")
Back to Journals » Infection and Drug Resistance » Volume 13
Genetic Diversity of the Flavohemoprotein Gene of Giardia lamblia: Evidence for High Allelic Heterozygosity and Copy Number Variation
Authors Saghaug CS , Klotz C, Kallio JP , Aebischer T , Langeland N , Hanevik K
Received 29 July 2020
Accepted for publication 29 October 2020
Published 18 December 2020 Volume 2020:13 Pages 4531—4545
DOI https://doi.org/10.2147/IDR.S274543
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sahil Khanna
Christina S Saghaug,1,2 Christian Klotz,3 Juha P Kallio,4 Toni Aebischer,3 Nina Langeland,1,2,5 Kurt Hanevik1
1Department of Clinical Science, University of Bergen, Bergen, Norway; 2Norwegian National Advisory Unit on Tropical Infectious Diseases, Department of Medicine, Haukeland University Hospital, Bergen, Norway; 3Department of Infectious Diseases, Unit 16 Mycotic and Parasitic Agents and Mycobacteria, Robert Koch-Institute, Berlin, Germany; 4Department of Biomedicine, University of Bergen, Bergen, Norway; 5Department of Medicine, Haraldsplass Deaconess Hospital, Bergen, Norway
Correspondence: Christina S Saghaug
Department of Clinical Science, University of Bergen, 8th Floor, Lab-Building, Bergen N-5021, Norway
Tel +47 90 13 24 14
Email [email protected]
Purpose: The flavohemoprotein (gFlHb) in Giardia plays an important role in managing nitrosative and oxidative stress, and potentially also in virulence and nitroimidazole drug tolerance. The aim of this study was to analyze the genetic diversity of gFlHb in Giardia assemblages A and B clinical isolates.
Methods: gFlHb genes from 20 cultured clinical Giardia isolates were subjected to PCR amplification and cloning, followed by Sanger sequencing. Sequences of all cloned PCR fragments from each isolate were analyzed for single nucleotide variants (SNVs) and compared to genomic Illumina sequence data. Identical clone sequences were sorted into alleles, and diversity was further analyzed. The number of gFlHb gene copies was assessed by mining PacBio de novo assembled genomes in eight isolates. Homology models for assessment of SNV’s potential impact on protein function were created using Phyre2.
Results: A variable copy number of the gFlHb gene, between two and six copies, depending on isolate, was found. A total of 37 distinct sequences, representing different alleles of the gFlHb gene, were identified in AII isolates, and 41 were identified in B isolates. In some isolates, up to 12 different alleles were found. The total allelic diversity was high for both assemblages (> 0.9) and was coupled with a nucleotide diversity of < 0.01. The genetic variation (SNVs per CDS length) was 4.8% in sub-assemblage AII and 5.4% in assemblage B. The number of non-synonymous (ns) SNVs was high in gFIHb of both assemblages, 1.6% in A and 3.0% in B, respectively. Some of the identified nsSNV are predicted to alter protein structure and possibly function.
Conclusion: In this study, we present evidence that gFlHb, a putative protective enzyme against oxidative and nitrosative stress in Giardia, is a variable copy number gene with high allelic diversity. The genetic variability of gFlHb may contribute metabolic adaptability against metronidazole toxicity.
Keywords: Giardia, genetic diversity, copy number variation, flavohemoprotein, oxidative stress, nitrosative stress, allele
Introduction
Giardia lamblia is a microaerophilic protozoan parasite that infects up to 280 million humans annually by causing giardiasis.1 This gastrointestinal infection is more common in developing countries, and may negatively affect growth properties and cognitive functions in children.2,3 In developed countries giardiasis is usually related to sporadic waterborne outbreaks, or seen in travelers returning from endemic areas.4,5 To treat giardiasis, the prodrug nitroimidazole antibiotic known as metronidazole (MTZ) is often used as first-line treatment.6 Over the past few years, treatment failures with MTZ have been reported more frequently, and 10–40% of the cases will not be eradicated after a 5–7 day course of MTZ treatment.7–9 The currently understood mode of action of MTZ depends on partial reduction (activation) resulting in highly reactive intermediates that further initiate damage to DNA and proteins, or on the other side, regeneration of MTZ, where oxygen radicals are created (‘futile cycling’).10–12 The enzymes in the detoxification system in Giardia must then be able to handle oxidative stress resulting from metabolizing MTZ, as well as being able to neutralize substances such as molecular oxygen (O2) and nitric oxide (NO) encountered in the gut habitat and released by the host.13,14 There are several known enzymes connected to free radical neutralization, including flavoprotein, desulfoferredoxin (SOR), NADH oxidase and flavohemoprotein (gFlHb).13,15 The gFlHb functions as a nitric oxide dioxygenase, responsible for catalyzing the formation of nitric oxide (NO) to nitrate (NO3−) by using O2 as a co-factor.16,17 The gFlHb enzyme may also possess NADH/NAD(P)H oxidase activities, similar to flavodiiron protein, by catalyzing O2 to H2O in low NO level conditions.18 The gFlHb gene was characterized a decade ago, and is likely to be an important detoxification enzyme in Giardia.15,18 It has been shown to be up-regulated during exposure to oxidative stresses caused by both O2 and H2O2, in addition to being upregulated during nitrosative stress.14,15,19 It was also recently shown that gFlHb protein levels were increased during MTZ exposure in an MTZ and nitazoxanide (NTZ) resistant Giardia isolate.20 In another anaerobic or microaerophilic pathogen, Trichomonas vaginalis, it has been observed that in vitro MTZ-resistant parasites can handle higher levels of oxygen than susceptible ones, probably linking resistance to increased tolerance or better mechanisms to handle oxidative stress.21 It has been proposed that refractory Giardia has a higher tolerance towards O2, as O2 will compromise the activation of MTZ through futile cycling, but also potentially through an O2 induced resistance mechanism.22 Because gFlHb uses O2 as a co-factor for converting NO to NO3−, it could also be relevant for increased MTZ tolerance in this way.
Activities of enzymes having similar properties as the gFlHb have been shown to be inhibited by bulky imidazoles (such as azoles; miconazole, econazole and ketoconazole) by binding to the heme pocket of the protein and generating reactive oxygen species (ROS), which may well mean that MTZ could potentially affect the function of the gFlHb enzyme, and link it to Giardia’s ability to handle and tolerate the toxic effects of MTZ.23,24 Giardia is a functionally tetraploid organism, with two diploid nuclei, each of them harboring two sets of its 5 chromosomes.25–27 However, aneuploidy with unequal distribution of chromosomes may also occur.25,28 Thus, even single-copy genes may occur in up to four versions, ie, alleles, per strain.25,28
The degree of allelic sequence heterozygosity (ASH), has been shown to differ between assemblages of G. lamblia. Current genome data suggests a comparably low ASH in sub-assemblage AI (<0.01%), a little higher in sub-assemblage AII (based on reference strain DH with an ASH of 0.04%), and highest in assemblage B isolates (cf GS reference strain’s ASH of 0.5%).25,29,30 Genetic diversity at the allele level, especially in assemblage B, has been analyzed mainly for typical genotyping genes or housekeeping genes (β-giardin (bg), glutamate dehydrogenase (gdh), elongation factor 1-alpha (ef-1), mlh1 (mlh), the FLORF-C4 (C4) and triosephosphate isomerase (tpi).29,31–34 The allelic forms, however, have rarely been determined by cloning.35–41 In a recent study on the genetic diversity of genes involved in MTZ induced oxidative and nitrosative stress management, we found indications that gFlHb genes may not only present allelic variation, but also be present at variable copy numbers per haploid genome.42 The current study is a follow-up study of this recent published article.42 As gFlHb may play a role in the ability of Giardia to tolerate MTZ, we performed this study to further explore the copy number variability and the allelic diversity of gFlHb in G. lamblia strains representing recent sub-assemblage AII and assemblage B isolates.
Materials and Methods
Giardia Lamblia Isolates
Trophozoite cultures of a recently established G. duodenalis biobank at the Robert Koch-Institute in Berlin, containing twelve Giardia sub-assemblage AII and eight Giardia assemblage B isolates, were cultured according to the methods of Keister.43 Collection, DNA extraction, concentration measurements and Illumina-based whole genome sequencing were carried out as previously stated.42 No clinical data have been collected from the clinical samples of Giardia.
