")
Back to Journals » OncoTargets and Therapy » Volume 13
Genetic Heterogeneity of Esophageal Squamous Cell Carcinoma with Inherited Family History
Authors He W , Leng X, Yang Y, Peng L, Shao Y, Li X, Han Y
Received 4 June 2020
Accepted for publication 8 August 2020
Published 3 September 2020 Volume 2020:13 Pages 8795—8802
DOI https://doi.org/10.2147/OTT.S262512
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Nicola Silvestris
Wenwu He,1,* Xuefeng Leng,1,* Yanyu Yang,2,* Lin Peng,1 Yang Shao,3,4 Xue Li,3 Yongtao Han1
1Department of Thoracic Surgery, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, School of Medicine, University of Electronic Science and Technology of China, Chengdu, Sichuan, People’s Republic of China; 2Department of Radiology, Dalian Medical University, Dalian, Liaoning, People’s Republic of China; 3Department of Medicine, Nanjing Geneseeq Technology, Inc, Nanjing, Jiangsu, People’s Republic of China; 4School of Public Health, Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yongtao Han Department of Thoracic Surgery, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, School of Medicine
University of Electronic Science and Technology of China, Chengdu, Sichuan, People’s Republic of China
Tel +86-28-85420229
Email [email protected]
Background: Esophageal squamous cell carcinoma (ESCC) is a common malignant tumor with significant geographical variation and familial aggregation. However, the potentially different mechanisms underlying tumorigenesis in patients with ESCC with and without a family history of the disease remain unclear. In this study, the genes mutated in familial and nonfamilial ESCC were analyzed. Further, we aimed to explore the genes related to ESCC and attempt to identify potential patients in families with a history of ESCC.
Methods: Next-generation sequencing technology was used to examine germline mutations and mutation profiles in 36 matched tumor-normal ESCC specimens. Additionally, tumor mutational burden (TMB) values were measured in two cohorts.
Results: We identified four novel germline mutations in patients with familial ESCC, in BAX (c.121dupG: p.E41G), CDKN2A (c.374dupA: p.D125E), TP53 (c.856G>A: p.E286K), and CHEK1 (c.923+1G>A). Mutation profiles revealed that patients with and without a family history of ESCC had similar high-frequency gene mutation profiles, among which TP53 was the most commonly mutated gene. Additionally, tumor-specific mutated genes in patients with a positive family history of ESCC were APC, AKT3, DPYD, EP300, NFE2L2, PPP2R1A, RUNX1, and VEGFA, while those in patients without a family history of ESCC were CXCR4, PIK3R2, SMARCA4, and TTF1. Moreover, patients with positive family history had significantly higher TMB values (7.8 ± 4.1 vs 5.0 ± 2.4, for patients with and without a family history, respectively; P = 0.038).
Conclusion: Our results identified mutation profiles in patients with familial and nonfamilial ESCC, and identified germline mutations in patients with positive history. TMB values may be informative for immunotherapy approaches in familial ESCC.
Keywords: esophageal squamous cell carcinoma, genetic heterogeneity, family heredity
Introduction
Esophageal cancer, of which esophageal squamous cell carcinoma (ESCC) is the major histological subtype, is the eighth leading cause of cancer-related death worldwide.1,2 The burden of ESCC incidence is closely related to geographical distribution and more than half of all ESCC cases worldwide are diagnosed in China.3,4 The significant geographical variation implies that environmental factors may play important roles in the development of ESCC. However, genetic factors may also contribute to the susceptibility to ESCC.5 Data from our hospital showed that individuals whose parents or siblings had ESCC account for 11.06% of all patients with ESCC. Despite improved treatments, including curative surgery, chemoradiotherapy, and other treatments, the long-term outcomes of ESCC have remained unsatisfactory.6,7
The development of new biochemical technologies, especially next-generation sequencing (NGS), has greatly advanced our understanding of the genomic features of ESCC. Previous genome-wide association or whole exon sequencing analyses have mainly compared patients with ESCC to those without ESCC to screen out mutated genes, including TP53, CDKN2A, NOTCH1, and PIK3CA.8,9 However, few studies have reported genetic mutations associated with familial ESCC and none have not concentrated on the different tumorigenesis mechanisms in familial and non-familial ESCC.10
In this study, we used NGS technology to explore the inheritance of genes related to ESCC and attempted to identify potential patients in families with a history of ESCC.
