")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 13
Genetic Influences in Breast Cancer Drug Resistance
Authors Daniyal A, Santoso I, Gunawan NHP, Barliana MI , Abdulah R
Received 29 September 2020
Accepted for publication 12 January 2021
Published 9 February 2021 Volume 2021:13 Pages 59—85
DOI https://doi.org/10.2147/BCTT.S284453
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Adhitiya Daniyal,1 Ivana Santoso,1 Nadira Hasna Putri Gunawan,1 Melisa Intan Barliana,2,3 Rizky Abdulah1,2
1Department of Pharmacology and Clinical Pharmacy, Faculty of Pharmacy, Universitas Padjadjaran, Jatinangor, Indonesia; 2Center of Excellence in Higher Education for Pharmaceutical Care Innovation, Universitas Padjadjaran, Jatinangor, Indonesia; 3Department of Biological Pharmacy, Faculty of Pharmacy, Universitas Padjadjaran, Jatinangor, Indonesia
Correspondence: Melisa Intan Barliana
Department of Biological Pharmacy, Faculty of Pharmacy, Universitas Padjadjaran, Jl. Raya Bandung Sumedang KM. 21, Jatinangor, 45363, Indonesia
Tel +622284288812
Fax +62-22-84288896
Email [email protected]
Abstract: Breast cancer is the most common cancer in adult women aged 20 to 50 years. The therapeutic regimens that are commonly recommended to treat breast cancer are human epidermal growth factor receptor 2 (HER2) targeted therapy, endocrine therapy, and systemic chemotherapy. The selection of pharmacotherapy is based on the characteristics of the tumor and its hormone receptor status, specifically, the presence of HER2, progesterone receptors, and estrogen receptors. Breast cancer pharmacotherapy often gives different results in various populations, which may cause therapeutic failure. Different types of congenital drug resistance in individuals can cause this. Genetic polymorphism is a factor in the occurrence of congenital drug resistance. This review explores the relationship between genetic polymorphisms and resistance to breast cancer therapy. It considers studies published from 2010 to 2020 concerning the relationship of genetic polymorphisms and breast cancer therapy. Several gene polymorphisms are found to be related to longer overall survival, worse relapse-free survival, higher pathological complete response, and increased disease-free survival in breast cancer patients. The presence of these gene polymorphisms can be considered in the treatment of breast cancer in order to shape personalized therapy to yield better results.
Keywords: breast cancer, genetic polymorphisms, resistance therapy
Introduction
Breast cancer is the most widespread cancer in women aged 20–50 years. Annually, approximately 2.1 million women are suffering from this disease, including those who have new diagnoses and received treatment.1 In 2018, a study estimated that 11.6% of cancer patients were classified as having breast cancer, with a mortality rate of 6.6% of all cancer-related deaths. Breast cancer has the highest rate of new cases among 154 countries and is the leading cause of mortality for 103 countries.2 It can be estimated that the incidence of breast cancer will increase by 26.1% by 2030, based on incident cases of the disease in 2018.3
Several medications are widely available for treating breast cancer. Characteristics of the tumor and its hormone receptor (HR) status, such as estrogen receptor (ER), progesterone receptor, and human epidermal growth factor receptor 2 (HER2) in the tumor, determine the recommendations for more specific treatment choices, such as systemic chemotherapy, endocrine therapy, or HER2-targeted therapy, to yield a better disease prognosis.4 According to the Clinical Practice Guidelines of Breast Cancer of the National Comprehensive Cancer Network, anthracycline and cyclophosphamide are usually chosen for a recommended chemotherapy regimen. The HER2-targeting drug trastuzumab suppresses the mitogen activated protein kinase (MAPK) and phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) pathways in cell cycle arrest5 and is also considered as an addition to the main chemotherapy regimens6 such as taxane,7–9 and thus increases early-stage breast cancer patient survival rate. Tamoxifen is usually given as monotherapy for early-stage breast cancer10–12 or as a replacement for an aromatase inhibitor (AI) regimen after 2 to 3 years.13,14 The use of AIs such as anastrozole, letrozole, and exemestane demonstrated better efficacy in lowering the risk of recurrence of breast cancer when compared with tamoxifen in postmenopausal women with HER2-positive breast cancer.15–17
Despite improvements in disease prognosis and the overall benefits of using chemotherapy and adjuvants, therapy in breast cancer often produces different results in selected populations. Such differences are a result of innate resistance to some of the drugs employed.18 Drug resistance is a major source of cancer therapy failure.19 The drug response differs from person to person mainly because of mutations in DNA that can alter drug efficacy.20 Resistance may be explained by different mechanisms, such as alteration of drug pharmacokinetics,21,22 amplification or reduction in cell signaling,23 changes in pharmacodynamic-related receptor numbers,24 and so on. It is highly relevant to explore further gene polymorphisms that may affect therapy responses in breast cancer, in order to identify drug resistance and provide information that enables development of personalized medicine.
Methodology
For this review, the PubMed database was searched for relevant literature. The search terms were “polymorphism breast cancer therapy” with added filters specific to articles that were published during the 10 years from 2010 to 2020. The search was made in May 2020, and scrutiny of eligible articles was conducted manually by excluding non-English studies, reviews, and unrelated studies, such as those not discussing breast cancer pharmacotherapy outcome and genetic polymorphisms. A flowchart for the literature search procedure is presented in Figure 1.
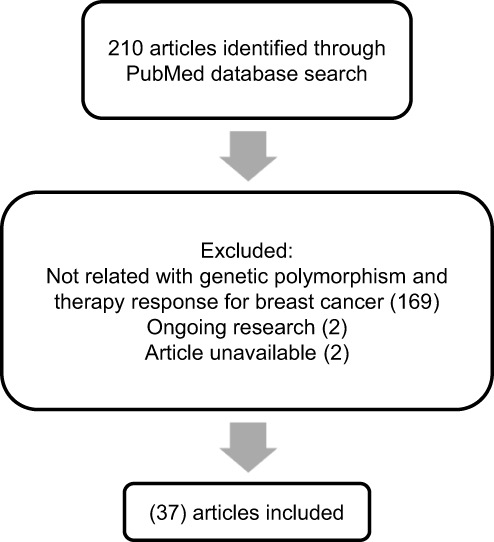 |
Figure 1 Flowchart representing the literature search process. |
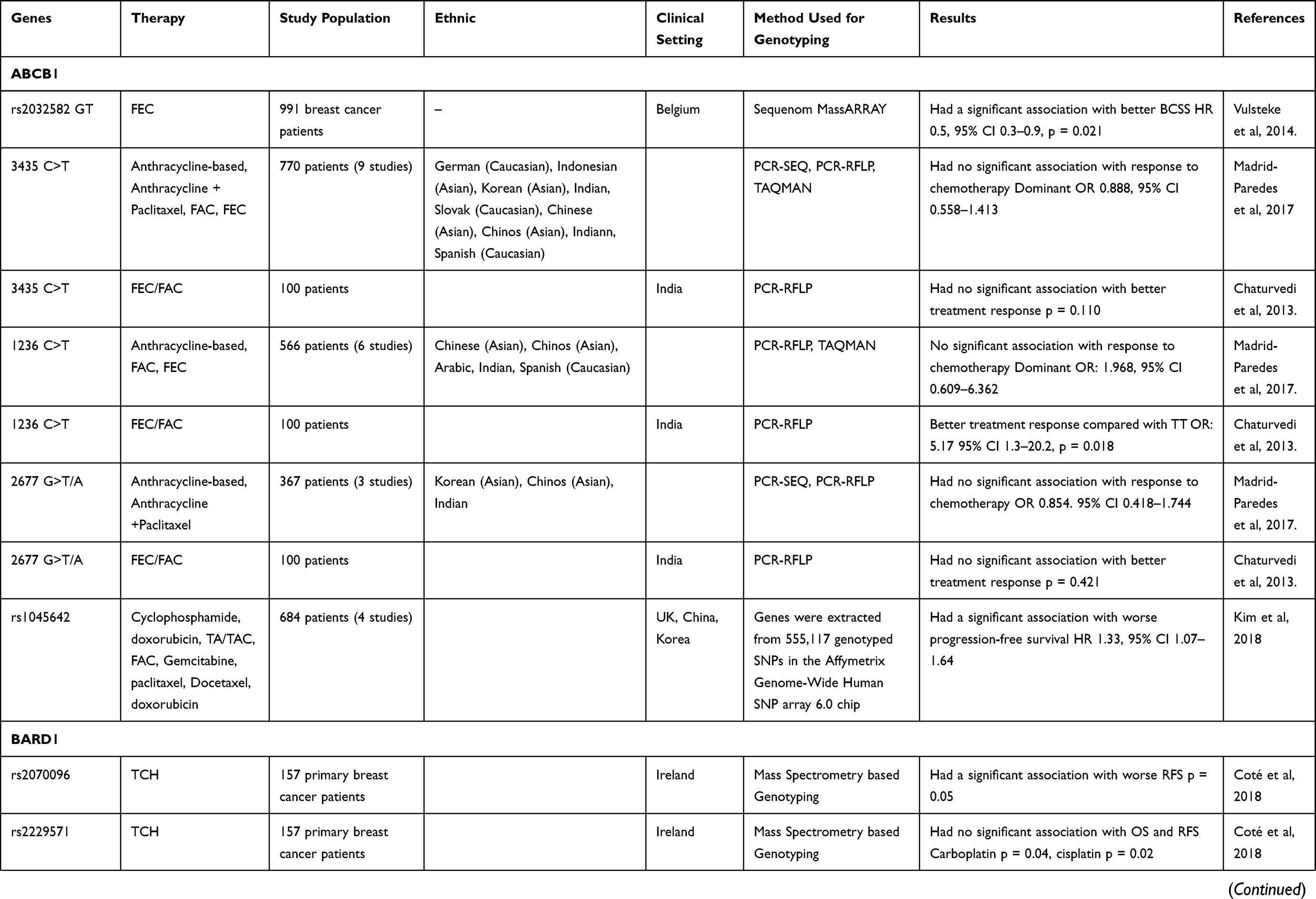
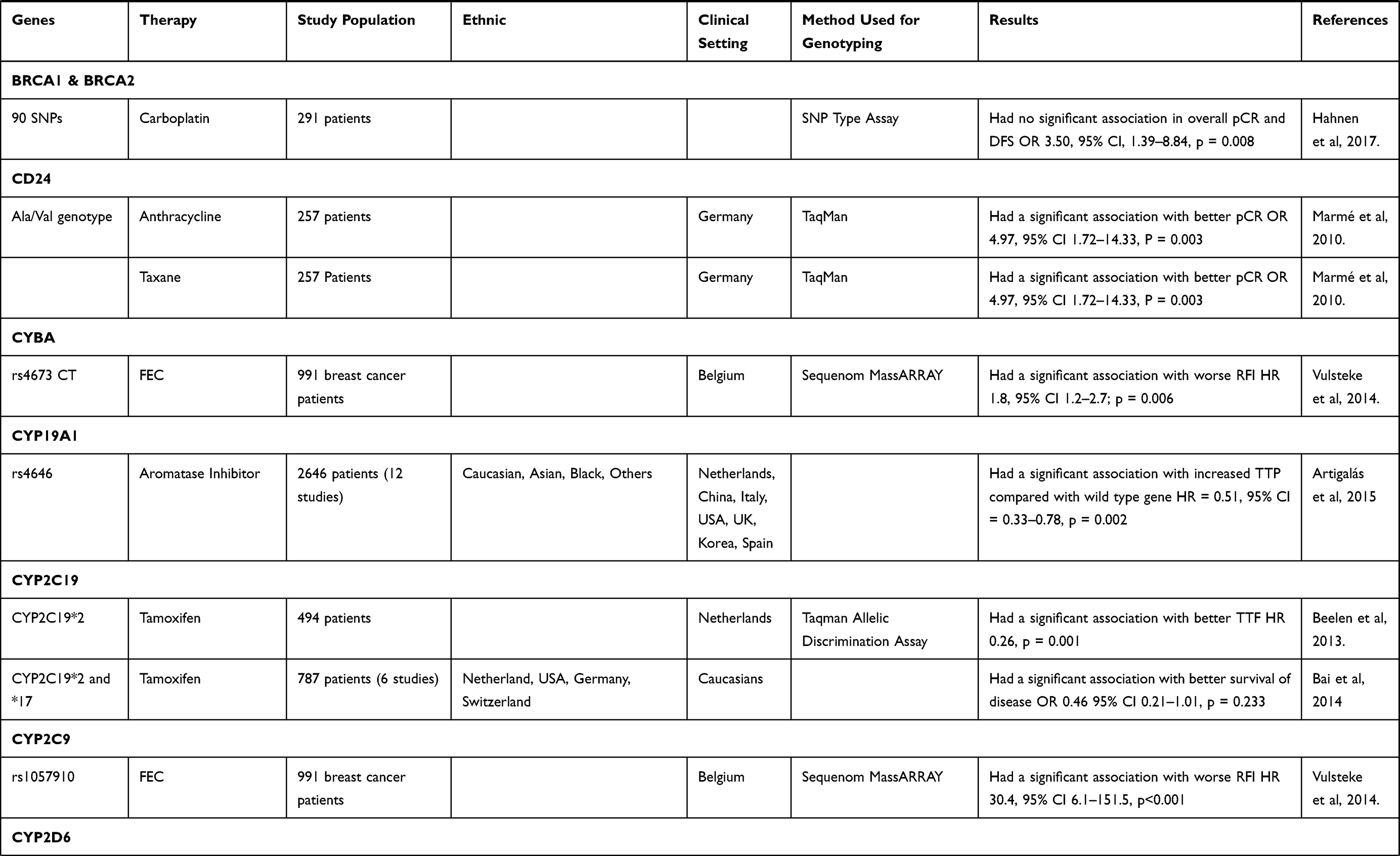
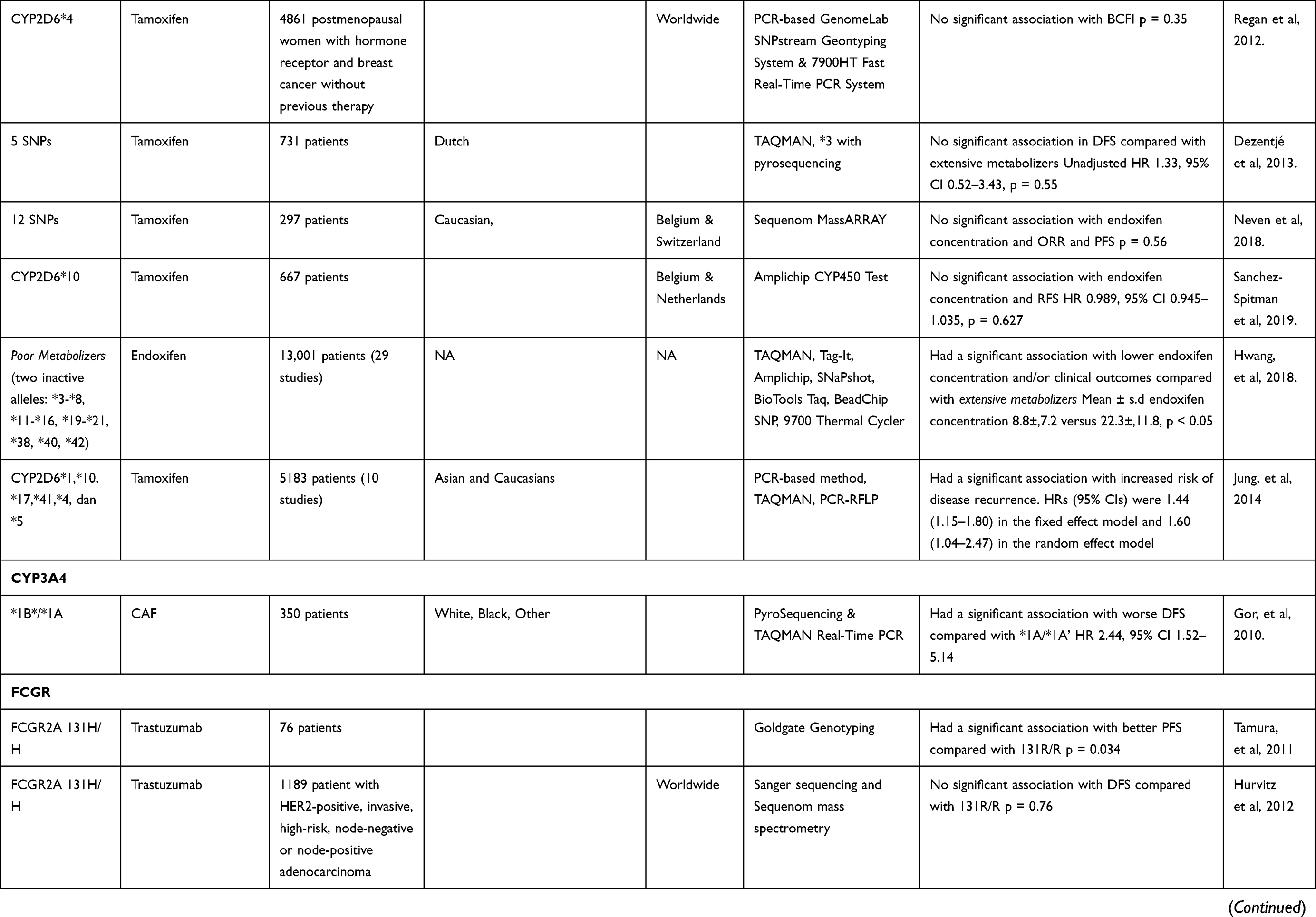
From the 210 articles identified in May 2020, this review evaluates the results of 36 studies18,25–60 that particularly focused on the pharmacogenetic influences in breast cancer drug resistance (Table 1). The data that discussed in this review article was extracted from each identified study. These studies reported an association with breast cancer drug resistance for several genes, including ABCB1, BARD1, BRCA1, BRCA2, CD24, CYBA, CYP19A1, CYP2C19, CYP2C9, CYP2D6, CYP3A4, FCGR2A, FCGR2B, FCGR3A, FGFR4, GSTM1, GSTP1, GSTT1, HER2, HER3, IL12B, KDR, MDM2, MEG3, SLC, TGFBR2, TP53, UGT1A8, UGT2B15, and UGT2B7.
