human Genome I. Springer; 2017:265–272.
23. Lim SH, Kim JH, Han KH, et al. Genotype and phenotype analyses in pediatric patients with HNF1B mutations. J Clin Med. 2020;9(7):2320. doi:10.3390/jcm9072320
24. Valkovicova T, Skopkova M, Stanik J, Gasperikova D. Novel insights into genetics and clinics of the HNF1A-MODY. Endocr Regul. 2019;53(2):110–134. doi:10.2478/enr-2019-0013
25. Nkonge KM, Nkonge DK, Nkonge TN. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin Diabetes Endocrinol. 2020;6(1):1–10. doi:10.1186/s40842-020-00112-5
26. Laimon W, El-Ziny M, El-Hawary A, et al. Genetic and clinical heterogeneity of permanent neonatal diabetes mellitus: a single tertiary centre experience. Acta Diabetol. 2021;58(12):1689–1700. doi:10.1007/s00592-021-01788-6
27. Lemelman MB, Letourneau L, Greeley SAW. Neonatal diabetes mellitus: an update on diagnosis and management. Clin Perinatol. 2018;45(1):41–59. doi:10.1016/j.clp.2017.10.006
28. Demirbilek H, Hatipoglu N, Gul U, et al. Permanent neonatal diabetes mellitus and neurological abnormalities due to a novel homozygous missense mutation in NEUROD1. Pediatr Diabetes. 2018;19(5):898–904. doi:10.1111/pedi.12669
29. Gopi S, Gowri P, Panda JK, et al. Insulin gene mutations linked to permanent neonatal diabetes mellitus in Indian population. J Diabetes Complications. 2021;35(12):108022. doi:10.1016/j.jdiacomp.2021.108022
30. Touati A, Errea-Dorronsoro J, Nouri S, et al. Transient neonatal diabetes mellitus and hypomethylation at additional imprinted loci: novel ZFP57 mutation and review on the literature. Acta Diabetol. 2019;56(3):301–307. doi:10.1007/s00592-018-1239-3
31. De Franco E, Flanagan SE, Houghton JA, et al. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet. 2015;386(9997):957–963. doi:10.1016/S0140-6736(15)60098-8
32. Greeley SAW, Tucker SE, Naylor RN, Bell GI, Philipson LH. Neonatal diabetes mellitus: a model for personalized medicine. Trends Endocrinol Metabol. 2010;21(8):464–472. doi:10.1016/j.tem.2010.03.004
33. Busiah K, Drunat S, Vaivre-Douret L, et al. Neuropsychological dysfunction and developmental defects associated with genetic changes in infants with neonatal diabetes mellitus: a prospective cohort study. Lancet Diabetes Endocrinol. 2013;1(3):199–207. doi:10.1016/S2213-8587(13)70059-7
34. Kamiya M, Judson H, Okazaki Y, et al. The cell cycle control gene ZAC/PLAGL1 is imprinted—a strong candidate gene for transient neonatal diabetes. Hum Mol Genet. 2000;9(3):453–460. doi:10.1093/hmg/9.3.453
35. Temple IK, Shield JP. 6q24 transient neonatal diabetes. Rev Endocr Metab Disord. 2010;11(3):199–204. doi:10.1007/s11154-010-9150-4
36. Støy J, Steiner DF, Park S-Y, Ye H, Philipson LH, Bell GI. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev Endocr Metab Disord. 2010;11(3):205–215.
37. Park S-Y, Ye H, Steiner DF, Bell GI. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem Biophys Res Commun. 2010;391(3):1449–1454. doi:10.1016/j.bbrc.2009.12.090
38. Singh VK, Saini A, Kalsan M, Kumar N, Chandra R. Describing the stem cell potency: the various methods of functional assessment and in silico diagnostics. Front Cell Develop Biol. 2016;4:134. doi:10.3389/fcell.2016.00134
39. Lin Y, Linask KL, Mallon B, et al. Heparin promotes cardiac differentiation of human pluripotent stem cells in chemically defined albumin‐free medium, enabling consistent manufacture of cardiomyocytes. Stem Cells Transl Med. 2017;6(2):527–538. doi:10.5966/sctm.2015-0428