Cloning of Flavohemoprotein
The coding regions and approximately 150 bp up/downstream of the gene flavohemoprotein, gFlHb, in sub-assemblage AII (DHA2_154000) and B (GSB_151570) were amplified from genomic DNA by PCR. The reference genomes for Giardia sub-assemblage AII and assemblage B (v.26) were downloaded from Giardia DB January 2016 versions 2013–11-25.44 Specific primers for the two orthologs of gFlHb of sub-assemblage AII and assemblage B were designed in Geneious Prime® 2019.0.3 (Biomatters Ltd., Auckland, New Zealand) (see Table 1) based on conserved pre-and post-CDS regions of the gFlHb gene represented in twelve assemblage A and eight assemblage B whole genome sequenced isolates previously presented in a former study.42 Each 25 µL PCR reaction contained 1X Q5 Reaction Buffer (cat. nr: B90276, New England BioLabs (NEB) Ipswich, MA, USA), 2 mM MgCl2 (included in buffer), 0.2 mM dNTP’s (catalog nr N0447L, NEB), 0.5 U Q5 High-Fidelity DNA Polymerase (catalog nr M0491L NEB) 0.2 µM each of forward and reverse primer, 1 µL template DNA and nuclease-free water up to 25 µL. Negative controls using master mix reactions with nuclease-free water were included in all PCR experiments. All PCR reactions included an initial denaturation at 98°C for 30s, followed by 35 cycles of denaturation at 98°C for 10 s, annealing for 30 s (55°C for sub-assemblage AII and 60°C for assemblage B), extension at 72°C for 45–60 s and final extension at 72°C for 3 minutes. Amplified PCR products were run on 1% Agarose gels stained with GelGreen® Nucleic Acid Stain (catalog nr: 41,005, Biotium, San Fransisco, CA, USA) and positive bands were cut from the gel using blue light illuminator (Serva, Heidelberg, Germany) and extracted using Wizard SV gel and PCR Clean-Up System (catalog nr: A9282, Promega, Madison, WI, USA) according to manufacturer’s descriptions, the only exception was using 70°C nuclease-free water in the last elution step. Concentrations of the PCR products were measured using Quantus™ Fluorometer (catalog Nr: E6150, Promega). DNA was available from 19 of the original Illumina sequenced samples, and two of the isolates were not included in the gFlHb gene cloning experiments due to unsuccessful amplification of the gene target.
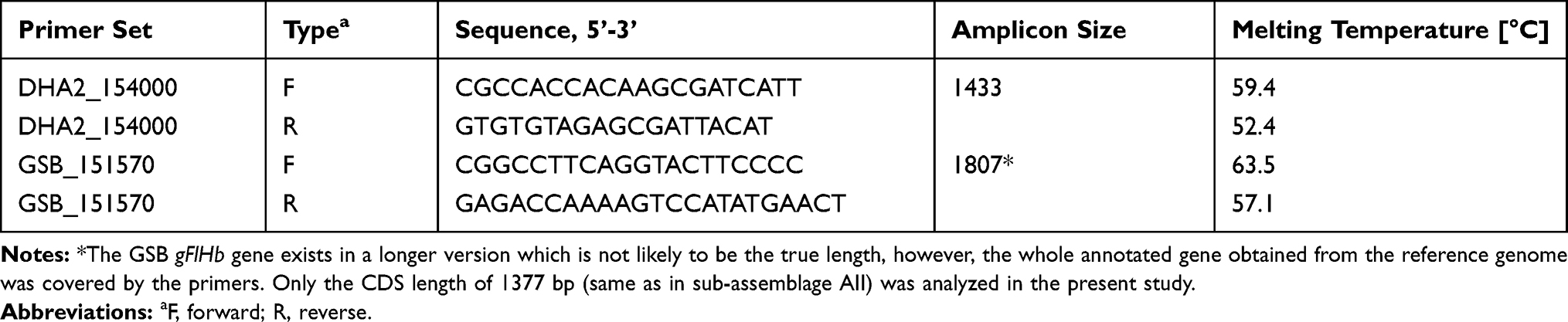 |
Table 1 Primers Designed for Gene-PCR of the gFlHb of Giardia lamblia Sub-Assemblage AII and Assemblage B All Primers Were Ordered from Eurofins Genomics (Ebersberg, Germany) |
The pJET 1.2/blunt cloning vectors (catalog nr: K1232, CloneJET PCR Cloning kit, Thermo Fisher Scientific, Waltham, MA, USA) were used in all of the cloning experiments. The blunt end protocol for ligation of PCR-product to vector was followed according to the manufacturer’s instructions. The ligation mixtures were used immediately or kept at −20°C until usage. The ligation mixtures were introduced into Escherichia coli DH5a competent cells by heat-shock transformation using standard protocols.45 The E. coli was plated on lysogeny broth (LB) agar plates containing ampicillin (100 µg/mL) (catalog nr: 10,835,269,001 (Sigma-Aldrich), Merck KGaA, Darmstadt, Germany) and cultured overnight at 37°C in a CO2 incubator. Approximately 20 clones from each Giardia isolate were picked for colony PCR. 14–20 positive clones were cultured overnight in a shaking incubator at 37°C in Falcon® 17x100 mm, 14 mL high-clarity polypropylene (PP) round bottom test tubes (Item nr: T7597-14F, Corning Life Sciences, NY, USA). The plasmids were purified from the overnight cultures using Zyppy plasmid miniprep kit (catalog nr: D4037, Zymo Research Corp., CA, USA) as advised by the manufacturer and DNA was eluted using nuclease-free water. The plasmid concentrations were measured using Quantus and sequenced using pJET1.2 F and R sequencing primers provided in the cloning kit together with the BigDye™ Terminator v3.1 Cycle Sequencing kit (catalog nr: 4,337,455, Thermo Fisher Scientific). The PCR products were Sanger sequenced at the Sequencing Laboratory of the Robert-Koch Institute, Berlin, Germany.
Data Analysis of Sequences and Single Nucleotide Variation
SNV-called Illumina sequences of gFlHb presented in a former study, accession numbers MK043521.1-MK043540.1 from Genbank (NCBI), were used in the present study to investigate SNVs.42 A SNV in the Illumina sequencing data was defined by a read coverage of minimum 10 and variant frequency above 0.1 (minimum 2 reads would need to have the SNV if the coverage was 10). The Illumina sequenced files with read data were used to compare and verify SNV positions in the cloned sequences in Geneious Prime® (Biomatters Ltd.). The forward and reverse sequences of one clone were aligned against the respective reference gene orthologs DHA2_154000 and GSB_151570 and trimmed at start and end of the sequences before further analysis. The chromatograms were inspected manually, and sequences with ambiguous base calling, or not covering the full CDS were not included. To manage potential random amplification or sequencing errors in the present study, we set criteria to exclude likely false SNVs by verifying SNVs found in clones with Illumina read data. SNVs found in the clones in the present project were termed high confidence (HC) and SNVs and included in the analysis if one or several of the following criteria were met:
- the SNV position was also found to be present in Illumina data for the same isolate.
- multiple clones would have the same SNV within one isolate.
- if the SNV was not found in Illumina data as a major nucleotide, the reads in the Illumina were checked for the presence of the SNV and the nucleotide had to be presented by two reads or more.
The SNVs that were not classified as HC were discarded from the analyses and termed low confidence (LC) SNVs. De novo assemblies were generated for eight of the 20 Giardia isolates at RKI using the software tools HGAP 2.0 (PacificBiosciences) as described elsewhere (Klotz et al 2020, manuscript in preparation). The gFlHb copy number was retrieved by mapping whole or partial sequences of the gFlHb AII and B reference sequences to de novo assemblies of PacBio consensus genomes of each isolate. To also identify partial genes or genes split between contigs, mapping of 20 bp parts of the start, middle and end of the reference genes were also performed. The identified gFlHb copies in PacBio consensus sequences were then extracted, and aligned together with the cloned allele sequences of the gFlHb gene separately for sub-assemblage AII and B. Further phylogenetic trees (Tamura-Nei with method UPGMA) were made. All sequence analysis work was carried out in Geneious Prime®. All nonsynonymous (ns) substitutions were analyzed using Geneious and DNA Sequence Polymorphism (DnaSP) v6.46,47 Nucleotide diversity (pi) was calculated as the average number of nucleotide differences in gene sequences for pairwise comparisons. Haplotype diversity, defined as allelic diversity in the present study, gives a measure of the uniqueness of a specific allele in a population, or the probability that two alleles differ from one another.