 |
 |
 |
 |
 |
 |
 |
Table 1 Association Between Genetic Polymorphism and Therapy Response |
Clinical endpoints included in this article are BCSS, breast-cancer-specific survival; BCFI, breast-cancer-free interval; DFS, disease-free survival; DSS, disease-specific survival CR, complete response; EFS, event-free survival; OR, overall response; ORR, overall response rate; OS, overall survival; pCR, progression complete response; PFS, progression-free survival; PR, partial response; RFS, recurrence-free survival; SD, stable disease; TTF, time-to-treatment failure; and TTP, time to progression.
ABCB1
ATP-binding cassette (ABC) is a transporter for various types of drug molecules, and among them are drugs for chemotherapy. By expending energy, this gene transporter helps drug molecules to pass through biological membranes. The subfamily of ABC is classified as ABCB, ABCG, ABCD, ABCF, ABCCI, and ABCCII.61 Polymorphism in the transporter gene can contribute to multidrug resistance because it may be responsible for changes that induce differences in therapy for different individuals.
The ABCB1 gene is located at chromosome 7 and expresses a 45-kB mRNA.62 It encodes an active transporter of drugs involved in secreting cytotoxic agents from cells.25 A study conducted by Vulsteke et al25 suggests that the ABCB1 GT genotype gene polymorphism gave better therapeutic effects in patients with early breast cancer treated with 5-fluorouracil (FU), eirenicon, and cyclophosphamide (FEC), compared with patients who had the ABCB1 GG/GA genotype. Another study reported different results, such that polymorphisms in the ABCB1 2677 GG genotype demonstrated resistance to paclitaxel and anthracycline treatment. It is possible that in metastatic breast cancer treatment this gene polymorphism contributes to cross-resistance between paclitaxel and anthracycline. In addition, cases of resistance were found for the ABCB1 3435 CT genotype, which resulted in shorter overall survival (OS) and lower disease control rate when using anthracycline treatment.63 Many studies had shown that the ABCB1 3435 C>T polymorphism is associated with anthracycline resistance, such as in a Chinese study where patients with the CT genotype were associated with poor prognosis64 and patients with the TT genotype were associated with worse clinical response.65
In studies by Zhang et al60 and Ji et al64 polymorphism at ABCB1 1236 C>T showed association with poor response to anthracycline regiment chemotherapy, which is about dose delay in patients. In response evaluation, 1236C > T polymorphism was significantly associated with treatment response for CT genotype [OR = 5.17 (1.3–20.2), P = 0.018] and in dominant model (CC vs CT + TT) [OR = 4.63 (1.25–17.0), P = 0.021] and the T allele of 1236C>T was found to be associated with grade 2–4 toxicity [OR 1.48 (1.00–2.20), P = 0.049]. This may due to the variant allele in ABCB1 gene may lead to P-gp lower expression and resulted accumulation of drugs inside the cell, thus altering the distribution profile of the chemotherapeutic drugs inside cells. Therefore, ABCB1 polymorphisms do exert significant effects on breast cancer chemotherapy responses.44 The meta-analysis results conducted by Kim et al45 polymorphism ABCB1 in rs1045642 (C>T) was associated with poor progression-free survival (PFS), especially in Asian patients (Hazard Ratio (HR) = 1.56, 95%, Confidence Interval (CI): 1.07–2.27). The association of rs1045642 with PFS was significant in observational studies (HR = 1.28, 95% CI: 1.05–1.56); however, this association was not significant in clinical trials (HR = 1.47, 95% CI: 0.96–2.27).
BARD1
Several genes may encode proteins that can interact with breast cancer gene-1 (BRCA1) and breast cancer gene-2 (BRCA2), thus inducing DNA and tumor suppressor damage. One such gene is BRCA1-associated RING domain 1 (BARD1) gene.66 The BARD1 gene produces a protein that is similar to the BRCA1 protein in terms of structure and function.67 BRCA1 and BARD1 can be transformed into homodimer and heterodimer structures, where the former can be constructed through interaction with the really interesting new gene (RING) finger domain in the N-terminal portion, and the latter is made stable with bonds of 26–119 amino acid residues from BARD1 and 1–109 amino acid residues from BRCA1.68 These interactions have an important function in the manifestation of breast cancer tumor suppression.67 Generally, BARD1 has a function of regulating the stability of genotype and phenotype, and also has a role in DNA repair and ubiquitination.
The gene BARD1 is located at chromosome region 2q34-35 with a size of 80 kB.66 Minor alleles of BARD1 (rs2229571) exhibit higher sensitivity to platinum-based treatments, such as carboplatin and cisplatin, in HER2 breast cancer patients. A study found no significant relationship between the polymorphism of BARD1 in rs2229571 and the response of patients using docetaxel, carboplatin, and trastuzumab (TCH) compared with non-TCH treatment, but it also proved that there is a significant association such that patients who carry SNP in BARD1 rs2070096 with minor alleles had worse relapse-free survival compared to patients who received non-TCH treatment. This thus suggests possible chemoresistance.18
BRCA1 and BRCA2
Mutation in the BRCA gene, which is classified as BRCA1 and BRCA2, is related to 20% of breast cancer cases.69 The main function of BRCA1 is to repair DNA through interaction with cell cycle regulators, tumor suppressors, and DNA repair proteins.70,71 BRCA1 contains the domain of BRCA C-Terminal and RING, which are known to suppress the initiation of breast and ovarian cancer,72 and therefore mutations at this domain are often observed in breast cancer patients. In contrast to BRCA1, BRCA2 has a major function in homologous recombination for repairing DNA damage.73 BRCA2 is directly involved in the DNA repair process by involving RAD51, and RAD51 is carried by BRCA2 to sites of double-strand breaks.74
The BRCA1 gene is located at chromosome region 17q21.3,75 functioning as a tumor suppressor gene in terms of the appearance of wild-type alleles that are somatically mutated.76 Mutations in this gene are more often found in triple-negative breast cancer (TNBC) patients.77 The BRCA2 gene is located at chromosome region 13q12-13.78 More than 1800 mutations are known in the BRCA2 gene, including insertion, frameshift deletion, and nonsense mutation.79 BRCA1 and BRCA2 have essential roles in the process of DNA repair in order to maintain genome integrity through the presence of homologous recombination. The presence of polymorphism in BRCA1 and BRCA2 genes can affect the efficacy of breast cancer treatment. It is known that TNBC patients with a variation in BRCA1 and BRCA2 genes did not show significant changes in overall part and disease-free survival (DFS) values between treatment regimens without carboplatin and with carboplatin. However, patients without a variation in BRCA1 and BRCA2 genes showed significant changes in the overall pathological complete response (pCR) value for treatment regimens without carboplatin compared to treatments with carboplatin. Therefore, patients with BRCA1 and BRCA2 gene mutations respond better if standard neoadjuvant therapy (paclitaxel, doxorubicin, and cyclophosphamide) is given.28 The lack of BRCA1 and BRCA2 proteins is associated with high sensitivity to DNA-damaging agents, so those TNBC patients who exhibit variations in the BRCA1 and BRCA2 genes exhibit more sensitivity to standard chemotherapy agents. This also means that TNBC patients with variations in the BRCA1 and BRCA2 genes have higher immune cell activity.80
The poly ADP ribose polymerase (PARP) inhibitors may be candidates for use in treatment of BRCA-mutated cancer patients.81 However, there have been therapeutic failures in clinical trials of the PARP inhibitor Iniparib. In TNBC patients, Iniparib failed to prolong survival in Phase III. This failure is known to be associated with a secondary BRCA2 mutation.82 Secondary mutations in BRCA1 or BRCA2 may also play a role in drug resistance to platinum therapy. This is caused by prolonged drug exposure, which exerts selection pressure and may lead to PARP inhibitors, as well as to resistance to platinum drugs. Mutation in c.9106 C>T translates into the BRCA2 protein without the C-terminal OB-fold and thus may impair binding with single-stranded (ss) DNA, as well as nuclear localization sequences and the TR2 RAD51-binding domain.83
CD24
The cluster of differentiation 24 (CD24) gene is located at chromosome region 6q21, encoding sialoglycoprotein, which can induce growth and signal differences in cells.84 CD24 is a protein on the cell surface, providing linkage to the cell membrane via glycosylphosphatidylinositol.85 Overexpressed CD24 protein has been found in cases of various cancers, including breast cancer. In breast cancer, CD24 expression was usually found in HER2-positive and luminal breast cancer cells.86 Prognosis in breast cancer patients is related to CD24 expression, which can regulate tumor cell proliferation87 and increase the likelihood of metastasis.88
So far, no convincing correlations have been found between differentiation of genotype and CD24 expression level. CD24 Val has been reported to be associated with higher susceptibility, more autoreactive immunity, and faster disease progression, and it contributes to chemotherapy response. In a previous study, CD24 Val demonstrated a high sensitivity to anthracycline-based and taxane-based therapy in primary breast cancer. The study showed that CD24 Ala/Val is the only single-factor predictor of pCR in breast cancer patients subsequent to neoadjuvant chemotherapy (NCT) treatment. CD24 Ala/Val may be able to modulate the antitumor immune response of the host so that this becomes more autoreactive. The response to NCT therapy is influenced by differences in the CD24 genotype, which was demonstrated by a significant relationship between CD24 Val/Val with intratumoral lymphocytic.37 However, a study conducted by Zhou et al89 showed that CD24 polymorphisms in rs3838646 and rs52812045 could not predict pCR in breast cancer patients who had received NCT treatment.
CYBA
The CYBA gene is used to produce cytochrome b-245 alpha chain (p22-phox), which is a subunit of proteins that can take part in constructing NADPH oxidase, an enzyme complex that had an essential role in the immune system, when bonded with a beta chain that is expressed by the CYBB gene. NADPH oxidase functions as a regulator of neutrophil activity, and its primary function in phagocytes is to produce reactive oxygen species.90 In cancer therapy, the presence of CYBA may be related to anthracycline metabolism.25
In a study conducted by Vulsteke et al,25 the T-allele carriers in rs4673 were significantly associated with a shorter recurrence-free interval (RFI), but the results were not significant for homozygous C-allele carriers. In this case, resistance may be caused by a missense mutation of His72Tyr that could cause decreased activity of the enzyme due to a change in the heme-binding site that is essential for protein stability, with further impaired reactive oxygen species (ROS) defense capacity and thus an increased ROS level.91 In several studies,92–94 the mutation was found to be caused by 242C>T. Hoffman et al95 suggested that 640A>G reduced the enzyme activity, but a contrary study conducted by Schirmer et al96 found an increase in nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity instead.
