")
Back to Journals » The Application of Clinical Genetics » Volume 14
Genetic and Proteomic Contributions to the Pathophysiology of Moyamoya Angiopathy and Related Vascular Diseases
Authors Dorschel KB , Wanebo JE
Received 20 March 2020
Accepted for publication 26 December 2020
Published 18 March 2021 Volume 2021:14 Pages 145—171
DOI https://doi.org/10.2147/TACG.S252736
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Kirsten B Dorschel,1,* John E Wanebo2,3,*
1Heidelberg University Medical School, Ruprecht-Karls-Universität Heidelberg, Heidelberg, Germany; 2Department of Neurosurgery, Barrow Neurological Institute, Phoenix, Arizona, USA; 3Department of Neuroscience, HonorHealth Research Institute, Scottsdale, AZ, USA
*These authors contributed equally to this work
Correspondence: John E Wanebo
c/o Neuroscience Publications; Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center, 350 W. Thomas Road, Phoenix, AZ, 85013, USA
Tel +1 602 406 3593
Fax +1 602 406 4104
Email [email protected]
Rationale: This literature review describes the pathophysiological mechanisms of the current classes of proteins, cells, genes, and signaling pathways relevant to moyamoya angiopathy (MA), along with future research directions and implementation of current knowledge in clinical practice.
Objective: This article is intended for physicians diagnosing, treating, and researching MA.
Methods and Results: References were identified using a PubMed/Medline systematic computerized search of the medical literature from January 1, 1957, through August 4, 2020, conducted by the authors, using the key words and various combinations of the key words “moyamoya disease,” “moyamoya syndrome,” “biomarker,” “proteome,” “genetics,” “stroke,” “angiogenesis,” “cerebral arteriopathy,” “pathophysiology,” and “etiology.” Relevant articles and supplemental basic science articles published in English were included. Intimal hyperplasia, medial thinning, irregular elastic lamina, and creation of moyamoya vessels are the end pathologies of many distinct molecular and genetic processes. Currently, 8 primary classes of proteins are implicated in the pathophysiology of MA: gene-mutation products, enzymes, growth factors, transcription factors, adhesion molecules, inflammatory/coagulation peptides, immune-related factors, and novel biomarker candidate proteins. We anticipate that this article will need to be updated in 5 years.
Conclusion: It is increasingly apparent that MA encompasses a variety of distinct pathophysiologic conditions. Continued research into biomarkers, genetics, and signaling pathways associated with MA will improve and refine our understanding of moyamoya’s complex pathophysiology. Future efforts will benefit from multicenter studies, family-based analyses, comparative trials, and close collaboration between the clinical setting and laboratory research.
Keywords: genetics, moyamoya, pathophysiology, proteomics, stroke
Introduction
Moyamoya angiopathy (MA) is a chronic, progressive, occlusive arteriopathy of the intracranial circulation that predominantly affects the major branches of the internal carotid artery (ICA); it is associated with dilated distal basal collateral arteries, reflecting aberrant angiogenesis, arteriogenesis, and vasculogenesis.1–5 Adults with MA typically present with ischemic (50–75%) or hemorrhagic (10–40%) stroke, whereas children may also present with transient ischemic attacks (including drop attacks).1,3,6–8 In 2013, Hishikawa et al reported that 9% of MA patients experience a type of stroke involving both ischemia and hemorrhage.9,10 In hemorrhagic MA, recurrent hemorrhages were found to appear with an average annual incidence of 4.5% and are associated with increased mortality; smoking and hypertension are modifiable risk factors.11 About 20% of MA patients, including pediatric and younger MA patients, may exhibit headaches of vascular origin with migrainous features; a decrease in cerebral blood flow or cerebrovascular reserve and spreading cortical depression are possible pathophysiological mechanisms for these features.9,12 Chorea, choreoathetosis, dyskinesia, dystonia, limb-shaking, epilepsia-partialis continua, and optic disk enlargement with concomitant retinovascular anomalies are observed in MA patients, with chorea being the characteristic movement pattern.1,9 Ischemia of the basal ganglia-thalamocortical circuits, increased sex hormones, and hyperthyroidism are possible pathophysiological mechanisms for these characteristics.9
MA was first reported in 195713 and initially defined according to radiographic criteria in 1969.14 It has since become increasingly evident that the angiographic findings of moyamoya represent a variety of distinct pathophysiological processes, manifesting a shared radiographic appearance.2 Accordingly, by definition, MA refers to an isolated bilateral arteriopathy. Moyamoya syndrome (MMS) is either a unilateral arteriopathy or an arteriopathy with an associated medical disorder.2 MMS encompasses a heterogeneous group of arteriopathies with variable penetrance of the cerebrovascular phenotype and various modes of inheritance.4 When the term moyamoya is used alone, independently of the cause, it refers exclusively to distinctive angiographic findings.1 By convention, the arteriopathies are collectively referred to as moyamoya disease or moyamoya angiopathy (the term used in this paper).2
Ideally, biomarkers should unequivocally identify the presence or absence of a disease with a high degree of sensitivity and specificity. A genetic mutation reproducibly linked to a distinct disease phenotype is an essential discovery in biomedical research. The association of specific genes with MA is complex and likely due to the heterogeneity of disorders that share a common phenotype.1,2 The initial efforts to elucidate the genetic markers of MA reflected this complexity, with a wide range of genes, chromosomes, and hereditary diseases reported to be potential markers (Table 1).1–6,9,15–119 The current literature on familial MA suggests a low penetrance, autosomal dominant inheritance at loci on chromosomes 3p24.2–p26, 6q25, 8q23, 10q23.31, 12p12, and 17q25 (Table 1).6,7,16,40,51,52,68,73,93 Polymorphisms of PDGFRB, MMP3, TIMP2, RNF213, TGFB1, RPTOR, and NOS3 genes may be associated with MA (Table 1).48 (See the Supplemental Data for definitions of gene symbols, proteins, and additional terminology.) Meyer et al120 reported a 2-year-old girl with MMS, pseudoxanthoma elasticum, and an ABCC6 (16p13.11) mutation.
