")
Back to Journals » The Application of Clinical Genetics » Volume 8
Genetics of inherited primary arrhythmia disorders
Received 2 December 2014
Accepted for publication 20 March 2015
Published 18 September 2015 Volume 2015:8 Pages 215—233
DOI https://doi.org/10.2147/TACG.S55762
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Danna A Spears, Michael H Gollob
Division of Cardiology – Electrophysiology, University Health Network, Toronto General Hospital, Toronto, ON, Canada
Abstract: A sudden unexplained death is felt to be due to a primary arrhythmic disorder when no structural heart disease is found on autopsy, and there is no preceding documentation of heart disease. In these cases, death is presumed to be secondary to a lethal and potentially heritable abnormality of cardiac ion channel function. These channelopathies include congenital long QT syndrome, catecholaminergic polymorphic ventricular tachycardia, Brugada syndrome, and short QT syndrome. In certain cases, genetic testing may have an important role in supporting a diagnosis of a primary arrhythmia disorder, and can also provide prognostic information, but by far the greatest strength of genetic testing lies in the screening of family members, who may be at risk. The purpose of this review is to describe the basic genetic and molecular pathophysiology of the primary inherited arrhythmia disorders, and to outline a rational approach to genetic testing, management, and family screening.
Keywords: long QT syndrome, catecholaminergic polymorphic ventricular tachycardia, Brugada syndrome, short QT syndrome, genetics
Introduction
In the general population, the incidence of sudden death ranges from 1/100,000 in adolescents to 1/1,000 in individuals aged 45–75 years.1 In young adults, there is some variability in the reported incidence of sudden death, with estimates ranging from 1/22,000 to 1/128,000 in nonathletes, and ranging from 1/250,000 in Italian athletes to 1/164,000 in US athletes.2 A sudden unexplained death is felt to be due to a primary arrhythmic disorder when no structural heart disease is found on autopsy and there is no preceding documentation of heart disease. In these cases, death is presumed to be secondary to a lethal and potentially heritable abnormality of cardiac ion channel function caused either by an aberration in the genes encoding these proteins, or genes encoding “accessory” proteins essential to ion channel function. These “channelopathies” include congenital long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), Brugada syndrome (BrS), and short QT syndrome (SQTS). It is believed that these disorders are responsible for 10%–15% of cases of sudden unexplained death in young adults and children.3 The management of a survivor of cardiac arrest typically culminates in implantation of an implantable cardioverter-defibrillator (ICD), as the risk of recurrent potentially lethal arrhythmias remains significant.4 An important component of the management of these individuals is a rigorous attempt at characterizing their arrhythmic substrate both for targeted management of arrhythmia risk and to direct genetic testing. In certain cases, genetic testing may have an important role in supporting a diagnosis of a primary arrhythmia disorder, and can also provide prognostic information, but by far the greatest strength of genetic testing lies in the screening of family members, who may be at risk. Post-mortem genetic evaluation, or “molecular autopsy” is becoming increasingly important in the evaluation of sudden unexplained death, particularly for identification of pathogenic mutations in genes underlying normal ion channel function, and also in cardiomyopathy genes in the setting of subclinical cardiomyopathy. This approach may have a significant impact on screening of family members at risk, but is not yet a routine component of post-mortem testing.5,6
A significant proportion of familial arrhythmia syndromes remain gene-elusive, even in the setting of an established phenotype with a clear pattern of familial inheritance. Historically, there are two main approaches to the identification of the culprit gene in these cases. The first is linkage analysis, which is used to identify the location of a disease gene within the chromosomes based on its relative location to established genetic markers.7 The second approach is candidate gene screening, which is based on the understanding of the ion channels and proteins involved in the generation of the cardiac action potential, and a focused analysis of the genes most likely to generate the observed arrhythmic phenotype. Genome wide association studies (GWAS), in which individuals are genotyped for the majority of common single nucleotide polymorphisms and the frequency of particular alleles in cases are compared with controls, have had some utility in the inherited arrhythmias.8 GWAS have been important, in certain cases, for identifying additional determinants of cardiac repolarization that would have been otherwise challenging to elucidate. Whole exome sequencing is emerging as an important tool for identifying culprit genes in inherited arrhythmia disorders, and has been useful in identifying candidate genes in arrhythmogenesis.9,10
The purpose of this review is to describe the basic genetic and molecular pathophysiology of the primary inherited arrhythmia disorders, and to outline a rational approach to genetic testing, management, and family screening.
Long QT syndrome
The hallmark of LQTS is the finding of QT prolongation on surface electrocardiography (ECG), in association with a risk of ventricular arrhythmias, syncope, or sudden death (Figures 1 and 2).11 Long QT syndrome was first described in 1957 by Jervell and Lange-Nielsen, when an association was made between congenital deafness, syncope, and QT interval prolongation on ECG in one family with an autosomal recessive form of the disease.12 Subsequently, the more common form of autosomal dominant LQTS was described by Romano and Ward.13,14 LQTS was the first inherited arrhythmia disorder in the absence of structural heart disease to be described. Population prevalence is thought to be in the range of 1/2,000.15 The QT interval reflects the total duration of cardiac depolarization and repolarization. QT prolongation typically is the result of prolongation of the plateau and final rapid phases of cardiac repolarization. A normal heart rate-corrected QT (QTc) is considered <450 msec for males and <460 msec for females.16,17 The diagnosis of LQTS is likely when cardiac symptoms (syncope or cardiac arrest) accompany a markedly prolonged QT interval: a resting QTc interval <470 msec in men and <480 msec in women is highly specific for LQTS in the absence of another cause for QT prolongation (eg, medication or electrolyte disturbance).18 However, up to 50% of patients with LQTS will not have a markedly prolonged QT,18 and silent gene carriers (ie, normal QTc at rest) constitute 36% of patients with LQT1, 19% with LQT2, and 10% with LQT3.19
 | Figure 1 Normal electrocardiogram. |
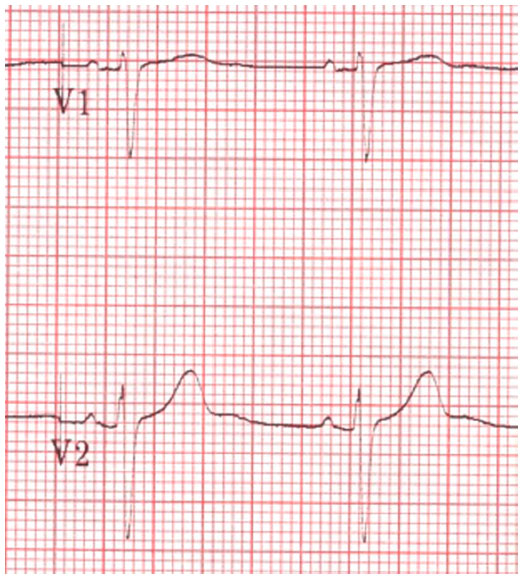
 | Figure 2 Electrocardiogram demonstrating significant QT prolongation (shown by arrow). |
The dominant repolarizing currents are IKr and IKs. Repolarization occurs with the time-dependent inactivation of ICaL and activation/reactivation of the inward rectifying currents, which include IKr and IKs. As a result of this, K is extruded from the cell, and the resting membrane potential is restored. The relative contribution of each of the ion currents to the action potential duration is influenced by adrenergic tone. Catecholamines activate adrenergic receptors, triggering increases in intracellular cAMP and activation of a complex cascade of protein phosphorylation. Increased cAMP directly results in an increased rate of depolarization in sinoatrial cells.20 Phosphorylation of calcium handling proteins, such as L-type calcium channels and ryanodine receptors, results in increased intracellular calcium and myocardial contractility. At rest, IKs likely has minimal activity.21 The activity of IKs is enhanced with adrenergic stimulation, which allows for shortening of the action potential duration with exercise.22 Impairment of IKs can prevent appropriate shortening of the action potential duration during exercise. This is the likely explanation for the observation that exercise is a common trigger for sudden death in LQTS due to defects in the gene encoding IKs.23 The effect of autonomic tone on IKr is less clear. In LQTS, prolongation of the action potential creates an electrophysiologic milieu that allows L-type calcium channels to recover from inactivation and to reactivate. This has the potential to trigger early afterdepolarizations that can lead to a form of ventricular arrhythmia known as torsades des points.
Genetics of LQTS
From the earliest descriptions of LQTS by Jervell, Lange-Nielsen, Romano, and Ward, it was apparent that LQTS, with its familial preponderance, was a genetic condition. In 1991, linkage analysis from a multigenerational family with many affected individuals demonstrated that the genetic defect mapped to the small arm of chromosome 11.24 Despite the unifying features of LQTS, namely syncope, torsades des points, and QT prolongation on ECG, there was significant heterogeneity in clinical presentation, arrhythmia triggers, and specific ECG findings of T wave morphology among unrelated individuals. Ultimately, linkage analysis in other cohorts demonstrated that not all kindreds with LQTS shared this locus.25 The genetic heterogeneity of LQTS was ultimately proven in 1994 when linkage analysis identified loci on chromosomes 7 and 3.26 This led to classification of LQTS into subtypes based on genetic loci. The first subtype was mapped to chromosome 11 and was classified as LQTS type 1 (LQTS1). LQTS types 2 and 3 were characterized next, and linked to loci on chromosomes 7 and 3, respectively.26 There are currently a total of 15 subtypes of LQTS, each with a unique genetic basis for an ion channelopathy that results in slowing of myocardial repolarization27 (Table 1). In general, across all genetic subtypes, the clinical penetrance of LQTS is in the range of 40%,28 with a broad range of 25%–100%.29,30 The gene at the LQTS1 locus on chromosome 11 is the KCNQ1 gene.31 This gene encodes the alpha-subunit of the voltage-gated potassium channel, which is responsible for the slow component of the delayed rectifier current (IKs).32 Mutations in this gene that impair channel function reduce IKs, causing prolongation of the repolarization phase of the action potential. Transmembrane mutations are related to longer QTc, more frequent cardiac events, and greater QTc prolongation with exercise.33–35 Cytoplasmic loop mutations that affect sites of adrenergically mediated phosphorylation are specifically associated with QT prolongation during exercise, while the QTc may be normal at rest.36 The gene at the LQTS2 locus on chromosome 7 is KCNH2, which encodes the alpha-subunit of the voltage-gated potassium channel and mediates the rapidly activating component of the delayed rectifying potassium current in the heart (IKr).37,38 Loss-of-function mutations result in a reduced IKr and delayed cardiac repolarization manifesting as a prolonged QT interval on ECG. Mutations in the pore region of this channel are more likely to be associated with cardiac events and sudden death.39,40 The gene at the LQTS3 locus on chromosome 3 is SCN5A, which encodes the voltage-gated sodium channel, Nav1.5.41 SCN5A gain-of-function mutations responsible for LQT3 result in an increased late inward Nav 1.5 current that slows cardiac repolarization, causing a prolonged QT interval.42 Mutations affecting KCNQ1, KCNH2, and SCN5A are responsible for over 90% of genetically defined cases of LQTS.43 Less common forms of LQTS have also been described, with heterogeneous molecular etiologies expressed as QT prolongation and risk of lethal ventricular arrhythmias. The genes involved typically encode an ion channel or an accessory protein that is critical to ion channel expression, function, or localization in the cell membrane. LQT13 arises as a result of a mutation in KCNJ5, which encodes the inwardly rectifying potassium channel, and results in reduced membrane expression of the protein.44 Three other forms of LQTS have been characterized by an electrophysiology alteration similar to LQT3, namely LQT9, 10, and 12. LQT10 has been attributed to a gain-of-function mutation within SCN4B, an auxiliary beta-subunit that modifies Nav 1.5 activity in a fashion similar to that observed with LQT3.45 LQT9 and 12 occur secondary to mutations within the structural proteins caveolin-3 (CAV3) and alpha1-syntrophin (SNTA1), respectively.46,33 Abnormalities in these proteins have the potential to modify Nav 1.5 activity in a manner that mirrors LQT3 through an increased late inward current.