Characterization of Amino Acid Changes
The bacterial genetic code was used for translation of the nucleotide codons in the gene sequences of gFlHb (DHA2_154000 and GSB_151570) in sub-assemblage AII and assemblage B. The abbreviations and characterization of the amino acid (aa) changes are based on the International Union of Pure and Applied Chemistry (IUPAC). As the crystal structure of the protein is not known, the estimation as to whether the aa change would affect the protein structure and function was based on homology models. Homology modeling was carried out using Protein Homology/analogy Recognition Engine V2.0 (Phyre2).48 Protein sequences Uniprot ID: E2RTZ4 and A0A482ESB4 were selected to create models for gFlHb assemblage A and B, respectively. The model was created using the single highest scoring template, a crystal structure of E. coli flavohemoglobin (PDB-ID 1GVH) sharing a 40% sequence identity with the gFlHb.49 Illustrations for the homology models were created using PyMol. A conservative replacement was identified as an aa substitution between two aa with rather small physicochemical distance, whilst a radical aa change would be considered as aa having large physicochemical characteristics, or potential rearrangement of the secondary structure.50
Results
gFlHb Copy Number Variation and Allele Diversity
Re-analysis of the mapping of Illumina sequencing data from the AII isolates onto the AII reference strain DH, showed that higher coverage for the gFlHb gene was present along the whole coding regions (CDS) of the gene and adjacent 0–230 bp upstream and 461–1271 bp downstream of the reference CDS.42 For the B isolates the coverage was found to be higher starting from 600 to 816 bp upstream of the GS reference CDS and extending to 90–913 bp downstream of it. PacBio consensus sequences derived from de novo assembly were available for eight isolates, five assemblage B isolates and three sub-assemblage AII isolates. More than one copy of the gFlHb gene was found in the PacBio sequences-derived assemblies of all three AII and five B isolates. In general, higher Illumina coverage fold difference corresponded to higher copy numbers in PacBio sequence assemblies. Isolate P344 had the same estimated copy number of gFlHb based on both analyses. Isolate P424 showed the highest relative coverage in Illumina sequence data with 5.1 fold the average and six copies were detected in the respective PacBio sequence derived assembly. The respective values for P392 were 3.6 and four (see Table 2 for more information). The gFlHb gene was successfully amplified by PCR from 17 of the 20 isolates (11 AII isolates and 6 B isolates). Between five and seventeen clones per isolate were obtained (Table 2). The cloned gFIHb genes were categorized into different alleles depending on their sequences whereby 37 alleles were identified in eleven sub-assemblage AII isolates, and 41 alleles were identified in six assemblage B isolates. In the AII isolates, the number of gFlHb alleles identified by cloning varied from two to eight, while in the B isolates two to twelve alleles per isolate were found (Table 2). The allelic diversity, the probability that two alleles are different from one another, for sub-assemblage AII was calculated to be 0.94 ± 0.01 and 0.95 ± 0.01 for assemblage B. To evaluate how alleles determined by cloning were represented in the gFlHb genes identified by de-novo assembled consensus genomes from PacBio sequenced isolates, we aligned all the CDS sequences and analyzed correspondence by creating phylogenetic trees (Figures 1 and 2). The phylogenetic tree of sub-assemblage AII in Figure 1 represents the relations between the alleles and PacBio copies found in the different isolates. Several allelic forms of gFlHb were found in different isolates, for example one allele was found to be present in four isolates represented by allele AA16 found in P033, P506, P034 and P064. A total of six alleles were found to be present in more than one isolate (table S4). A total of nine PacBio consensus sequences of gFlHb were available from three AII isolates, representing the five, three and two copies found in isolates P392, P407 and P064, respectively. These sequences were aligned to the cloned sub-assemblage AII alleles in order to check whether cloned alleles would match these consensus sequence copies. Three PacBio copies were found to be identical to alleles in their respective isolates (table S4), whereas the P407 AA37 allele and P407 PacBio copy 3 were identical with the exception of three, likely artefactual, indels in the PacBio consensus sequence. P392 PacBio copy 1 and 3 and P407 PacBio copy 1 and 2, were not identical to alleles from the same isolate, but closely related (2–6 SNVs difference), and were likely to be part of the alleles resulting in the consensus PacBio copy. Intriguingly, two PacBio copies, P064 copy 2 and P407 copy 2 were identical to alleles found in other isolates, suggesting that sequencing of more clones would be necessary to pick up all possible alleles of each isolate. The relations between the 41 alleles and PacBio copies that were discovered in assemblage B are presented in phylogenetic tree in Figure 2. Fewer alleles were shared between the assemblage B isolates than in the sub-assemblage AII isolates, where only three alleles were found in more than one isolate, such as AB01 in isolates P424, P458 and reference sequence GSB_151570, and alleles AB05 and AB13 in isolates P424 and P413 (see table S5).
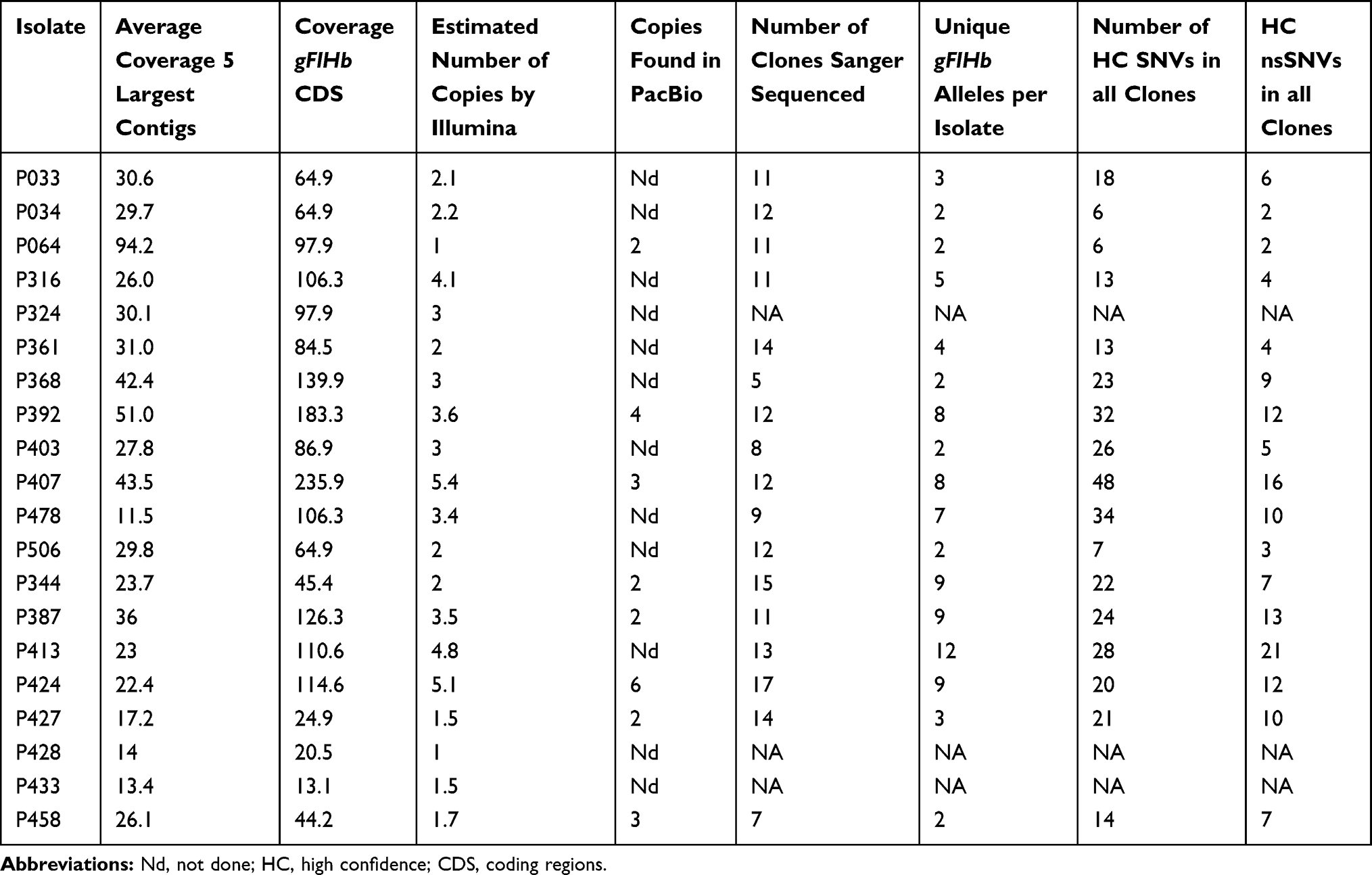 |
Table 2 Estimates of gFlHb Copy Numbers in PacBio and Illumina Sequencing Data, Number of Clones, and Identified Alleles and SNVs in Cloned Sequences |
 |