Methods
Study Subjects
A total of 40 patients with ESCC were recruited from Sichuan Cancer Hospital from February 2013 to February 2017. All diagnoses were confirmed by independent experienced pathologists. At the time of recruitment, study subjects were interviewed in detail about whether their parents, siblings, or children had ever been diagnosed with ESCC. For patients whose relatives had a history of cancer, we gathered information about their age at cancer diagnosis, physical condition, or age at death. We collected the vital status of patients with relatives with no history of cancer. The inclusion criteria were: 1) histological diagnosis of resectable ESCC in patients over the age of 18; 2) availability of tumor tissue samples by operation or puncture and corresponding adjacent non-tumor tissues (located more than 3 cm from the tumors); and 3) availability of blood samples. Patients with other severe systemic disease, metastasis, or any preoperative neoadjuvant therapy were excluded from the study. This study was approved by the Ethics Committee of Sichuan Cancer Hospital (SCCHEC-02-2017-043). All participants signed informed consent, in accordance with the Declaration of Helsinki, for specimen collection and genetic testing following a detailed description of the purpose of the study.
DNA Extraction and Quantification
Formalin-fixed and paraffin-embedded (FFPE) blocks were de-paraffinized twice with xylene and DNA was extracted using a QIAamp DNA FFPE Tissue Kit (Qiagen) following the manufacturer’s instructions. Unstained FFPE sections (n = 5–8) with an area greater than 0.5 × 0.5 cm and thickness of 6–10 μm were used for sequencing. More sections were used if the tissue area was less than 0.5 cm. In this study, the total amount of DNA extracted from samples ranged from 88 to 17,300 ng. All extracted samples met the required DNA yield for quality control, which is 50 ng. Extracted DNA was purified, qualified using a Nanodrop2000 (Thermo), and quantified by Qubit3.0 (Life Technology) using a dsDNA HS Assay Kit (Life Technology) following the manufacturer’s protocols.
Library Preparation and Sequencing
DNA libraries were subjected to polymerase chain reaction (PCR) amplification and purification before targeted enrichment. Libraries from different samples were marked with unique indices during library preparation and up to 2 μg of different libraries were pooled together for targeted enrichment. Human cot-1 DNA (Life Technologies) and xGen Universal Blocking Oligos (Integrated DNA Technologies) were added to block nonspecific binding of library DNA to targeted probes. Customized xGen lockdown probe panels (Integrated DNA Technologies) were used for targeted enrichment of 425 predefined genes. The hybridization reaction was performed using NimbleGen SeqCap EZ Hybridization and Wash Kit (Roche). Dynabeads M-270 (Life Technologies) were used to capture probe-bound fragments. Then, the library was amplified with Illumina p5 and p7 primers in KAPA HiFi HotStart ReadyMix (KAPA Biosystems) and purified using Agencourt AMPure XP beads. Library quantification was achieved using the KAPA Library Quantification kit (KAPA Biosystems). The size distribution was measured using Agilent Technologies 2100 Bioanalyzer (Agilent Technologies). Enriched libraries were sequenced on HiSeq 4000 NGS platforms (Illumina) to coverage depths of at least 100 x and 300 x for normal tissue and tumor, respectively, after removing PCR duplicates.
Bioinformatics Analysis
Original HiSeq 4000 sequencing platform data were transferred by base calling analysis into raw sequence data containing both sequence and sequencing quality information. Single-nucleotide variants (SNVs) and short insertions/deletions (indels) were identified using VarScan2 with the minimum variant allele frequency threshold set at 0.01, and the p-value threshold for calling variants set at 0.05 to generate Variant Call Format (VCF) files. All SNVs/indels were annotated with ANNOVAR, and each SNV/indel was manually checked on the Integrative Genomics Viewer (IGV). Copy number variations (CNVs) were detected using software developed in-house.