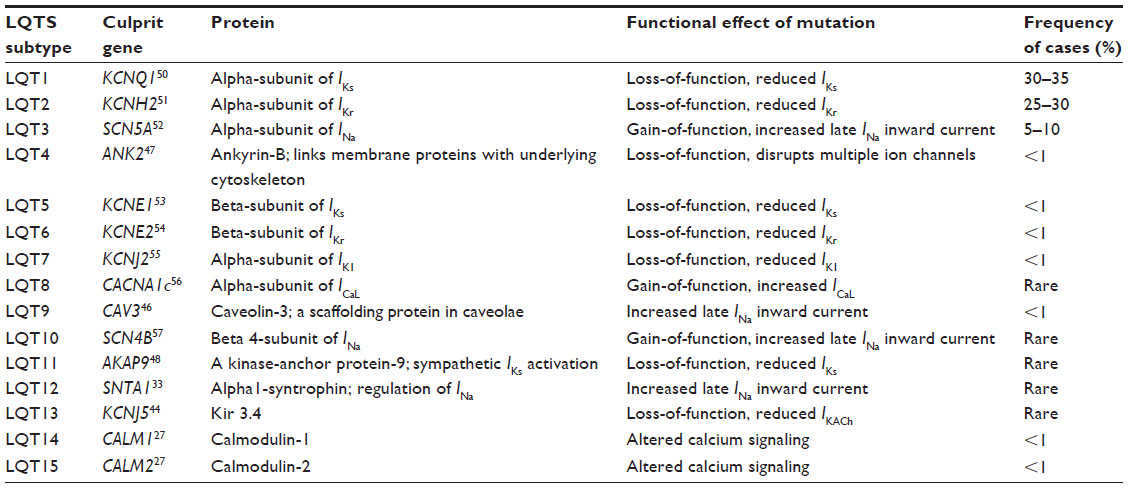 | Table 1 Long QT syndrome by genetic subtype |
Other structural proteins have been implicated in LQTS. Ankyrin-B (ANK2) was identified in the course of linkage analysis of a large family with LQTS.47 LQT4 is characterized by a mutation within ANK2, the protein product of which is a structural protein (ankyrin-B) responsible for coordinated assembly of the sodium/calcium exchanger, the sodium/potassium ATPase pump, and the inositol triphosphate receptor.47 Screening of candidate genes in LQTS cohorts with no mutation in any of the more common LQTS genes has identified the remainder. These include CAV3, A kinase-anchor protein-9 (AKAP9), and SNTA1, which appear to modify ion channel function.33,46,48 In the case of LQT11, a mutation within AKAP9, which encodes an A kinase-anchoring protein responsible for facilitating phosphorylation of KCNQ1 by protein kinase A, impairs IKs augmentation, leading to a clinical phenotype similar to that for LQT1 and LQT5.48 LQT8, or Timothy’s syndrome, occurs secondary to gain-of-function mutations within CACNA1c, which encodes an L-type calcium channel responsible for a depolarizing calcium current (ICa).49 This current contributes to prolongation of the plateau phase within the cardiac action potential, effectively prolonging repolarization and accounting for the resultant long QT interval. Abnormalities of calcium signaling have recently been implicated in LQTS. Recently, with the use of whole exome sequencing, specific mutations in CALM1 and CALM2 have been identified. These result in dysfunctional calmodulin, which may prolong repolarization by more than one mechanism.27 Impaired Ca2+-dependent inactivation of L-type calcium channels or dysregulation of voltage-gated sodium channels may lead to an increased depolarizing current during the plateau phase of the cardiac action potential.
The phenotypic expression of LQTS can also be influenced by modifier genes that may be common genetic variants in the general population but can have an important impact on QT interval and possibly the clinical outcome. Some of these modifiers are single nucleotide polymorphisms in genes encoding signaling proteins, as in NOS1AP.58–60 Others are variants within the LQT genes themselves that may have an impact on the QT interval in addition to an identified pathogenic mutation in any of the other, or same, LQT gene(s). Examples of these variants have been reported in KCNE1,61 KCNH2,62 and SCN5A.63
GWAS have provided further insight into the genetic determinants of cardiac repolarization. It was by this method that the first nucleotide polymorphisms in genes encoding signaling proteins as in NOS1AP were revealed.58–60 Subsequently, GWAS was useful in identifying other genetic loci, which, collectively, confer significant inherited variability in the QT interval.64,65 In addition, certain single nucleotide polymorphisms that are present in 1%–3% of the population may lead to acquired LQTS and events in the setting of low potassium, calcium, magnesium, and certain QT prolonging drugs.66,67
There are distinct phenotypes that reflect the genetic heterogeneity of LQTS, although the final common characteristic is prolonged repolarization and risk of arrhythmia. There are unique ECG characteristics that typify the most common LQT subtypes.68 In LQT1, the T wave is characteristically broad-based, while the T wave in LQT2 is typically bifid and of low amplitude. In contrast, in LQT3, prolongation of the ST segment accounts for QT prolongation and is typically followed by a relatively small-amplitude T wave. In addition, certain forms of LQTS are accompanied by extracardiac manifestations, including periodic paralysis in Andersen–Tawil syndrome (LQT7), and syndactyly, immune deficiency, and autism in Timothy’s syndrome (LQT8).49,55
The Jervell and Lange-Nielsen syndrome, with sensorineural deafness in association with QT prolongation and arrhythmia, arises secondary to homozygous or compound heterozygous mutations in either KCNQ1 or KCNE1, the genes encoding the alpha-subunits and beta-subunits of IKs, respectively.69–71 The cardiac manifestations are most often inherited in an autosomal dominant fashion, while the sensorineural deafness requires mutations in both allele copies, accounting for the original characterization of the syndrome as autosomal recessive.72
The influence of genotype, sex, and age on the LQT phenotype have been well described by the International LQT Registry research group.73–75 The most common LQT subtypes have distinct arrhythmia triggers.23 Episodes of torsades des points in LQT1 typically occur at elevated heart rates, such as with exercise, while events during sleep are less common.23,76 This can be understood in the context of the normal function of IKs. This current becomes critical for shortening of the action potential at high heart rates, and its activity is increased under conditions of increased adrenergic tone, providing a link between exercise-induced arrhythmia and loss-of-function mutations in KCNQ1.77 IKr is responsible for repolarization reserve at lower heart rates, and serves to shorten the QT interval at resting heart rates and in the early phases of heart rate elevation in response to emotional or physical stress, prior to IKs activation. Patients with LQT2 appear to be susceptible to events during both sleep and exercise, while having a particular vulnerability to abrupt auditory stimuli, such as alarm clocks.23,78 Conversely, arrhythmic events in the context of LQT3 occur rarely during exercise and most frequently during sleep.23
There is significant variability in the event rates in LQTS, even within the same genetic subgroup. Because of the implications for clinical management, efforts have been made to identify clinical and genetic variables that may contribute to the risk stratification of patients. Clinically, the QTc has been shown to be an independent predictor for clinical events when >500 msec.11,19 The onset of arrhythmic events in childhood is an important predictor of risk, with individuals having their first event in infancy being at very high risk of future events.79–81 A risk stratification model has been proposed for predicting a first cardiac event, and divides LQTS patients into low-risk, intermediate-risk, and high-risk groups on the basis of symptom history, QTc, and age.82
Sex has also been shown to correlate with event rates in an age-dependent manner. Affected males have an increased event rate during childhood; however, in adolescence and early adulthood, the trend reverses.73,75,83,84 In fact, analysis of affected individuals 40 years of age or older from the International LQTS Registry reveals that affected females continue to be at an increased risk of arrhythmic death relative to their male counterparts.11 Analysis of sex in relation to genetic subtype has revealed that females with LQT2 and males with LQT3 are at particularly high risk for sudden death or first cardiac arrest.85 In genotype-positive individuals, a family history of sudden death in a first-degree relative has not been shown to be associated with a greater risk of death in LQTS.86
The prognosis of LQTS is also related to the underlying genetic abnormality. Observations from the International LQTS Registry suggested that the frequency of clinical events prior to initiation of beta-blocker therapy from birth to 40 years of age was significantly higher in LQT2 (46%) and LQT3 (42%) patients relative to those with LQT1 (30%).85 There is also evidence that events in LQT3 are more likely to be lethal.87 In one study of patients prescribed beta-blocker therapy over an average of 5 years, cardiac event rates also differed according to subtype. Events such as syncope or cardiac arrest were reported to occur in 10% of LQT1 patients, but at higher rates in LQT2 (23%) and LQT3 (32%) patients.88 Although LQT1 and to a certain extent LQT2 patients appear to respond to beta-blockers, this therapy is of less obvious efficacy in LQT3. Mutations that result in amino acid changes in specific regions of the ion channel also confer an increased arrhythmia risk. For example, in LQT1, mutations in the cytoplasmic loops of the KCNQ1 protein89 or mutations with dominant-negative ion current effects39 are associated with a worse prognosis, especially when compared with mutations affecting the C-terminal regions of the protein. Similarly, in LQT2, mutations in the pore region of the KCNH2 protein result in a greater risk of arrhythmia, especially in males.39
Accuracy in the diagnosis of LQTS is further confounded by the observation that LQTS patients may have QTc values within normal limits even if they are genotype-positive, or may show QT prolongation only under specific circumstances, resulting in the potential for significant overlap in QTc between healthy controls and LQTS patients.90 Scoring systems have been proposed,24,91 but lack sensitivity when compared with analysis of QTc alone. In an effort to improve diagnostic accuracy, strategies have been developed to determine QTc values at rest and also to characterize the dynamic changes that occur on a daily basis and with physiological stress. The first step involves serial ECG testing or acquisition of previously acquired ECGs in order to take account of the considerable temporal variability of the QTc in LQTS.90 Given the autosomal dominant inheritance pattern of the condition, examination of the ECGs of family members may be helpful if the phenotype is present in a first-degree relative. Exercise stress testing has been used extensively in the past to identify parameters of repolarization that may be indicative of LQTS. When stress testing is inconclusive, intravenous administration of epinephrine followed by analysis of the QT response in relation to heart rate92,93 may be useful in differentiating LQTS patients from healthy controls. Collectively, these relatively simple techniques may help guide the clinician in reaching a diagnostic conclusion.