Figure 1 Phylogenetic tree of all the alleles of gFlHb found in the sub-assemblage AII isolates and gFlHb copies found in the PacBio sequencing data. A is abbreviated for allele, and the number of clones representing each allele is listed in the parenthesis. |
 |
Figure 2 Phylogenetic tree of all the alleles found in the assemblage B isolates and gFlHb copies found by PacBio sequencing. A is an abbreviation of allele, and the number of clones representing each allele is listed in the parenthesis. |
Fifteen PacBio consensus sequences of gFlHb were obtained from five assemblage B isolates, and the copies were checked for similarity to the cloned alleles. Eight of the copies, all in P458 and P424, were found to match cloned alleles in the same isolates. For the isolates P344, P387 the PacBio copies were not identified in the clones, however the alleles from isolate P427 were highly similar to the two identical PacBio copies found in the isolate (1 SNV difference). The PacBio copies found in isolate P387 had 14 bp shorter sequences, probably due to assembly artefacts. Isolate P344 had two identical PacBio copies and the closest allele, P344 AB33, had a total of four SNVs different from the PacBio sequences. Some of the gFlHb PacBio copies in assemblage B were also found to be identical, such as P458 copy 1 and 2, P387 copy 1 and 2, P424 copy 4 and 5 (table S5). See supplementary box 1 for more information about the PacBio copies.
The copy number found in the PacBio data was compared to the number of cloned alleles. For the three sub-assemblage AII isolates with available PacBio data, more alleles were identified in isolates with a higher copy number (P064 two copies and two alleles, P392 four copies and eight alleles and P407 three copies and eight alleles). Among the assemblage B isolates, two isolates with two identified PacBio copies (P344 and P387) both had a total of nine identified alleles, thus higher than the theoretical maximum for a tetraploid organism with two gene copies. Isolates P427 and P458, both with two identified PacBio copies, had three and two alleles, respectively. In the isolate with the highest number of identified copies, P424, with six copies, nine alleles were identified.
Sequence Variation of the gFlHb Gene in Giardia Assemblages A and B
The identified SNVs in the cloned sequences were compared against Illumina sequencing data and categorized into high confidence (HC) and low confidence (LC) SNVs using the algorithm described in methods. LC SNVs were discarded and not used in the analysis, see Supplementary Table S1 for full overview of HC and LC SNVs. Sequence analysis showed a higher proportion of nsSNVs/SNVs in assemblage B isolates (53%) than sub-assemblage AII isolates (38%) (Table S1). The number of SNV positions found in the respective cloned gFlHb CDS for each isolate varied from 6 to 48 for sub-assemblage AII and 14–28 for assemblage B sequences (Table 2). One AII isolate, P407, had the most SNV positions of all isolates with 48 SNV positions found, while the numbers of SNVs were more congruent among the B isolates with approx. 20 SNVs per isolate. A total of 4.8% and 5.4% of all positions of the gFIHb CDS showed some variation in AII and B, respectively (see Tables S2–S5 and S6-S7). The total number of nsSNV positions per CDS was lower for AII isolates (22 positions, 1.6%), than for B isolates (42 positions, 3.1%). The average nucleotide diversity, pi, between cloned alleles was calculated to be 0.007 in sub-assemblage AII isolates’ sequences and 0.009 in assemblage B isolates. Generally, there were more nsSNVs found in just one or a few isolates in assemblage B isolates, compared to sub-assemblage AII, where numerous nsSNV positions were common for several isolates (see Tables S3-S4).
Amino Acid Changes and Relation to Predicted gFlHb Protein Structure
Based on homology modelling gFlHb shares the same domain structures as the homologous FlHb from bacteria49,51 and yeast.52 It is a protein formed of 3 structural domains: (1) heme binding globin domain; (2) a FAD-binding domain; (3) a C-terminal NAD binding domain. Homology models for gFlHb for assemblages A and B were generated and are shown in Figure 3, where the detected nsSNVs that induced aa mutations are visualized. As noted before, assemblage B has a considerably higher number of aa substitutions than sub-assemblage AII. Most of the mutations are predicted to concern residues at the protein surface and, more accurately, those located in loop regions. Also, the majority is considered to only moderately affect the physicochemical properties, eg, simply altering the size of a hydrophobic side chain. Some mutations, however, will cause a reversal of charge, hence affect local electrostatic properties of the protein surface. Some of the changes are predicted to be in near vicinity of the heme or FAD-binding sites and could have an effect for accommodating these co-factors. These changes are marked in red on Figure 3. The most drastic change amongst sub-assemblage AII alleles is a premature stop codon at position 49 in alleles AA24 and AA25 in isolate P478 that likely results in early truncation within the first domain. In addition, there was a change of aromatic Y at position 211 to basic and positively charged H in several alleles (AA07, AA10, AA11, AA12, AA13 and AA15) observed in isolates P316, P361, P368, P392 and P407. This mutation is located in a putative FAD-binding region of domain 2. For assemblage B, the most different allelic forms are obviously those with the deletion Del-75HEL77 that is located to the helix above the heme binding site, and the E80G variant is located on the same helix. Both the deletion and the E80G mutation are potential helix breakers that could disturb the secondary structure and affect heme binding.