40. Takahashi K, Tanabe MO, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;2007(11):1–12.
41. Ilic D, Ogilvie C. Concise review: human embryonic stem cells—what have we done? What are we doing? Where are we going? Stem Cells. 2017;35(1):17–25. doi:10.1002/stem.2450
42. King NM, Perrin J. Ethical issues in stem cell research and therapy. Stem Cell Res Ther. 2014;5(4):1–6. doi:10.1186/scrt474
43. Jaiswal MK. Therapeutic opportunities and challenges of induced pluripotent stem cells-derived motor neurons for treatment of amyotrophic lateral sclerosis and motor neuron disease. Neural Regen Res. 2017;12(5):723. doi:10.4103/1673-5374.206635
44. McKinney CE. Using induced pluripotent stem cells derived neurons to model brain diseases. Neural Regen Res. 2017;12(7):1062. doi:10.4103/1673-5374.211180
45. Memon B, Abdelalim EM. Stem cell therapy for diabetes: beta cells versus pancreatic progenitors. Cells. 2020;9(2):283. doi:10.3390/cells9020283
46. Memon B, Karam M, Al-Khawaga S, Abdelalim EM. Enhanced differentiation of human pluripotent stem cells into pancreatic progenitors co-expressing PDX1 and NKX6. Stem Cell Res Ther. 2018;9(1):1–15.
47. Nostro MC, Sarangi F, Yang C, et al. Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Rep. 2015;4(4):591–604. doi:10.1016/j.stemcr.2015.02.017
48. Boroviak T, Loos R, Bertone P, Smith A, Nichols J. The ability of inner-cell-mass cells to self-renew as embryonic stem cells is acquired following epiblast specification. Nat Cell Biol. 2014;16(6):513–525. doi:10.1038/ncb2965
49. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. cell. 2006;126(4):663–676. doi:10.1016/j.cell.2006.07.024
50. Burgos JI, Vallier L, Rodríguez-Seguí SA. Monogenic diabetes modeling: in vitro pancreatic differentiation from human pluripotent stem cells gains momentum. Front Endocrinol. 2021;12:692596.
51. Karagiannis P, Takahashi K, Saito M, et al. Induced pluripotent stem cells and their use in human models of disease and development. Physiol Rev. 2019;99(1):79–114. doi:10.1152/physrev.00039.2017
52. Teo AKK, Wagers AJ, Kulkarni RN. New opportunities: harnessing induced pluripotency for discovery in diabetes and metabolism. Cell Metab. 2013;18(6):775–791. doi:10.1016/j.cmet.2013.08.010
53. Rezania A, Bruin JE, Arora P, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;32(11):1121–1133. doi:10.1038/nbt.3033
54. Teo AK, Windmueller R, Johansson BB, et al. Derivation of human induced pluripotent stem cells from patients with maturity onset diabetes of the young*[S]. J Biol Chem. 2013;288(8):5353–5356. doi:10.1074/jbc.C112.428979
55. Shang L, Hua H, Foo K, et al. β-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes. 2014;63(3):923–933. doi:10.2337/db13-0717
56. Urano F. Wolfram syndrome: diagnosis, management, and treatment. Curr Diab Rep. 2016;16(1):6. doi:10.1007/s11892-015-0702-6
57. Ng NHJ, Jasmen JB, Lim CS, et al. HNF4A haploinsufficiency in MODY1 abrogates liver and pancreas differentiation from patient-derived induced pluripotent stem cells. Iscience. 2019;16:192–205. doi:10.1016/j.isci.2019.05.032
58. Boj SF, Petrov D, Ferrer J. Epistasis of transcriptomes reveals synergism between transcriptional activators Hnf1α and Hnf4α. PLoS Genet. 2010;6(5):e1000970. doi:10.1371/journal.pgen.1000970
59. Balboa D, Saarimäki-Vire J, Borshagovski D, et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. Elife. 2018;7:e38519. doi:10.7554/eLife.38519
60. Ma S, Viola R, Sui L, Cherubini V, Barbetti F, Egli D. Beta cell replacement after gene editing of a neonatal diabetes-causing mutation at the insulin locus. Stem Cell Rep. 2018;11(6):1407–1415. doi:10.1016/j.stemcr.2018.11.006
61. Khan SH. Genome-editing technologies: concept, pros, and cons of various genome-editing techniques and bioethical concerns for clinical application. Mol Ther Nucleic Acids. 2019;16:326–334. doi:10.1016/j.omtn.2019.02.027
62. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11(9):636–646. doi:10.1038/nrg2842
63. Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188(4):773–782. doi:10.1534/genetics.111.131433
64. Paschon DE, Lussier S, Wangzor T, et al. Diversifying the structure of zinc finger nucleases for high-precision genome editing. Nat Commun. 2019;10(1):1–12. doi:10.1038/s41467-019-08867-x