Results
Patient Characteristics
A total of 40 ESCC patients were included in the study. One cancer tissue specimen and one adjacent non-tumor tissue specimen were obtained from each patient for sequencing. Four patients were excluded because of unqualified sequencing and quality control. Sequencing information was obtained and analyzed from 36 patients, including 18 cases with family history and 18 cases without. Patient characteristics are presented in Table 1. Statistical differences were observed in age, gender, stage, tumor location, and smoking status between the two cohorts (p < 0.05 for each).
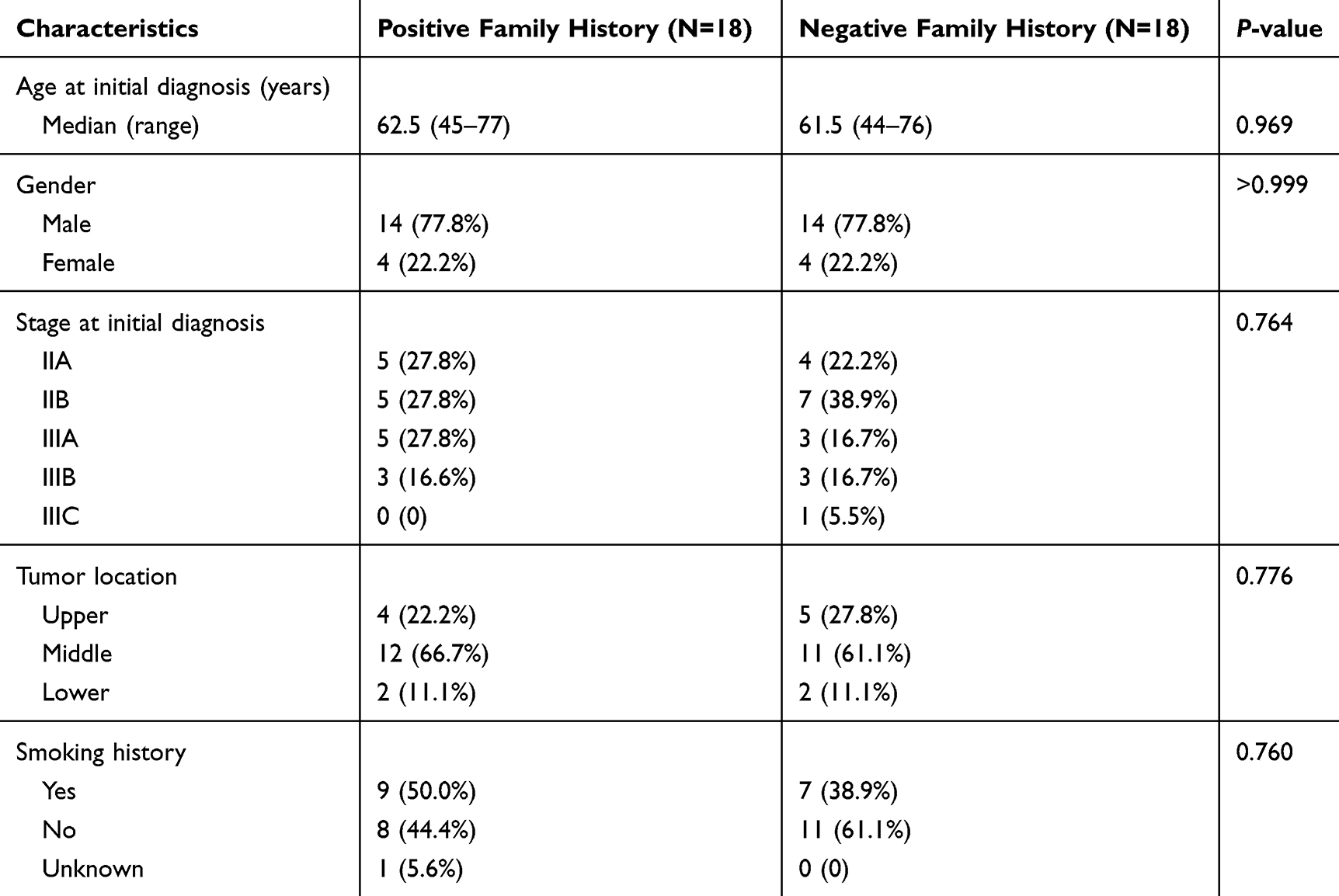 |
Table 1 Clinical Characteristics of Esophageal Squamous Cell Carcinoma Patients |
Germline Mutations Detected in Patients with Positive Family History
Of the 36 patients with ESCC, four patients with family history (11.11%, 4/36) harbored germline mutations. The CHEK1 mutation in patient M10-F was in an intron, while the remaining three germline mutations identified, BAX E41G in patient M09-F, TP53 E286K in patient M14-F, and CDKN2A D125E in patient F02-F fell within exons (Table 2). We did not detect germline mutation in patients without a family history of ESCC. The parents of three of these four patients suffered from ESCC and the sibling of one patient had ESCC. Fisher exact tests were used to evaluate the correlation between pathogenic germline mutation and family history of ESCC risk. The odds ratio (OR) of co-occurring germline mutation and low risk of familial ESCC was 0 (95% confidence interval (CI), 0–1.4, P = 0.104). This implies that germline mutation may result in a greater risk of familial ESCC. However, this observation was not statistically significant, possibly because of the limited number of patients included in the study.
 |
Table 2 Summary of Pathogenic Germline Mutations Detected in Esophageal Squamous Cell Carcinoma Patients |
ESCC Tumor-Specific Mutation Gene Profile