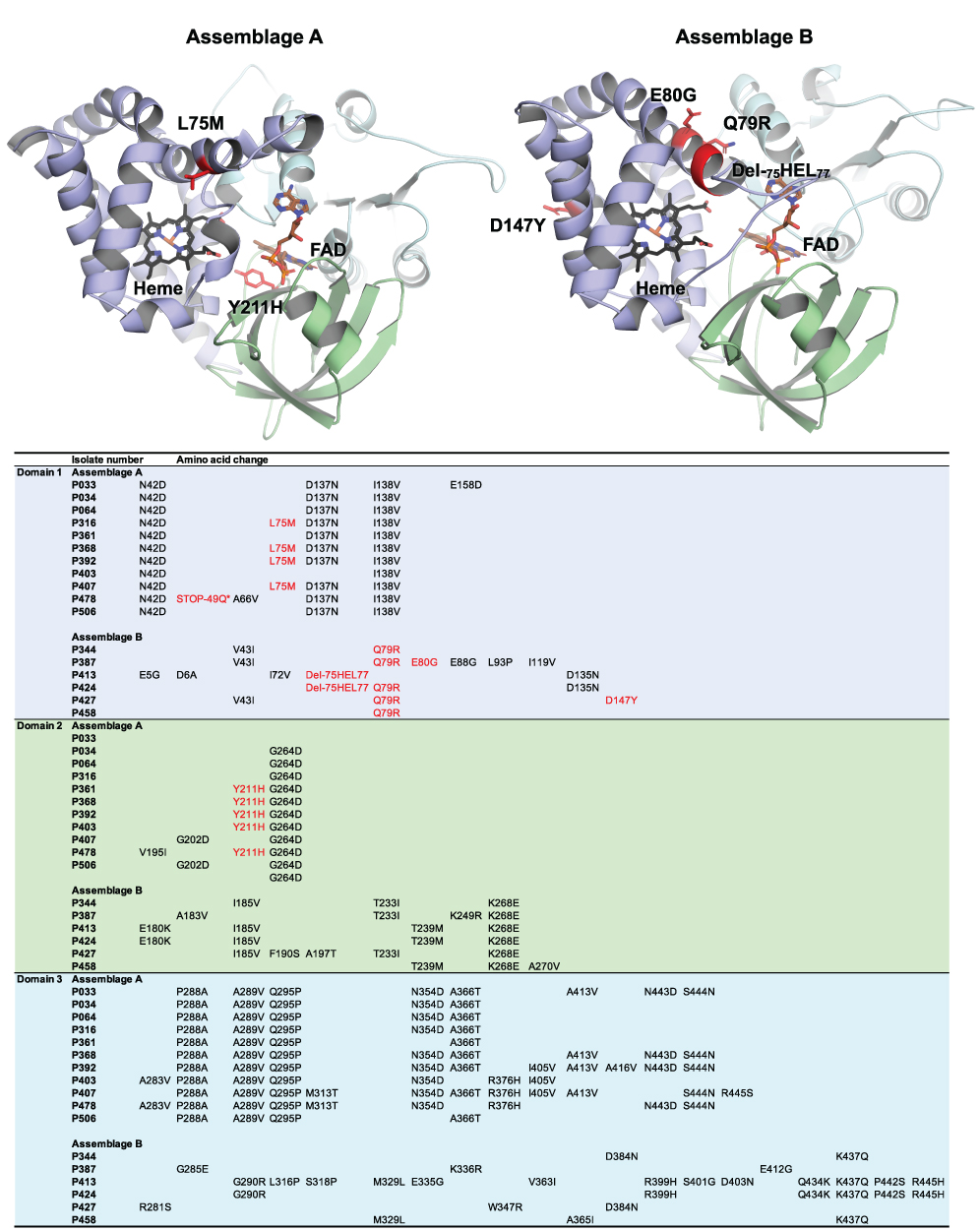 |
Figure 3 Cartoon representation of homology models created for gFlHb from assemblage A and B. Heme binding globing domain is presented in purple, FAD-binding domain in green and C-terminal FAD- binding domain in cyan. Heme and FAD were fitted to the homology model and are presented by stick representation. Detected nsSNV-induced mutations to the amino acid sequences for individual isolates are presented in the table. Mutations possibly affecting directly to protein function by disturbing the heme or FAD binding are indicated with red in both cartoon models and in the table. The mutation marked as STOP-49Q introduces a stop codon into the sequence interrupting the protein synthesis. |
Discussion
In this study, the allelic diversity of gFlHb was investigated through combined analyses of cloned sequences, genomic Illumina sequencing data, and de-novo assembled genomes derived from PacBio sequencing.
Sequencing Methods, PCR, Cloning and SNVs
Studies using a clone-based approach to identify SNPs in Giardia genes have been conducted earlier.37,38,41 In some of these studies a Taq-based polymerase was used for the PCR-amplification, and may have caused an inflated high diversity as the error rate of standard Taq is around 1 in every 3500 bp.53 However, some of the studies have used Q5 or other high-fidelity polymerases that have a lower error rate.36,39,40,54 When identifying allele sequences it is important to utilize high-fidelity polymerases to minimize the chance of introducing errors during amplification.40 The error rate of the Q5 polymerase used in this study has earlier been reported to be 1 per million bp.53 Still, some SNVs identified in the clones could potentially be caused by amplification or sequencing errors, specifically within reads in the beginning or end of the Sanger sequences. The 35 cycles used in the initial gene-PCR reactions could introduce amplification errors leading to false-positive SNVs in the clones. Validation of SNVs therefore becomes important. Indeed, identification of HC SNVs, and excluding LC SNVs from the analysis reduced the number of SNVs from an average of 23 to 21 in eleven sub-assemblage AII isolates and from 37 to 22 for six assemblage B isolates (table S1). We, therefore, consider the reported number of alleles and diversity as a conservative interpretation of the data. We could see that the majority of the SNVs identified in the clones were also identified in Illumina data for the same isolate. The clones had generally more unique SNVs than the number identified in Illumina data. However, for one isolate (P344), two different batches of DNA (trophozoites cultured an additional time to obtain enough DNA) were used for the Illumina sequencing data and the cloning experiments. Although derived by limiting dilution, P344 may represent a mixed isolate with variable contribution to, and dominance of, co-existing lineages between batches, and possibly introduction of new mutations. These could all be reasons why this isolate had the highest number of LC SNVs, and gFlHb alleles matching other alleles in the B assemblage isolates.
General Features of gFlHb Genetic Diversity
Our analysis of SNVs in the CDS and copy number variation of the two ortholog gFlHb genes, DHA2_154000 and GSB_151570 in Giardia sub-assemblage AII and assemblage B reference strains DH and GS, respectively, show a high degree of genetic variation (number of variable SNV positions per CDS length) for both assemblages compared to the set of 29 MTZ-metabolizing and other metabolism genes analyzed in a previous study.42 One interesting finding in the present study is that sub-assemblage AII isolates, shown to generally have less genetic variation than assemblage B, possessed a higher number of SNVs than assemblage B isolates in gFlHb.30,38,55,56 In addition, a relatively high allelic heterozygosity of the gFlHb gene coupled with nucleotide diversity values <0.01 was found in both assemblages. Other studies looking at nucleotide diversity in single copy genes glutamate dehydrogenase gene (gdh), beta-giardin (bg) and triosephosphate isomerase (tpi), that are often used for genotyping, have found similar nucleotide diversity ranging from 0.003 to 0.02.36,39 It is challenging to culture clinical isolates of Giardia, especially for assemblage B parasites.57,58 Giardia strains that are able to grow in culture could have an adapted metabolism of MTZ and detoxification processes of free radicals and O2 and may represent a selected version of the isolates causing disease in humans. The isolates in the present study could therefore be biased with regard to gFlHb copy numbers or be the reason for the low nucleotide diversity and many different, but similar, alleles of gFlHb. With this caveat in mind, the alleles and copy number variation identified in the present study will nevertheless enable future studies and analysis of gFIHb in non-culturable isolates, as it provides a start of a respective database of gene variants.
Alleles of the gFlHb Gene
In the present study, several alleles of the gFlHb gene were identified and PacBio sequencing confirmed that the number of copies of gFlHb in each isolate is variable. As several copies of gFlHb are sometimes found on the same contig, we use the term allele rather than the term haplotype which has been used in previous studies of Giardia single-copy gene variants.39,40,54 The phylogenetic trees in Figures 1 and 2 represent all the identified alleles in each isolate from sub-assemblages AII and assemblage B. Assemblage B isolates had less available samples and clones than AII, but had more unique alleles (41 vs 37). No sub-assemblage AII derived allele was found in assemblage B isolates, or vice versa, thus indicating no inter-assemblage recombination. However, it is difficult to define full tetraploid gFlHb genotypes with the number of clones available for each isolate, further complicated by the presence of copy number variation (CNV) which allows for more possible combinations of gFlHb alleles. Phylogenetic trees for each assemblage, on the other hand, are consistent with the occurrence of intra-assemblage recombination and genetic exchange although mixed isolate infections cannot completely be ruled out. Some alleles were common and found to be present in more than one isolate, while the same isolates also harbor several other, and different, alleles only occurring in one isolate (see tables S4 and S5). The high allelic diversity indicates that the overall diversity in the population may be much larger. As gFlHb is a variable-copy gene, the maximum number of alleles one tetraploid Giardia strain may harbor is four times the number of copies. In sub-assemblage AII isolates, higher copy numbers correlated with higher numbers of identified alleles but no breach to said rule was observed. In previous studies investigating haplotypes of single copy genes such as (bg, gdh and mlh, tpi), more than the expected maximum four haplotypes per paralog were encountered.36–39 At present, we cannot rule out that this could be due to unidentified additional copies of the gene hidden in the current versions of de novo assembled genomes. Also, mixed infections consisting of multiple Giardia strains as demonstrated by previous studies, cannot be discounted completely (see above).29,33,37–39,59 However, we favor the idea that supernumerary alleles reflect genome variations that have occurred during culture causing co-existence of daughter lineages as described by Choy et al.31 Remarkable genetic variation due to selective pressure during axenization has been recently described also for other parasites such as Leishmania donovani.60–62 In both assemblages, some of the identified alleles were not matching the identified PacBio copies. Minor alleles, representing 1:4 of the possible alleles in a tetraploid organism, are likely to be missed in consensus sequences as nucleotides of the major allele would be decisive. This, in addition to potential sequencing errors in combination with correction routines or assembly errors in the PacBio genomes might be the reason why not all copies are represented by the identified alleles.