CYP
Cytochrome P450 (CYP450) is an enzyme that serves as a xenobiotic metabolizer by drawing the xenobiotic into an oxidation reaction that changes the drug into its metabolites. In breast cancer therapy, this process is important in treatments using drugs such as tamoxifen, where the drug must be subsequently converted into a more active metabolite such as 4-hydroxy-tamoxifen (4-HT) and endoxifen by CYP450 3A4 and CYP2D6, thus developing a higher binding affinity for the ER.97
In a study conducted by Artigalás et al46 finds that rs4646 polymorphism in the CYP19A1 may be a predictive factor in aromatase inhibitor (AI) therapy. Among metastatic BC patients treated with AI, SNP rs4646 were associated with increased time to progression (TTP) compared with the wild-type gene (hazard ratio (HR) = 0.51 [95% confidence interval (CI), 0.33–0.78], P = 0.002). Furthermore, Liu et al98 reported a statistically significant association between rs4646 T alleles (G/T or T/T) and increased OS in women with metastatic BC (HR, 2.37 [95% CI, 1.20–4.65], P = 0.001). However, Miron et al99 did not find any significant association with OS in the same SNP. Henry et al100 also did not find any statistically significant association between 127 SNPs in CYP19A1 related to estrogen metabolism and modulation of breast density. These data suggest that CYP19A1 genotypes may be associated with OS in BC patients treated with AIs. However, this association appears very variable between patients.
Gor et al50 conducted a retrospective cohort study to determine chemoresistance caused by CYP3A4 polymorphisms, and found that patients having at least of CYP3A4 *1B variant allele had a significant association with worse disease-free survival (DFS) compared with those having a wild-type *1A/*1A. The mechanism underlying this chemoresistance is that *1B polymorphism leads to reduced Phase I enzyme activity and thus having suboptimal 4-hydroxy-cyclophosphamide concentration. Due to the nature of cyclophosphamide pharmacokinetics, it needs to be activated to 4-hydroxy-cyclophosphamide to be able to diffuse into cancer cells through Phase I metabolisms CYP enzymes and one of them is 3A4.
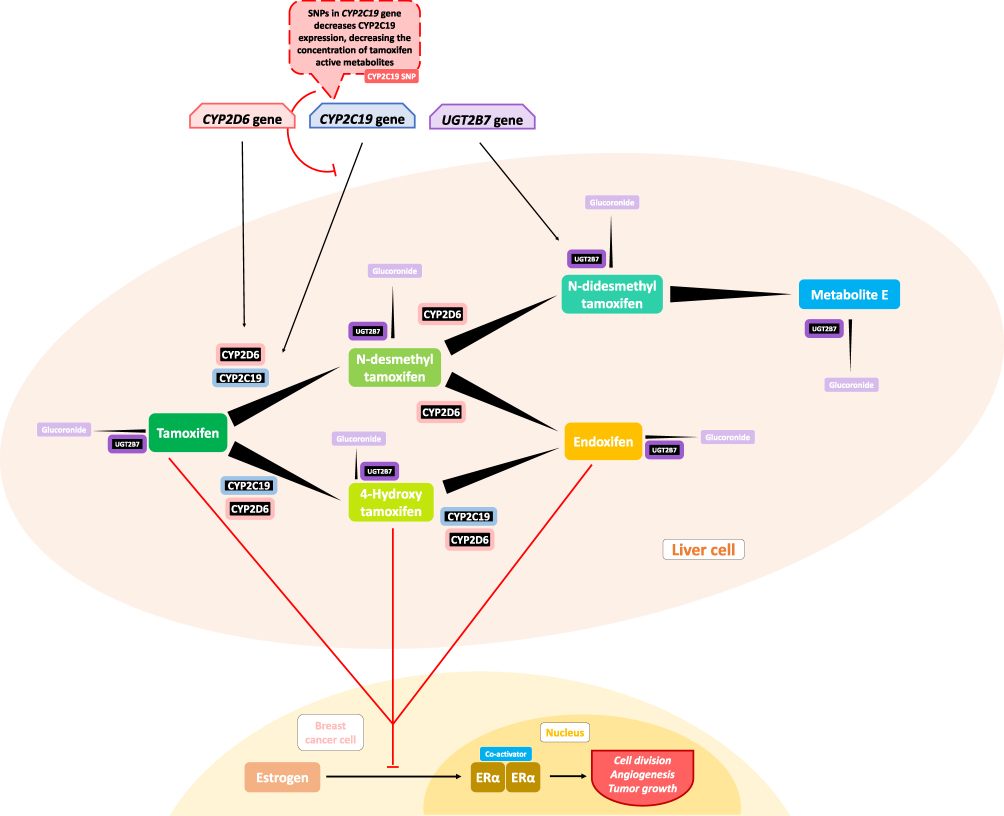
Previous studies by Beelen et al38 showed a significant relation between CYP2C19 variant alleles and time to treatment failure (TTF) in patients using tamoxifen, where CYP2C19*2 carriers were associated with longer TTF, and those who had the CYP2C19*17 allele showed a shorter TTF, but not to a statistically significant degree. The inhibition of CYP2C19 effectively influences tamoxifen metabolism, where conversion to its active metabolites such as endoxifen is seen (shown later in Figure 2). The tamoxifen resistance mechanism may be related to lower concentrations of tamoxifen and trans-4-OH-tamoxifen that were triggered by isomerization to the cis isomer, and this isomerization may also occur for endoxifen.101 Vulsteke et al25 also suggested that resistance was caused by CYP2C9 rs1057910 polymorphism in his study, where there was a significantly worse RFI, but the C-allele variant carrier was only present in 3 subjects, and thus this suggestion needs further research.


Regan et al39 did not find any association with tamoxifen therapy for differences in CYP2D6 phenotype metabolism. Endoxifen, a tamoxifen metabolite with higher affinity for ER, is suspected to be related to disease control, while the polymorphism of CYP2D6, an enzyme that could metabolize tamoxifen into its active metabolite, was hypothesized to be related to lower endoxifen concentrations and thus worse disease control and higher side effects. Regan et al39 indicated that CYP2D6 metabolism phenotype failed to predict tamoxifen efficacy and that there was thus a need for further study regarding tamoxifen metabolism and its mechanism of disease control. Dezentjé et al40 suspected incorrect interpretation in that study and replicated it while considering whether the loss of heterozygosity (LOH) could explain a Hardy–Weinberg equilibrium deviation that might exclude false genotype by LOH in tumor tissues. However, their study failed to find any association between CYP2D6 genotype differences and tamoxifen efficacy. Studies by Neven et al41 and Sanchez-Spitman et al42 also support these findings, reporting that there were no associations for low-activity CYP2D6 genotypes and low concentrations of endoxifen with clinical outcomes.
Two meta-analysis related with the impact of CYP2D6 polymorphisms on therapy effectiveness are included in this article. Hwang et al48 found that poor endoxifen metabolizers, stated as having two inactive alleles of *3-*8, *11-*16, *19-*21, *38, *40, and *42 was found to have a significant association with lower endoxifen concentration compared with those having extensive metabolizers (p < 0.05). Jung et al49 also found that those having alleles of *1, *10, *17, *41, *4, and *5 had a significantly increased risk of disease recurrence. These studies suggest that endoxifen dose may need to be adjusted for those with poor metabolizer alleles to have optimal efficacy.
FCGR
One mechanism of action for trastuzumab in treating breast cancer cells is known as antibody-dependent cell-mediated cytotoxicity (ADCC) or antibody-dependent cellular phagocytosis (ADCP). Both mechanisms include the Fc fragment of IgG receptor (FCGR) on its process. In ADCC, the FCGR located on natural killer (NK) cells binds to the Fc part of trastuzumab and triggers release of a factor such as interferon-γ (IFN-γ) or one of the perforins or granzymes, which could induce apoptosis of tumor cells, while ADCP is initiated when the FCGR on a macrophage binds instead with trastuzumab and induces phagocytosis of tumor cells.56
Gavin et al56 found that patients who had the 158F/F genotype had better prognosis when treated with the doxorubicin–cyclophosphamide–paclitaxel (ACT) regimen and less benefit when trastuzumab was added, while patients with 158F/V or V/V gained more benefit from adding trastuzumab to ACT. These findings suggest that changes in ADCC mechanism may alter the efficacy of trastuzumab. Furthermore, FCGR2A-131 polymorphism showed no evidence of differential trastuzumab treatment effects because of the lack of expression of FCGR2A on natural killer (NK) cells, which are the main effectors of ADCC. These findings may reflect the mechanism of FCGR3A-158V, not FCGR3A-158F, as FCGR3A-158V has been found to bind at low concentration to immunoglobulin (Ig) G1 immune complexes.102
Norton et al55 found no association between the FCGR2A and FCGR3A genes in relation to DFS, but demonstrated that FCGR2B I/I patients had better DFS when trastuzumab was added in the therapy combination. The results differed for patients having FCGR2B with T alleles, as they did not show any improvement in DFS when adding trastuzumab. Immune response inhibition by FCGR2B may be reduced in the minor allele (232T) and increased in response to infection and autoimmunity; it is possible that the T-allele may be related to this escalation of immunity response to tumor mechanism like the one that was triggered by trastuzumab, and thus that the T-allele carrier may have better survival but less response to trastuzumab. Hurvitz et al47 also did not find any significant correlation between FCGR3A and FCGR2A genotype differences with DFS.
In a study conducted by Tamura et al,36 there was a significant association of the FCGR2A-131H/H genotype with greater tumor response and longer FPS, whereas the FCGR3A-158V/V genotype was usually correlated with tumor response after trastuzumab was given. The metastatic cancer patient’s immune system is usually suppressed and therefore the trastuzumab-induced immune response was decreased in such patients. Roca et al57 also found that in breast cancer patients treated with trastuzumab the FCGR2A-131R/R genotype is significantly associated with worse event-free survival (EFS), and found that considering FCGR3A genotype polymorphism yielded no predictive value toward clinical outcome.
The different outcomes in multiple studies may be influenced by distinctions in intrinsic to the populations or in the chemotherapy regimens conducted, by different levels of aggressiveness of the disease, and by different sample sizes, sampling bias, and methodologies. However, these lead to conclusions that substitution of valine into phenylalanine in FCGR3A at position 158 may amplify ADCC activity due to stronger IgG1 binding compared with the wild-type (F)103,104 and that the change from histidine to arginine in FCGR2A at position 131 causes less efficient binding to IgG2, hence causing therapy resistance.105
FGFR4
The fibroblast growth factor receptors (FGFRs) are classified as tyrosine kinase receptors, which are growth-stimulating transducers and play decisive roles in regulation of cell growth. The FGFR family consists of more than 20 ligands that are important in cell cycle processes such as cell migration, cell differentiation, and tumorigenesis.106
Normally, fibroblast growth factors (FGFs) signaling takes part in multiple biological processes such as angiogenesis, inflammation, and regeneration of cells. The release of FGFs in wound repair may be triggered during wound creation by endothelial cells in response to mechanical force as a stimulus. FGF-1 and FGF-4 stimulate the production of inflammatory regulators such as interleukin-2 (IL-2) and megakaryocyte progenitor cells.107
FGF may activate many transduction cascades, which could promote cell cycle progression and halt the cell death process. A disruption of any of this regulation process may result in uncontrollable cell growth. The exact tumor growth-promoting mechanism resulting from mutation in the gene expressing FGFR4 is unknown. However, it may be related to autocrine FGF signaling, as FGF is usually observed alongside FGFR in FGFR overexpression. FGFs may be secreted by tumor cells or neighboring stromal cells and could act on either of these sources.107
An SNP, the transmembrane domain missense mutation from glycine to arginine at codon 388, is associated with breast cancer disease outcome. This polymorphism occurs in one of every two persons. There is speculation that the FGFR4 Arg388 genotype is not involved in tumor induction, as FGFR4 alleles are homogeneously distributed. FGFR4 Arg388 is overexpressed in node-positive breast cancer patients, but there is no evidence of it being significantly associated with DFS.108 Another study confirmed that FGFR4 Arg388 could be used as a disease progression predictor and suggests that it could also be used to predict chemotherapy resistance.24
In a study conducted by Marmé et al,58 the FGFR4 Gly388Arg polymorphism showed application as a specific predictive factor for therapy response to doxorubicin–cyclophosphamide–docetaxel (AC-Doc) as NCT with an odds ratio of 3.79, and there were no significant associations of pCR rates between patients with different HR status using AP-Doc treatment (42.9% versus 7.8% to 17.8% versus 15.6%). This thus suggests that regimens of drugs may affect two biological subgroups differently. The study also showed that FGFR4 Arg388 carriers have a higher risk of breast cancer involving the axillary lymph nodes, and thus supports a previous report linking the FGFR4 Arg388 allele with worse disease progression but better responses to NCT.58 The exact molecular mechanism that leads to FGFR4 Arg388 being a more hostile phenotype is not yet clearly understood. There may be a linkage disequilibrium with other mutations that could affect breast cancer prognosis. There was no observation of elevated tyrosine phosphorylation in FGFR4 Arg388 compared with Gly388 in tumor cells, which further indicated that any change in the kinase activity may be too minuscule to be detected.24
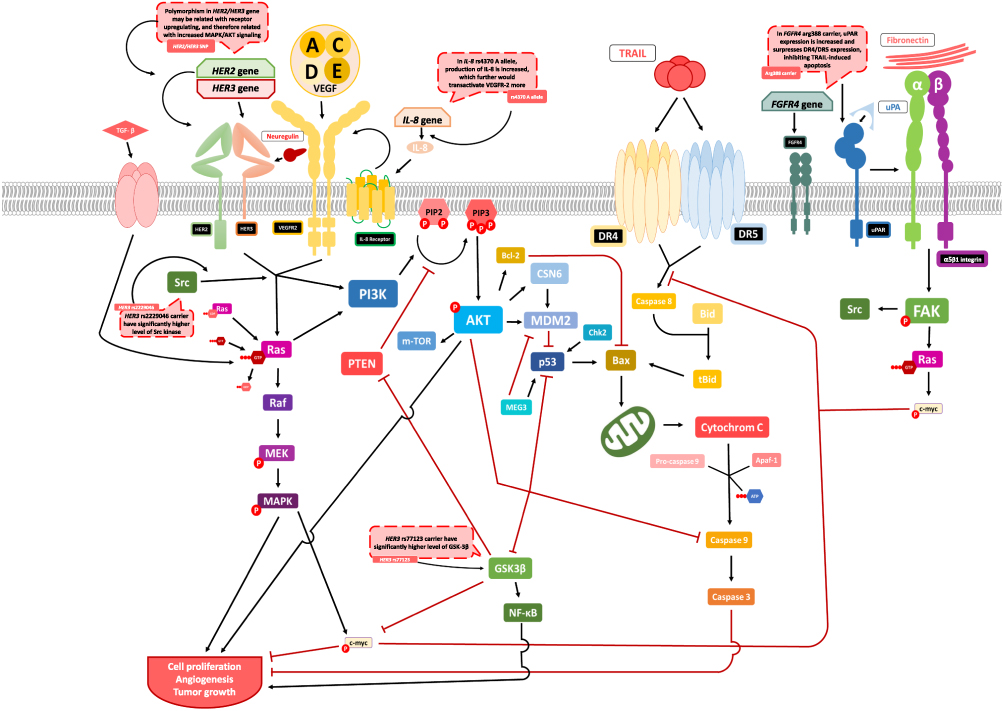
A contrary result was found in a study conducted by Thussbas et al,24 who reported that the FGFR4 Arg388 allele is significantly associated with worse DFS and poorer overall survival (OS). Chemotherapy failure may result from tumor cells resisting the induction of apoptosis. Urokinase-type plasminogen activator-receptor (uPAR) downregulation increases the susceptibility of tumor cells in chemotherapy-induced apoptosis; thus, it could be that because uPAR expression is escalated in cells producing FGFR4 Arg388 allele compared with Gly388, this increases the release of anti-apoptotic factors or downregulates proapoptotic factors in cells expressing the Gly388 allele.24 uPAR increases miR-17-5p/20a, a microRNA involved in inhibition of apoptosis by suppressing death receptor 4 (DR4) and death receptor 5 (DR5). If mechanism applies, upregulation of c-myc by uPAR may further increase miR-17-5p/20a expression. If c-myc is suppressed, uPAR concentration would decrease and the expression of DR4 and DR5 would be enhanced, activating TRAIL-induced apoptosis. These findings suggest that miR-17-5p/20a may offer a potential target therapy for breast cancer treatment and should be considered in preventing chemoresistance (Figure 3).109

GST
The glutathione S-transferases (GSTs) are a superfamily of dimeric Phase II metabolic enzymes. The family plays a vital role in cell defense by catalyzing the conjugation reaction of oncogenic substances with glutathione and thus preventing cellular damage.60 Any mutation in a gene expressing this enzyme could change the catalysis process, which in turn could alter the bioavailability of the drug and may amplify or decrease drug efficacy and toxicity.60
Genetic variability in GSTP1 is significantly associated with therapy effectiveness. Zhang et al60 conducted a study that revealed in patients with GSTP1 105Val/Val genotype a statistically significant relationship with resistance of breast cancer chemotherapy, especially epirubicin. This mutation is known to occur via an SNP in the coding sequence of GSTP1 (1578 A>G), which then gives rise to Ile105Val substitutions in the substrate-binding site of GSTP1. This was supported by a study that demonstrates that the 105Val variant carrier is correlated with more thermolabile and altered catalytic activity compared with those having 105Ile, and concludes that the homozygous isoleucine carrier is associated with the highest GSTP1 activity, with that activity decreasing as more valine was substituted. The reduced GSTP1 activity was also associated with increased toxicity from chemotherapy. As the chemotherapy mechanism needs to be activated by GST and other hepatic enzymes, the decreased GSTP1 activity may suggest an inefficient metabolism and less active metabolite concentration in such patients’ bodies.60
Another study conducted by Romero et al59 suggested that breast cancer patients treated with doxorubicin and carrying homologous G alleles in GSTP1 had a lower risk of chemoresistance, shown with polymorphism GSTP1 c.313A>G as a main cause, but no association was found between any GST genotype and the response outcome in patients treated with docetaxel. The different responses might suggest that there is specialization within GSTs activity in catalyzing the conjugation of reduced glutathione. This activity is related to how doxorubicin acts in cancer cells, where it can generate superoxide as a reactive oxygen species (ROS) when the semiquinolone in doxorubicin’s active metabolite is converted into quinine. The ROS then forms propenal, which can be detoxified by GSTP1.59
In contrary to those findings, Yao et al26 suggested that there were no associations between polymorphism in GSTP1 genes and treatment outcomes in a patient who received cyclophosphamide. This may strengthen the hypothesis that the relation between polymorphism of GSTP1 genes and breast cancer therapy is drug specific and may vary in terms of affinity and activity for different drugs.