NGS results revealed that patients with familial and non-familial ESCC had similar high-frequency mutation gene profiles. The most frequent mutations observed were missense mutations (Figure 1). Patients with familial ESCC had mutation frequencies more than 20% in TP53 (94.4%), MCL1 (44.4%), CCND1 (38.9%), PIK3CA (38.9%), MYC (33.3%), CDKN2A (27.8%), FGF19 (27.8%), SOX2 (27.8%), and NOTCH1 (22.2%). Patients with non-familial ESCC had mutation frequencies more than 20% in TP53 (88.9%), MCL1 (66.7%), MYC (38.9%), PIK3CA (33.3%), CCND1 (33.3%), SOX2 (33.3%), FGF19 (27.8%), TERC (27.8%), NOTCH1 (22.2%), and CDK6 (22.2%). The most commonly mutated gene observed in both cohorts was TP53. In both cohorts, mutated genes were mainly observed in the NOTCH1 and PI3K pathways. In patients with familial ESCC, the special high-frequency mutated genes, with a mutation burden more than 10%, were APC, AKT3, DPYD, EP300, NFE2L2, PPP2R1A, RUNX1, and VEGFA, and in patients with non-familial ESCC these genes were CXCR4, PIK3R2, SMARCA4, and TTF1 (Figure 2). Most of these genes had missense mutations. These two groups of genes are mainly involved in the WNT and VEGF signaling pathways.
 |
Figure 1 Tumor-specific mutation landscape in esophageal squamous cell carcinoma. The oncoprint presents the most frequent tumor-specific mutations accounting for more than 10% of patients with familial and non-familial ESCC. |
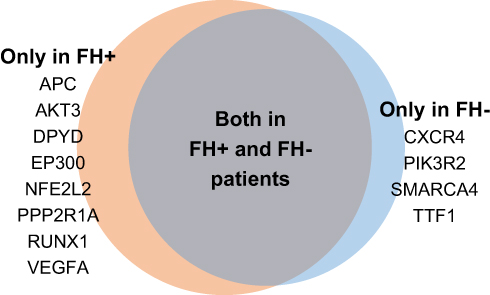 |
Figure 2 Mutually exclusive tumor-specific gene mutations in patients with familial (FH+) and non-familial (FH-). The summary of mutual exclusive genes only includes genes with mutation frequencies higher than 10% in patients with positive family history or negative family history of ESCC. |
Patients with Familial ESCC Had Higher Tumor Mutational Burden (TMB)
Comparisons of TMB values between patients with familial and non-familial ESCC are shown in Figure 3. The TMB values in patients with familial ESCC (TMB: 7.8 ± 4.1 Muts/Mb) were significantly higher than those of patients with non-familial (TMB: 5.0 ± 2.4 Muts/Mb, P=0.038). Therefore, patients with familial ESCC harbored more mutations in gene coding regions within ESCC tumors.