Copy Number Variation
gFlHb is an important enzyme in the detoxification processes in Giardia, to eliminate the harmful effects of NO. One study showed that this was the sole enzyme that was upregulated in several different stress-exposures such as O2 and H2O2, although there was no correlation between transcription responses to H2O2 and MTZ.14 Our finding of a variable number of gFlHb gene copies may be important in relation to tackling the oxidative stress caused by MTZ treatment. A recent study by Müller et al, reported upregulated gFlHb protein levels in an MTZ and nitazoxanide resistant isolate.20 One can hypothesize that it is beneficial for the parasite to have more gFlHb gene copies, as it may promote survival in an environment with higher O2 levels, where activation of MTZ is lower, or in the oxidative stress condition induced by MTZ. Interestingly, a major antibiotic function of MTZ is to cause oxidative stress, and it is tempting to speculate whether gFlHb could be involved in neutralizing the harmful effects of the drug as Giardia lacks conventional antioxidant enzymes.63 In a previous study, the nitro-drug resistant line C4 of Giardia was compared to its corresponding wild-type WBC6 isolate with respect to mRNA expression, O2 consumption and functional assays.66 It was found that the C4 line exhibited lower nitroreductase activity, while mRNA levels of gFlHb were not significantly different before and after drug exposure while reduction of nitro-drug activation was found to be lower.66 In another study by Ansell et al 2017, strand-specific RNA analysis was carried out in three resistant laboratory lines.67 gFlHb was found to be strongly induced in one of the lines (713-r), and the authors suggested that gFlHb could play the role of an alternative MTZ detoxification enzyme.67 This may be assay dependent, but is more likely to be strain specific, especially as different Giardia isolates harbor a variable number of gFlHb copies. Taken together these findings indicate that different Giardia isolates could have different MTZ resistance formation strategies, ie, isolates with a higher gFlHb copy number may be more prone to use an active detoxifying MTZ resistance strategy, while other isolates may adopt a strategy of reduced MTZ activation.66 For higher eukaryotes, such as the insects Anopheles gambiae and Culex pipiens, increased CNV has been linked to insecticide resistance.64 Aneuploidy causing increased copy numbers of several genes in fungi has also been linked with drug resistance.65 Although several copies of the gFlHb gene in some isolates were found, we must acknowledge that we do not know how the copies, and their various alleles would be expressed or regulated in an MTZ exposed Giardia isolate in need of more potent free radical neutralization. Further studies combining single-cell DNA and RNA sequencing are needed to elucidate the effect of having multiple gFlHb copies on the ability to overcome MTZ toxicity or improve tolerance of oxidative stress. In the current reference genome of sub-assemblage AII there are two shorter paralogs of gFlHb (DHA2_152971 and DHA2_153995) that could potentially result in falsely higher coverage for the gFlHb gene. There is no reason to believe that these shorter fragments contribute to the higher coverage found in the Illumina sequence data, and this was shown in a previous study.42 Furthermore, the PacBio consensus sequences were used to determine the CNV in the gFlHb gene, and the shorter paralogs could not affect the results, as full-length genes were investigated.
Putative Effect of SNV-Induced Mutations on gFlHb Proteins
The gFlHb gene in both sub-assemblage AII and assemblage B was found to have a high number of nsSNVs that would alter the protein’s amino acid sequence. Some previous studies have also reported high numbers of nsSNPs in house-keeping genes, especially for assemblage B isolates, but these SNVs were commonly only causing conservative aa changes, unlikely to affect protein function or structure.37,40 The crystal structure of gFlHb protein is not yet known, and structural mapping of the location of the mutated amino acids was consequently based on homology models. The premature stop codon found in two alleles in sub-assemblage AII clones will cause a truncated protein without its normal function. Interpretation of the mutations that seem to be located on the surface of the protein is more challenging in terms of predicting the effect. Altering the surface charge could potentially have an effect on inter-molecular interactions. It is clear that further research is needed to investigate whether the aa changes can potentially affect the function of the respective protein variants. Specifically locating the exact site for NAD binding would help us investigate the effect of the mutations in the domain 3, the NAD binding domain. However, some of the mutations described might indeed affect heme and FAD binding, therefore altering protein function.
Limitations
For some of the isolates, few clones were obtained, and rare alleles may have been missed in some isolates. For isolate P064, one of the two gFlHb copies found in the PacBio consensus sequence, was not found among the clones obtained in this study, showing that several more alleles may exist for this isolate. One other explanation for missing alleles, is that the PCR primers targeting gFlHb in the initial amplification may not have matched all alleles and did not bind to the template DNA and amplify these. However, PacBio consensus copies may not be identical to the clones due to sequencing errors, or due to PacBio sequence correction routines. The study population in this study is rather small and future studies of non-cultured isolates are likely to extend information about the number of copies and genetic variation of the gene. The SNVs analyzed in the present study were all limited to the CDS of the gFlHb of assemblage A and B isolates. There have been previous studies linking SNPs in intergenic areas to MTZ resistance in T. vaginalis, specifically 12 SNPs, that potentially could be markers of resistance.68 SNVs in the up- or downstream regions of the gFlHb CDS could affect the transcription and therefore be of potential interest for investigating MTZ susceptibility and oxidative stress responses markers in future studies.
Conclusion
In this study, we show evidence that the gFlHb gene in Giardia sub-assemblage AII and assemblage B, is a variable copy number gene with a high allelic diversity. The high genetic diversity seems to be due to both high copy number variation, many similar but unique, alleles per isolate, and a high number of SNV positions among both Giardia sub-assemblage AII and assemblage B isolates. Relatively high percentages of nonsynonymous SNVs were identified in both assemblages and some changes could potentially affect the protein function. The variable copy number nature of the gFlHb gene may allow some Giardia strains to better adapt to nitrosative and oxidative stress and could thereby potentially play a role in MTZ susceptibility.
Abbreviations
CDS, coding region; gFlHb, Giardia lamblia Flavohemoprotein; HC, High Confidence; LC, Low confidence; MTZ, Metronidazole; ns, nonsynonymous; SNV, single nucleotide polymorphism; s, synonymous.
Data Sharing Statement
The sequences for the identified gFlHb alleles are available in NCBI genbank with accession numbers: MT713149-MT713229, MT713231-MT713243, MT713245-MT713263, MT713267, MT713269.
Ethics Statement
No patient data was collected and used in the present study. Any link between individual parasite data and patient information had been removed before WGS of trophozoites and cloning experiments were carried out.
Acknowledgments
The personnel at the Genomics Core Facility at Oslo University Hospital (oslo.genomics.no) should be acknowledged due to their work with the Illumina whole-genome sequencing of Giardia DNA. We would also like to thank the personnel at the Department of Clinical Science, University of Bergen, for providing technical assistance with the sequencing and assembly of genome data. We acknowledge Prof Dr. Ralf Ignatius from Institute of Tropical Medicine and International Health, Charité, Berlin, Germany for kindly providing biological samples. We would also like to thank Simone Caccio for support in the de-novo PacBio sequencing of the Giardia isolates. We would also like to thank Petra Gosten-Heinrich, Robert Koch-Institute Berlin for providing excellent technical assistance during culturing experiments and sequencing clones of gFIHb.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study has been funded by Norwegian Surveillance System for Antimicrobial Drug Resistance (NORM), Centre for Pharmacy, University of Bergen, Helse-Vest (grant number 912245), and Norwegian PhD School of Pharmacy (NFIF). All of the experiments, data analysis and evaluation of the results in the present study were performed independently by the authors, without any interference from any of the funding institutions.