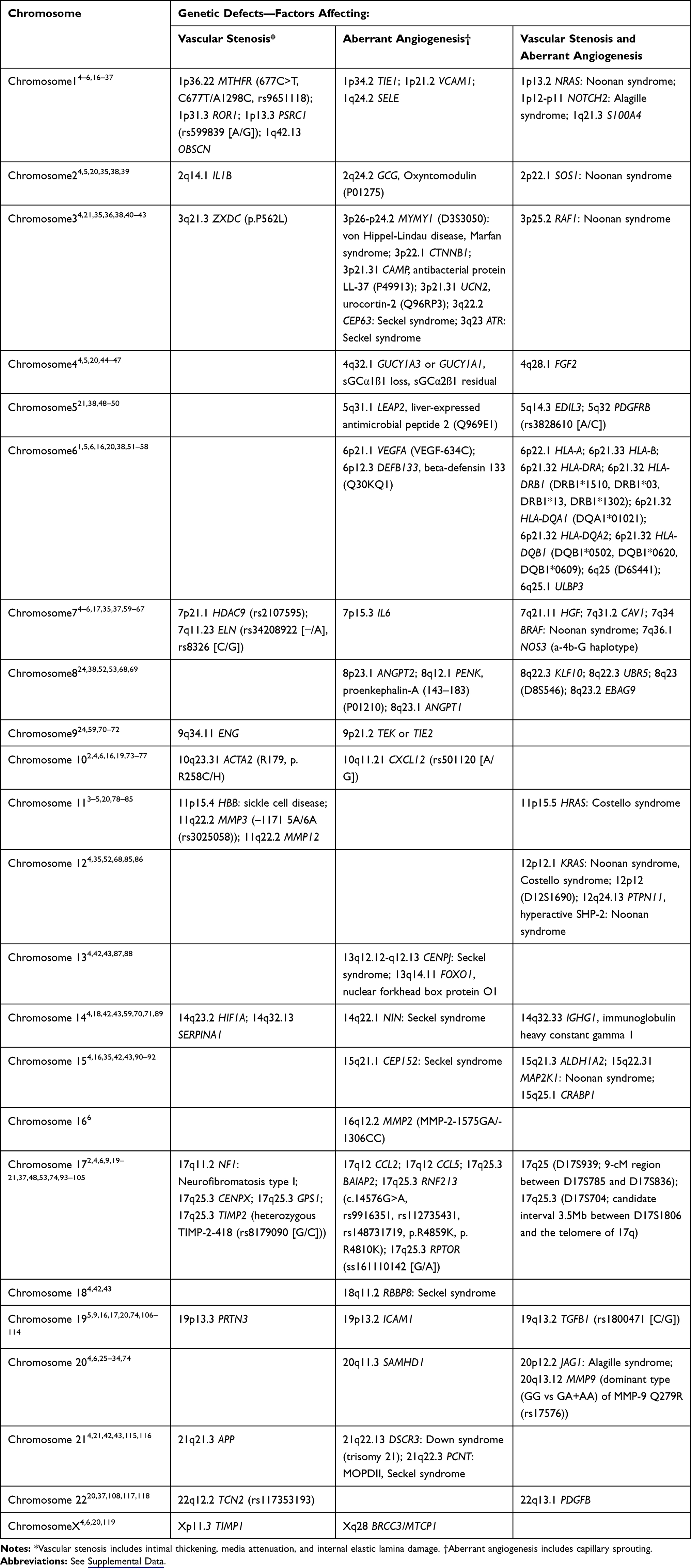 |
Table 1 Chromosomes and Genetic Defects Related to Moyamoya Angiopathy |
Whether a putative biomarker is uniquely causative of MA or is a secondary by-product of the arteriopathy can be difficult to ascertain.2,16 Two major approaches to biomarker discovery have been used. In hypothesis-driven discovery, specific molecules are selected a priori because of suspected roles in pathophysiological processes. In proteomic screening, the entire proteome of specimens from MA patients is compared to matched controls for differences in expression.2 In searching for candidate biomarkers for MA, both approaches have been applied. Proteome analysis of the cerebrospinal fluid (CSF) has been used by many research groups worldwide.2 Categorizing subgroups of molecules by their function is an additional method of organizing biomarker discovery. The broad classes of proteins linked to MA as a result of hypothesis-driven biomarker discovery include enzymes, growth factors, transcription factors, adhesion molecules, inflammation and coagulation peptides, immune-related factors, biomarker candidates, and gene mutation products (Table 2).2,5,6,9,15–24,38,39,45–47,49,50,52,54–56,59–71,76–79,87–92,106,108–115,117,118,121–149 Houkin et al150 proposed a double-hit hypothesis in which, in addition to the RNF213 mutation, environmental factors such as inflammation and/or infection, autoimmune disease, ischemia, and hemodynamic stress are essential in inducing the first step of MA pathophysiology (Figure 1).