 |
Figure 3 TMB distribution by family history status in esophageal squamous cell carcinoma. The top and bottom of the boxes show the upper and lower and quartiles and the middle line is median. Mann–Whitney test was used for inter-group comparison and the P value calculation was two-tailed. Abbreviations: FH+, patients with positive family history; FH-, patients with negative family history. |
Discussion
ESCC seriously endangers human health. Numerous studies have shown that the incidence of ESCC varies geographically and is observed in family groups. In our study, we used NGS technology to explore germline mutations, gene mutation profiles, and their frequencies in patients with familial and non-familial ESCC. Additionally, TMB values for the two cohorts revealed that those with familial TMB had a higher tumor mutation burden. To our knowledge, this is the first study to explore mutations in patients with ESCC in western China. These results may reveal the molecular mechanisms of ESCC, and how familial and non-familial ESCC differ at the molecular level.
We identified four genes with germline mutations in patients with familial ESCC. The protein encoded by the BAX gene belongs to the BCL2 protein family, whose expression is regulated by the TP53 tumor suppressor. CDKN2A contains an alternate open reading frame (ARF) whose product functions as a stabilizer of TP53. One combined analysis of TP53, BAX, and CDKN2A showed that patients with ESCC and high levels of BAX and CDKN2A expression had a good prognosis. In our study, we identified mutations in BAX (c.121dupG: p.E41G fs*33) and CDKN2A (c.374dupA: p.D125E fs*17). However, no published studies about these alterations indicate how they could influence disease development in patients with ESCC at the mechanistic level. Interestingly, Clemons et al detected TP53 mutation (c.856G>A: p.E286K) in a novel esophageal adenocarcinoma cell line, ONAC1, which was derived from a Barrett’s-associated EA.11 Creemers et al also found a TP53 mutation (c.856G>A: p.E286K) in a series of adrenocortical carcinoma case reports.12 Of note, we identified a novel donor splice site mutation (c.923+1G>A) in CHEK1, which might affect the canonical splice sequence and result in abnormal CHEK1 proteins.
Our study highlights the high-frequency gene mutation profiles of patients with familial and non-familial ESCC, and highlights the differences between the two cohorts. Similar mutation profiles suggest that ESCC related mutations are more frequent in the NOTCH1 and PI3K pathways in both cohorts, which is consistent with previous reports.8,9,13 This implies that additional factors, such as the environment, also play a major role in the development of ESCC. A strong cumulative effect of genetic risk factors has been described in familial ESCC, and the identification of the same SNPs in familial and non-familial ESCC can be explained by environmental factors.10
Herein, we observed some special high-frequency mutated genes in patients with familial ESCC, including APC, AKT3, DPYD, EP300, NFE2L2, PPP2R1A, RUNX1, and VEGFA. Deng et al showed that EP300 and NFE2L2 had high mutation rates in Asian patients with ESCC.14 EP300 encodes the adenovirus E1A-associated cellular p300 transcriptional co-activator protein, which plays an important role in cell proliferation and differentiation. Previous studies have demonstrated that EP300 mutation predicted a shorter survival time in patients with ESCC.15 NFE2L2 (also known as NRF2) encodes a transcription factor that plays a key role in cellular defense against oxidative stress and can be induced by radiotherapy through the production of reactive oxygen species in cancer cells. NRF2 mutations caused patients with ESCC to respond poorly to chemoradiation therapy, resulting in a poor prognosis.16
We also observed mutations in the WNT (APC, EP300, and PPP2R1A) and VEGF (AKT3 and VEGFA) signaling pathways. These molecular alterations that may contribute to the development of familial ESCC may represent the basis of novel drug development. However, Salem et al showed that gene mutations in the WNT pathway (APC and RNF43) were present in low frequencies in patients with ESCC.13 This inconsistency may be due to methodological differences in the studies. PPP2R1A encodes the subunit of protein phosphatase 2A (PP2A), which is implicated in the negative control of cell growth and division. In a previous study, PPP2R1A was shown to promote the growth of malignant cells in uterine cancer,17 but no studies have described the role of PPP2R1A in ESCC. Our study suggests that PPP2R1A may be a new candidate gene for facilitating our understanding of ESCC and establishing therapeutic targets.