In cyclophosphamide metabolism, GSTs had a role as inactivator. 4-hydroxy-cyclophosphamide are metabolized through Phase II metabolism to be conjugated with thiol or sulfate by GSTT. Gor et al50 conducted a study to measure chemoresistance relationship with polymorphism of GSTT1 and found that those with null genotype of GSTT1 have significantly better DFS and OS compared with those without due to having higher concentration of circulating active drug. Kong et al51 also found similar results in GSTM1 null genotype for patients using anthracycline-based therapy in his meta-analysis, strengthening the association between polymorphism of GST and chemoresistance.
HER2 and HER3
Human epidermal growth factor receptor 2 (HER2), also known as erb-B2 receptor tyrosine kinase 2 (ERBB2), is one of the 20 known tyrosine kinase receptor families that are well known to be mutated in diseases that involve uncontrolled proliferation. It is also known to be an oncogenic driver.110 The HER2 gene is located at chromosome region 17q21 and can encode transmembrane tyrosine kinase GFR. It is usually expressed in the epithelial cells of breast tissue.111 HER2 may interact with tyrosine kinase binding partners even while not having any ligand. This is of concern, as when HER2 is overexpressed, it exists in open conformation that can interact freely with any available tyrosine kinase, and leads to dimerization and promotes neoplastic transformation of cells.110
About one in five breast cancer patients have an overexpression of HER2, and it is also associated with worse disease prognosis. A monoclonal antibody, like trastuzumab, is used to directly target HER2 specifically. In 2015, Falchook et al27 reported that with trastuzumab–lapatinib–bevacizumab combination therapy there are no associations between six SNPs in HER2 (rs1810132 STR C>T, −1985 G>T, −3444C>T, P1170A C>G, rs1810132 STR C>T, I655A A>G) and stable disease (SD) ≥6 months/partial response (PR)/complete response (CR) rate, nor with TTF. This result contradicts previous findings that HER2 SNPs had an association with the risk of developing breast cancer.112 Falchook et al27 also found that an escalated concentration of circulating HER2 extracellular domain (ECD) in plasma was significantly associated with SD ≥6 months/PR rate and TTF, consistent with previous studies.113
These results contradict a study conducted by Han et al114 that found a resistance to trastuzumab accompanying Ile655Val HER2 polymorphism, where HER2-positive patients with the Val/Ile and Val/Val genotype had a significantly worse DFS score compared with those with the Ile/Ile genotype. This may be caused by a decrease in tyrosine kinase activity when Val is substituted into Ile at codon 655,115 and the combination showed a lower apoptosis rate and higher growth capacity in an in vitro study.116
Recently, data suggested that SNPs in epidermal growth factor genes may affect relapse-free survival or OS; this includes the HER3 gene. Previous studies had shown that mutation in HER-family genes may activate the PI3K/AKT signaling pathway, and monoclonal antibody-based drugs are made to inhibit this activation by stopping downstream signaling of HER2 activation.110
Coté et al18 found that patients who are treated with TCH and who have the minor allele of the HER3 SNPs (rs2229046 and rs77123) had a higher risk of worse relapse-free survival compared with patients not using TCH. The data suggested that patients with SNP in rs2229046 had a heightened concentration of Src kinase, while those with rs77123 have significantly elevated glycogen synthase kinase-3 beta (GSK-3β) phosphorylation (Figure 3). The increase in PI3K/AKT signaling may potentially indicate unresponsiveness of the TCH-based regimen in selected patients. Despite not having a mechanism specific to cancer susceptibility, both of the SNPs have been related with alternative splicing, and there are not enough data to determine their specific action on signaling.23
IL12B
Interleukin 12 (IL12) is an immune modulator that has characteristics as a connector between acquired and innate immune response. Produced by macrophages, dendritics and monocytes, IL-12 consists of two polypeptide chains that bind to disulfide p35 or p40 to encode the IL12A and IL12B genes, respectively. IL12A is located on chromosome 3p12-q13.2 and IL12B is located on chromosome 5q31-33.117 IL12 is known to have antitumor activity because it can induce cytotoxic T cell (CTL) activation, NK cell activation and differentiation of naïve cluster of differentiation 4 (CD4+) cells into T helper 1 (Th1) cells so that it can increase cytotoxic T lymphocyte response.118,119 This is supported by a study where the systemic administration of IL12 can prevent tumor growth in transgenic HER2/neu oncogene mice.120 Therefore, giving IL12 could have potential in the treatment of breast cancer in humans. However, giving IL12 can also form autoimmunity. One example, excess production of IL12 is found in autoimmune diseases such as rheumatoid arthritis and type 1 diabetes mellitus.121,122
IL12B encodes IL12 p40 which is a subunit of the IL12 and IL23 heterodimeric structures that have an important role in immune cytokines in cell-mediated immunity. IL12 and IL 23 have a mechanism to convert naïve T cells into Th1 and T-helper 17 (Th17) and maintain a balance between Treg cells and Th17 cells in maintaining a normal immune response.123 IL12B plays a major role in the initiation of the IL-12 activation signaling cascade.124 Polymorphisms that occur in the IL12A and IL12B genes are known to play a role in cancer development. Polymorphisms will change the expression of the IL12 gene and reduce IL12 protein synthesis so that it can lead to immune system dysfunction and the development of malignant tumors.125
ER-negative breast cancer patients have a high number of lymphocytes infiltrating the tumor. Tumor infiltration by immune cells, such as Treg cells and myeloid-derived suppressor cells (MDSCs) is involved in the prognosis of cancer patients after chemotherapy. The presence of polymorphisms in genes involved in the immunosuppressive pathway is known to modulate the response to given therapy.123 One of them is evidenced by studies that reported IL12B SNPs have a relationship with OS in ER-Negative breast cancer patients after chemotherapy. There are two results obtained, namely IL12B rs2546892 (G> A) had a significant association with poorer OS (HR 1.50 (95% CI 1.21 to 1.86), P = 1.81 × 10−4) and IL12B rs2853694 (A> C) had a significant association with improved OS (HR 0.73 (95% CI 0.61 to 0.87), P = 3.67 × 10−4).53
KDR/VEGFR2
Kinase insert domain receptor (KDR), also referred as vascular endothelial protein receptor 2 (VEGFR2), is a tyrosine kinase receptor that regulates growth, survival, and endothelial cell movement through paracrine signaling by producing autocrine signal and can be expressed in tumor cells.126,127 It is located at chromosome region 4q11–q12.128 Studies about KDR expression with prognostic implication in carcinoma demonstrated that SNPs on the receptor genes may affect the VEGF signaling, which in turn influences the carcinoma prognosis and the treatment response. However, this information remains controversial.129–133
In 2018, Babyshkina et al30 reported that the −604T>C (rs2071559) mutation may be a functional polymorphism within the KDR gene promoter region and may be able to change potential transcription of KDR, leading to reduced expression of KDR.128 The value of pCR was higher in patients using the cyclophosphamide–doxorubicin–capecitabine (CAX) regimen than in those who used the fluorouracil–doxorubicin–cyclophosphamide (FAC) chemotherapy regimen. Therapy for those younger than 50 years carrying the −604TT genotype of rs2071559 gave results significantly correlated with pCR within the CAX-treated patients. However, there was no clear confirmation that the pCR rate correlates with KDR rs2305948 within the two treatment groups. KDR expression and polymorphism of KDR gene usually act as additional predictive markers of pCR in breast cancer patients.
Allegrini et al29 suggested that KDR gene interacts with the IL-8 gene and may affect the efficacy of bevacizumab therapy. In tumor progression, KDR has a significant role in promoting tumor angiogenesis.134,135 The phosphorylation of KDR may be transactivated by IL-8 due to physical interactions between KDR and the IL-8 receptors, and it has been shown that this activity may occur in the presence of VEGF such as CBO-P-11 at the site (Figure 3). These findings may explain the failure of tumor angiogenesis inhibition when treating with drugs such as bevacizumab, as the overexpression of IL-8 in the presence of the SNP would lead to more transactivation of KDR. Added to the mutation of KDR, the upregulation of the receptor supports the angiogenic process. These findings were determined in a case-control study comparing patients treated with and without bevacizumab as a first-line chemotherapy; the study revealed decreased values of progression-free survival and OS in patients carrying SNP KDR rs11133360 and IL-8 rs4073, suggesting a resistance to bevacizumab therapy.
MDM2
The mouse double minute-2 (MDM2) homolog is a promoter that suppresses p53 transcriptional activity136 through direct binding, ubiquitination, and degradation.137 In previous studies, overexpression of MDM2 has been studied as another mechanism for suppressing protein p53 (Figure 3), and MDM2 protein levels in the body may also be interpreted as prognostic biomarker of human breast cancer.52,138
Overexpression of MDM2 was suggested to be related to drug resistance in targeted cancer therapy, such as in chemotherapy and radiotherapy through the MDM2–p53 loop dependent pathway and epithelial–mesenchymal transition (EMT) pathway. In the EMT pathway, MDM2 overexpression induces the EMT process in tumor cells, resulting in resistance to the chemotherapeutic drug.139 MDM2 overexpression was reported to inhibit the sensitivity to cisplatin, with potential for leading to cisplatin-based therapy resistance.140 Also, overexpression of MDM2 was associated with resistance in trastuzumab regimens in HER2-positive breast cancer.141
The MDM2 SNPs at T309G may decrease the activity of protein wild-type p53 and thus increase the chance of developing cancer cells. In a recent study, polymorphism in MDM2 (SNP309T>G, rs2279744) was associated with increased risk of various cancer development through its association with an increased MDM2 mRNA level.142,143 The effect of SNP 309G aligns with the mechanism of MDM2 that suppresses p53 protein activity.142,144,145 Polymorphism in the 309G allele enhances MDM2 activity, so it may substitute for TP53 mutation in similar patient cohorts, yet the importance of SNP309 in familial breast cancer remains unclear.146 In a study conducted by Chrisanthar et al,35 genotype differences of MDM2 showed no association with treatment response to epirubicin or paclitaxel, and there was no effect on relapse-free survival value. In multivariate analysis, SNP309 TG/GG persisted as a poor prognostic factor by excluding ER status from the analysis.