Disclosure
Some of the data in this paper has been published as an abstract and was presented at the VIIth International Giardia and Cryptosporidium Conference (IGCC), 2019, as a conference talk with interim findings. The abstract was published in the “Conference abstracts on USB Key” and is available from [https://en.rouentourisme.com/wp-content/uploads/2019/06/Programme-d%C3%A9finitif-modifi%C3%A9-200619.pdf].
The authors report no conflicts of interest for this work.
References
1. Lane S, Lloyd D. Current trends in research into the waterborne parasite Giardia. Crit Rev Microbiol. 2002;28(2):123–147. doi:10.1080/1040-840291046713
2. Berkman DS, Lescano AG, Gilman RH, Lopez SL, Black MM. Effects of stunting, diarrhoeal disease, and parasitic infection during infancy on cognition in late childhood: a follow-up study. Lancet. 2002;359(9306):564–571. doi:10.1016/S0140-6736(02)07744-9
3. Rogawski ET, Platts-Mills JA, Seidman JC, et al. Early antibiotic exposure in low-resource settings is associated with increased weight in the first two years of life. J Pediatr Gastroenterol Nutr. 2017;65(3):350–356. doi:10.1097/MPG.0000000000001640
4. Craun GF, Calderon RL, Craun MF. Outbreaks associated with recreational water in the United States. Int J Environ Health Res. 2005;15(4):243–262. doi:10.1080/09603120500155716
5. Farthing MJ. Giardiasis. Gastroenterol Clin North Am. 1996;25(3):493–515. doi:10.1016/S0889-8553(05)70260-0
6. Gardner TB, Hill DR. Treatment of giardiasis. Clin Microbiol Rev. 2001;14(1):114–128.
7. Munoz Gutierrez J, Aldasoro E, Requena A, et al. Refractory giardiasis in Spanish travellers. Travel Med Infect Dis. 2013;11(2):126–129. doi:10.1016/j.tmaid.2012.10.004
8. Requena-Mendez A, Goni P, Rubio E, et al. The use of quinacrine in nitroimidazole-resistant giardia duodenalis: an old drug for an emerging problem. J Infect Dis. 2017;215(6):946–953. doi:10.1093/infdis/jix066
9. Nabarro LE, Lever RA, Armstrong M, Chiodini PL. Increased incidence of nitroimidazole-refractory giardiasis at the hospital for tropical diseases, London: 2008–2013. Clin Microbiol Infect. 2015;21(8):791–796. doi:10.1016/j.cmi.2015.04.019
10. Dingsdag SA, Hunter N. Metronidazole: an update on metabolism, structure-cytotoxicity and resistance mechanisms. J Antimicrob Chemother. 2018;73(2):265–279. doi:10.1093/jac/dkx351
11. Leitsch D. A review on metronidazole: an old warhorse in antimicrobial chemotherapy. Parasitology. 2017;1–12.
12. Church DL, Laishley EJ. Reduction of metronidazole by hydrogenase from clostridia. Anaerobe. 1995;1(2):81–92. doi:10.1006/anae.1995.1002
13. Mastronicola D, Falabella M, Forte E, Testa F, Sarti P, Giuffre A. Antioxidant defence systems in the protozoan pathogen Giardia intestinalis. Mol Biochem Parasitol. 2016;206(1–2):56–66. doi:10.1016/j.molbiopara.2015.12.002
14. Ma’ayeh SY, Knorr L, Svard SG. Transcriptional profiling of Giardia intestinalis in response to oxidative stress. Int J Parasitol. 2015;45(14):925–938. doi:10.1016/j.ijpara.2015.07.005
15. Mastronicola D, Testa F, Forte E, et al. Flavohemoglobin and nitric oxide detoxification in the human protozoan parasite Giardia intestinalis. Biochem Biophys Res Commun. 2010;399(4):654–658. doi:10.1016/j.bbrc.2010.07.137
16. Gardner PR, Gardner AM, Martin LA, Salzman AL. Nitric oxide dioxygenase: an enzymic function for flavohemoglobin. Proc Natl Acad Sci U S A. 1998;95(18):10378–10383.
17. Gardner PR. Nitric oxide dioxygenase function and mechanism of flavohemoglobin, hemoglobin, myoglobin and their associated reductases. J Inorg Biochem. 2005;99(1):247–266. doi:10.1016/j.jinorgbio.2004.10.003
18. Rafferty S, Luu B, March RE, Yee J. Giardia lamblia encodes a functional flavohemoglobin. Biochem Biophys Res Commun. 2010;399(3):347–351. doi:10.1016/j.bbrc.2010.07.073
19. Ansell BR, McConville MJ, Baker L, et al. Divergent transcriptional responses to physiological and xenobiotic stress in giardia duodenalis. Antimicrob Agents Chemother. 2016;60(10):6034–6045. doi:10.1128/AAC.00977-16
20. Müller J, Braga S, Heller M, Müller N. Resistance formation to nitro drugs in Giardia lamblia: no common markers identified by comparative proteomics. Int J Parasitol Drugs Drug Resist. 2019;9:112–119. doi:10.1016/j.ijpddr.2019.03.002
21. Rasoloson D, Tomkova E, Cammack R, Kulda J, Tachezy J. Metronidazole-resistant strains of Trichomonas vaginalis display increased susceptibility to oxygen. Parasitology. 2001;123(\(Pt1)):45–56. doi:10.1017/S0031182001008022
22. Ansell BR, McConville MJ, Ma’ayeh SY, et al. Drug resistance in Giardia duodenalis. Biotechnol Adv. 2015;33(6 Pt 1):888–901. doi:10.1016/j.biotechadv.2015.04.009
23. Forrester MT, Foster MW. Protection from nitrosative stress: a central role for microbial flavohemoglobin. Free Radic Biol Med. 2012;52(9):1620–1633. doi:10.1016/j.freeradbiomed.2012.01.028
24. Helmick RA, Fletcher AE, Gardner AM, et al. Imidazole antibiotics inhibit the nitric oxide dioxygenase function of microbial flavohemoglobin. Antimicrob Agents Chemother. 2005;49(5):1837–1843. doi:10.1128/AAC.49.5.1837-1843.2005
25. Yu LZ, Birky CW
26. Bernander R, Palm JE, Svard SG. Genome ploidy in different stages of the Giardia lamblia life cycle. Cell Microbiol. 2001;3(1):55–62. doi:10.1046/j.1462-5822.2001.00094.x
27. Kabnick KS, Peattie DA. In situ analyses reveal that the two nuclei of Giardia lamblia are equivalent. J Cell Sci. 1990;95(Pt 3):353–360.
28. Tumova P, Uzlikova M, Jurczyk T, Nohynkova E. Constitutive aneuploidy and genomic instability in the single-celled eukaryote Giardia intestinalis. MicrobiologyOpen. 2016;5(4):560–574. doi:10.1002/mbo3.351
29. Ankarklev J, Svard SG, Lebbad M. Allelic sequence heterozygosity in single Giardia parasites. BMC Microbiol. 2012;12:65. doi:10.1186/1471-2180-12-65
30. Franzen O, Jerlstrom-Hultqvist J, Castro E, et al. Draft genome sequencing of giardia intestinalis assemblage B isolate GS: is human giardiasis caused by two different species? PLoS Pathog. 2009;5(8):e1000560.
31. Choy SH, Mahdy MA, Al-Mekhlafi HM, Low VL, Surin J. Population expansion and gene flow in Giardia duodenalis as revealed by triosephosphate isomerase gene. Parasit Vectors. 2015;8:454. doi:10.1186/s13071-015-1084-y
32. Caccio SM, Ryan U. Molecular epidemiology of giardiasis. Mol Biochem Parasitol. 2008;160(2):75–80. doi:10.1016/j.molbiopara.2008.04.006
33. Hussein AI, Yamaguchi T, Nakamoto K, Iseki M, Tokoro M. Multiple-subgenotype infections of Giardia intestinalis detected in Palestinian clinical cases using a subcloning approach. Parasitol Int. 2009;58(3):258–262.