The RUNX1 transcription factor is a differentiation regulator in hematopoietic cells, and RUNX1 mutation often results in the development of leukemia. Jiang et al found that RUNX1 can promote cell proliferation in ESCC.18 Additionally, DPYD is a rate-limiting enzyme in the catabolism of 5-fluorouracil (5-fu) and can transform 5-fu into inactive metabolites and reduce its side effects.19 Our findings suggest that RUNX1 and DPYD might be further used as therapeutic targets to guide chemoradiotherapy in patients with familial ESCC, and could improve the safety of treatment.
The special high-frequency mutated genes, CXCR4, PIK3R2, SMARCA4, and TTF1 were observed in patients with non-familial ESCC. CXCR4 is a chemokine receptor which plays a pivotal role in the regulation of cell migration. Lu et al reported that CXCR4-positive ESCC cells showed strong migration ability, especially to lymph nodes.20 In a recent study by Song et al, the activation of the CXCL12/CXCR4 axis in vascular endothelial cells was shown to stimulate angiogenesis by upregulating MAPK/ERK, PI3K/AKT, and Wnt/β-catenin pathways.21 PIK3R2 is also involved in the MAPK/ERK and PI3K/AKT pathways. Additionally, in the present study, we found that TTF1 mutation frequency was higher in patients with non-familial ESCC. Previously, TTF1 was associated with esophageal small cell carcinoma, which is more aggressive than ESCC and differs in response to therapy and prognosis.22
Finally, we analyzed TMB values in the two cohorts (Figure 3). TMB refers to the total amount of somatic non-synonymous mutations in the coding region of tumor genes. The higher the TMB value is, the more mutation-related new antigens are produced, leading to greater T lymphocytes infiltration, and a stronger anti-tumor immune response.23 Previous studies have shown that high TMB, in combination with PD-L1 and MSI-H/dMMR, can be used to select patients who will benefit from immunotherapy.24,25 Song et al reported that patients with ESCC and high TMB can benefit from PD-1 inhibitor treatment, but TMB, as a single predictor, may not accurately estimate the efficacy of immunotherapy.26 In our study, patients with familial ESCC generally had higher TMB values than did those with non-familial ESCC, which could imply the former may benefit more from immunotherapy. Of note, Parikh et al reported that the TMB value in patients with frameshift mutations was significantly higher than that in patients with any other mutation type.27 Our findings were consistent with this research. In the two cohorts, frameshift mutations were observed in FAT1 and ATR in patient F04-F, DPYD in patient M11-F, CDKN2A in patient M06, and NOTCH1 in patient M01 and these patients had higher TMB values. Interestingly, these mutations were identified to have durable immunotherapy benefits.28–31 Therefore, it is of interest to further explore the relationship between frameshift mutations in specific genes and the conferred immunotherapy benefits.
Our study has some limitations. First, because of the strict enrollment conditions and funding constraints, we are unable to include more patients. Further studies with larger cohorts should be performed to validate our results and elaborate upon them. Secondly, TMB value may be related to smoking status. Due to small sample size in this study, subgroup analyses were not performed to investigate the relationship between TMB values and smoking status.