 |
Table 2 Molecular Biomarkers of Moyamoya Angiopathy by Protein Class |
 |
Figure 1 Potential disease mechanisms in MA. Red indicates that the factor affects vascular stenosis (intimal thickening, media attenuation, and internal elastic lamina damage). Blue indicates that the factor affects aberrant angiogenesis (capillary sprouting). Green indicates that the factor affects both vascular stenosis and aberrant angiogenesis. Abbreviations: See Supplemental Data. Notes: Used with permission from Barrow Neurological Institute, Phoenix, AZ, USA. |
Heterogeneity in clinical presentation, progression, and response to therapy has encouraged research to define MA more precisely and focus on developing techniques to improve diagnosis and prognosis.2 Identifying genes, proteins, cells, and signaling pathways involved in MA may provide insights into pathophysiological models and allow the discovery of potential candidates for targeted treatment strategies (Figure 1–4 and Table 1).2,4
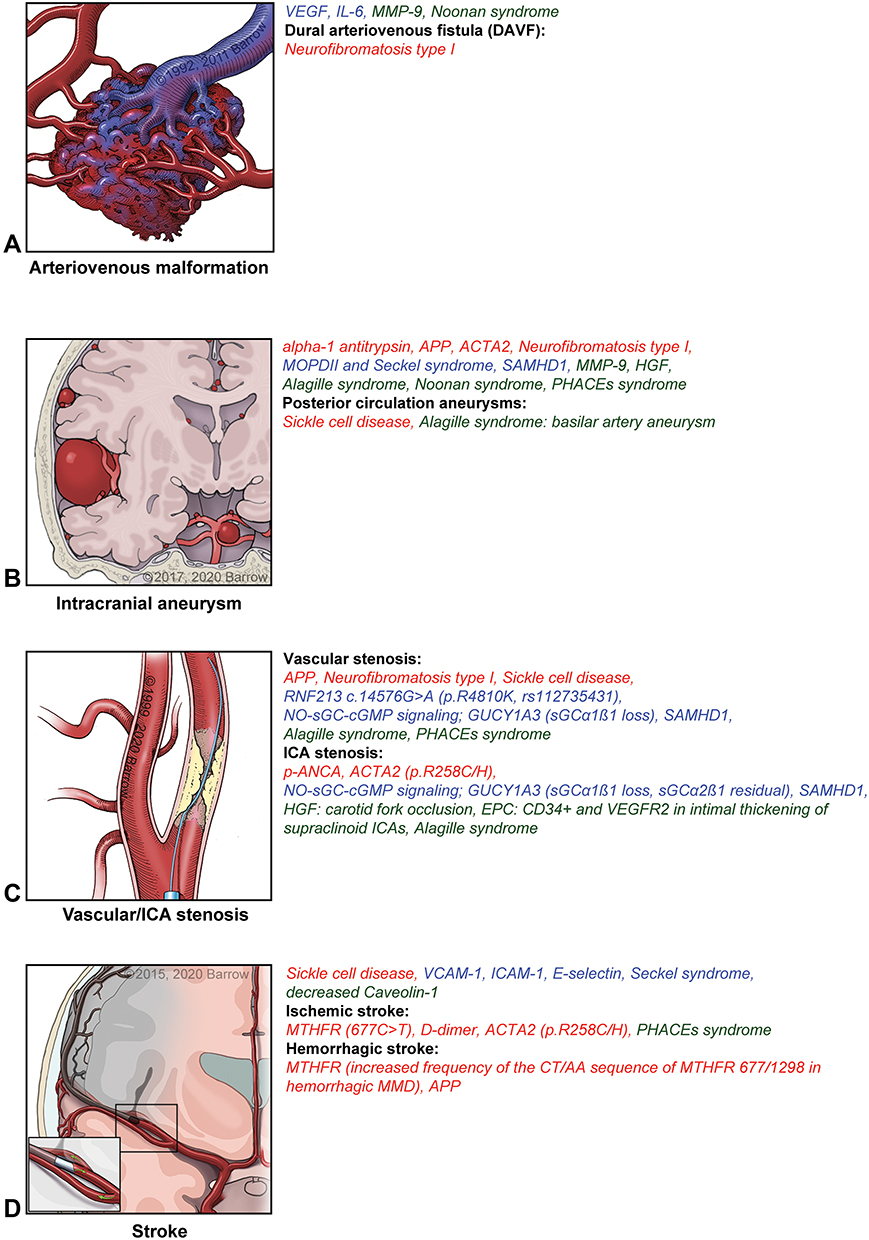 |
Figure 2 Proteins, cells, genes, and signaling pathways related to cerebral angiopathy characteristics in MA and associated disorders. (A) Arteriovenous malformation. (B) Intracranial aneurysm. (C) Vascular/internal carotid artery stenosis. (D) Stroke. Red indicates that the factor affects vascular stenosis (intimal thickening, media attenuation, and internal elastic lamina damage). Blue indicates that the factor affects aberrant angiogenesis (capillary sprouting). Green indicates that the factor affects both vascular stenosis and aberrant angiogenesis. Abbreviations: See Supplemental Data. Notes: Used with permission from Barrow Neurological Institute, Phoenix, AZ, USA. |
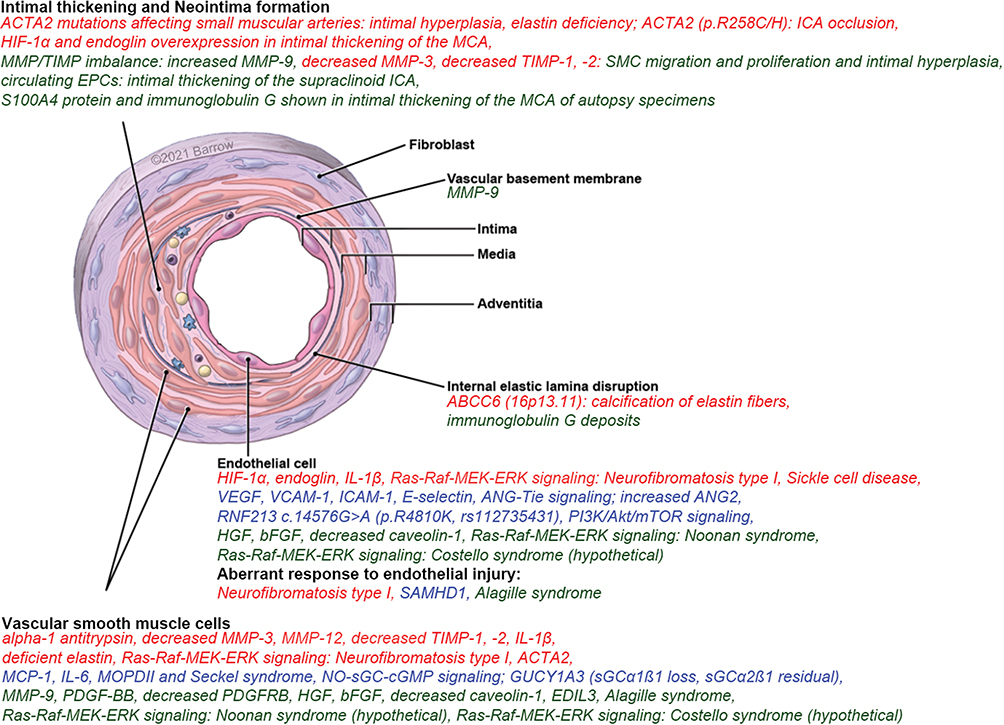 |
Figure 3 Schematic representation of an artery depicting macroscopic active sites of proteins, signaling pathways, genes, and cells in MA and associated disorders. Red indicates that the factor affects vascular stenosis (intimal thickening, media attenuation, and internal elastic lamina damage). Blue indicates that the factor affects aberrant angiogenesis (capillary sprouting). Green indicates that the factor affects both vascular stenosis and aberrant angiogenesis. Abbreviations: See Supplemental Data. Notes: Used with permission from Barrow Neurological Institute, Phoenix, AZ, USA. |