Genetic testing in LQTS
The diagnosis of LQTS is made on clinical grounds, but incorporation of genetic testing in the evaluation of LQTS can be a powerful supportive tool, particularly as the results may assist in risk stratification, guide therapy, and facilitate family screening.94 Genetic testing for LQTS should not preclude careful clinical evaluation. Interpretation of clinical genetic testing results is not always straightforward. In particular, the clinical significance of novel genetic variants is frequently unclear. An analysis of culprit mutations in LQTS types 1–3 has revealed important features that provide a framework to differentiate between pathogenic and benign variants. For example, although non-missense mutations account for less than 5% of mutations, their likelihood of being pathogenic, regardless of their location within the ion channel, is greater than 99%.95 KCNQ1 and KCNH2 missense mutations within the transmembrane, linker, or pore regions also have a high likelihood of being pathogenic, and in the case of KCNH2 may also be predictive of cardiac events.34,95 In contrast, the significance of missense mutations in the interdomain linkers of SCN5A is less clear.95
Management of LQTS
Beta-blocker therapy is the first-line treatment for LQTS.96 However, when genotype-specific management of LQTS is considered, not all beta-blockers are equivalent. The four beta-blockers studied have equal efficacy in preventing first cardiac events in LQTS1, but in LQTS2, nadolol is the only beta-blocker that offers a significant risk reduction.97 Propranolol has been shown, at high concentrations, to also block IKr, which may explain its inferior protective capacity in LQTS2.98 An adjunctive therapy to be considered in LQT2 is long-term oral potassium supplementation.99,100 There are limited data regarding the use of beta-blockers in the less common LQTS subtypes. In LQTS3, beta-blockers appear to have much less of a protective effect,19,74 but do have some efficacy in reducing event rates101 and should be considered first-line therapy. There is some evidence that propranolol may have a specific advantage in this subtype,102 an effect likely mediated by its weak sodium channel properties.103 Adjunctive therapies such as mexiletine104 or ranolazine can be considered in very select cases of LQTS3.105
Lifestyle advice is of particular importance in the management of LQTS.106 Individuals with any LQTS subtype must avoid QT-prolonging drugs (www.crediblemeds.org). In LQT1, high-intensity exercise is restricted, especially swimming. In LQT2, the high rate of nonexercise triggers warrants avoidance of arousal sources such as loud telephones or alarms. Patients with very long QTc intervals (>500 msec) may shorten their QT interval with potassium supplements and/or spironolactone.107 In the postpartum period, compliance with beta-blockers and caution regarding sudden auditory arousal are essential.108
In LQTS, ICD implantation is reserved for patients with recurrent events despite treatment, in whom a contraindication to medical therapy exists or in whom the calculated risk of sudden cardiac death (SCD) is very high. Left sympathetic denervation also reduces the risk of SCD in high-risk LQTS. It is useful for patients in whom recurrent events occur despite use of medication or after implantation of an ICD.109
Short QT syndrome
SQTS is characterized by an abbreviated QT interval and a risk of both atrial and ventricular arrhythmias (Figure 3). Because it is a rare condition, there are limited data on its prevalence and demographics. A review of 61 reported cases found a male preponderance (75%). The median age of symptom onset was 21 years. SCD was reported in 33% and atrial fibrillation in 18%.110 The initial description of SQTS included a QTc <300 msec.111 The distribution of QT intervals in the general population, however, follows a bell-shaped distribution. The lower limit of normal for the QT interval is approximately 350 msec for males and 360 msec for females.112 A diagnostic scoring system has been proposed, which incorporates several features of the condition to aid in identifying SQTS and subsequent decision-making (Table 2).110,113 PQ segment depression is frequently seen in SQTS, and may be an additional unique ECG feature characteristic of this condition.114 The current guideline consensus is that the case definition should be a corrected QT interval <330 msec.115 However, this represents an extreme of QT shortening and may lead to missing the diagnosis in more moderate cases. In the proposed diagnostic scorecard reported by Gollob et al,110 a corrected QT interval of <370 msec may still satisfy a diagnosis of SQTS. The mechanism of arrhythmogenesis in SQTS likely involves increased dispersion of repolarization within the myocardium. Analogous to LQTS, gain-of-function repolarizing currents cause variable degrees of shortening of the action potential.112
 | Figure 3 Electrocardiogram characteristic of short QT syndrome. |
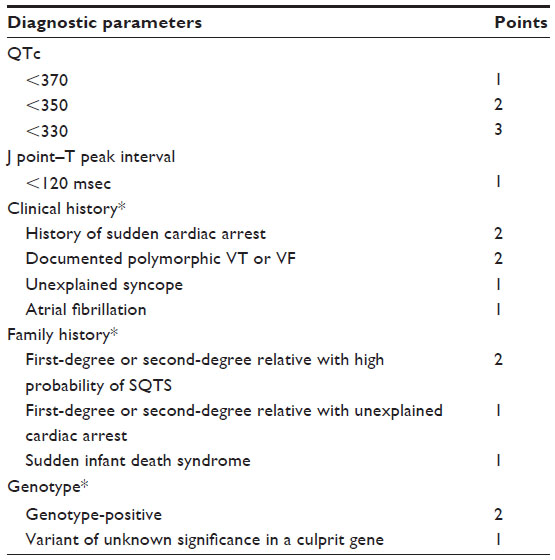 | Table 2 Short QT syndrome diagnostic scorecard |
Genetics of SQTS
Genetic subtypes of SQTS have been identified (Table 3). In SQT1, a gain-of-function mutation in KCNH2 impairs the voltage-dependent inactivation of IKr. This gain-of-function mutation causes increased current flow through the channel and shortens the action potential duration and QT interval. The first mutation described was N588K.116,117 This remains the most prevalent genetic subtype of SQTS.110 Other mutations in this gene have subsequently been identified. Gain-of-function mutations in KCNQ1 are the underlying genetic defect in SQT2. Similarly, these defects increase the repolarizing current, shortening the QT interval.118,119 SQT3 is caused by a gain-of-function mutation in KCNJ2, which encodes the alpha-subunit of the inward rectifier potassium current (IK1).120
 | Table 3 Short QT syndrome by genetic subtype |
A reduction in depolarizing currents can also shorten the QT interval. Loss-of-function mutations in the L-type calcium channel (CACNA1C, alpha-subunit, and CACNB2b, alpha-subunit) are associated with a shortening of the QT interval and precordial ST elevation reminiscent of BrS.121 BrS is the dominant phenotype in these patients, and we believe they should be classified as such, thus leaving KCNH2, KCNQ1, and KCNJ2 as the principal genetic causes of SQTS. There are limited data regarding the overall penetrance of SQTS, but it may be as high as 100%.122
Genetic testing in SQTS
SQTS is a clinical diagnosis based on ECG findings and clinical features, as described in the diagnostic scorecard or an absolute QTc of <330 msec in isolation. Individuals with an intermediate to high probability of having SQTS may be screened for mutations in KCNH2, KCNQ1, and KCNJ2, and a positive finding may guide medical therapy and family screening. The yield of genetic testing in SQTS ranges from 18% to 40% in the literature.110,123 However, even in carefully selected patients, the yield of genetic testing is likely to be much lower because of a publication bias for more severe cases.
Management of SQTS
Early approaches to medical therapy in SQTS included use of QT-prolonging drugs. Sotalol, a prototypical QT-prolonging drug, is ineffective in patients with KCNH2 mutations (SQTS1), the most common subtype.116 It was subsequently recognized that most QT-prolonging drugs have the highest affinity to the inactivated state of IKr.124 Given that SQTS1 typically is the result of a mutation in KCNH2, which impairs inactivation of IKr, the relative resistance to these medications is understood.125,126 Quinidine, which has similar affinity to the open and inactivated states of IKr, is effective therapy for SQT1.125 Quinidine is reported to reduce event rates from approximately 4.9% per year to 0% per year, although current data are derived from very small cohorts with only a moderate follow-up duration. An ICD is recommended in patients with resuscitated SCD and those who suffer cardiac events on quinidine.115
Catecholaminergic polymorphic ventricular tachycardia
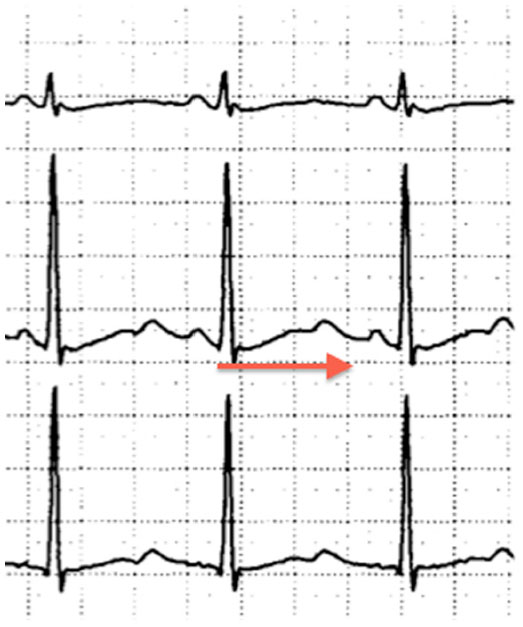
CPVT was first described in 1978.127 The hallmark of CPVT is the onset of ventricular arrhythmias with physical exercise, emotional stress, or catecholamine administration, in the absence of structural heart disease. There is typically an orderly progression of severity, beginning with premature ventricular beats and ventricular bigeminy, followed by bidirectional or polymorphic ventricular tachycardia that ultimately results in ventricular fibrillation (Figure 4).128,129 Symptoms typically begin in childhood or adolescence, but there are cases with onset in adulthood.130 Syncope during exercise is typically the presenting symptom.128 Untreated, this condition has very high mortality, with reports of up to 50% by the age of 30 years.