Conclusion
Our results show that patients with familial and non-familial ESCC had similar high-frequency mutation gene profiles, but different harbored tumor-specific mutated genes. Moreover, we detected special germline mutations and higher TMB values in patients with familial ESCC, which suggest that they may have a greater benefit from immunotherapy. More importantly, these findings might provide novel insights into the molecular alterations in ESCC and assist in the identification of potential patients in families with familial ESCC. These results might facilitate the development of precision targeted therapies for ESCC.
Disclosure
Yang Shao and Xue Li are employees of Nanjing Geneseeq Technology, Inc. The authors report no other potential conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Arnold M, Soerjomataram I, Ferlay J, Forman D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut. 2015;64(3):381–387. doi:10.1136/gutjnl-2014-308124
3. Liang H, Fan JH, Qiao YL. Epidemiology, etiology, and prevention of esophageal squamous cell carcinoma in China. Cancer Biol Med. 2017;14(1):33–41. doi:10.20892/j.issn.2095-3941.2016.0093
4. Malhotra GK, Yanala U, Ravipati A, Follet M, Vijayakumar M, Are C. Global trends in esophageal cancer. J Surg Oncol. 2017;115(5):564–579. doi:10.1002/jso.24592
5. Chen T, Cheng H, Chen X, et al. Family history of esophageal cancer increases the risk of esophageal squamous cell carcinoma. Sci Rep. 2015;5:16038. doi:10.1038/srep16038
6. Leng X, He W, Yang H, et al. Prognostic impact of postoperative lymph node metastases after neoadjuvant chemoradiotherapy for locally advanced squamous cell carcinoma of esophagus: from the results of NEOCRTEC5010, a randomized multicenter study. Ann Surg. 2019. doi:10.1097/SLA.0000000000003727
7. Yang H, Liu H, Chen Y, et al. Neoadjuvant chemoradiotherapy followed by surgery versus surgery alone for locally advanced squamous cell carcinoma of the esophagus (NEOCRTEC5010): a Phase III multicenter, randomized, open-label clinical trial. J clin oncol. 2018;36(27):2796–2803. doi:10.1200/JCO.2018.79.1483
8. Yang JW, Choi YL. Genomic profiling of esophageal squamous cell carcinoma (ESCC)-basis for precision medicine. Pathol Res Pract. 2017;213(7):836–841. doi:10.1016/j.prp.2017.02.021
9. Sasaki Y, Tamura M, Koyama R, Nakagaki T, Adachi Y, Tokino T. Genomic characterization of esophageal squamous cell carcinoma: insights from next-generation sequencing. World J Gastroenterol. 2016;22(7):2284–2293. doi:10.3748/wjg.v22.i7.2284
10. Suo C, Qing T, Liu Z, et al. Differential cumulative risk of genetic polymorphisms in familial and nonfamilial esophageal squamous cell carcinoma. Cancer Epidemiol Biomark Prev. 2019;28(12):2014–2021. doi:10.1158/1055-9965.EPI-19-0484
11. Sturm I, Petrowsky H, Volz R, et al. Analysis of p53/BAX/p16 ink4a/CDKN2 in esophageal squamous cell carcinoma: high BAX and p16 ink4a/CDKN2 identifies patients with good prognosis. J clin oncol. 2001;19(8):2272–2281. doi:10.1200/JCO.2001.19.8.2272
12. Clemons NJ, Do H, Fennell C, et al. Characterization of a novel tumorigenic esophageal adenocarcinoma cell line: OANC1. Dig Dis Sci. 2014;59(1):78–88. doi:10.1007/s10620-013-2882-8
13. Creemers SG, Korpershoek E, Atmodimedjo PN, et al. Identification of mutations in cell-free circulating tumor DNA in adrenocortical carcinoma: a case series. J Clin Endocrinol Metab. 2017;102(10):3611–3615. doi:10.1210/jc.2017-00174
14. Salem ME, Puccini A, Xiu J, et al. Comparative molecular analyses of esophageal squamous cell carcinoma, esophageal adenocarcinoma, and gastric adenocarcinoma. The Oncologist. 2018;23(11):1319–1327. doi:10.1634/theoncologist.2018-0143