34. Lalle M, Pozio E, Capelli G, Bruschi F, Crotti D, Cacciò SM. Genetic heterogeneity at the beta-giardin locus among human and animal isolates of Giardiaduodenalis and identification of potentially zoonotic subgenotypes. Int J Parasitol. 2005;35(2):207–213. doi:10.1016/j.ijpara.2004.10.022
35. Gabin-Garcia LB, Bartolome C, Abal-Fabeiro JL, Mendez S, Llovo J, Maside X. Strong genetic structure revealed by multilocus patterns of variation in Giardia duodenalis isolates of patients from Galicia (NW-Iberian Peninsula). Infect Genet Evol. 2017;48:131–141. doi:10.1016/j.meegid.2016.12.014
36. Kosuwin R, Putaporntip C, Pattanawong U, Jongwutiwes S. Clonal diversity in Giardia duodenalis isolates from Thailand: evidences for intragenic recombination and purifying selection at the beta giardin locus. Gene. 2010;449(1–2):1–8. doi:10.1016/j.gene.2009.09.010
37. Siripattanapipong S, Leelayoova S, Mungthin M, et al. Clonal diversity of the glutamate dehydrogenase gene in Giardia duodenalis from Thai isolates: evidence of genetic exchange or mixed infections? BMC Microbiol. 2011;11:206. doi:10.1186/1471-2180-11-206
38. Lasek-Nesselquist E, Welch DM, Thompson RC, Steuart RF, Sogin ML. Genetic exchange within and between assemblages of Giardia duodenalis. J Eukaryot Microbiol. 2009;56(6):504–518. doi:10.1111/j.1550-7408.2009.00443.x
39. Mizuno T, Matey EJ, Bi X, Songok EM, Ichimura H, Tokoro M. Extremely diversified haplotypes observed among assemblage B population of Giardia intestinalis in Kenya. Parasitol Int. 2020;75:102038. doi:10.1016/j.parint.2019.102038
40. Lecova L, Tumova P, Nohynkova E. Clone-based haplotyping of Giardia intestinalis assemblage B human isolates. Parasitol Res. 2019;118(1):355–361. doi:10.1007/s00436-018-6161-7
41. Aguiar JM, Silva SO, Santos VA, et al. Evidence of heterozygosity and recombinant alleles in single cysts of Giardia duodenalis. Revista Brasileira De Parasitologia Veterinaria. 2016;25(2):187–195. doi:10.1590/S1984-29612016031
42. Saghaug CS, Klotz C, Kallio JP, et al. Genetic variation in metronidazole metabolism and oxidative stress pathways in clinical Giardia lamblia assemblage A and B isolates. Infect Drug Resist. 2019;12:1221–1235. doi:10.2147/IDR.S177997
43. Keister DB. Axenic culture of Giardia lamblia in TYI-S-33 medium supplemented with bile. Trans R Soc Trop Med Hyg. 1983;77(4):487–488. doi:10.1016/0035-9203(83)90120-7
44. Aurrecoechea C, Brestelli J, Brunk BP, et al. GiardiaDB and TrichDB: integrated genomic resources for the eukaryotic protist pathogens Giardia lamblia and Trichomonas vaginalis. Nucleic Acids Res. 2009;37(Databaseissue):D526–530. doi:10.1093/nar/gkn631
45. Inoue H, Nojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96(1):23–28. doi:10.1016/0378-1119(90)90336-P
46. Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34(12):3299–3302. doi:10.1093/molbev/msx248
47. Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19(18):2496–2497. doi:10.1093/bioinformatics/btg359
48. Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. doi:10.1038/nprot.2015.053
49. Ilari A, Bonamore A, Farina A, Johnson KA, Boffi A. The X-ray structure of ferric Escherichia coli flavohemoglobin reveals an unexpected geometry of the distal heme pocket. J Biol Chem. 2002;277(26):23725–23732. doi:10.1074/jbc.M202228200
50. Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185(4154):862–864. doi:10.1126/science.185.4154.862
51. Ermler U, Siddiqui RA, Cramm R, Friedrich B. Crystal structure of the flavohemoglobin from Alcaligenes eutrophus at 1.75 A resolution. EMBO J. 1995;14(24):6067–6077. doi:10.1002/j.1460-2075.1995.tb00297.x
52. El Hammi E, Warkentin E, Demmer U, Marzouki NM, Ermler U, Baciou L. Active site analysis of yeast flavohemoglobin based on its structure with a small ligand or econazole. FEBS J. 2012;279(24):4565–4575. doi:10.1111/febs.12043
53. Potapov V, Ong JL. Examining sources of error in PCR by single-molecule sequencing. PLoS One. 2017;12(1):e0169774. doi:10.1371/journal.pone.0169774
54. Teodorovic S, Braverman JM, Elmendorf HG. Unusually low levels of genetic variation among Giardia lamblia isolates. Eukaryotic Cell. 2007;6(8):1421–1430. doi:10.1128/EC.00138-07
55. Adam RD, Dahlstrom EW, Martens CA, et al. Genome sequencing of Giardia lamblia genotypes A2 and B isolates (DH and GS) and comparative analysis with the genomes of genotypes A1 and E (WB and Pig). Genome Biol Evol. 2013;5(12):2498–2511. doi:10.1093/gbe/evt197
56. Morrison HG, McArthur AG, Gillin FD, et al. Genomic minimalism in the early diverging intestinal parasite Giardia lamblia. Science. 2007;317(5846):1921–1926. doi:10.1126/science.1143837
57. Benere E, Geurden T, Robertson L, Van Assche T, Cos P, Maes L. Infectivity of Giardia duodenalis Assemblages A and E for the gerbil and axenisation of duodenal trophozoites. Parasitol Int. 2010;59(4):634–637. doi:10.1016/j.parint.2010.08.001
58. Binz N, Thompson RC, Lymbery AJ, Hobbs RP. Comparative studies on the growth dynamics of two genetically distinct isolates of Giardia duodenalis in vitro. Int J Parasitol. 1992;22(2):195–202. doi:10.1016/0020-7519(92)90101-P
59. Caccio SM, Sprong H. Giardia duodenalis: genetic recombination and its implications for taxonomy and molecular epidemiology. Exp Parasitol. 2010;124(1):107–112. doi:10.1016/j.exppara.2009.02.007
60. Bussotti G, Gouzelou E, Côrtes Boité M, et al. Leishmania genome dynamics during environmental adaptation reveal strain-specific differences in gene copy number variation, karyotype instability, and telomeric amplification. mBio. 2018;9:6. doi:10.1128/mBio.01399-18
61. Imamura H, Downing T, Van den Broeck F, et al. Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. Elife. 2016;5.
62. Domagalska MA, Imamura H, Sanders M, et al. Genomes of Leishmania parasites directly sequenced from patients with visceral leishmaniasis in the Indian subcontinent. PLoS Negl Trop Dis. 2019;13(12):e0007900. doi:10.1371/journal.pntd.0007900
63. Argüello-García R, Leitsch D, Skinner-Adams T, Ortega-Pierres MG. Drug resistance in giardia: mechanisms and alternative treatments for giardiasis. Adv Parasitol. 2020;107:201–282.
64. Weetman D, Djogbenou LS, Lucas E. Copy number variation (CNV) and insecticide resistance in mosquitoes: evolving knowledge or an evolving problem? Current Opinion Insect Sci. 2018;27:82–88. doi:10.1016/j.cois.2018.04.005
65. Wertheimer NB, Stone N, Berman J. Ploidy dynamics and evolvability in fungi. Philos Trans R Soc Lond B Biol Sci. 2016;371:1709.
66. Müller J, Hemphill A, Müller N. Physiological aspects of nitro drug resistance in Giardia lamblia. Int J Parasitol Drugs Drug Resist. 2018;8(2):271–277. doi:10.1016/j.ijpddr.2018.04.008
67. Ansell BR, Baker L, Emery SJ, et al. Transcriptomics indicates active and passive metronidazole resistance mechanisms in three seminal giardia lines. Front Microbiol. 2017;8:398. doi:10.3389/fmicb.2017.00398
68. Bradic M, Warring SD, Tooley GE, et al. Genetic indicators of drug resistance in the highly repetitive genome of trichomonas vaginalis. Genome Biol Evol. 2017;9(6):1658–1672. doi:10.1093/gbe/evx110
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.