 |
Figure 4 Microscopic active sites of proteins, cells, genes, and signaling pathways in MA and associated disorders. (A) Bone marrow. (B) Blood/immune system. (C) Cerebrospinal fluid. Red indicates that the factor affects vascular stenosis (intimal thickening, media attenuation, and internal elastic lamina damage). Blue indicates that the factor affects aberrant angiogenesis (capillary sprouting). Green indicates that the factor affects both vascular stenosis and aberrant angiogenesis. Abbreviations: See Supplemental Data. Notes: Used with permission from Barrow Neurological Institute, Phoenix, AZ, USA. |
Methods
References were identified using a systematic computerized search of the medical literature from January 1, 1957, through August 4, 2020, conducted by the authors, using the PubMed, EMBASE, BIOSIS Previews, and Medline databases and various combinations of the key words “moyamoya disease,” “moyamoya syndrome,” “genetics,” “proteomics,” “biomarker,” “stroke,” “angiogenesis,” “cerebral arteriopathy,” “pathophysiology,” and “etiology.” Relevant articles on MA and supplemental basic science articles published in English were included. Reference lists of relevant articles and supplemental articles were reviewed for additional sources, and 206 articles were included in the final manuscript. The review reports by Smith2 and by Guey et al4 served as substantial references to this manuscript. Biomarkers involved in the pathophysiology of MA may be classified as genetic biomarkers of MA and molecular biomarkers of MA.2 In this literature review, each biomarker has been described and specified according to its relation to MA. As some aspects of MA have been studied more thoroughly than others, some topics receive more attention than others. Despite substantial progress in the study of MA in recent years, much of the literature remains descriptive. Continued basic and clinical research is essential if we are to better understand the etiology of MA, which may result in a further differentiated diagnosis and treatment of patients with MA.