MEG3
The maternally expressed 3 (MEG3) gene is located at chromosome region 14q32.3 in humans147 and is involved in growth and development of cell. Reexpression of MEG3 suppressed proliferation of tumor cells in vitro (Figure 3)148–151 and reduced the growth of gliomas, tumor volume, and the expression of Ki67.152
Cao et al153 reported that SNP in MEG3 can increase cancer development risk and toxicity of chemotherapy in other type of cancers.154 Peng et al155 reported that in ER-positive breast cancer, MEG3 was downregulated, which in turn inhibited cell growth and thus induced apoptosis through ER stress activation, nuclear factor κB (NF-κB), and p53 pathways.156
Polymorphism in MEG3 was associated with regulation of cells in breast cancer. In 2019, Bayarmaa et al31 showed that SNP in MEG3 rs10132552 was significantly associated with response to cisplatin-containing chemotherapy in breast cancer patients, such that a patient carrying the rs10132552 TT genotype had significantly worse DFS, and there was a higher level of Ki67 in patients who had the T-allele in the rs10132552 phenotype.
SLC
When chronically exposed to selective chemotherapy, cancer cells often regulate drug efflux transporters that may result in development of drug resistance. The change of transporter may be initiated when cancer cells demand more nutrients to support their rapid growth and gather these nutrients via plasma membrane transporters.157 The solute carrier (SLC) genes were classified into 65 subfamilies. The main function of SLC genes is to encode the transporters of endogenous and exogenous compounds.158–160 Most SLC transporters are equilibrative. This trait is beneficial in facilitating substrate uptake into the cell by regulating the electrochemical and concentration gradients. Polymorphisms in SLC genes have been affiliated with efficacy and toxicity outcomes of drugs.157
A recent study conducted by Okazaki et al161 reported that SLC28A3 rs7867504 polymorphism was significantly associated with toxicity in pancreatic cancer patients who received gemcitabine. SNPs in SLC28A3 (rs7867504) and SLC29A1 with the GA haplotype were associated with OS in metastatic breast cancer patients receiving a paclitaxel–gemcitabine combination. SLC29A1 (rs747199 and rs760370) with the GA haplotype resulted in a significantly shorter OS, while SLC28A3 (rs7867504) with the CC and CT genotypes was associated with a longer OS compared with the TT genotype. These findings suggested that the efficacy of paclitaxel–gemcitabine treatment may be influenced by the transport of gemcitabine.32 Also, these results fall in line with earlier pharmacogenetic studies in other type of cancers that received gemcitabine as chemotherapy. SNPs in SLC29A1, SLC28A1, and SLC28A3 (rs7867504) were associated with gemcitabine metabolite clearance in solid tumors.162
The solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene has a common polymorphism as SLCO1B1*5 at rs4149056. Patients carrying this SNP had higher estrogen levels prior to treatment with AI163 and showed a higher exemestane level during treatment.164 SLCO1B1 SNP rs10841753 carriers are also known to have decreased estrogens prior to AI treatment, as they increased expression of the organic anion-transporting polypeptide 1B1 (OATP1B1) transporter.163 A study conducted by Dempsey et al33 showed that patients carrying the SLCO1B1*5 allele (rs4149056) may have had worse outcomes when receiving AI treatment because they were at higher risk for having a higher concentration of detectable estrone, yet patients with SNP rs10841753 had a lower concentration of estrone during the first 3 months from the initiation of AI treatment. Those who had SLCO1B1*5 rs4149056 SNP were associated with increased levels of estrone sulfate during pretreatment of AI chemotherapy, while rs10841753 carriers were associated with lower levels instead. However, there is no direct evidence associating suppression in estrogen with treatment effectiveness. Estrone is the most abundant estrogen in postmenopausal women.165 The lack of association of SLCO1B1*5 or rs10841753 polymorphism with risk of breast cancer development in a large genome-wide association study suggested that estrone and estrone sulfate levels do not have any clinical consequence in predicting the effectiveness of breast cancer therapy.166
TGFBR2
Transforming growth factor beta receptor II (TGFBR2) is an important cytokine in the tumor microenvironment and included as a ligand binding receptor for the TGF β family (TGF-1, −2, −3), this gene is located on chromosome 3 locus 3p22.167 TGFBR2 encodes the TGF-β receptor II which is the transmembrane serine/threonine protein kinase receptor in the TGF-β signaling pathway.168 After binding to the ligand, TGFBR2 will induce phosphorylation of solvated metal atom dispersion (SMAD) 2/3 through activation of TGFBR1. This SMAD 2/3 induction will form hetero-oligomers with SMAD 4 and accumulate in the nucleus. In addition, TGFBR2 can induce intracellular pathways with non-SMAD signaling pathways via Src, PI3K/AKT, p42/44 and p38 MAPK.169,170 TGFβ is known to have two roles depending upon the cellular context, namely as tumor suppression at the initial stage and invasion and metastatic tumors in later stage cancers, specifically TGFβ as a stimulator in Treg cell proliferation and immune prevention.171
The presence of overexpression of TGFBR2 in ER-negative breast cancer can be a poor prognostic indicator of patient survival.172 Excessive TGFBR2 expression is associated with an overactive PI3K/AKT signaling pathway. AKT activation will mediate FAF1 phosphorylation and activate pro-metastatic function in cancer cells because it increases the stability of TGFBR2 on the cell surface.170 This is proven by the association of TGFβ on lung metastases in patients with ER-negative breast cancer.173 TGFBR2 gene polymorphisms may be a prognostic indicator and predictor of breast cancer therapy by looking inhibition of TGFβ signaling. This is shown by a study that found SNPs TGFBR2 rs1367610 (G> C) had a significant association with poorer OS in ER-negative patients who received chemotherapy (P = 3.08 × 10−4).53 In addition, another study showed a low number of TGFBR2 expression in ER-positive patients on tamoxifen therapy to have a significant association with shortened recurrence-free survival (RFS) (HR: 0.312, 95% CI, 0.131–0.742; P = 0.008).174
TP53 and CHEK2
The tumor protein (TP53) gene is the most common mutated gene in human cancer. Its presence in more than 50% of the whole cancer patient cohort implies that the TP53 gene has some action related to the formation of cancer.175 p53 is involved in processes such as growth, DNA repair, and apoptosis of cells.176 In DNA repair activity, p53 gave signals to halt the cell cycle and gave the cell time to repair, resulting in revived genome stability. Additionally, p53 is directly involved in the activity of various DNA repair systems.177
The most common mutations in TP53 are of the missense type, leading to diverse changes in amino acid positions.178 Most of the time, mutations occurred more often in higher stages of cancers or in aggressive behavior subtypes such as triple-negative or HER2-related.179–181 In patients with the wild-type of TP53, several tumors were confirmed to exhibit chemoresistance. Findings to date suggest that tumors may accommodate mutations in the checkpoint kinase 2 (CHEK2) gene, which expresses the Chk2 protein that phosphorylates p53.137
The CHEK2 gene is located at chromosome region 22q12.1 and can be activated by Thr68 phosphorylation via ataxia-telangiectasia mutated. CHEK2 has a role in regulating the cell cycle. Mutation in the CHEK2 gene will affect the function and expression of the Chk2 protein.182 In addition, mutation in the CHEK2 gene can influence the activity of p53,35 as this may be phosphorylated by various type of kinases, including Chk2. This process is important in the mechanism of antitumor agents when responding to DNA damage in breast cancer.183 The nonfunctional Chk2 protein can affect drug sensitivity by altering the p53 activation process.184 When mutations in CHEK2 and TP53 genes are compared, the role of Chk2 can be indirectly identified in chemoresistance (Figure 3).185
Previous studies reported that mutations within the TP53 gene are related to resistance to anthracycline therapy in carcinoma patients.184,186,187 In vitro studies showed that taxane sensitivity is related to p53 function.188,189 However, a clinical study has shown that there is no correlation between TP53 status and paclitaxel sensitivity.187
Chrisanthar et al184 found that TP53 and CHEK2 mutations may predict resistance to paclitaxel treatment, but not in patients receiving epirubicin as first-line therapy. Mutations of TP53 are related to poor prognosis in carcinoma patients who are not using any adjuvant therapy.190 These effects probably are due to the inclusion of patients with paclitaxel as a second-line treatment. CHEK2 nonsense mutations were previously shown to affect Chk2 activity and may be used to predict resistance to anthracycline treatment.184
UGT
The uridine 5ʹdiphospho-glucuronosyltransferase (UDP-glucuronosyltransferase, UGT) gene in mammals is known to have four families: UGT1, UGT2, UGT3, and UGT8.61 This superfamily usually encodes enzymes that can place glycosyl groups on a lipophilic substrate.191 The UGT1 gene is located at chromosome region 2q37.192 It is known to encode nine types of enzymes related with glucuronidation process. UGT2 genes are classified further into two subfamilies, UGT2A and UGT2B. The latter are encoded by different genes such as UGT2B4, UGT2B7, UGT2B10, UGT2B11, UGT2B15, and UGT2B17.193
Various UGT gene isoforms exhibited different selectivity and sensitivity roles in every process of drug glucuronidation. Many types of drugs are metabolized by the UGT gene. Epirubicin is an anticancer drug in the anthracycline group. Like other anthracyclines, epirubicin undergoes metabolism in the liver by interacting with aldo-ketoreductase to form epirubicinol194 or undergoes glucuronidation to form EPI-glucuronide.195 Epirubicinol and EPI-glucuronide are inactive forms of epirubicin, and EPI-glucuronide had a faster excretion rate than epirubicinol and was noncardiotoxic.196 The epirubicin glucuronidation process is carried out by UGT, specifically UGT2B7 in the liver.195 UGT2B7 gene is located at chromosome region 4q13.2.197 The presence of polymorphisms in the UGT2B7 gene may disrupt the inactivation process for epirubicin. One study showed that breast cancer patients which carry the G-allele homozygous UGT2B7 gene on rs3924194 experienced a worse recurrence-free interval (RFI) when treated with the fluorouracil, epirubicin, cyclophosphamide (FEC) based regimen.25
In addition to epirubicin, tamoxifen is also often used in breast cancer therapy as a selective ER modulator and goes through a metabolic process catalyzed by UGTs.34 Tamoxifen that had passed through the metabolism stage, which is catalyzed by UGTs, was found to add glucuronide groups and to produce 4-HT and endoxifen, which may deactivate antiestrogenic effects (Figure 2).198 Variants in the UGT2B15, UGT2B7, and UGT1A8 genes are not correlated with breast cancer recurrence in tamoxifen treatment.34 Dezentjé et al40 found a contradictory result, reporting that UGT2B15*2 may be associated with worse DFS in his exploratory study, but this result requires further investigation.
Conclusion and Future Prospects
After exploring through studies related with breast cancer chemoresistance caused by gene polymorphisms, we have reached a conclusion that some of the molecular changes that are caused by upregulation or downregulation due to different genetic activity, and some may lead to increase efficacy of the drug while the other halts the drug activity. Genes that suggesting chemoresistance due to having significant association with decreased drug efficacy and may be studied further to determine its exact mechanism are ABCB1 rs1045642, BARD1 rs2070096, CYBA rs4673 CT, CYP19A1 rs4646, CYP2C9 rs1057910, CYP2D6 poor metabolizers, CYP3A4 *1B*/*1A, FCGR3A 158V/V, GSTP1 105Val/Val genotype, GSTM null genotype, HER3 rs2229046 and rs77123, KDR rs11133360 (T>C) for patients carrying SNP IL-8 rs4073, IL12B rs2546892 (G>A), MEG3 rs10132552 TT genotype, SLC rs4149056, TGFBR2 rs1367610 (G>C), TP53, UGT2B15 *2, and UGT2B7 rs3924194. While some studies, which are included in the study or not, may have conflicting results caused by different clinical setting or chemotherapy used and other factors, these studies strengthen the importance of exploring polymorphism and its impact on genes related with breast cancer.
Genetic polymorphisms in patients with breast cancer are related to variation in therapeutic responses in patients using the same drug. This review examines the relationship between genetic polymorphisms and breast cancer therapy resistance. There are several gene polymorphisms that produce differences in results in terms of OS, relapse-free survival, pathological CR, DFS, and other parameters. Moreover, many studies suggest that polymorphism in genes may be assessed as a predictive and prognostic biomarker for identifying breast cancer. Although conflicting results remain to be understood, in the future these polymorphisms may become considerations in developing personalized medicines that yield better results for each individual and in predicting the clinical outcome of breast cancer therapies.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work
Disclosure
All authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. DeSantis CE, Ma J, Gaudet MM, et al. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69(6):438–451. doi:10.3322/caac.21583
3. Cancer Tomorrow. Available from: https://gco.iarc.fr/tomorrow/graphic-isotype?type=0&type_sex=0&mode=population&sex=2&populations=900&cancers=20&age_group=value&apc_male=0&apc_female=0&single_unit=500000&print=0.
4. McDonald ES, Clark AS, Tchou J, Zhang P, Freedman GM. Clinical diagnosis and management of breast cancer. J Nucl Med. 2016;57(Supplement_1):9S–16S. doi:10.2967/jnumed.115.157834
5. Junttila TT, Akita RW, Parsons K, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15(5):429–440. doi:10.1016/j.ccr.2009.03.020
6. Arnould L, Gelly M, Penault-Llorca F, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer. 2006;94(2):259–267. doi:10.1038/sj.bjc.6602930
7. Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol. 2014;32(33):3744–3752. doi:10.1200/JCO.2014.55.5730
8. Gianni L, Pienkowski T, Im Y-H, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, Phase 2 trial. Lancet Oncol. 2012;13(1):25–32. doi:10.1016/S1470-2045(11)70336-9
9. Schneeweiss A, Chia S, Hickish T, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann Oncol. 2013;24(9):2278–2284. doi:10.1093/annonc/mdt182
10. Wells BG, DiPiro JT, Schwinghammer TL, DiPiro CV. Pharmacotherapy Handbook.
11. Shagufta AI, Ahmad I. Tamoxifen a pioneering drug: an update on the therapeutic potential of tamoxifen derivatives. Eur J Med Chem. 2018;143:515–531. doi:10.1016/j.ejmech.2017.11.056
12. Drǎgǎnescu M, Carmocan C. Hormone therapy in breast cancer. Chir. 2017;112(4):413–417. doi:10.21614/chirurgia.112.4.413
13. Burstein HJ, Temin S, Anderson H, et al. Adjuvant endocrine therapy for women with hormone receptor-positive breast cancer: American Society of Clinical Oncology clinical practice guideline focused update. J Clin Oncol. 2014;32(21):2255–2269. doi:10.1200/JCO.2013.54.2258
14. Senkus E, Kyriakides S, Ohno S, et al. Primary breast cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26:v8–v30. doi:10.1093/annonc/mdv298
15. Geisler J, Haynes B, Anker G, Dowsett M, Lønning PE. Influence of letrozole and anastrozole on total body aromatization and plasma estrogen levels in postmenopausal breast cancer patients evaluated in a randomized, cross-over study. J Clin Oncol. 2002;20(3):
16. Geisler J, King N, Anker G. In vivo inhibition of aromatization by exemestane, a novel irreversible aromatase inhibitor, in postmenopausal breast cancer patients. Clin Cancer Res. 1998;4(9):2089–2093.