65. Jabalameli HR, Zahednasab H, Karimi-Moghaddam A, Jabalameli MR. Zinc finger nuclease technology: advances and obstacles in modelling and treating genetic disorders. Gene. 2015;558(1):1–5. doi:10.1016/j.gene.2014.12.044
66. Christian M, Cermak T, Doyle EL, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186(2):757–761. doi:10.1534/genetics.110.120717
67. Miller JC, Tan S, Qiao G, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29(2):143–148. doi:10.1038/nbt.1755
68. Chandrasegaran S, Carroll D. Origins of programmable nucleases for genome engineering. J Mol Biol. 2016;428(5):963–989. doi:10.1016/j.jmb.2015.10.014
69. Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326(5959):1501. doi:10.1126/science.1178817
70. Cong L, Zhou R, Kuo Y-C, Cunniff M, Zhang F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat Commun. 2012;3(1):1–6. doi:10.1038/ncomms1962
71. Streubel J, Blücher C, Landgraf A, Boch J. TAL effector RVD specificities and efficiencies. Nat Biotechnol. 2012;30(7):593–595. doi:10.1038/nbt.2304
72. Guilinger JP, Pattanayak V, Reyon D, et al. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Methods. 2014;11(4):429–435. doi:10.1038/nmeth.2845
73. Juillerat A, Pessereau C, Dubois G, et al. Optimized tuning of TALEN specificity using non-conventional RVDs. Sci Rep. 2015;5(1):1–7. doi:10.1038/srep08150
74. Yang J, Zhang Y, Yuan P, et al. Complete decoding of TAL effectors for DNA recognition. Cell Res. 2014;24(5):628–631. doi:10.1038/cr.2014.19
75. Gaj T, Sirk SJ, Shui S-l S-L, Liu J. Genome-editing technologies: principles and applications. Cold Spring Harb Perspect Biol. 2016;8(12):a023754. doi:10.1101/cshperspect.a023754
76. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. science. 2012;337(6096):816–821. doi:10.1126/science.1225829
77. Makarova KS, Haft DH, Barrangou R, et al. Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol. 2011;9(6):467–477. doi:10.1038/nrmicro2577
78. Wright AV, Nuñez JK, Doudna JA. Biology and applications of CRISPR systems: harnessing nature’s toolbox for genome engineering. Cell. 2016;164(1–2):29–44. doi:10.1016/j.cell.2015.12.035
79. Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482(7385):331–338. doi:10.1038/nature10886
80. Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi:10.1126/science.1232033
81. Ming S, Tian-Rui X, Ce-Shi C. The big bang of genome editing technology: development and application of the CRISPR/Cas9 system in disease animal models. Zoo Res. 2016;37(4):191. doi:10.13918/j.issn.2095-8137.2016.4.191
82. Ceasar SA, Rajan V, Prykhozhij SV, Berman JN, Ignacimuthu S. Insert, remove or replace: a highly advanced genome editing system using CRISPR/Cas9. Biochim Biophys Acta Mol Cell Res. 2016;1863(9):2333–2344. doi:10.1016/j.bbamcr.2016.06.009
83. Jiang F, Doudna JA. CRISPR–Cas9 structures and mechanisms. Annu Rev Biophys. 2017;46(1):505–529. doi:10.1146/annurev-biophys-062215-010822
84. Mei Y, Wang Y, Chen H, Sun ZS, Ju X-D. Recent progress in CRISPR/Cas9 technology. J Genet Genomics. 2016;43(2):63–75. doi:10.1016/j.jgg.2016.01.001
85. Deng L, Luo M, Velikovsky A, Mariuzza RA. Structural insights into the evolution of the adaptive immune system. Annu Rev Biophys. 2013;42(1):191–215. doi:10.1146/annurev-biophys-083012-130422
86. Tiyaboonchai A, Cardenas-Diaz FL, Ying L, et al. GATA6 plays an important role in the induction of human definitive endoderm, development of the pancreas, and functionality of pancreatic β cells. Stem Cell Rep. 2017;8(3):589–604. doi:10.1016/j.stemcr.2016.12.026
87. Zeng H, Guo M, Zhou T, et al. An isogenic human ESC platform for functional evaluation of genome-wide-association-study-identified diabetes genes and drug discovery. Cell Stem Cell. 2016;19(3):326–340. doi:10.1016/j.stem.2016.07.002
88. Shi Z-D, Lee K, Yang D, et al. Genome editing in hPSCs reveals GATA6 haploinsufficiency and a genetic interaction with GATA4 in human pancreatic development. Cell Stem Cell. 2017;20(5):675–88. e6. doi:10.1016/j.stem.2017.01.001
89. Zhu Z, González F, Huangfu D. The iCRISPR platform for rapid genome editing in human pluripotent stem cells. Methods Enzymol. 2014;546:215–250.