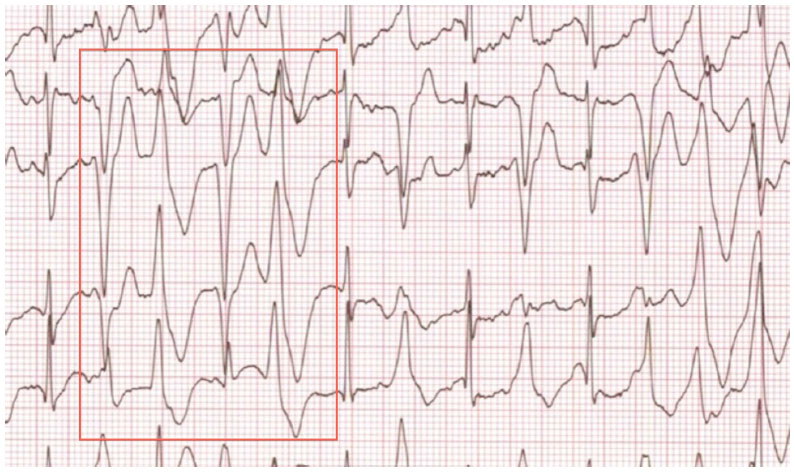 | Figure 4 Arrhythmias characteristic of catecholaminergic polymorphic ventricular tachycardia, ie, frequent premature ventricular complexes, ventricular bigeminy, and bidirectional ventricular tachycardia. |
Genetics of CPVT
In 2001, Priori et al first identified mutations in the gene encoding the cardiac ryanodine receptor (RyR2) in four of 12 probands presenting with polymorphic ventricular tachycardia reproducibly initiated by exercise or isoproterenol.131 The cardiac ryanodine receptor is located on the sarcoplasmic reticulum and controls intracellular calcium release and cardiac muscle contraction. Mutations in RyR2 are found in approximately 60% of individuals with a diagnosis of CPVT.132 In the past decade, over 100 different mutations in RyR2 have been associated with CPVT.133 Inheritance follows both autosomal dominant and, uncommonly, autosomal recessive inheritance patterns.131 Mutations in the calsequestrin 2 gene (CASQ2) were subsequently identified in a kindred with an autosomal recessive inheritance pattern of CPVT (CPVT2).131 Other reports of CASQ2 alterations have consistently shown an autosomal recessive inheritance pattern, requiring either homozygous CASQ2 mutations, or compound heterozygosity.134,135 In CPVT, overall penetrance is 78%.136 Genome-wide scanning has revealed another locus at chromosome 7p22–p14 (homozygous) in a family with CPVT and an autosomal recessive pattern of inheritance.137
The mechanism of arrhythmia in CPVT is related to instability in the RyR2 Ca(2+) release channel complex of the sarcoplasmic reticulum, which results in spontaneous Ca(2+) release through abnormal RyR2 channels leading to delayed after depolarization, triggered activity, and ventricular arrhythmias (Figure 3).138 Alterations in other calcium handling genes have been identified in CPVT (Table 4). Genome-wide linkage analysis has implicated calmodulin 1 (CALM1) in CPVT4.139 Candidate gene screening has identified mutations in triadin (TRDN) as a cause of CPVT5.140 Finally, SCN5A has been implicated in exercise-induced polymorphic ventricular arrhythmias, with the recent identification of a novel missense mutation in a highly conserved region of the gene in a large family with a CPVT phenotype.141
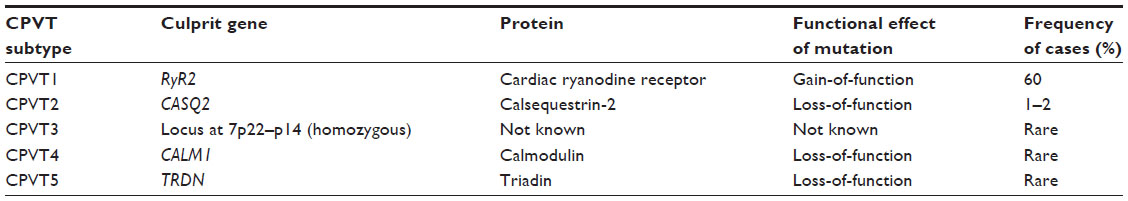 | Table 4 Catecholaminergic polymorphic ventricular tachycardia by genetic subtype |
Genetic testing for CPVT
Genetic testing is recommended in the evaluation of an individual with documented exercise-induced bidirectional VT or polymorphic VT, and when these arrhythmias occur in the context of emotional stress.142 Consideration may also be given to genetic testing in the evaluation of cardiac arrest in the setting of exertion or emotional stress. In sporadic cases, or when examination of the family pedigree suggests autosomal dominant inheritance, sequencing of RyR2 is recommended. CASQ2 sequencing should be considered in sporadic cases when there is a family history of consanguinity or when an autosomal recessive pattern of inheritance is suspected in a large pedigree. CASQ2 mutations are identified in only 1%–2% of all patients with CPVT.132 After identification of a pathogenic mutation, genetic testing can be extended to first-degree relatives.
Management of CPVT
The cornerstone of management of CPVT is restriction of moderate-intensity to high-intensity exercise and administration of high-dose beta-adrenergic blockade.115 Although verapamil has been given previously for breakthrough arrhythmia on beta-blockers,128,143 flecainide may be more effective.144 The antiarrhythmic activity of flecainide may have more than one mechanism, as it may confer direct RyR2 channel block145 or have a direct effect on voltage-gated sodium channels to raise the threshold for delayed after depolarization-induced triggered activity.146 Left cardiac sympathetic denervation may provide excellent long-term rhythm control and can be protective in cases of noncompliance, which are not uncommon in the chronic administration of beta-blockers in younger individuals.147 When arrhythmia persists despite medical therapy, an ICD is recommended. However, avoidance of ICD shocks with adequate medication is paramount in CPVT because the adrenergic outflow following a shock may provoke further arrhythmia, and there is a significant risk of arrhythmic death despite ICD implantation.148–150
Gene therapy for this condition is not available for human use, but a recent study in a murine model of CPVT has demonstrated successful gene therapy, with achievement of sustained infection using a viral vector carrying the wild-type CASQ2 gene.151
Brugada syndrome
In 1992, Pedro and Josep Brugada described a small cohort of patients with ECG findings of J point and ST segment elevation and a history of cardiac arrest due to ventricular fibrillation.152 BrS shows a marked male predominance (male-to-female, 8:1) and significant population variance according to ethnicity. In European and North American populations, the prevalence varies from 0.012% to 0.26%, whereas in Japanese populations the prevalence varies from 0.7% and 1.0%.143,153 Cardiac events typically occur at rest or during sleep at night.