17. Dowsett M, Cuzick J, Ingle J, et al. Meta-analysis of breast cancer outcomes in adjuvant trials of aromatase inhibitors versus tamoxifen. J Clin Oncol. 2010;28(3):509–518. doi:10.1200/JCO.2009.23.1274
18. Coté D, Eustace A, Toomey S, et al. Germline single nucleotide polymorphisms in ERBB3 and BARD1 genes result in a worse relapse free survival response for HER2-positive breast cancer patients treated with adjuvant based docetaxel, carboplatin and trastuzumab (TCH). PLoS One. 2018;13(8):1–17. doi:10.1371/journal.pone.0200996
19. Polgar O, Bates SE. ABC transporters in the balance: is there a role in multidrug resistance?. Biochem Soc Trans. 2005;33(1):241–245. doi:10.1042/BST0330241
20. Longacre M, Snyder N, Sarkar S. Drug resistance in cancer: an overview. Cancer. 2014;1769–1792. doi:10.3390/cancers6031769
21. Gandhi N, Das G. Metabolic reprogramming in breast cancer and its therapeutic implications. Cells. 2019;8(89):1–33. doi:10.3390/cells8020089
22. Muley H, Fadó R, Rodríguez-rodríguez R, Casals N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem Pharmacol. 2020;177(January):113959. doi:10.1016/j.bcp.2020.113959
23. Raghav D, Sharma V. An in silico evaluation of deleterious nonsynonymous single nucleotide polymorphisms in the ErbB3 oncogene. Biores Open Access. 2013;2(3):206–211. doi:10.1089/biores.2013.0007
24. Thussbas C, Nahrig J, Streit S, et al. FGFR4 Arg388 allele is associated with resistance to adjuvant therapy in primary breast cancer. J Clin Oncol. 2006;24(23):3747–3755. doi:10.1200/JCO.2005.04.8587
25. Vulsteke C, Pfeil AM, Schwenkglenks M, et al. Impact of genetic variability and treatment-related factors on outcome in early breast cancer patients receiving (neo-) adjuvant chemotherapy with 5-fluorouracil, epirubicin and cyclophosphamide, and docetaxel. Breast Cancer Res Treat. 2014;147(3):557–570. doi:10.1007/s10549-014-3105-5
26. Yao S, Barlow WE, Albain KS, et al. Gene polymorphisms in cyclophosphamide metabolism pathway, treatment-related toxicity, and disease-free survival in SWOG 8897 clinical trial for breast cancer. Clin Cancer Res. 2010;16(24):6169–6176. doi:10.1158/1078-0432.CCR-10-0281
27. Falchook GS, Moulder S, Naing A, et al. A phase I trial of combination trastuzumab, lapatinib, and bevacizumab in patients with advanced cancer. Invest New Drugs. 2015;33(1):177–186. doi:10.1007/s10637-014-0173-7
28. Hahnen E, Lederer B, Hauke J, et al. Germline mutation status, pathological complete response, and disease-free survival in triple-negative breast cancer: secondary analysis of the GeparSixto randomized clinical trial. JAMA Oncol. 2017;3(10):1378–1385. doi:10.1001/jamaoncol.2017.1007
29. Allegrini G, Coltelli L, Orlandi P, et al. Pharmacogenetic interaction analysis of VEGFR-2 and IL-8 polymorphisms in advanced breast cancer patients treated with paclitaxel and bevacizumab. Pharmacogenomics. 2014;15(16):1985–1999. doi:10.2217/pgs.14.140
30. Babyshkina N, Zavyalova M, Tarabanovskaya N, et al. Predictive value of vascular endothelial growth factor receptor type 2 in triple-negative breast cancer patients treated with neoadjuvant chemotherapy. Mol Cell Biochem. 2018;444(1–2):197–206. doi:10.1007/s11010-017-3244-1
31. Bayarmaa B, Wu Z, Peng J, et al. Association of LncRNA MEG3 polymorphisms with efficacy of neoadjuvant chemotherapy in breast cancer. BMC Cancer. 2019;19(1):877. doi:10.1186/s12885-019-6077-3
32. Lee SY, Im SA, Park YH, et al. Genetic polymorphisms of SLC28A3, SLC29A1 and RRM1 predict clinical outcome in patients with metastatic breast cancer receiving gemcitabine plus paclitaxel chemotherapy. Eur J Cancer. 2014;50(4):698–705. doi:10.1016/j.ejca.2013.11.028
33. Dempsey JM, Kidwell KM, Gersch CL, et al. Effects of SLCO1B1 polymorphisms on plasma estrogen concentrations in women with breast cancer receiving aromatase inhibitors exemestane and letrozole. Pharmacogenomics. 2019;20(8):571–580. doi:10.2217/pgs-2019-0020
34. Ahern TP, Christensen M, Cronin-fenton DP, et al. Functional polymorphisms in UDP-glucuronosyl transferases and recurrence in tamoxifen-treated breast cancer survivors. Cancer Epidemiol Biomarkers Prev. 2011;20(9):1937–1943. doi:10.1158/1055-9965.EPI-11-0419
35. Chrisanthar R, Knappskog S, Løkkevik E, et al. Predictive and prognostic impact of TP53 mutations and MDM2 promoter genotype in primary breast cancer patients treated with epirubicin or paclitaxel. PLoS One. 2011;6(4):e19249. doi:10.1371/journal.pone.0019249
36. Tamura K, Shimizu C, Hojo T, et al. FcγR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann Oncol. 2011;22(6):1302–1307. doi:10.1093/annonc/mdq585
37. Marmé F, Werft W, Walter A, et al. CD24 ala57Val polymorphism predicts pathologic complete response to sequential anthracycline- and taxane-based neoadjuvant chemotherapy for primary breast cancer. Breast Cancer Res Treat. 2012;132(3):819–831. doi:10.1007/s10549-011-1759-9
38. Beelen K, Opdam M, Severson TM, et al. CYP2C19*2 predicts substantial tamoxifen benefit in postmenopausal breast cancer patients randomized between adjuvant tamoxifen and no systemic treatment. Breast Cancer Res Treat. 2013;139(3):649–655. doi:10.1007/s10549-013-2568-0
39. Regan MM, Leyland-Jones B, Bouzyk M, et al. CYP2D6 genotype and tamoxifen response in postmenopausal women with endocrine-responsive breast cancer: the breast international group 1–98 trial. J Natl Cancer Inst. 2012;104(6):441–451. doi:10.1093/jnci/djs125
40. Dezentjé VO, Van Schaik RHN, Vletter-Bogaartz JM, et al. CYP2D6 genotype in relation to tamoxifen efficacy in a Dutch cohort of the tamoxifen exemestane adjuvant multinational (TEAM) trial. Breast Cancer Res Treat. 2013;140(2):363–373. doi:10.1007/s10549-013-2619-6
41. Neven P, Jongen L, Lintermans A, et al. Tamoxifen metabolism and efficacy in breast cancer: a prospective multicenter trial. Clin Cancer Res. 2018;24(10):2312–2318. doi:10.1158/1078-0432.CCR-17-3028
42. Sanchez-Spitman A, Dezentjé V, Dezentjé D, et al. Tamoxifen pharmacogenetics and metabolism: results from the prospective CYPTAM study. J Clin Oncol. 2019;37(8):636–646. doi:10.1200/JCO.18
43. Madrid-Paredes A, Cañadas-Garre M, Sánchez-Pozo A, Expósito-Ruiz M, Calleja-Hernández MÁ. ABCB1 gene polymorphisms and response to chemotherapy in breast cancer patients: a meta-analysis. Surg Oncol. 2017;26(4):473–482. doi:10.1016/j.suronc.2017.09.004
44. Chaturvedi P, Tulsyan S, Agarwal G, et al. Influence of ABCB1 genetic variants in breast cancer treatment outcomes. Cancer Epidemiol. 2013;37(5):754–761. doi:10.1016/j.canep.2013.04.012
45. Kim JE, Choi J, Park JY, et al. Associations between genetic polymorphisms of membrane transporter genes and prognosis after chemotherapy: meta-analysis and finding from Seoul breast cancer study (SEBCS). Pharmacogenomics J. 2018;18(5):633–645. doi:10.1038/s41397-018-0016-6
46. Artigalás O, Vanni T, Hutz MH, Ashton-Prolla P, Schwartz IV. Influence of CYP19A1 polymorphisms on the treatment of breast cancer with aromatase inhibitors: a systematic review and meta-analysis. BMC Med. 2015;13(1):1–10. doi:10.1186/s12916-015-0373-9
47. Hurvitz SA, Betting DJ, Stern HM. Analysis of Fcγ receptor IIIa and IIa polymorphisms: lack of correlation with outcome in trastuzumab-treated breast cancer patients. Clin Cancer Res. 2012;18(12):3478–3486. doi:10.1158/1078-0432.CCR-11-2294
48. Hwang GS, Bhat R, Crutchley RD, Trivedi MV. Impact of CYP2D6 polymorphisms on endoxifen concentrations and breast cancer outcomes. Pharmacogenomics J. 2018;18(2):201–208. doi:10.1038/tpj.2017.36
49. Jung JA, Lim HS. Association between CYP2D6 genotypes and the clinical outcomes of adjuvant tamoxifen for breast cancer: a meta-analysis. Pharmacogenomics. 2014;15(1):49–60. doi:10.2217/pgs.13.221
50. Gor PP, Su HI, Gray RJ, et al. Cyclophosphamide-metabolizing enzyme polymorphisms and survival outcomes after adjuvant chemotherapy for node-positive breast cancer: a retrospective cohort study. Breast Cancer Res. 2010;12(3):1–10. doi:10.1186/bcr2570
51. Kong X, Li Z, Li X. GSTP1, GSTM1, and GSTT1 polymorphisms as predictors of response to chemotherapy in patients with breast cancer: a meta-analysis. Cancer Chemother Pharmacol. 2016;78(6):1163–1173. doi:10.1007/s00280-016-3173-9
52. Bartel F, Meye A, W¨url PW, et al. Amplification of the MDM2 gene, but not expression of splice variants of MDM2 MRNA, is associated with prognosis in soft tissue sarcoma. Int J Cancer. 2001;95(3):168–175. doi:10.1002/1097-0215(20010520)95:3<168::AID-IJC1029>3.0.CO;2-A
53. Lei J, Rudolph A, Moysich KB, et al. Assessment of variation in immunosuppressive pathway genes reveals TGFBR2 to be associated with prognosis of estrogen receptor-negative breast cancer after chemotherapy. Breast Cancer Res. 2015;17(1). doi:10.1186/s13058-015-0522-2
54. Bai L, He J, He G, He J, Xu F, Xu G. Association of CYP2C19 polymorphisms with survival of breast cancer patients using tamoxifen: results of a meta-analysis. Asian Pac J Cancer Prev. 2014;15(19):8331–8335. doi:10.7314/APJCP.2014.15.19.8331
55. Norton N, Olson RM, Pegram M, et al. Association studies of Fcγ receptor polymorphisms with outcome in HER2+ breast cancer patients treated with trastuzumab in NCCTG (alliance) trial N9831. Cancer Immunol Res. 2014;2(10):962–969. doi:10.1158/2326-6066.CIR-14-0059
56. Gavin PG, Song N, Rim Kim S, et al. Association of polymorphisms in FCGR2A and FCGR3A with degree of trastuzumab benefit in the adjuvant treatment of ERBB2/HER2-positive breast cancer analysis of the NSABP B-31 trial. JAMA Oncol. 2017;3(3):335–341. doi:10.1001/jamaoncol.2016.4884
57. Roca L, Diéras V, Roché H, et al. Correlation of HER2, FCGR2A, and FCGR3A gene polymorphisms with trastuzumab related cardiac toxicity and efficacy in a subgroup of patients from UNICANCER-PACS04 trial. Breast Cancer Res Treat. 2013;139(3):789–800. doi:10.1007/s10549-013-2587-x
58. Marmé F, Werft W, Benner A, et al. FGFR4 Arg388 genotype is associated with pathological complete response to neoadjuvant chemotherapy for primary breast cancer. Ann Oncol. 2010;21(February):1636–1642. doi:10.1093/annonc/mdq017
59. Romero A, Martín M, Oliva B, et al. Glutathione S-transferase P1 c.313A > G polymorphism could be useful in the prediction of doxorubicin response in breast cancer patients. Ann Oncol. 2012;23(7):1750–1756. doi:10.1093/annonc/mdr483
60. Zhang BL, Sun T, Zhang BN, et al. Polymorphisms of GSTP1 is associated with differences of chemotherapy response and toxicity in breast cancer. Chin Med J (Engl). 2011;124(2):199–204. doi:10.3760/cma.j.issn.0366-6999.2011.02.008
61. Mackenzie P, Owens I, Burchell B, et al. The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence. Pharmacogenetics. 1997;7(4):255–269. doi:10.1097/00008571-199708000-00001
62. Fojo A, Lebo R, Shimizu N, et al. Localization of multidrug resistance-associated DNA sequences to human chromosome 7. Somat Cell Mol Genet. 1986;12(4):415–420. doi:10.1007/BF01570737
63. Chang H, Rha SY, Jeung HC, et al. Association of the ABCB1 gene polymorphisms 2677G>T/A and 3435C>T with clinical outcomes of paclitaxel monotherapy in metastatic breast cancer patients. Ann Oncol. 2009;20(2):272–277. doi:10.1093/annonc/mdn624
64. Ji M, Tang J, Zhao J, Xu B, Qin J, Lu J. Polymorphisms in genes involved in drug detoxification and clinical outcomes of anthracycline-based neoadjuvant chemotherapy in Chinese Han breast cancer patients. Cancer Biol Ther. 2012;13(5):264–271. doi:10.4161/cbt.18920
65. Wu H, Kang H, Liu Y, et al. Roles of ABCB1 gene polymorphisms and haplotype in susceptibility to breast carcinoma risk and clinical outcomes. J Cancer Res Clin Oncol. 2012;138(9):1449–1462. doi:10.1007/s00432-012-1209-z
66. Suszynska M, Kluzniak W, Wokolorczyk D, et al. Bard1 is a low/moderate breast cancer risk gene: evidence based on an association study of the Central European p.q564x recurrent mutation. Cancers (Basel). 2019;11(6):740. doi:10.3390/cancers11060740
67. Hashizume R, Fukuda M, Maeda I, et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001;276(18):14537–14540. doi:10.1074/jbc.C000881200
68. Meza JE, Brzovic PS, King M-C, Klevit RE, Mapping the functional domains of BRCA1: interaction of the RING finger domains of BRCA1 and BARD1. J Biol Chem. 1999;274(9):5659–5665. doi:10.1074/jbc.274.9.5659
69. Noh JM, Choi DH, Baek H, et al. Associations between BRCA mutations in high-risk breast cancer patients and familial cancers other than breast or ovary. J Breast Cancer. 2012;15(3):283–287. doi:10.4048/jbc.2012.15.3.283
70. Huen MSY, Sy SMH, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol. 2010;11(2):138–148. doi:10.1038/nrm2831
71. Deng C-X, Brodie SG. Roles of BRCA1 and its interacting proteins. Bioessays. 2000;22(8):728–737. doi:10.1002/1521-1878(200008)22:8<728::AID-BIES6>3.0.CO;2-B
72. Shattuck-Eidens D, McClure M, Simard J, et al. A collaborative survey of 80 mutations in the BRCA1 breast and ovarian cancer susceptibility gene. Implications for presymptomatic testing and screening. JAMA. 1995;273(7):535–541.
73. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12(1):68–78. doi:10.1038/nrc3181
74. Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25(43):5864–5874. doi:10.1038/sj.onc.1209874
75. Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science (80-). 1994;266(5182):66–71.
76. Petrucelli N, Daly MB, Feldman GL. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet Med. 2010;12(5):245–259. doi:10.1097/GIM.0b013e3181d38f2f
77. Godet I, Gilkes DM. BRCA1 and BRCA2 mutations and treatment strategies for breast cancer. Integr Cancer Sci Ther. 2017;4(1):1–7.
78. Wooster R, Neuhausen S, Mangion J, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Proc Natl Acad Sci USA. 1984;265(5181):2088–2090.
79. Jiang T, Shi W, Wali VB, et al. Predictors of chemosensitivity in triple negative breast cancer: an integrated genomic analysis. PLoS Med. 2016;13(12):1–23. doi:10.1371/journal.pmed.1002193
80. Gorodetska I, Kozeretska I, Dubrovska A. BRCA genes: the role in genome stability, cancer stemness and therapy resistance. J Cancer. 2019;10(9):2109–2127. doi:10.7150/jca.30410
81. Livraghi L, Garber JE. PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med. 2015;13(1). doi:10.1186/s12916-015-0425-1
82. Barber LJ, Sandhu S, Chen L, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(3):422–429. doi:10.1002/path.4140
83. Mylavarapu S, Das A, Roy M. Role of BRCA mutations in the modulation of response to platinum therapy. Front Oncol. 2018;8(FEB). doi:10.3389/fonc.2018.00016
84. CD24 gene - genetics home reference - NIH. Available from: https://ghr.nlm.nih.gov/gene/CD24#location.
85. Schabath H, Runz S, Joumaa S, Altevogt P. CD24 affects CXCR4 function in pre-B lymphocytes and breast carcinoma cells. J Cell Sci. 2006;119(2):314–325. doi:10.1242/jcs.02741
86. Kwon MJ, Han J, Seo JH, et al. CD24 overexpression is associated with poor prognosis in luminal a and triple-negative breast cancer. PLoS One. 2015;10(10):1–21. doi:10.1371/journal.pone.0139112
87. Smith SC, Oxford G, Wu Z, et al. The metastasis-associated gene CD24 is regulated by Ral GTPase and is a mediator of cell proliferation and survival in human cancer. Cancer Res. 2006;66(4):1917–1922. doi:10.1158/0008-5472.CAN-05-3855
88. Baumann P, Cremers N, Kroese F, et al. CD24 expression causes the acquisition of multiple cellular properties associated with tumor growth and metastasis. Cancer Res. 2005;65(23):10783–10793. doi:10.1158/0008-5472.CAN-05-0619
89. Zhou X. CD24 polymorphisms cannot predict pathologic complete response to anthracycline- and taxane-based neoadjuvant chemotherapy in breast cancer. Clin Breast Cancer. 2014;14(2):e33–e40. doi:10.1016/j.clbc.2013.11.001
90. CYBA gene - genetics home reference - NIH. Available from: https://ghr.nlm.nih.gov/gene/CYBA.
91. Wojnowski L, Kulle B, Schirmer M, et al. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation. 2005;112(24):3754–3762. doi:10.1161/CIRCULATIONAHA.105.576850
92. Guzik TJ, West NEJ, Black E, et al. Functional impact of genetic polymorphisms in NAD(P)H oxidase p22phox subunit on vascular superoxide production in atherosclerosis. Heart. 2000;83(SUPPL.1):1744–1747.
93. Wyche KE, Wang SS, Griendling KK, et al. C242T CYBA polymorphism of the NADPH oxidase is associated with reduced respiratory burst in human neutrophils. Hypertension. 2004;43(6):1246–1251. doi:10.1161/01.HYP.0000126579.50711.62
94. Shimo-Nakanishi Y, Hasebe T, Suzuki A, et al. Functional effects of NAD(P)H oxidase p22phox C242T mutation in human leukocytes and association with thrombotic cerebral infarction. Atherosclerosis. 2004;175(1):109–115. doi:10.1016/j.atherosclerosis.2004.01.043
95. Hoffmann M, Schirmer MA, Tzvetkov MV, et al. A functional polymorphism in the NAD(P)H oxidase subunit CYBA is related to gene expression, enzyme activity, and outcome in non-Hodgkin lymphoma. Cancer Res. 2010;70(6):2328–2338. doi:10.1158/0008-5472.CAN-09-2388
96. Schirmer M, Hoffmann M, Kaya E, Tzvetkov M, Brockmöller J. Genetic polymorphisms of NAD(P)H oxidase: variation in subunit expression and enzyme activity. Pharmacogenomics J. 2008;8(4):297–304. doi:10.1038/sj.tpj.6500467
97. Kim H, Ellis SW, Lennurd MS, Tucker GT. Variable contribution of cytochromes I ’ 450 2D6, 2C9 and 3A4 to the 4-hydroxylation of tamoxifen by human liver microsomes. Biochem Pharmacol. 1997;53(2):171–178. doi:10.1016/S0006-2952(96)00650-8
98. Liu L, Bai YX, Zhou JH, et al. A polymorphism at the 3ʹ-UTR region of the aromatase gene is associated with the efficacy of the aromatase inhibitor, anastrozole, in metastatic breast carcinoma. Int J Mol Sci. 2013;14(9):18973–18988. doi:10.3390/ijms140918973
99. Miron L, Negurǎ L, Peptanariu D, Marinca M. Research on aromatase gene (CYP19A1) polymorphisms as a predictor of endocrine therapy effectiveness in breast cancer. Rev Med Chir Soc Med Nat Iasi. 2012;116(4):997–1004.
100. Henry NL, Chan HP, Dantzer J, et al. Aromatase inhibitor-induced modulation of breast density: clinical and genetic effects. Br J Cancer. 2013;109(9):2331–2339. doi:10.1038/bjc.2013.587
101. Johnston SRD, Haynes BP, Smith IE, Sacks NPM, Ebbs SR, Dowsett M. Acquired tamoxifen resistance in human breast cancer and reduced intra-tumoral drug concentration. Lancet. 1993;342(8886–8887):1521–1522. doi:10.1016/S0140-6736(05)80088-1
102. Bruhns P, Iannascoli B, England P, et al. Specificity and affinity of human Fc γ receptors and their polymorphic variants for human IgG subclasses specificity and affinity of human Fc ␥receptors and their polymorphic variants for human IgG subclasses. Blood. 2014:3716–3725. doi:10.1182/blood-2008-09-179754.
103. Wu J, Edberg JC, Redecha PB, et al. A novel polymorphism of FcγRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. 1997;100(5):1059–1070. doi:10.1172/JCI119616
104. Shields RL, Namenuk AK, Hong K, et al. High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J Biol Chem. 2001;276(9):6591–6604. doi:10.1074/jbc.M009483200
105. Koene BHR, Kleijer M, Algra J, et al. Fc g RIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc g RIIIa, independently of the Fc g RIIIa-48L/R/H phenotype. Blood. 1997;90(3):3–8. doi:10.1182/blood.V90.3.1109
106. Burke D, Wilkes D, Blundell TL, Malcolm S. Fibroblast growth factor receptors: lessons from the genes. Trends Biochem Sci. 1998;23(2):59–62. doi:10.1016/S0968-0004(97)01170-5
107. Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7(3):165–197. doi:10.1677/erc.0.0070165
108. Bange J, Prechtl D, Cheburkin Y, et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg388 allele. Cancer Res. 2002;62(3):840–847.
109. Li X, Wu B, Chen L, Ju Y, Li C, Meng S. Urokinase-type plasminogen activator receptor inhibits apoptosis in triple-negative breast cancer through miR-17/20a suppression of death receptors 4 and 5. Oncotarget. 2017;8(51):88645–88657. doi:10.18632/oncotarget.20435
110. Elster N, Collins DM, Toomey S, Crown J, Eustace AJ, Hennessy BT. HER2-family signalling mechanisms, clinical implications and targeting in breast cancer. Breast Cancer Res Treat. 2015;149(1):5–15. doi:10.1007/s10549-014-3250-x
111. Dipiro J, Talbert R, Yee G, Matzke G, Wells B. Pharmacotherapy: A Pathophysiologic Approach.
112. Kallel I, Kharrat N, Al-fadhly S. HER2 polymorphisms and breast cancer. Genet Test Mol Biomarkers. 2010;14(1):29–35. doi:10.1089/gtmb.2009.0069
113. Köstler WJ, Schwab B, Singer CF, et al. Monitoring of serum Her-2/neu predicts response and progression-free survival to trastuzumab-based treatment in patients with metastatic breast cancer. Clin Cancer Res. 2004;10(5):1618–1624. doi:10.1158/1078-0432.CCR-0385-3
114. Han X, Diao L, Xu Y, et al. Association between the HER2 Ile655Val polymorphism and response to trastuzumab in women with operable primary breast cancer. Ann Oncol. 2014;25(6):1158–1164. doi:10.1093/annonc/mdu111
115. Fleishman SJ, Schlessinger J, Ben-Tal N. A putative molecular-activation switch in the transmembrane domain of erbB2. Proc Natl Acad Sci U S A. 2002;99(25):15937–15940. doi:10.1073/pnas.252640799
116. Beauclair S, Formento P, Fischel JL, et al. Role of the HER2 [Ile655Val] genetic polymorphism in tumorogenesis and in the risk of trastuzumab-related cardiotoxicity. Ann Oncol. 2007;18(8):1335–1341. doi:10.1093/annonc/mdm181
117. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3(2):133–146. doi:10.1038/nri1001
118. Del Vecchio M, Bajetta E, Canova S, et al. Interleukin-12: biological properties and clinical application. Clin Cancer Res. 2007;13(16):4677–4685. doi:10.1158/1078-0432.CCR-07-0776
119. He XZ, Wang L, Zhang YY. An effective vaccine against colon cancer in mice: use of recombinant adenovirus interleukin-12 transduced dendritic cells. World J Gastroenterol. 2008;14(4):532–540. doi:10.3748/wjg.14.532
120. Nanni P, Nicoletti G, De Giovanni C, et al. Combined Allogeneic Tumor Cell Vaccination and Systemic Interleukin 12 Prevents Mammary Carcinogenesis in HER-2/Neu Transgenic Mice. Vol. 194. Rockefeller University Press; 2001.
121. Morita Y, Yamamura M, Nishida K, et al. Expression of Interleukin-12 in Synovial Tissue from Patients with Rheumatoid Arthritis. Vol. 41. 1998.
122. Morahan G, Huang D, Ymer SI, et al. Linkage disequilibrium of a type 1 diabetes susceptibility locus with a regulatory IL12B allele. Nat Genet. 2001;27(2):219–221. doi:10.1038/84872
123. Yu X, Wang C, Luo J, Zhao X, Wang L, Li X. Combination with methotrexate and cyclophosphamide attenuated maturation of dendritic cells: inducing treg skewing and Th17 suppression in vivo. Clin Dev Immunol. 2013;2013:1–12. doi:10.1155/2013/238035
124. Klinke DJ. The ratio of P40 monomer to dimer is an important determinant of IL-12 bioactivity. J Theor Biol. 2006;240(2):323–335. doi:10.1016/j.jtbi.2005.09.022
125. Zheng Y, Wang M, Tian T, et al. Role of Interleukin-12 Gene Polymorphisms in the Onset Risk of Cancer: A Meta-Analysis. Vol. 8. 2017.