90. Nostro MC, Keller G. Generation of beta cells from human pluripotent stem cells: potential for regenerative medicine. In: Seminars in Cell & Developmental Biology. Elsevier; 2012.
91. Pagliuca FW, Millman JR, Gürtler M, et al. Generation of functional human pancreatic β cells in vitro. Cell. 2014;159(2):428–439. doi:10.1016/j.cell.2014.09.040
92. Balboa D, Saarimäki‐Vire J, Otonkoski T. Concise review: human pluripotent stem cells for the modeling of pancreatic β‐cell pathology. Stem Cells. 2019;37(1):33–41. doi:10.1002/stem.2913
93. Gupta N, Susa K, Yoda Y, Bonventre JV, Valerius MT, Morizane R. CRISPR/Cas9‐based targeted genome editing for the development of monogenic diseases models with human pluripotent stem cells. Curr Protoc Stem Cell Biol. 2018;45(1):e50. doi:10.1002/cpsc.50
94. Zhu Z, Li QV, Lee K, et al. Genome editing of lineage determinants in human pluripotent stem cells reveals mechanisms of pancreatic development and diabetes. Cell Stem Cell. 2016;18(6):755–768. doi:10.1016/j.stem.2016.03.015
95. Brissova M, Blaha M, Spear C, et al. Reduced PDX-1 expression impairs islet response to insulin resistance and worsens glucose homeostasis. Am J Physiol Endocrinol Metab. 2005;288(4):E707–E14. doi:10.1152/ajpendo.00252.2004
96. Johnson JD, Ahmed NT, Luciani DS, et al. Increased islet apoptosis in Pdx1+/–mice. J Clin Invest. 2003;111(8):1147–1160. doi:10.1172/JCI200316537
97. Cardenas-Diaz FL, Osorio-Quintero C, Diaz-Miranda MA, et al. Modeling monogenic diabetes using human ESCs reveals developmental and metabolic deficiencies caused by mutations in HNF1A. Cell Stem Cell. 2019;25(2):273–89. e5. doi:10.1016/j.stem.2019.07.007
98. Chia CY, Madrigal P, Denil SL, et al. GATA6 cooperates with EOMES/SMAD2/3 to deploy the gene regulatory network governing human definitive endoderm and pancreas formation. Stem Cell Rep. 2019;12(1):57–70. doi:10.1016/j.stemcr.2018.12.003
99. Braverman-Gross C, Nudel N, Ronen D, Beer NL, McCarthy MI, Benvenisty N. Derivation and molecular characterization of pancreatic differentiated MODY1-iPSCs. Stem Cell Res. 2018;31:16–26. doi:10.1016/j.scr.2018.06.013
100. Colclough K, Bellanne‐Chantelot C, Saint‐Martin C, Flanagan SE, Ellard S. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity‐onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat. 2013;34(5):669–685. doi:10.1002/humu.22279
101. Pearson ER, Boj SF, Steele AM, et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007;4(4):e118. doi:10.1371/journal.pmed.0040118
102. Yamagata K, Oda N, Kaisaki PJ, et al. Mutations in the hepatocyte nuclear factor-1α gene in maturity-onset diabetes of the young (MODY3). Nature. 1996;384(6608):455–458. doi:10.1038/384455a0
103. Aqel YWA, Ali G, Elsayed AK, Al-Khawaga S, Hussain K, Abdelalim EM. Generation of two human iPSC lines from patients with maturity-onset diabetes of the young type 2 (MODY2) and permanent neonatal diabetes due to mutations in the GCK gene. Stem Cell Res. 2020;48:101991. doi:10.1016/j.scr.2020.101991
104. Vionnet N, Stoffel M, Takeda J, et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356(6371):721–722. doi:10.1038/356721a0
105. Low BSJ, Lim CS, Ding SSL, et al. Decreased GLUT2 and glucose uptake contribute to insulin secretion defects in MODY3/HNF1A hiPSC-derived mutant β cells. Nat Commun. 2021;12(1):1–20. doi:10.1038/s41467-021-22843-4