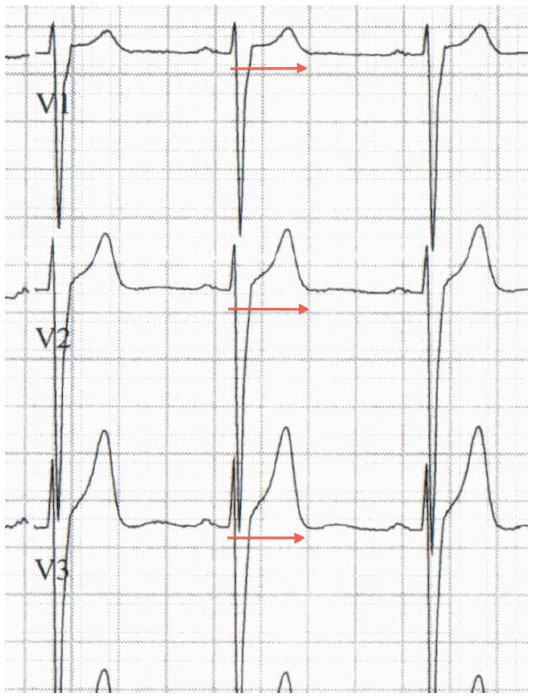
The characteristic ECG pattern of BrS (Figure 5) consists of a >0.2 mV coved ST segment elevation followed by a negative T wave in more than one right precordial ECG lead.154 This is called the type 1 Brugada ECG pattern. A type 1 ECG pattern may be exposed by provocative testing with sodium channel-blocking agents or during febrile illness. The mechanism of ST segment changes in the setting of fever is likely secondary to further compromise of INa due to accelerated inactivation of the channel with higher body temperature,155 or reduced channel conductance. Sensitivity for ECG detection of this pattern can be increased by placing the right precordial ECG leads in the second or third intercostal spaces.156,157 Other ECG findings may be suggestive of a Brugada pattern, specifically the type 2 and 3 Brugada ECG patterns. These are characterized by a “saddleback” pattern of the ST segment with and without ST elevation, respectively. These patterns are not diagnostic for BrS, but should prompt further evaluation with provocative testing or serial ECG acquisition.154 A blunter, broader R wave in V1 or V2 predicts conversion of both types to a type 1 pattern with sodium channel-blocking drugs.158
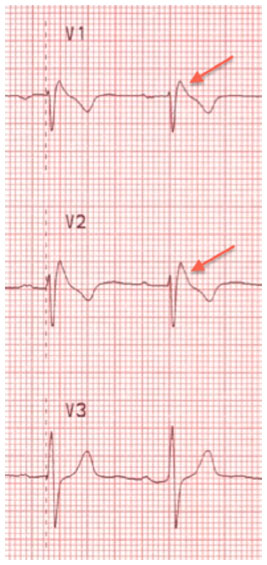 | Figure 5 Brugada electrocardiogram pattern with >2 mm of joint elevation, with coved ST segment elevation in V1 and V2 (arrows). |
The pathophysiology underlying the Brugada ECG and risk of arrhythmia is incompletely understood, but three main theories have been proposed. Briefly, these may be summarized as: the “depolarization” hypothesis, whereby slow conduction into the right ventricular outflow tract predisposes to electrical re-entry;159 the “repolarization” hypothesis, in which an exaggerated phase 1 notch in the epicardium of the right ventricular outflow tract provokes a transmural dispersion of repolarization favoring phase 2 re-entry;153 and the “developmental” hypothesis, in which abnormal expression of cardiac neural crest cells in the right ventricular outflow tract leads to abnormal connexin expression (Cx43) and combined depolarization–repolarization abnormalities favoring arrhythmia.160
Genetics of BrS
Several genes have been implicated in BrS, but in most clinical cases (~65%) no gene mutation is identified. In 1998, screening of the SCN5A gene in a small number of families and individuals with idiopathic ventricular fibrillation and an ECG characteristic for BrS revealed missense, frameshift, and splice donor mutations. In the 11%–28% of individuals with positive genetic testing, the most frequent genetic alterations identified were loss-of-function mutations in SCN5A.161,162 In this subtype of BrS, clinical findings that correlate with the finding of a pathogenic mutation are longer PR interval on the resting ECG and an exaggerated increase in QRS duration with sodium channel-blocking agents.163 Although SCN5A mutations are strongly associated with BrS, genetic background appears to play an important role in expression of the ECG phenotype.164
Mutations have been identified in several other genes. Mutations in GPD1L (BrS2) were identified initially by linkage analysis in a large kindred,165 and then in victims of sudden infant death syndrome.166 In all cases, cellular models demonstrated that the mutations were associated with a significant reduction in sodium current. Candidate gene screening later revealed loss-of-function mutations in CACNA1C (BrS3) and CACNB2 (BrS4) in patients with BrS associated with short QT intervals (three of seven probands with a QTc <360 msec).121 In a later screening study, two mutations in CACNA2D1 were found in four BrS probands who were gene-negative for SCN5A mutations.167
Candidate gene screening has revealed numerous other genetic causes of BrS. These include mutations in SCN1B which result in reduced sodium current (BrS5),168 KCNE3 mutations which lead to increased Ito (BrS6),169 SCN3B mutations which result in reduced sodium current (BrS7),170 HCN4 mutations which result in reduced repolarizing If current (BrS8),171 and lead to action potential prolongation during bradycardia. More recently, SCN10A, which encodes Nav1.8, has been identified as a BrS susceptibility gene, with identified mutations causing a significant reduction in normal sodium channel current.172 The estimated clinical penetrance based on analysis of individuals carrying pathogenic sodium channel mutations is 16%.173 Table 5 contains a detailed listing of these genes.
 | Table 5 Brugada syndrome by genetic subtype |
Genetic testing in BrS
Targeted genetic screening of SCN5A can be useful in patients with a type 1 Brugada ECG pattern, and if a pathogenic mutation is identified, mutation-specific testing is recommended in first-degree relatives.115 The diagnosis of BrS is based only on clinical history and ECG findings. The finding of a genetic mutation is an indicator of potential for development of the clinical phenotype. Such patients should be monitored carefully for the spontaneous appearance of a type 1 Brugada ECG or onset of clinical symptoms (syncope, in particular). Advice for the treatment of fever and avoidance of certain medications should be given to mutation carriers whether or not they display the ECG phenotype.
Management of BrS
There is no effective medical treatment for BrS. The only method proven to prevent sudden death is ICD implantation.174,175 However, not all patients are at a level of risk that justifies this intervention. A history of cardiac arrest or unexplained syncope predicts a high rate of recurrent events176,177 and is an indication for ICD implantation.115 The management of asymptomatic patients with a type 1 Brugada ECG is more challenging, as there are conflicting data regarding the importance of a family history of sudden death, inducibility of ventricular arrhythmias on an electrophysiological study, and presence of a spontaneous Brugada type 1 pattern on ECG.176–179 An important finding common to many risk stratification studies is that one risk factor in isolation is probably not a strong enough indication for ICD implantation, and that a comprehensive approach with evaluation of multiple risk factors is likely to result in more accurate risk assessment. Novel markers of risk that require more rigorous study include the finding of QRS fragmentation and a ventricular refractory period of <200 msec on an electrophysiological study.177
All patients with a Brugada ECG should be taught to aggressively treat any episodes of fever, to seek ECG monitoring in the event of refractory fever, and to avoid drugs known to exacerbate the condition (http://www.brugadadrugs.org). In addition, all individuals with this condition should carry their ECG when seeking emergency medical care to prevent a false diagnosis of acute myocardial infarction and inappropriate care.
In the setting of refractory arrhythmias, isoproterenol, which increases the L-type calcium current, can be useful as an acute antiarrhythmic.180 Quinidine, a class Ia antiarrhythmic that blocks Ito and IKr, can also be used in the ambulatory setting for arrhythmia control and to reduce ICD shocks.181 Neither drug has been tested against ICD implantation, so medical therapy cannot at this time be recommended as an alternative to ICD implantation in high-risk individuals.
Early repolarization syndrome
Early repolarization is defined as >1 mm of J-point elevation in any two contiguous ECG leads, with the exception of the right precordial (V1–V3) leads. The right precordial leads are excluded from the diagnostic definition to avoid inclusion of individuals with Brugada type 1 pattern on ECG. There is, however, some clinical and pathophysiologic overlap between early repolarization (ER) syndrome (ERS) and BrS, and it has even been suggested that the two entities are part of a spectrum of “J-wave syndromes”.182,183 Early repolarization can also be manifest as slurring (a smooth transition from the QRS segment to the ST segment) or notching (a positive J deflection inscribed on the S wave).
Early repolarization was historically considered a normal physiologic variant,184 with a prevalence of 1%–13% in the general population185–188 along with a higher incidence in children,189,190 males,185,190 athletes,191,192 and African-Americans.193 However, it has more recently been associated with arrhythmic events, particularly idiopathic ventricular fibrillation185–187,194,195 and ventricular fibrillation complicating myocardial infarction.196–198 When ER is seen in the context of prior cardiac arrest, the rate of recurrence of ventricular fibrillation is much higher than that seen in survivors of idiopathic ventricular fibrillation who do not have ECG evidence of ER.194 Conversely, when ER is noted incidentally in an asymptomatic individual, the risk of adverse events is very low. Features that may be associated with increased risk include J point amplitude (>0.2 mV) and J-point elevation in the inferior lead or both the inferior and lateral leads.185,187,199 Additional ECG findings, such as a horizontal or downward sloping ST segment, may also be associated with arrhythmic mortality.191,200
The diagnosis of ERS is made when the ECG features of ER are noted in an individual with a history of idiopathic ventricular fibrillation or polymorphic ventricular tachycardia (Table 6).115 Other conditions known to cause J-point elevation, such as ischemia,201 hypokalemia,202 hypercalcemia,203 and hypothermia,204 need to be excluded.
 | Table 6 Recommendations for the diagnosis and management of ER syndrome |
J-point elevation is felt to reflect the transmural differences in the early phases of the action potential.182,183 The relative prominence of Ito in the epicardium accentuates the notching of the action potential, and data from perfused myocardial wedge preparations have suggested that this is the origin of the inscribed J point.205 Augmentation of the gradient created by Ito may occur secondary to a relative increase in outward currents (Ito, IKATP) or a reduction in the opposing inward currents (ICa, INa).
Genetics of ERS
ERS is a clinical diagnosis, and the contributory role of genetic testing has not been established.206 Candidate gene studies have identified rare variants associated with ERS. These include KCNJ8,207,208 ABCC9,209 SCN5A,210 CACNA1C, CACNB2B, and CACNA2D1.167 Recently, a rare variant was found in the KCND2 gene encoding for the Kv4.2 channel in a patient with J waves in the right precordial leads.211 However, all of these variants were detected in single patients and only in a minority of the ERS cases studied.
Management of ERS
A proposed approach to the clinical assessment and management of ER is detailed below. There is an emphasis on identification of clinical predictors of risk rather than on ECG features alone to guide management decisions. ICD implantation should be recommended in any individual with a history of resuscitated cardiac arrest.212 Consideration can be given to ICD implantation in an individual with ER and a history of non-neurocardiogenic syncope or a strong family history of SCD.96 Arrhythmias occurring in the setting of ER can be managed with quinidine, which has been shown to be effective in normalizing the ECG.207,213,214 Isoproterenol can be used in the acute management of ventricular arrhythmias.207,213 Other commonly used antiarrhythmic medications, such as amiodarone, mexiletine, verapamil, beta-blockers, and class Ic antiarrhythmics have little role in the management of arrhythmias in ERS.
Conclusion
Genetic testing has a very important role in the evaluation of individuals with inherited arrhythmia disorders, particularly in screening of potentially affected family members. This is a complex and evolving field, and genetic counseling should be initiated early in the patient encounter, prior to genetic testing, to prepare the patient and their family for possible outcomes. As our understanding of the genetics underlying these primary arrhythmia disorders grows, the interpretation of test results may change, and an ongoing relationship with a genetic counselor over time is essential.
In certain conditions, such as LQTS, genotyping can also provide valuable prognostic information, and can guide therapy. In most cases, however, the greatest role of genetic evaluation lies in streamlining the process of family screening when a pathogenic mutation is found in an affected individual. In the evaluation of a victim of sudden unexplained death, post-mortem genetic analysis has been recommended as part of standard practice in parts of Canada and the European Union.215,216 This is an important first step in identifying living family members who may be at risk. Guidelines for the appropriate handling of post-mortem tissue and blood samples for future clinical and research gene testing is critical as our understanding of the molecular basis of these diseases evolves. In the short-term, while a frustrating proportion of these cases remain gene-elusive, we must rely on careful proband phenotyping and clinical screening of family members.