126. Deng Y, Yang Y, Yao B, et al. RETRACTED: paracrine signaling by VEGF-C promotes non-small cell lung cancer cell metastasis via recruitment of tumor-associated macrophages. Exp Cell Res. 2018;364(2):208–216. doi:10.1016/j.yexcr.2018.02.005
127. Dai X, Mei Y, Chen X, Cai D. ANLN and KDR are jointly prognostic of breast cancer survival and can be modulated for triple negative breast cancer control. Front Genet. 2019;10:790. doi:10.3389/fgene.2019.00790
128. Wang Y, Zheng Y, Zhang W, et al. Polymorphisms of KDR gene are associated with coronary heart disease. J Am Coll Cardiol. 2007;50(8):760–767. doi:10.1016/j.jacc.2007.04.074
129. Dales JP, Garcia S, Carpentier S, et al. Prediction of metastasis risk (11 year follow-up) using VEGF-R1, VEGF-R2, Tie-2/Tek and CD105 expression in breast cancer (n = 905). Br J Cancer. 2004;90(6):1216–1221. doi:10.1038/sj.bjc.6601452
130. Ghosh S, Sullivan CAW, Zerkowski MP, et al. High levels of vascular endothelial growth factor and its receptors (VEGFR-1, VEGFR-2, neuropilin-1) are associated with worse outcome in breast cancer. Hum Pathol. 2008;39(12):1835–1843. doi:10.1016/j.humpath.2008.06.004
131. Dhakal HP, Naume B, Synnestvedt M, et al. Expression of vascular endothelial growth factor and vascular endothelial growth factor receptors 1 and 2 in invasive breast carcinoma: prognostic significance and relationship with markers for aggressiveness. Histopathology. 2012;61(3):350–364. doi:10.1111/j.1365-2559.2012.04223.x
132. Jansson S, Bendahl PO, Grabau DA, et al. The three receptor tyrosine kinases c-KIT, VEGFR2 and PDGFRα, closely spaced at 4q12, show increased protein expression in triple-negative breast cancer. PLoS One. 2014;9(7):e102176. doi:10.1371/journal.pone.0102176
133. Yan JD, Liu Y, Zhang ZY, et al. Expression and prognostic significance of VEGFR-2 in breast cancer. Pathol Res Pract. 2015;211(7):539–543. doi:10.1016/j.prp.2015.04.003
134. Shibuya M. Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem. 2013;153(1):13–19. doi:10.1093/jb/mvs136
135. Claesson L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013;273(2):114–127. doi:10.1111/joim.12019
136. Momand J, Zambetti GP, Olson DC, George D, Levine’ AJ. The MDM-2 oncogene product forms a complex with the 53 protein and inhibits p53-mediated transactivation. Cell. 1992;69(7):1237–1245. doi:10.1016/0092-8674(92)90644-R
137. Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Mono-versus polyubiquitination: differential control of p53 fate by MDM2. Science (80-). 2003;302(5652):1972–1975. doi:10.1126/science.1091362
138. Park HS, Park JM, Park S, Cho J, Kim S
139. Hou H, Sun D, Zhang X. The role of MDM2 amplification and overexpression in therapeutic resistance of malignant tumors. Cancer Cell Int. 2019;19:216. doi:10.1186/s12935-019-0937-4
140. Kondo S, Barnett GH, Hara H, Morimura T, Takeuchi J. MDM2 protein confers the resistance of a human glioblastoma cell line to cisplatin-induced apoptosis. Oncogene. 1995;10(10):2001–2006.
141. Yu Z-H, Qu Z-L, Zhou S, Xiong J. MDM2 overexpression predicts trastuzumab resistance in HER-2 positive breast cancer tissues. Cell Res. 2015;22:1813–1818.
142. Bond GL, Hu W, Bond EE, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119(5):591–602. doi:10.1016/j.cell.2004.11.022
143. Hu Z, Jin G, Wang L, Chen F, Wang X, Shen H. MDM2 promoter polymorphism SNP309 contributes to tumor susceptibility: evidence from 21 case-control studies. Cancer Epidemiol Biomarkers Prev. 2007;16(12):2717–2723. doi:10.1158/1055-9965.EPI-07-0634
144. Yap DBS, Hsieh JK, Lu X. Mdm2 inhibits the apoptotic function of p53 mainly by targeting it for degradation. J Biol Chem. 2000;275(47):37296–37302. doi:10.1074/jbc.M004359200
145. Chène P. Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nat Rev Cancer. 2003;3(2):102–109. doi:10.1038/nrc991
146. Haupt S, Vijayakumaran R, Miranda PJ, Burgess A, Lim E, Haupt Y. The role of MDM2 and MDM4 in breast cancer development and prevention. J Mol Cell Biol. 2017;9(1):53–61. doi:10.1093/jmcb/mjx007
147. Zhou Y, Zhang X, Klibanski A. MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol. 2012;48(3):45–53. doi:10.1530/JME-12-0008
148. Zhang X, Gejman R, Mahta A, et al. Maternally expressed gene 3, an imprinted noncoding RNA gene, is associated with meningioma pathogenesis and progression. Cancer Res. 2010;70(6):2350–2358. doi:10.1158/0008-5472.CAN-09-3885
149. Zhou Y, Zhong Y, Wang Y, et al. Activation of p53 by MEG3 non-coding RNA. J Biol Chem. 2007;282(34):24731–24742. doi:10.1074/jbc.M702029200
150. Braconi C, Kogure T, Valeri N, et al. MicroRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene. 2011;30(47):4750–4756. doi:10.1038/onc.2011.193
151. Wang P, Ren Z, Sun P. Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J Cell Biochem. 2012;113(6):1868–1874. doi:10.1002/jcb.24055
152. Zhang L, Liang X, Li Y. Long non-coding RNA MEG3 inhibits cell growth of gliomas by targeting miR-93 and inactivating PI3K/AKT pathway. Oncol Rep. 2017;38(4):2408–2416. doi:10.3892/or.2017.5871
153. Cao X, Zhuang S, Hu Y, et al. Associations between polymorphisms of long non-coding RNA MEG3 and risk of colorectal cancer in Chinese. Oncotarget. 2016;7(14):19054–19059. doi:10.18632/oncotarget.7764
154. Gong WJ, Peng JB, Yin JY, et al. Association between well-characterized lung cancer lncRNA polymorphisms and platinum-based chemotherapy toxicity in Chinese patients with lung cancer. Acta Pharmacol Sin. 2017;38(4):581–590. doi:10.1038/aps.2016.164
155. Peng J, Zhang L, Yuan C, et al. Expression profile analysis of long noncoding RNA in ER-positive subtype breast cancer using microarray technique and bioinformatics. Cancer Manag Res. 2017;9:891–901. doi:10.2147/CMAR.S151120
156. Zhang Y, Wu J, Jing H, Huang G, Sun Z, Xu S. Long noncoding RNA MEG3 inhibits breast cancer growth via upregulating endoplasmic reticulum stress and activating NF-κB and p53. J Cell Biochem. 2019;120(4):6789–6797. doi:10.1002/jcb.27982
157. Schaller L, Lauschke VM. The genetic landscape of the human solute carrier (SLC) transporter superfamily. Hum Genet. 2019;138(11–12):1359–1377. doi:10.1007/s00439-019-02081-x
158. Fredriksson R, Nordström KJV, Stephansson O, Hägglund MGA, Schiöth HB. The solute carrier (SLC) complement of the human genome: phylogenetic classification reveals four major families. FEBS Lett. 2008;582(27):3811–3816. doi:10.1016/j.febslet.2008.10.016
159. Höglund PJ, Nordström KJV, Schiöth HB, Fredriksson R. The solute carrier families have a remarkably long evolutionary history with the majority of the human families present before divergence of bilaterian species. Mol Biol Evol. 2011;28(4):1531–1541. doi:10.1093/molbev/msq350
160. Schlessinger A, Yee SW, Sali A, Giacomini KM. SLC classification: an update. Clin Pharmacol Ther. 2013;94(1):19–23. doi:10.1038/clpt.2013.73
161. Okazaki T, Javle M, Tanaka M, Abbruzzese JL, Li D. Single nucleotide polymorphisms of gemcitabine metabolic genes and pancreatic cancer survival and drug toxicity. Clin Cancer Res. 2010;16(1):320–329. doi:10.1158/1078-0432.CCR-09-1555
162. Mitra AK, Kirstein MN, Khatri A, et al. Pathway-based pharmacogenomics of gemcitabine pharmacokinetics in patients with solid tumors. Pharmacogenomics. 2012;13(9):1009–1021. doi:10.2217/pgs.12.81
163. Dudenkov TM, Ingle JN, Buzdar AU, et al. SLCO1B1 polymorphisms and plasma estrone conjugates in postmenopausal women with ER+ breast cancer: genome-wide association studies of the estrone pathway. Breast Cancer Res Treat. 2017;164(1):189–199. doi:10.1007/s10549-017-4243-3
164. Gregory BJ, Chen SM, Murphy MA, Atchley DH, Kamdem LK. Impact of the OATP1B1 c.521T>C single nucleotide polymorphism on the pharmacokinetics of exemestane in healthy post-menopausal female volunteers. J Clin Pharm Ther. 2017;42(5):547–553. doi:10.1111/jcpt.12569
165. Smiley DA, Khalil RA. Estrogenic compounds, estrogen receptors and vascular cell signaling in the aging blood vessels. Curr Med Chem. 2000;16(15):1863–1887. doi:10.2174/092986709788186093
166. Wang X, Pankratz VS, Fredericksen Z, et al. Common variants associated with breast cancer in genome-wide association studies are modifiers of breast cancer risk in BRCA1 and BRCA2 mutation carriers. Hum Mol Genet. 2010;19(14):2886–2897. doi:10.1093/hmg/ddq174
167. Seijo ER, Song H, Lynch MA, et al. Identification of Genetic Alterations in the TGF Type II Receptor Gene Promoter. Vol. 483. 2001.
168. Derynck R, Akhurst RJ. TGF-β signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129.
169. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-b family signalling. Nature. 2003;425:577–584. doi:10.1038/nature02006
170. Xie F, Jin K, Shao L, et al. FAF1 phosphorylation by AKT accumulates TGF-β type II receptor and drives breast cancer metastasis. Nat Commun. 2017;8(1). doi:10.1038/ncomms15021
171. Ferretti G, Felici A, Cognetti F, Mandala M. Transforming growth factor-signaling and regulatory T cells. J Clin Oncol. 2007;25(29):4693–4695. doi:10.1200/JCO.2007.13.4007
172. Buck MB, Fritz P, Dippon J, Zugmaier G, Knabbe C. Prognostic significance of transforming growth factor β receptor II in estrogen receptor-negative breast cancer patients. Clin Cancer Res. 2004;10(2):491–498. doi:10.1158/1078-0432.CCR-0320-03
173. Padua D, Zhang H-F, Wang Q. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133(1):66–77. doi:10.1016/j.cell.2008.01.046
174. Busch S, Sims AH, Stal O, Ferno M, Landberg G. Loss of TGFβ receptor type 2 expression impairs estrogen response and confers tamoxifen resistance. Cancer Res. 2015;75(7):1457–1469. doi:10.1158/0008-5472.CAN-14-1583
175. Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther. 2013;7:57–67. doi:10.2147/OTT.S53876
176. Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2(8):594–604. doi:10.1038/nrc864
177. Williams AB, Schumacher B. p53 in the DNA-damage-repair process. Cold Spring Harb Perspect Med. 2016;6(5):1–15. doi:10.1101/cshperspect.a026070
178. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008–a001008. doi:10.1101/cshperspect.a001008
179. Wang Y, Helland Å, Holm R, et al. TP53 mutations in early-stage ovarian carcinoma, relation to long-term survival. Br J Cancer. 2004;90(3):678–685. doi:10.1038/sj.bjc.6601537
180. Wang Y, Kringen P, Kristensen GB, et al. Effect of the codon 72 polymorphism (c.215G>C, p.Arg72Pro) in combination with somatic sequence variants in the TP53 gene on survival in patients with advanced ovarian carcinoma. Hum Mutat. 2004;24(1):21–34. doi:10.1002/humu.20055
181. Langerød A, Zhao H, Borgan Ø, et al. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 2007;9(3). doi:10.1186/bcr1675
182. Apostolou P, Papasotiriou I. Current perspectives on CHEK2 mutations in breast cancer. Breast Cancer. 2017;9:331–335. doi:10.2147/BCTT.S111394
183. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6(12):909–923. doi:10.1038/nrc2012
184. Chrisanthar R, Knappskog S, Løkkevik E, et al. CHEK2 mutations affecting kinase activity together with mutations in TP53 indicate a functional pathway associated with resistance to epirubicin in primary breast cancer. PLoS One. 2008;3(8):1–15. doi:10.1371/journal.pone.0003062
185. Geisler S, Lønning E, Aas T, et al. Influence of TP53 gene alterations and C-ErbB-2 expression on the response to treatment with doxorubicin in locally advanced breast cancer. Cancer Res. 2001;61(6):2505–2512.
186. Geisler S, Børresen-Dale A-L, Johnsen H, et al. Gene mutations predict the response to neoadjuvant treatment with 5-fluorouracil and mitomycin in locally advanced breast cancer. J Clin Oncol. 2003;20(3):751–757.
187. Kandioler-Eckersberger D, Ludwig C, Rudas M, et al. TP53 mutation and p53 overexpression for prediction of response to neoadjuvant treatment in breast cancer patients. Clin Cancer Res. 2000;6(1):50–56.
188. Lanni JS, Lowe SW, Licitra EJ, Liu JO, Jacks T, p53-independent apoptosis induced by paclitaxel through an indirect mechanism. Proc Natl Acad Sci USA. 1997;94(18):9679–9683. doi:10.1073/pnas.94.18.9679
189. Wahl AF, Donaldson KL, Faircnild C, et al. Loss of normal p53 function confers sensitization to Taxol by increasing G2/M arrest and apoptosis. Nat Med. 1996;2(1):72–79. doi:10.1038/nm0196-72
190. Bergh J, Norberg T, Sjögren S, Lindgren A, Holmberg L. Complete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in relation to adjuvant systemic therapy and radiotherapy. Nat Med. 1995;1(10):1029–1034. doi:10.1038/nm1095-1029
191. Guillemette C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J. 2003;3(3):136–158. doi:10.1038/sj.tpj.6500171
192. Gong Q-H, Cho Aã JW, Huang T, et al. Thirteen UDPglucuronosyltransferase genes are encoded at the human UGT1 gene complex locus. Pharmacogenet. 2001;11:367–368.
193. Turgeon D, Carrier J-S, Levesque E, Hum DW, Belanger A. Relative enzymatic activity, protein stability, and tissue distribution of human steroid-metabolizing UGT2B subfamily members this work was supported by the medical research council of canada, the fonds de la recherche en sante du quebec, and endorecherche. Endocrinology. 2001;142(2):778–787. doi:10.1210/endo.142.2.7958
194. Jin Y, Penning TM. Aldo-keto reductases and bioactivation/detoxication. Annu Rev Pharmacol Toxicol. 2007;47(1):263–292. doi:10.1146/annurev.pharmtox.47.120505.105337
195. Hu DG, Rogers A, Mackenzie PI. Epirubicin upregulates UDP glucuronosyltransferase 2B7 expression in liver cancer cells via the p53 pathway. Mol Pharmacol. 2014;85(6):887–897. doi:10.1124/mol.114.091603
196. Ormrod D, Holm K, Goa K, Spencer C, Ackland SP. Epirubicin A review of its efficacy as adjuvant therapy and in the treatment of metastatic disease in breast cancer. Drugs Aging. 1999;15(5):389–416. doi:10.2165/00002512-199915050-00006
197. UGT2B7 UDP glucuronosyltransferase family 2 member B7 [Homo sapiens (human)] - Gene - NCBI. Available from: https://www.ncbi.nlm.nih.gov/gene/7364.
198. Zheng Y, Sun D, Sharma AK, Chen G, Amin S, Lazarus P. Elimination of antiestrogenic effects of active tamoxifen metabolites by glucuronidation. Drug Metab Dispos. 2007;35(10):1942–1948. doi:10.1124/dmd.107.016279
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.