Pathology
MA is related to angioarchitecture and hemodynamics; it involves continuous vascular remodeling, including vascular stenosis and aberrant angiogenesis.2,9 Different pathogenetic mechanisms may underlie pathologic vessel changes in MA, and mounting evidence indicates that MA is an acquired proliferative disease of the intima.16,151,152
In MA, progressive stenosis involves the supraclinoid ICA, distal ICA bifurcation (C1), and, less frequently, the proximal anterior cerebral artery (ACA) (A1), middle cerebral artery (MCA) sphenoidal segment (M1), the posterior cerebral artery (PCA), and rarely the vertebral artery, posterior inferior cerebellar artery (PICA), and proximal basilar artery (BA).1–3 A significant correlation between the anterior and posterior circulation is present in MA. Hishikawa et al9,153 reported that MA stenotic and occlusive ICA lesions ipsilateral to PCA lesions might be significantly advanced compared to ICA lesions ipsilateral to PCAs without lesions. An early and delayed form of interaction between the anterior (ICA, MCA, and ACA) and posterior circulation (PCA) are evident. In early interaction, PCA involvement is correlated with a less advanced anterior circulation involvement, causing symptoms during childhood. In delayed interaction, PCA involvement is correlated with an advanced anterior circulation involvement, causing adult-onset MA. These findings potentially explain the pathophysiology of the biphasic onset age in MA.9 As indicated by Suzuki in 1969,14 MA converts the cerebral vascular supply from the ICA to the external carotid artery (ECA). “ECA–ICA conversion” may result in intracranial hemorrhage and cerebral ischemia due to inadequate collaterals. Extracranial–intracranial bypass surgery may complement ECA–ICA conversion.14,154
Previous studies demonstrate various associations of MA with atherosclerotic and autoimmune diseases,17,151,152,155–158 including a potentially higher autoimmune disease prevalence in unilateral MA.151 The association between MA and a single nucleotide polymorphism located in PSRC1 (1p13.3), rs599839, previously demonstrated to be associated with coronary artery disease in a European cohort, suggests a common pathophysiological pathway between MA and atherosclerotic disease.17 Atherosclerotic and juvenile-onset autoimmune diseases are associated with both pediatric and adult MA. Adult-onset autoimmune disease is associated with pediatric MA but not with adult MA.158 Both pediatric and adult MA are associated with inflammatory disease clusters. In a subset of MA patients, pathologic vessel changes may thus be a sequela of systemic inflammation.18,152,158
Histopathological studies show that vessel occlusion results from a combination of intimal fibrocellular thickening, intimal hyperplasia, luminal thrombosis, noninflammatory lipid deposits, and media attenuation with irregular, tortuous, stratified, ruptured, straight, or multilayered internal elastic lamina, and a decrease of the outer vascular diameter.1,16,17,53 Intimal hyperplasia is promoted by endothelial proliferation, vascular smooth muscle cell (SMC) proliferation, SMC migration from the media to the intima, angiogenesis, and production of vascular matrix proteins such as elastin, encoded by ELN (7q11.23), and collagen.17,59 Intimal thickening, which is less prominent in pediatric MA,159 may result from malfunctioning endothelial and smooth muscle progenitor cells during vascular repair and maintenance.135 Vessel exposure to blood constituents may facilitate intimal thickening.5 Lin et al18 showed that the S100A4 protein was detected in the thickened intima and media of the MCA from autopsy specimens and that cerebrovascular SMCs in different parts of the brain showed vacuolar degeneration. These investigators also stated that histopathological findings showed a narrowed lumen due to intimal fibrous thickening without significant inflammatory cell infiltration. Masuda et al160 in a study of six autopsy samples of stenotic or occlusive intracranial lesions of MA patients, showed the infiltration of human alveolar macrophage 56-positive cells, presumably macrophages, and ubiquitin carboxy-terminal hydrolase L1-positive cells, presumably T cells. These results should be interpreted with caution as the autopsy samples were obtained from patients with subarachnoid hemorrhage or intracerebral hemorrhage, which may induce inflammation. Detection of macrophage and T cell infiltrates in stenotic segments supports the hypothesis that microthrombi formation may result from chronic inflammation.5,160 Aberrant vascular endothelial growth factor (VEGF), transforming growth factor (TGF)-β1, or matrix metalloproteinases (MMP) may be released in response to inflammatory stimuli or may activate downstream regulators of inflammatory signaling cascades.5 A proinflammatory milieu is essential for angiogenesis and collateralization.5 Thinning or duplication of the internal elastic lamina creates a neointima, an intimal fibrocellular layer consisting of migrated SMCs.17,18 TGF-β and bone morphogenetic protein 4 promote the vascular SMC contractile phenotype, characterized by decreased migration and proliferation. Platelet-derived growth factor (PDGF) induces aspects of the vascular SMC synthetic phenotype, characterized by increased migration and proliferation. A vascular SMC phenotypic switch from a differentiated/contractile to a proinflammatory/promatrix remodeling dedifferentiated/synthetic phenotype causes myointimal hyperplasia, vessel wall degeneration, matrix degradation, and inflammation.19,161,162 Subsequent vascular SMC apoptosis leads to a hypocellular vessel wall in intracranial aneurysms with increased susceptibility of rupture.161 Caspase-dependent apoptosis is involved in the disintegration of the arterial wall.1