The current knowledge gap in our understanding of molecular cardiology has encouraged alternative approaches to genetic testing, as well as investigation of gene–gene interactions and environmental contributions to phenotype expression. GWAS and whole exome sequencing have expanded the potential for genetic diagnostics, and have been contributed to furthering our understanding of molecular electrophysiology. This is a time of exponential growth and discovery in the field of cardiac genetics. The gains made in the last decade are paving the way for the next era of cardiac genetics, ie, genotype-specific therapies, novel drug development, and potentially curative gene therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Myerburg RJ. Sudden cardiac death: exploring the limits of our knowledge. J Cardiovasc Electrophysiol. 2001;12(3):369–381. | |
Harmon KG, Drezner JA, Wilson MG, Sharma S. Incidence of sudden cardiac death in athletes: a state-of-the-art review. Heart. 2014;100(16):1227–1234. | |
Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation. 2012;125(4):620–637. | |
Wilber DJ, Garan H, Finkelstein D, et al. Out-of-hospital cardiac arrest. Use of electrophysiologic testing in the prediction of long-term outcome. N Engl J Med. 1988;318(1):19–24. | |
Kauferstein S, Kiehne N, Jenewein T, et al. Genetic analysis of sudden unexplained death: a multidisciplinary approach. Forensic Sci Int. 2013; 229(1–3):122–127. | |
Loporcaro CG, Tester DJ, Maleszewski JJ, Kruisselbrink T, Ackerman MJ. Confirmation of cause and manner of death via a comprehensive cardiac autopsy including whole exome next-generation sequencing. Arch Pathol Lab Med. 2014;138(8):1083–1089. | |
Li L, Wang QK, Rao S. LINKAGE programs: linkage analysis for monogenic cardiovascular diseases. Methods Mol Med. 2006;128:41–60. | |
Bush WS, Moore JH. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822. | |
Marsman RF, Barc J, Beekman L, et al. A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J Am Coll Cardiol. 2014;63(3):259–266. | |
Chugh SS, Huertas-Vazquez A. Inherited arrhythmia syndromes: exome sequencing opens a new door to diagnosis. J Am Coll Cardiol. 2014;63(3):267–268. | |
Goldenberg I, Moss AJ, Bradley J, et al. Long-QT syndrome after age 40. Circulation. 2008;117(17):2192–2201. | |
Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54(1):59–68. | |
Romano C, Gemme G, Pongiglione R. [Rare cardiac arrythmias of the pediatric age. II. Syncopal attacks due to paroxysmal ventricular fibrillation. (Presentation of 1st case in Italian pediatric literature)]. Clin Pediatr (Bologna). 1963;45:656–683. Italian. | |
Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103–106. | |
Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120(18):1761–1767. | |
Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm. 2009;6(5):652–657. | |
Mason JW, Ramseth DJ, Chanter DO, Moon TE, Goodman DB, Mendzelevski B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J Electrocardiol. 2007;40(3):228–234. | |
Sy RW, van der Werf C, Chattha IS, et al. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation. 2011;124(20):2187–2194. | |
Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348(19):1866–1874. | |
DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. 2010;106(3):434–446. | |
Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K(+) channels: structure, function, and clinical significance. Physiol Rev. 2012;92(3):1393–1478. | |
Terrenoire C, Clancy CE, Cormier JW, Sampson KJ, Kass RS. Autonomic control of cardiac action potentials: role of potassium channel kinetics in response to sympathetic stimulation. Circ Res. 2005; 96(5):e25–e34. | |
Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103(1):89–95. | |
Keating M, Atkinson D, Dunn C, Timothy K, Vincent GM, Leppert M. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science. 1991;252(5006):704–706. | |
Towbin JA, Li H, Taggart RT, et al. Evidence of genetic heterogeneity in Romano-Ward long QT syndrome. Analysis of 23 families. Circulation. 1994;90(6):2635–2644. | |
Jiang C, Atkinson D, Towbin JA, et al. Two long QT syndrome loci map to chromosomes 3 and 7 with evidence for further heterogeneity. Nat Genet. 1994;8(2):141–147. | |
Crotti L, Johnson CN, Graf E, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127(9):1009–1017. | |
Berge KE, Haugaa KH, Früh A, et al. Molecular genetic analysis of long QT syndrome in Norway indicating a high prevalence of heterozygous mutation carriers. Scand J Clin Lab Invest. 2008;68(5):362–368. | |
Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation. 1999;99(4):529–533. | |
Viadero MT, Rubín E, Amigo T, González-Lamuño D. Three generations of hereditary long-QT syndrome with complete penetrance caused by the p.G316E KCNQ1 mutation. Pediatr Cardiol. 2011;32(1):102–104. | |
Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12(1):17–23. | |
Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384(6604):78–80. | |
Ueda K, Valdivia C, Medeiros-Domingo A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105(27):9355–9360. | |
Moss AJ. What duration of the QTc interval should disqualify athletes from competitive sports? Eur Heart J. 2007;28(23):2825–2826. | |
Shimizu W, Horie M, Ohno S, et al. Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in the LQT1 form of congenital long QT syndrome: multicenter study in Japan. J Am Coll Cardiol. 2004;44(1):117–125. | |
Bartos DC, Giudicessi JR, Tester DJ, et al. A KCNQ1 mutation contributes to the concealed type 1 long QT phenotype by limiting the Kv7.1 channel conformational changes associated with protein kinase A phosphorylation. Heart Rhythm. 2014;11(3):459–468. | |
Gianulis EC, Trudeau MC. Rescue of aberrant gating by a genetically encoded PAS (Per-Arnt-Sim) domain in several long QT syndrome mutant human ether-á-go-go-related gene potassium channels. J Biol Chem. 2011;286(25):22160–22169. | |
Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81(2):299–307. | |
Shimizu W, Moss AJ, Wilde AA, et al. Genotype-phenotype aspects of type 2 long QT syndrome. J Am Coll Cardiol. 2009;54(22):2052–2062. | |
Moss AJ, Zareba W, Hall WJ, et al; Multicenter Automatic Defibrillator Implantation Trial II Investigators. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346(12):877–883. | |
Abriel H. [Cardiac sodium channel Nav1.5 and its associated proteins]. Arch Mal Coeur Vaiss. 2007;100(9):787–793. French. | |
Bennett PB, Yazawa K, Makita N, George AL. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376(6542):683–685. | |
Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102(10):1178–1185. | |
Yang Y, Yang Y, Liang B, et al. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am J Hum Genet. 2010;86(6):872–880. | |
Medeiros-Domingo A, Kaku T, Tester DJ, et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation. 2007;116(2):134–142. | |
Vatta M, Ackerman MJ, Ye B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114(20):2104–2112. | |
Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421(6923):634–639. | |
Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A. 2007;104(52):20990–20995. | |
Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31. | |
Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12(1):17–23. | |
Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80(5):795–803. | |
Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80(5):805–811. | |
Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17(3):338–340. | |
Abbott GW, Sesti F, Splawski I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97(2):175–187. | |
Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105(4):511–519. | |
Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31. | |
Li RG, Wang Q, Xu YJ, et al. Mutations of the SCN4B-encoded sodium channel β4 subunit in familial atrial fibrillation. Int J Mol Med. 2013;32(1):144–150. | |
Crotti L, Monti MC, Insolia R, et al. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120(17):1657–1663. | |
Tomás M, Napolitano C, De Giuli L, et al. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J Am Coll Cardiol. 2010;55(24):2745–2752. | |
Arking DE, Pfeufer A, Post W, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38(6):644–651. | |
Nishio Y, Makiyama T, Itoh H, et al. D85N, a KCNE1 polymorphism, is a disease-causing gene variant in long QT syndrome. J Am Coll Cardiol. 2009;54(9):812–819. | |
Crotti L, Lundquist AL, Insolia R, et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005; 112(9):1251–1258. | |
Makielski JC, Ye B, Valdivia CR, et al. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ Res. 2003;93(9):821–828. | |
Newton-Cheh C, Eijgelsheim M, Rice KM, et al. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. 2009;41(4):399–406. | |
Pfeufer A, Sanna S, Arking DE, et al. Common variants at ten loci modulate the QT interval duration in the QTSCD study. Nat Genet. 2009;41(4):407–414. | |
Yang P, Kanki H, Drolet B, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105(16):1943–1948. | |
Ramirez AH, Shaffer CM, Delaney JT, et al. Novel rare variants in congenital cardiac arrhythmia genes are frequent in drug-induced torsades de pointes. Pharmacogenomics J. 2013;13(4):325–329. | |
Zhang L, Timothy KW, Vincent GM, et al. Spectrum of ST-T-wave patterns and repolarization parameters in congenital long-QT syndrome: ECG findings identify genotypes. Circulation. 2000;102(23):2849–2855. | |
Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Pathophysiological mechanisms of dominant and recessive KVLQT1 K+ channel mutations found in inherited cardiac arrhythmias. Hum Mol Genet. 1997;6(11):1943–1949. | |
Tyson J, Tranebjaerg L, Bellman S, et al. IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum Mol Genet. 1997;6(12):2179–2185. | |
Wang Z, Li H, Moss AJ, et al. Compound heterozygous mutations in KvLQT1 cause Jervell and Lange-Nielsen syndrome. Mol Genet Metab. 2002;75(4):308–316. | |
Neyroud N, Tesson F, Denjoy I, et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15(2):186–189. | |
Locati EH, Zareba W, Moss AJ, et al. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: findings from the International LQTS Registry. Circulation. 1998;97(22):2237–2244. | |
Zareba W, Moss AJ, Schwartz PJ, et al. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339(14):960–965. | |
Zareba W, Moss AJ, Locati EH, et al; International Long QT Syndrome Registry. Modulating effects of age and gender on the clinical course of long QT syndrome by genotype. J Am Coll Cardiol. 2003;42(1):103–109. | |
Moss AJ, Robinson JL, Gessman L, et al. Comparison of clinical and genetic variables of cardiac events associated with loud noise versus swimming among subjects with the long QT syndrome. Am J Cardiol. 1999;84(8):876–879. | |