106. Teo AKK, Lau HH, Valdez IA, et al. Early developmental perturbations in a human stem cell model of MODY5/HNF1B pancreatic hypoplasia. Stem Cell Rep. 2016;6(3):357–367. doi:10.1016/j.stemcr.2016.01.007
107. Horikawa Y, Iwasaki N, Hara M, et al. Mutation in hepatocyte nuclear factor–1β gene (TCF2) associated with MODY. Nat Genet. 1997;17(4):384–385. doi:10.1038/ng1297-384
108. Bouillet B, Crevisy E, Baillot-Rudoni S, et al. Whole-exome sequencing identifies the first French MODY 6 family with a new mutation in the NEUROD1 gene. Diabetes Metab. 2020;46(5):400–402. doi:10.1016/j.diabet.2020.03.001
109. Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Nat Acad Sci. 2005;102(13):4807–4812. doi:10.1073/pnas.0409177102
110. Johansson BB, Torsvik J, Bjørkhaug L, et al. Diabetes and pancreatic exocrine dysfunction due to mutations in the carboxyl ester lipase gene-maturity onset diabetes of the young (CEL-MODY): a protein misfolding disease. J Biol Chem. 2011;286(40):34593–34605. doi:10.1074/jbc.M111.222679
111. de Medeiros Abreu G, CdAPD S, Tarantino RM, et al. Identification of the first PAX4-MODY family reported in Brazil. Diabetes Metab Syndr Obes. 2020;13:2623. doi:10.2147/DMSO.S256858
112. Plengvidhya N, Kooptiwut S, Songtawee N, et al. PAX4 mutations in Thais with maturity onset diabetes of the young. J Clin Endocrinol Metab. 2007;92(7):2821–2826. doi:10.1210/jc.2006-1927
113. Borowiec M, Liew CW, Thompson R, et al. Mutations at the BLK locus linked to maturity onset diabetes of the young and β-cell dysfunction. Proc Nat Acad Sci. 2009;106(34):14460–14465. doi:10.1073/pnas.0906474106
114. Lin L, Quan H, Chen K, Chen D, Lin D, Fang T. ABCC8-related maturity-onset diabetes of the young (MODY12): a report of a Chinese family. Front Endocrinol (Lausanne). 2020;11:645. doi:10.3389/fendo.2020.00645
115. Ovsyannikova AK, Rymar OD, Shakhtshneider EV, et al. ABCC8-related maturity-onset diabetes of the young (MODY12): clinical features and treatment perspective. Diabetes Ther. 2016;7(3):591–600. doi:10.1007/s13300-016-0192-9
116. Mlynarski W, Tarasov AI, Gach A, et al. Sulfonylurea improves CNS function in a case of intermediate DEND syndrome caused by a mutation in KCNJ11. Nat Clin Pract Neurol. 2007;3(11):640–645. doi:10.1038/ncpneuro0640
117. Prudente S, Jungtrakoon P, Marucci A, et al. Loss-of-function mutations in APPL1 in familial diabetes mellitus. Am J Hum Genet. 2015;97(1):177–185. doi:10.1016/j.ajhg.2015.05.011
118. Støy J, Edghill EL, Flanagan SE, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Nat Acad Sci. 2007;104(38):15040–15044. doi:10.1073/pnas.0707291104
119. Allen HL, Flanagan SE, Shaw-Smith C, et al. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2012;44(1):20–22. doi:10.1038/ng.1035
120. Bonnefond A, Sand O, Guerin B, et al. GATA6 inactivating mutations are associated with heart defects and, inconsistently, with pancreatic agenesis and diabetes. Diabetologia. 2012;55(10):2845–2847. doi:10.1007/s00125-012-2645-7
121. Senée V, Vattem KM, Delépine M, et al. Wolcott-Rallison Syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes. 2004;53(7):1876–1883. doi:10.2337/diabetes.53.7.1876
122. Gupta S, McGrath B, Cavener DR. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes. 2010;59(8):1937–1947. doi:10.2337/db09-1064