Moyamoya collateral vascular networks are usually dilated medium or small muscular-perforating arteries that are regarded as a combination of preexisting and newly evolved vessels.1 Moyamoya collaterals demonstrate evidence of stress associated with increased flow, an irregular-shaped lumen with intimal thickening or thinning, fibrin deposits in the vessel wall, a fragmented elastic lamina, attenuated media, and saccular and dissecting intracranial microaneurysms—this stress potentially explains hemorrhagic events in MA.1,16,152,163 Other moyamoya vessels are secondarily collapsed with concentric intimal thickening and luminal thrombosis caused by loose fibrous connective tissue, which potentially elucidates the cause of ischemic symptoms.1,163
Moyamoya cortical microvascularization may be characterized by increased microvascular diameter and density, resulting in an increased microvascular surface, accompanied by aberrant microvascular hemodynamics, such as a chronically decreased cortical perfusion and cerebral blood flow, reflecting cerebrovascular hemodynamic insufficiency.16,152,164 Vascular anastomotic networks in pediatric MA may be described as a composition of four anastomotic networks with distinct angioarchitectures.165 Moyamoya basal and cortical vessels may represent either collaterals compensating for reduced cerebral blood flow or aberrant active angiogenic neovascularization occurring before vascular occlusion due to the proliferation of endothelial cells or SMCs.16,166
The pathology of stenotic segments in MA, including coexistence of proliferation and shrinkage and an unknown biology of neovascularization, suggests an aberrant versus a compensatory process that creates a complex MA pathophysiology.16 Given that several pathologic processes in the formation and potential coexistence of intracranial aneurysms and cerebral arteriovenous malformations (AVM) overlap with MA and MMS—such as vascular stenosis, media thinning, internal elastic lamina damage, angiogenesis, aberrant microvascular hemodynamics, and vascular remodeling—signaling pathways and inflammatory and/or genetic biomarkers may also overlap.40,60,72,105,161,167,168
Genetic Biomarkers of Moyamoya Angiopathy
Specific Mutations Linked to Moyamoya Angiopathy
The E3 ubiquitin-protein ligase RNF213 (RNF213) (14576 G>A) is encoded by ring finger protein 213 (RNF213) (17q25.3). RNF213 encodes a protein-harboring RING (really interesting new gene) finger motif that functions as a RING-class E3 ubiquitin ligase.169 RNF213 is a cytosolic gene finger protein, with a Walker motif, a structural component of the AAA+ ATPase, and a RING finger domain.99,100 Specific mutations in RNF213 may improve prognosis. The homozygous RNF213 missense variant c.14576G>A (p.R4810K, rs112735431) increases susceptibility to MA in East Asians and predicts a severe, early-onset form of MA, including severe ischemia, stenosis, and PCA occlusion.2,9,101,102,170 In one study, rs9916351 in RNF213 showed a stronger genetic effect on early-onset than late-onset MA.37 The RNF213 p.Arg4810Lys variant causes a classical MA when present in a heterozygous state, but the same variant results in MA and systemic vascular diseases when present in a homozygous state in a gene-dosage–dependent manner. In addition, RNF213 homozygous patients show a distinctive pattern of diffuse narrowing of the aorta and iliofemoral arteries, along with stenosis of the renal, celiac, or peripheral pulmonary arteries.171 Parallel appearance of the homozygous and heterozygous variant of c.14576G>A demonstrates a variable clinical course and severity of MA in siblings.103 In unilateral MA, bilateral progression may be associated with the number of RNF213 risk alleles.104 In a genome-wide association study of 785,720 single-nucleotide polymorphisms (SNPs), Kamada et al99 compared 72 Japanese MA patients with 45 Japanese controls, resulting in a strong association of chromosome 17q25-ter with an increased risk of MA. The research group confirmed this result through a locus-specific association study with 335 SNPs in the 17q25-ter region. The RNF213 (p.R4859K, rs112735431) missense variant is linked to an increased risk of MA in Japanese people. RNF213 polymorphisms rs112735431 and rs148731719 may be associated with MA in Chinese Hans.48 RNF213 mutations are also associated with susceptibility to MA in the Chinese Han population. Ischemic MA is particularly related to the R4810K mutation. Hemorrhagic MA is primarily associated with the A4399T polymorphism. Identification of novel variants in RNF213 further emphasizes the genetic heterogeneity in MA.169 In a study using a Chinese population, rs9916351 and rs8074015 in RNF213 were demonstrated to be significantly associated with MA.172 The RNF213 (p.R4810K) variant may predispose individuals of Asian descent living in the United States to MA.173 Zhang et al174 identified 27 rare RNF213 missense variants in 255 Chinese MA patients not found in 300 healthy controls. No possible disease-causing mutations were identified in ACTA2, BRCC3, or GUCY1A3. In comparison to patients without rare RNF213 variants, p.R4810K heterozygous patients were younger at diagnosis and showed an increased percentage of familial cases, ischemia, and PCA involvement.174 Downregulation of Securin by RNF213 R4810K (rs112735431, G>A) reduces angiogenic activity of induced pluripotent stem cell-derived vascular endothelial cells from MA patients, suggesting that it may be a promising in vitro model for MA.175 The MA susceptibility variant RNF213 R4810K (rs112735431) induces mitotic abnormality and increases genomic instability.176 Fujimura et al hypothesized that RNF213 deficiency might cause vulnerability to hemodynamic stress and vascular fragility, leading to MA.15 In their 2019 review article, Mikami et al170 hypothesized that RNF213 may not be directly involved in the pathophysiology of