Marx SO, Kurokawa J, Reiken S, et al. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295(5554):496–499. | |
Wilde AA, Jongbloed RJ, Doevendans PA, et al. Auditory stimuli as a trigger for arrhythmic events differentiate HERG-related (LQTS2) patients from KVLQT1-related patients (LQTS1). J Am Coll Cardiol. 1999;33(2):327–332. | |
Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292(11):1341–1344. | |
Schwartz PJ, Spazzolini C, Crotti L. All LQT3 patients need an ICD: true or false? Heart Rhythm. 2009;6(1):113–120. | |
Spazzolini C, Mullally J, Moss AJ, et al. Clinical implications for patients with long QT syndrome who experience a cardiac event during infancy. J Am Coll Cardiol. 2009;54(9):832–837. | |
Schwartz PJ, Spazzolini C, Priori SG, et al. Who are the long-QT syndrome patients who receive an implantable cardioverter-defibrillator and what happens to them? Data from the European Long-QT Syndrome Implantable Cardioverter-Defibrillator (LQTS ICD) Registry. Circulation. 2010;122(13):1272–1282. | |
Hobbs JB, Peterson DR, Moss AJ, et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296(10):1249–1254. | |
Sauer AJ, Moss AJ, McNitt S, et al. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49(3):329–337. | |
Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348(19):1866–1874. | |
Goldenberg I, Horr S, Moss AJ, et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol. 2011;57(1):51–59. | |
Zareba W, Moss AJ, Schwartz PJ, et al. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339(14):960–965. | |
Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292(11):1341–1344. | |
Migdalovich D, Moss AJ, Lopes CM, et al. Mutation and gender-specific risk in type 2 long QT syndrome: implications for risk stratification for life-threatening cardiac events in patients with long QT syndrome. Heart Rhythm. 2011;8(10):1537–1543. | |
Goldenberg I, Mathew J, Moss AJ, et al. Corrected QT variability in serial electrocardiograms in long QT syndrome: the importance of the maximum corrected QT for risk stratification. J Am Coll Cardiol. 2006;48(5):1047–1052. | |
Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88(2):782–784. | |
Shimizu W, Noda T, Takaki H, et al. Epinephrine unmasks latent mutation carriers with LQT1 form of congenital long-QT syndrome. J Am Coll Cardiol. 2003;41(4):633–642. | |
Vyas H, Hejlik J, Ackerman MJ. Epinephrine QT stress testing in the evaluation of congenital long-QT syndrome: diagnostic accuracy of the paradoxical QT response. Circulation. 2006;113(11):1385–1392. | |
Gollob MH, Blier L, Brugada R, et al. Recommendations for the use of genetic testing in the clinical evaluation of inherited cardiac arrhythmias associated with sudden cardiac death: Canadian Cardiovascular Society/Canadian Heart Rhythm Society joint position paper. Can J Cardiol. 2011;27(2):232–245. | |
Kapa S, Tester DJ, Salisbury BA, et al. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120(18):1752–1760. | |
Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Expert consensus statement on inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013:e75–e106. | |
Abu-Zeitone A, Peterson DR, Polonsky B, McNitt S, Moss AJ. Efficacy of different beta-blockers in the treatment of long QT syndrome. J Am Coll Cardiol. 2014;64(13):1352–1358. | |
Kawakami K, Nagatomo T, Abe H, et al. Comparison of HERG channel blocking effects of various beta-blockers – implication for clinical strategy. Br J Pharmacol. 2006;147(6):642–652. | |
Compton SJ, Lux RL, Ramsey MR, et al. Genetically defined therapy of inherited long-QT syndrome. Correction of abnormal repolarization by potassium. Circulation. 1996;94(5):1018–1022. | |
Etheridge SP, Compton SJ, Tristani-Firouzi M, Mason JW. A new oral therapy for long QT syndrome: long-term oral potassium improves repolarization in patients with HERG mutations. J Am Coll Cardiol. 2003;42(10):1777–1782. | |
Wilde A. Sodium channel mutations, risk of cardiac events, and efficacy of beta-blocker therapy in type 3 long QT syndrome. Heart Rhythm. 2012;9(5):S321. | |
Calvillo L, Spazzolini C, Vullo E, Insolia R, Crotti L, Schwartz PJ. Propranolol prevents life-threatening arrhythmias in LQT3 transgenic mice: implications for the clinical management of LQT3 patients. Heart Rhythm. 2014;11(1):126–132. | |
Matthews JC, Baker JK. Effects of propranolol and a number of its analogues on sodium channels. Biochem Pharmacol. 1982;31(9):1681–1685. | |
Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation. 2007;116(10):1137–1144. | |
Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol. 2008;19(12):1289–1293. | |
Priori SG, Wilde AA, Horie M, et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace. 2013;15(10):1389–1406. | |
Etheridge SP, Compton SJ, Tristani-Firouzi M, Mason JW. A new oral therapy for long QT syndrome: long-term oral potassium improves repolarization in patients with HERG mutations. J Am Coll Cardiol. 2003;42(10):1777–1782. | |
Khositseth A, Tester DJ, Will ML, Bell CM, Ackerman MJ. Identification of a common genetic substrate underlying postpartum cardiac events in congenital long QT syndrome. Heart Rhythm. 2004; 1(1):60–64. | |
Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation. 2004;109(15):1826–1833. | |
Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol. 2011;57(7):802–812. | |
Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: a familial cause of sudden death. Circulation. 2003;108(8):965–970. | |
Patel C, Yan GX, Antzelevitch C. Short QT syndrome: from bench to bedside. Circ Arrhythm Electrophysiol. 2010;3(4):401–408. | |
Villafañe J, Atallah J, Gollob MH, et al. Long-term follow-up of a pediatric cohort with short QT syndrome. J Am Coll Cardiol. 2013;61(11):1183–1191. | |
Tülümen E, Giustetto C, Wolpert C, et al. PQ segment depression in patients with short QT syndrome: a novel marker for diagnosing short QT syndrome? Heart Rhythm. 2014;11(6):1024–1030. | |
Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10(12):1932–1963. | |
Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109(1):30–35. | |
Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: a familial cause of sudden death. Circulation. 2003;108(8):965–970. | |
Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109(20):2394–2397. | |
Schimpf R, Wolpert C, Gaita F, Giustetto C, Borggrefe M. Short QT syndrome. Cardiovasc Res. 2005;67(3):357–366. | |
Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96(7):800–807. | |
Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):442–449. | |
Mazzanti A, Kanthan A, Monteforte N, et al. Novel insight into the natural history of short QT syndrome. J Am Coll Cardiol. 2014;63(13):1300–1308. | |
Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol. 2011;58(6):587–595. | |
Perrin MJ, Kuchel PW, Campbell TJ, Vandenberg JI. Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-à-go-go-related gene channels. Mol Pharmacol. 2008;74(5):1443–1452. | |
Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43(8):1494–1499. | |
Perrin MJ, Kuchel PW, Campbell TJ, Vandenberg JI. Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-à-go-go-related gene channels. Mol Pharmacol. 2008;74(5):1443–1452. | |
Coumel P. Catecholaminergic induced severe ventricular arrhythmias with Adams-Stokes syndrome in children: report of four cases. Br Heart J. 1978;40 Suppl:28–37. | |
Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106(1):69–74. | |
Bhuiyan ZA, Hamdan MA, Shamsi ET, et al. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J Cardiovasc Electrophysiol. 2007;18(10):1060–1066. | |
Arakawa J, Hamabe A, Aiba T, et al. A novel cardiac ryanodine receptor gene (RyR2) mutation in an athlete with aborted sudden cardiac death: a case of adult-onset catecholaminergic polymorphic ventricular tachycardia. Heart Vessels. Epub 2014 Aug 5. | |
Lahat H, Pras E, Olender T, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69(6):1378–1384. | |
Cerrone M, Napolitano C, Priori SG. Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated to impaired Ca(2+) regulation. Heart Rhythm. 2009;6(11):1652–1659. | |
Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J Am Coll Cardiol. 2009;54(22):2065–2074. | |
di Barletta MR, Viatchenko-Karpinski S, Nori A, et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2006;114(10):1012–1019. | |
Al-Hassnan ZN, Tulbah S, Al-Manea W, Al-Fayyadh M. The phenotype of a CASQ2 mutation in a Saudi family with catecholaminergic polymorphic ventricular tachycardia. Pacing Clin Electrophysiol. 2013;36(5):e140–e142. | |
Postma AV, Denjoy I, Kamblock J, et al Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet. 2005;42(11):863–870. | |
Bhuiyan ZA, Hamdan MA, Shamsi ET, et al. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J Cardiovasc Electrophysiol. 2007;18(10):1060–1066. | |
Watanabe H, Knollmann BC. Mechanism underlying catecholaminergic polymorphic ventricular tachycardia and approaches to therapy. J Electrocardiol. 2011;44(6):650–655. | |
Nyegaard M, Overgaard MT, Søndergaard MT, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91(4):703–712. | |
Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21(12):2759–2767. | |
Swan H, Amarouch MY, Leinonen J, et al. A gain-of-function mutation of the SCN5A gene causes exercise-induced polymorphic ventricular arrhythmias. Circ Cardiovasc Genet. 2014;7(6):771–781. | |
Ackerman MJ, Priori SG, Willems S; European Heart Rhythm Association. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011;13(8):1077–1109. | |
Miyasaka Y, Tsuji H, Yamada K, et al. Prevalence and mortality of the Brugada-type electrocardiogram in one city in Japan. J Am Coll Cardiol. 2001;38(3):771–774. | |
van der Werf C, Zwinderman AH, Wilde AA. Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: state of the art and future developments. Europace. 2012;14(2):175–183. | |
Hilliard FA, Steele DS, Laver D, et al. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol. 2010;48(2):293–301. | |
Liu N, Denegri M, Ruan Y, et al. Short communication: flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity. Circ Res. 2011;109(3):291–295. | |
Schneider M. Cellular quality control: a two-edged sword in Brugada syndrome. Heart Rhythm. 2014;11(4):635–636. | |
Mohamed U, Gollob MH, Gow RM, Krahn AD. Sudden cardiac death despite an implantable cardioverter-defibrillator in a young female with catecholaminergic ventricular tachycardia. Heart Rhythm. 2006;3(12):1486–1489. | |
Sy RW, Gollob MH, Klein GJ, et al Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2011;8(6):864–871. | |
Roses-Noguer F, Jarman JW, Clague JR, Till J. Outcomes of defibrillator therapy in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2014;11(1):58–66. | |