123. Osbak KK, Colclough K, Saint‐Martin C, et al. Update on mutations in glucokinase (GCK), which cause maturity‐onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30(11):1512–1526. doi:10.1002/humu.21110
124. Sellick GS, Barker KT, Stolte-Dijkstra I, et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36(12):1301–1305. doi:10.1038/ng1475
125. Weedon MN, Cebola I, Patch A-M, et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet. 2014;46(1):61–64. doi:10.1038/ng.2826
126. Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–21. doi:10.1038/83713
127. Senée V, Chelala C, Duchatelet S, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. 2006;38(6):682–687. doi:10.1038/ng1802
128. Nicolino M, Claiborn KC, Senée V, Boland A, Stoffers DA, Julier C. A novel hypomorphic PDX1 mutation responsible for permanent neonatal diabetes with subclinical exocrine deficiency. Diabetes. 2010;59(3):733–740. doi:10.2337/db09-1284
129. Sansbury F, Flanagan S, Houghton J, et al. SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia. 2012;55(9):2381–2385. doi:10.1007/s00125-012-2595-0
130. Labay V, Raz T, Baron D, et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat Genet. 1999;22(3):300–304. doi:10.1038/10372
131. Rubio-Cabezas O, Jensen JN, Hodgson MI, et al. Permanent neonatal diabetes and enteric anendocrinosis associated with biallelic mutations in NEUROG3. Diabetes. 2011;60(4):1349–1353. doi:10.2337/db10-1008
132. Ketola I, Otonkoski T, Pulkkinen M-A, et al. Transcription factor GATA-6 is expressed in the endocrine and GATA-4 in the exocrine pancreas. Mol Cell Endocrinol. 2004;226(1–2):51–57. doi:10.1016/j.mce.2004.06.007
133. Naya FJ, Huang H-P, Qiu Y, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11(18):2323–2334. doi:10.1101/gad.11.18.2323
134. Rubio-Cabezas O, Minton JA, Kantor I, Williams D, Ellard S, Hattersley AT. Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes. 2010;59(9):2326–2331. doi:10.2337/db10-0011
135. Pinney SE, Oliver-Krasinski J, Ernst L, et al. Neonatal diabetes and congenital malabsorptive diarrhea attributable to a novel mutation in the human neurogenin-3 gene coding sequence. J Clin Endocrinol Metab. 2011;96(7):1960–1965. doi:10.1210/jc.2011-0029
136. Flanagan SE, De Franco E, Allen HL, et al. Analysis of transcription factors key for mouse pancreatic development establishes NKX2-2 and MNX1 mutations as causes of neonatal diabetes in man. Cell Metab. 2014;19(1):146–154. doi:10.1016/j.cmet.2013.11.021
137. Smith SB, Qu H-Q, Taleb N, et al. Rfx6 directs islet formation and insulin production in mice and humans. Nature. 2010;463(7282):775–780. doi:10.1038/nature08748
138. Valenzuela I, Boronat S, Martínez-Sáez E, et al. Microcephaly with simplified gyral pattern, epilepsy and permanent neonatal diabetes syndrome (MEDS). A new patient and review of the literature. Eur J Med Genet. 2017;60(10):517–520. doi:10.1016/j.ejmg.2017.07.007
139. Bonnefond A, Vaillant E, Philippe J, et al. Transcription factor gene MNX1 is a novel cause of permanent neonatal diabetes in a consanguineous family. Diabetes Metab. 2013;39(3):276–280. doi:10.1016/j.diabet.2013.02.007
140. Greeley SAW, Naylor RN, Philipson LH, Bell GI. Neonatal diabetes: an expanding list of genes allows for improved diagnosis and treatment. Curr Diab Rep. 2011;11(6):519–532. doi:10.1007/s11892-011-0234-7
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.