MA, suggesting that inflammation, based on an autoimmune response, may affect RNF213, leading to MA progression. To evaluate the role of RNF213 in the etiology of MA, Sonobe et al177 generated RNF213-deficient mice and investigated whether they developed MA in comparison to wild-type littermates. Their results indicate that a functional loss of RNF213 did not sufficiently induce MA, suggesting that additional insults, such as inflammation and/or infection, autoimmune disease, ischemia, and radiation, may be required for MA development. The RNF213 c.14576G>A variant in MA patients may not represent a loss of function mutation. Suppression of vascular remodeling in RNF213-deficient mice requires further study.177 In their 2015 study with homozygous RNF213-knockin mice and wild-type littermates, Kanoke et al178 demonstrated through imaging findings and the anatomy of the circle of Willis that RNF213 knockin mice did not spontaneously develop MA. The research group suggested that, in addition to genetic factors, multiple secondary insults, eg, environmental factors, may contribute to the onset of MA. Whether the RNF213 polymorphism in MA is a gain-of function or loss-of-function mutation remains to be elucidated.178 In 2016, Kanoke et al179 demonstrated that RNF213 deficiency and administration of immunological adjuvants such as muramyl dipeptide (MDP)-Lys (L18) or complete Freund’s adjuvant did not sufficiently induce MA. The ratio of regulatory T cells after administration of MDP-Lys (L18) was shown to be significantly decreased in RNF213-deficient mice, suggesting an involvement of RNF213 in regulatory T cell differentiation. RNF213 may compromise immunological self-tolerance, thus contributing to MA pathogenesis.179
Smooth muscle aortic alpha-actin 2 (ACTA2) is encoded by ACTA2 (10q23.31).2,6,16 In 2009, Guo et al73 performed a linkage analysis and association studies, phenotyping 127 individuals of 20 families with heterozygous ACTA2 mutations and 192 ethnically matched controls, indicating that mutation carriers may have a variety of premature vascular diseases. Heterozygous ACTA2 missense mutations may predispose patients to various diffuse and diverse vascular diseases, including familial thoracic aortic aneurysms and dissections, premature coronary artery disease, premature ischemic strokes, and MA.73,180 The ACTA2 p.R258C/H mutation is associated with ICA occlusion and early-onset ischemic stroke.73 The ACTA2 R179H de novo heterozygous missense mutation is related to an MMS, which might be caused by an autosomal dominant disorder and is associated with early-onset severe cerebrovascular disease due to smooth muscle dysfunction and with nonfamilial MA in European patients.2,74,75,181 ACTA2 mutations have been identified through DNA sequencing of the ACTA2 gene, according to a previously described protocol.180,181 ACTA2 mutations affecting small muscular arteries cause increased or abnormal vascular SMC proliferation, leading to intimal hyperplasia, occlusive disease, and elastin deficiency. ACTA2 mutations affecting large elastic arteries result in actin changes that decrease contractility, leading to aneurysmal disease, arterial dilatation, or dolichoectasia.16,19,74,75 Cerebral angiopathy characteristics show diverse vascular phenotypes, including a bilateral MMS with proximal ICA ectasia, terminal ICA stenosis, bilateral dolichoectatic ICA, intracranial arteries with a straight course, absence of basal moyamoya collaterals, and potential posterior circulation involvement.4,73–75
Lys-63-specific deubiquitinase BRCC36 (BRCC3)/Protein p13 MTCP-1 (MTCP1) is associated with MMS, which may be caused by a recessive X-linked disorder. Deletion of BRCC3/MTCP1 (Xq28) leads to loss of BRCC3/MTCP1 gene expression. BRCC3 is a ubiquitously expressed K63-specific deubiquitinating enzyme containing a JAMM (JAB1/MPN/MOV34 metalloenzyme) domain. BRCC3 encodes for two protein complexes, the nuclear BRCA1/Rap80/Abraxas/Merit40/BRCC45 DNA repair complex and cytoplasmic BRISC complex.4,119 Cerebral angiopathy characteristics include bilateral anterior MA.4
Guanylate cyclase soluble subunit alpha-1 (GUCY1A3) is related to an MMS potentially caused by an autosomal recessive disorder. GUCY1A3 (4q32.1) encodes the α1 subunit of the heterodimeric protein sGC, the primary receptor for nitric oxide (NO). Loss-of-function mutations cause a NO pathway alteration in SMCs. The NO-sGC-cyclic guanosine monophosphate (cGMP) pathway is a key signal transduction pathway controlling vascular smooth-muscle relaxation, vascular tone, and vascular remodeling. In 2014, Herve et al44 suggested that homozygous loss-of-function mutations in GUCY1A3 may be linked to the loss of the main sGC isoform (sGCα1ß1) in SMCs and inadequate response of SMCs to NO. Herve et al stated that the intima-media thickness in carotid arteries of healthy subjects and the neointimal growth in pathological conditions, including moyamoya, are inversely associated with shear stress. In arterial bifurcations, shear stress is modified, and the main function of NO signaling is to normalize shear stress.44 Factors specific to a particular vascular territory, such as the residual level of α2ß1 sGC, may explain the predominance of MA at ICA bifurcations.4,44 Cerebral angiopathy characteristics include MMS or MA, posterior circulation involvement, isolated MCA and ACA stenosis, and early onset before 5 years of age.4
SAM domain and HD domain-containing protein 1 (SAMHD1) (20q11.23) is associated with an MMS potentially caused by an autosomal recessive disorder. SAMHD1 is involved in cerebral vascular homeostasis and immunoregulation. SAMHD1 loss-of-function mutations may be associated with intracerebral large artery disease.4,74 Cerebral angiopathy characteristics include MA, distal ICA or proximal MCA/ACA stenosis (or both), and saccular aneurysms.4