Denegri M, Bongianino R, Lodola F, et al. Single delivery of an adeno-associated viral construct to transfer the CASQ2 gene to knock-in mice affected by catecholaminergic polymorphic ventricular tachycardia is able to cure the disease from birth to advanced age. Circulation. 2014; 129(25):2673–2681. | |
Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20(6):1391–1396. | |
Antzelevitch C. Role of transmural dispersion of repolarization in the genesis of drug-induced torsades de pointes. Heart Rhythm. 2005;2 (2 Suppl):S9–S15. | |
Priori SG, Wilde AA, Horie M, et al. Executive Summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm. 2013;10(12):e85–e108. | |
Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85(9):803–809. | |
Shimeno K, Takagi M, Maeda K, Tatsumi H, Doi A, Yoshiyama M. Usefulness of multichannel Holter ECG recording in the third intercostal space for detecting type 1 Brugada ECG: comparison with repeated 12-lead ECGs. J Cardiovasc Electrophysiol. 2009;20(9):1026–1031. | |
Savastano S, Rordorf R, Vicentini A, et al. A comprehensive electrocardiographic, molecular and echocardiographic study of the Brugada syndrome: validation of the 2013 diagnostic criteria. Heart Rhythm. 2014;11(7):1176–1183. | |
Chevallier S, Forclaz A, Tenkorang J, et al. New electrocardiographic criteria for discriminating between Brugada types 2 and 3 patterns and incomplete right bundle branch block. J Am Coll Cardiol. 2011;58(22):2290–2298. | |
Wilde AA, Postema PG, Di Diego JM, et al. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J Mol Cell Cardiol. 2010;49(4):543–553. | |
Elizari MV, Levi R, Acunzo RS, et al. Abnormal expression of cardiac neural crest cells in heart development: a different hypothesis for the etiopathogenesis of Brugada syndrome. Heart Rhythm. 2007;4(3):359–365. | |
Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392(6673):293–296. | |
Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7(1):33–46. | |
Smits JP, Eckardt L, Probst V, et al. Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J Am Coll Cardiol. 2002;40(2):350–356. | |
Probst V, Wilde AA, Barc J, et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet. 2009;2(6):552–557. | |
Weiss R, Barmada MM, Nguyen T, et al. Clinical and molecular heterogeneity in the Brugada syndrome: a novel gene locus on chromosome 3. Circulation. 2002;105(6):707–713. | |
Van Norstrand DW, Valdivia CR, Tester DJ, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. 2007;116(20):2253–2259. | |
Burashnikov E, Pfeiffer R, Barajas-Martinez H, et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7(12):1872–1882. | |
Watanabe H, Koopmann TT, Le Scouarnec S, et al. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008;118(6):2260–2268. | |
Delpón E, Cordeiro JM, Núñez L, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1(3):209–218. | |
Hu D, Barajas-Martinez H, Burashnikov E, et al. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2(3):270–278. | |
Ueda K, Hirano Y, Higashiuesato Y, et al. Role of HCN4 channel in preventing ventricular arrhythmia. J Hum Genet. 2009;54(2):115–121. | |
Hu D, Barajas-Martínez H, Pfeiffer R, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. 2014;64(1):66–79. | |
Priori SG, Napolitano C, Gasparini M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: a prospective evaluation of 52 families. Circulation. 2000;102(20):2509–2515. | |
Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105(11):1342–1347. | |
Sarkozy A, Boussy T, Kourgiannides G, et al. Long-term follow-up of primary prophylactic implantable cardioverter-defibrillator therapy in Brugada syndrome. Eur Heart J. 2007;28(3):334–344. | |
Probst V, Veltmann C, Eckardt L, et al. Long-term prognosis of patients diagnosed with Brugada syndrome: results from the FINGER Brugada Syndrome Registry. Circulation. 2010;121(5):635–643. | |
Priori SG, Gasparini M, Napolitano C, et al. Risk stratification in Brugada syndrome: results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J Am Coll Cardiol. 2012;59(1):37–45. | |
Delise P, Allocca G, Marras E, et al. Risk stratification in individuals with the Brugada type 1 ECG pattern without previous cardiac arrest: usefulness of a combined clinical and electrophysiologic approach. Eur Heart J. 2011;32(2):169–176. | |
Delise P, Allocca G, Sitta N, DiStefano P. Event rates and risk factors in patients with Brugada syndrome and no prior cardiac arrest: a cumulative analysis of the largest available studies distinguishing ICD-recorded fast ventricular arrhythmias and sudden death. Heart Rhythm. 2014;11(2):252–258. | |
Maury P, Hocini M, Haïssaguerre M. Electrical storms in Brugada syndrome: review of pharmacologic and ablative therapeutic options. Indian Pacing Electrophysiol J. 2005;5(1):25–34. | |
Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110(13):1731–1737. | |
Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm. 2010;7(4):549–558. | |
Antzelevitch C. J wave syndromes: molecular and cellular mechanisms. J Electrocardiol. 2013;46(6):510–518. | |
Rautaharju PM, Surawicz B, Gettes LS, et al; and Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society: endorsed by the International Society for Computerized Electrocardiology. Circulation. 2009;119(10):e241–e250. | |
Tikkanen JT, Anttonen O, Junttila MJ, et al. Long-term outcome associated with early repolarization on electrocardiography. N Engl J Med. 2009;361(26):2529–2537. | |
Sinner MF, Reinhard W, Müller M, et al. Association of early repolarization pattern on ECG with risk of cardiac and all-cause mortality: a population-based prospective cohort study (MONICA/KORA). PLoS Med. 2010;7(7):e1000314. | |
Rollin A, Maury P, Bongard V, et al. Prevalence, prognosis, and identification of the malignant form of early repolarization pattern in a population-based study. Am J Cardiol. 2012;110(9):1302–1308. | |
Olson KA, Viera AJ, Soliman EZ, Crow RS, Rosamond WD. Long-term prognosis associated with J-point elevation in a large middle-aged biracial cohort: the ARIC study. Eur Heart J. 2011; 32(24):3098–3106. | |
Sager SJ, Hoosien M, Junttila MJ, Tanawuttiwat T, Perry AC, Myerburg RJ. Comparison of inferolateral early repolarization and its electrocardiographic phenotypes in pre- and postadolescent populations. Am J Cardiol. 2013;112(3):444–448. | |
Panicker GK, Manohar D, Karnad DR, Salvi V, Kothari S, Lokhandwala Y. Early repolarization and short QT interval in healthy subjects. Heart Rhythm. 2012;9(8):1265–1271. | |
Tikkanen JT, Junttila MJ, Anttonen O, et al. Early repolarization: electrocardiographic phenotypes associated with favorable long-term outcome. Circulation. 2011;123(23):2666–2673. | |
Noseworthy PA, Weiner R, Kim J, et al. Early repolarization pattern in competitive athletes: clinical correlates and the effects of exercise training. Circ Arrhythm Electrophysiol. 2011;4(4):432–440. | |
Uberoi A, Jain NA, Perez M, et al. Early repolarization in an ambulatory clinical population. Circulation. 2011;124(20):2208–2214. | |
Haïssaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358(19):2016–2023. | |
Junttila MJ, Tikkanen JT, Kenttä T, et al. Early repolarization as a predictor of arrhythmic and nonarrhythmic cardiac events in middle-aged subjects. Heart Rhythm. 2014;11(10):1701–1706. | |
Tikkanen JT, Wichmann V, Junttila MJ, et al. Association of early repolarization and sudden cardiac death during an acute coronary event. Circ Arrhythm Electrophysiol. 2012;5(4):714–718. | |
Naruse Y, Tada H, Harimura Y, et al. Early repolarization is an independent predictor of occurrences of ventricular fibrillation in the very early phase of acute myocardial infarction. Circ Arrhythm Electrophysiol. 2012;5(3):506–513. | |
Park YM, Kang WC, Suh SY, et al. Early repolarization is associated with atrial and ventricular tachyarrhythmias in patients with acute ST elevation myocardial infarction undergoing primary percutaneous coronary intervention. Int J Cardiol. 2014;176(2):327–332. | |
Haruta D, Matsuo K, Tsuneto A, et al. Incidence and prognostic value of early repolarization pattern in the 12-lead electrocardiogram. Circulation. 2011;123(25):2931–2937. | |
Adler A, Rosso R, Viskin D, Halkin A, Viskin S. What do we know about the “malignant form” of early repolarization? J Am Coll Cardiol. 2013;62(10):863–868. | |
Yagihara N, Sato A, Furushima H, Chinushi M, Hirono T, Aizawa Y. Ischemia-induced prominent J waves in a patient with Brugada syndrome. Intern Med. 2010;49(18):1979–1982. | |
Myojo T, Sato N, Nimura A, et al. Recurrent ventricular fibrillation related to hypokalemia in early repolarization syndrome. Pacing Clin Electrophysiol. 2012;35(8):e234–e238. | |
Otero J, Lenihan DJ. The “normothermic” Osborn wave induced by severe hypercalcemia. Tex Heart Inst J. 2000;27(3):316–317. | |
Osborn JJ. Experimental hypothermia; respiratory and blood pH changes in relation to cardiac function. Am J Physiol. 1953;175(3):389–398. | |
Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93(2):372–379. | |
Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8(8):1308–1339. | |
Haïssaguerre M, Chatel S, Sacher F, et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. J Cardiovasc Electrophysiol. 2009;20(1):93–98. | |
Medeiros-Domingo A, Tan BH, Crotti L, et al. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm. 2010;7(10):1466–1471. | |
Hu D, Barajas-Martínez H, Terzic A, et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol. 2014;171(3):431–442. | |
Watanabe H, Nogami A, Ohkubo K, et al. Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fibrillation associated with early repolarization. Circ Arrhythm Electrophysiol. 2011;4(6):874–881. | |
Perrin MJ, Adler A, Green S, et al. Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J wave syndrome associated with sudden cardiac death. Circ Cardiovasc Genet. 2014;7(6):782–789. | |
Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10(12):1932–1963. | |
Nam GB, Kim YH, Antzelevitch C. Augmentation of J waves and electrical storms in patients with early repolarization. N Engl J Med. 2008;358(19):2078–2079. | |
Sacher F, Derval N, Horlitz M, Haïssaguerre M. J wave elevation to monitor quinidine efficacy in early repolarization syndrome. J Electrocardiol. 2014;47(2):223–225. | |
Gollob M. Canada’s first cardiac autopsy guidelines for unexplained sudden deaths. The Beat, University of Ottawa Heart Institute. 2008;3(2):1. Available from: http://www.ottawaheart.ca/content_documents/TheBeat-v3i2-Eng.pdf. Accessed April 1, 2015. | |
Basso C, Carturan E, Pilichou K, Rizzo S, Corrado D, Thiene G. Sudden cardiac death with normal heart: molecular autopsy. Cardiovasc Pathol. 2010;19(6):321–325. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.