15. Deng J, Chen H, Zhou D, et al. Comparative genomic analysis of esophageal squamous cell carcinoma between Asian and Caucasian patient populations. Nat Commun. 2017;8(1):1533. doi:10.1038/s41467-017-01730-x
16. Bi Y, Kong P, Zhang L, et al. EP300 as an oncogene correlates with poor prognosis in esophageal squamous carcinoma. J Cancer. 2019;10(22):5413–5426. doi:10.7150/jca.34261
17. Shibata T, Kokubu A, Saito S, et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia. 2011;13(9):864–873. doi:10.1593/neo.11750
18. Haesen D, Abbasi Asbagh L, Derua R, et al. Recurrent PPP2R1A mutations in uterine cancer act through a dominant-negative mechanism to promote malignant cell growth. Cancer Res. 2016;76(19):5719–5731. doi:10.1158/0008-5472.CAN-15-3342
19. Jiang YY, Lin DC, Mayakonda A, et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut. 2017;66(8):1358–1368. doi:10.1136/gutjnl-2016-311818
20. van Kuilenburg AB. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur J Cancer. 2004;40(7):939–950. doi:10.1016/j.ejca.2003.12.004
21. Lu CL, Guo J, Gu J, et al. CXCR4 heterogeneous expression in esophageal squamous cell cancer and stronger metastatic potential with CXCR4-positive cancer cells. Dis Esophagus. 2014;27(3):294–302. doi:10.1111/dote.12100
22. Song ZY, Wang F, Cui SX, Qu XJ. Knockdown of CXCR4 inhibits CXCL12-induced angiogenesis in HUVECs through downregulation of the MAPK/ERK and PI3K/AKT and the Wnt/beta-catenin pathways. Cancer Invest. 2018;36(1):10–18. doi:10.1080/07357907.2017.1422512
23. Yamamoto J, Ohshima K, Ikeda S, Iwashita A, Kikuchi M. Primary esophageal small cell carcinoma with concomitant invasive squamous cell carcinoma or carcinoma in situ. Hum Pathol. 2003;34(11):1108–1115. doi:10.1053/j.humpath.2003.07.010
24. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–2608. doi:10.1158/1535-7163.MCT-17-0386
25. Zang YS, Dai C, Xu X, et al. Comprehensive analysis of potential immunotherapy genomic biomarkers in 1000 Chinese patients with cancer. Cancer Med. 2019;8(10):4699–4708. doi:10.1002/cam4.2381
26. Yang C, Zhang J, Ding M, et al. Ki67 targeted strategies for cancer therapy. Clin Transl Oncol. 2018;20(5):570–575. doi:10.1007/s12094-017-1774-3
27. Song W, Wang H, Tian Y, et al. Refractory solitary cervical lymph node metastasis after esophageal squamous cell carcinoma surgery and its successful treatment with immune checkpoint inhibitor: a case report and literature review. Medicine. 2020;99(10):e19440. doi:10.1097/MD.0000000000019440
28. Parikh AR, He Y, Hong TS, et al. Analysis of DNA damage response gene alterations and tumor mutational burden across 17,486 tubular gastrointestinal carcinomas: implications for therapy. The Oncologist. 2019;24(10):1340–1347. doi:10.1634/theoncologist.2019-0034
29. Fang W, Ma Y, Yin JC, et al. Comprehensive genomic profiling identifies novel genetic predictors of response to anti-PD-(L)1 therapies in non-small cell lung cancer. Clin Cancer Res. 2019;25(16):5015–5026. doi:10.1158/1078-0432.CCR-19-0585
30. Ren W, Sun Q, Wu PY, et al. Profiles of genomic alterations in primary esophageal follicular dendritic cell sarcoma: a case report. Medicine. 2018;97(48):e13413. doi:10.1097/MD.0000000000013413
31. Ferrando A. Can one target T-cell ALL? Best Pract Res Clin Haematol. 2018;31(4):361–366. doi:10.1016/j.beha.2018.10.001
32. Sun LL, Yang RY, Li CW, et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am J Cancer Res. 2018;8(7):1307–1316.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.