Shoemaker et al36 showed that ZXD family zinc finger C (ZXDC) (3q21.3) (p.P562L), a gene involved in MHC Class II activation, was the most enriched variant in White patients and non-RNF213 founder mutation cases. Using collapsing variant methodology, the research group also showed that obscurin, cytoskeletal calmodulin, and titin-interacting (OBSCN) (1q42.13), a gene involved in myofibrillogenesis, was most enriched in White patients and non-RNF213 founder mutation cases.36
Another SNP associated with MA, rs2107595 in histone deacetylase 9 (HDAC9) (7p21.1), may be involved in large-vessel disease.37
Genetic Associations with Moyamoya Angiopathy
MYMY1 (3p26–p24.2) is associated with moyamoya. Proposed MA genes on chromosome 3 code for von Hippel-Lindau (VHL) disease, suggesting that proteins encoded by VHL (3p25.3) are involved in vascular wall homeostasis.40 Low oxygen and a mutated VHL gene are linked to a decreased ability of the VHL disease tumor suppressor (pVHL) protein to downregulate HIF-1α, which allows pVHL to dimerize with stable HIF-1β subunits and cause overexpression of HIF-target genes, including the genes for VEGF, PDGF-BB, TGF-β, TNF-α, and erythropoietin.41 Chromosome 6 encodes human leukocyte antigens (HLAs).51,52 HLA1 (6p22.1) and HLA2 are linked to MA. HLAB35 (6p21.33) may impact inflammatory and autoimmune processes and have been found in older Korean women with MA.1,57 HLA-DR (6p21.32) and HLA-DQ (6p21.32) genes are associated with familial MA.58 The frequency of the HLA allele DRB1*0405 is lower, and that of the DRB1*1510-DQA1*01021-DQB1*0620 haplotype is higher compared to controls. The D6S441 microsatellite marker on chromosome 6q25 showed linkage with MA in 20 affected sibling pairs.51 Two loci on chromosome 8 encode MA candidate genes, KLF10 (8q22.3) and ANGPT1 (8q23.1). KLF10 is involved in TGF-β1 expression. ANGPT1 predominantly acts on Tie2. Receptor-binding cancer antigen expressed on SiSo cells, encoded by EBAG9 (8q23.2) and E3 ubiquitin-protein ligase UBR5, encoded by UBR5 (8q22.3), may regulate angiogenesis through VEGF expression.52,68 Sakurai et al68 provided significant genetic evidence for linkage to chromosome 8q23 and suggestive evidence for linkage to chromosome 12p12. Yamauchi et al93 linked familial MA to chromosome 17q25. Mineharu et al97 analyzed large families with an autosomal dominant inheritance pattern of MA and narrowed down the linkage region to chromosome 17q25.3. Based on these results, Liu and colleagues from the same research group101 identified RNF213 as a major susceptibility gene for MA. In 2008, Mineharu et al97 stated that brain-specific angiogenesis inhibitor 1-associated protein 2, encoded by BAIAP2 (17q25.3), interacts with brain-specific angiogenesis inhibitor-1, which might be an inhibitor of basic fibroblast growth factor (bFGF)-induced angiogenesis.97 The ss161110142 polymorphism, located in position −1480 from the transcription site of RPTOR (17q25), with evidence for linkage to chromosome 17q25, may be associated with MA among East Asians.93,96–98
Neurofibromatosis type I (NF-1) is an autosomal dominant disorder that may be a cause of MMS.4 NF1 (17q11.2) is a tumor-suppressor gene that inhibits cell cycle progression.74 Neurofibromin is a regulatory protein involved in blood vessel maintenance and repair that inhibits the Ras-Raf-MEK-ERK pathway.94 Loss-of-function mutations in NF1 cause increased mitogenic signaling, resulting in proliferation and differentiation of endothelial cells and hyperplastic vascular SMCs, leading to vascular wall thickening and stenosis due to absent, dysfunctional, or nonfunctional neurofibromin expression.4,19,94,95 NF-1 vasculopathy may be due to failure of the endothelium to suppress increased migration and proliferation of SMCs or due to an altered SMC response to normal endothelial signals.95 Cerebral angiopathy of intracranial arteries includes MA, stenosis, occlusion, ectasia, aneurysms, arteriovenous fistula, involvement of the anterior or posterior circulation (or both), and the potential involvement of extracranial carotid or vertebral arteries, with a mean onset age of 5 to 11 years.4
Sickle-cell disease is an autosomal recessive disorder, encoded by HBB (11p15.4), which may be a cause of MMS. A pain crisis in the disease leads to changes in the formation of factor XIIIa-crosslinked fibrin or fibrinogen, which increases D-dimer levels (a hypercoagulable state).80 This hypercoagulable state increases IL-6, E-selectin, and von Willebrand factor, resulting in tissue factor and thrombin generation.80 Repeated vascular injury from sickling, abnormal regulation of vasoconstriction, and intimal hyperplasia result in an increased incidence of stroke.3,81–83 MMS screening by noninvasive imaging leads to improved neurologic outcomes.82,84 Cerebral angiopathy characteristics include distal ICA, proximal ACA or MCA stenosis (or both), moyamoya collaterals, and posterior circulation involvement, with posterior circulation intracranial aneurysms.4
Down syndrome (trisomy 21) is an aneuploidy disorder with the karyotype (47, XY + 21) in males, (47, XX + 21) in females, or mosaicism, featuring increased prevalence of autoimmune disorders and autoantibodies, including antiphospholipid antibodies. Chromosome 21 encodes the alpha chains of collagen type VI. Abnormal collagen VI expression is associated with an increased risk of cerebrovascular diseases. Inappropriate expression of proteins encoded by chromosome 21, including cystathionine-β-synthetase, interferon-γ receptor, and superoxide dismutase 1, is linked to structural vascular defects.4,116 Cerebral angiopathy characteristics resemble MA.4
Posterior fossa malformations, hemangiomas, arterial anomalies, cardiac defects, eye abnormalities, sternal cleft, and supraumbilical raphe syndrome (PHACEs) is a neurocutaneous disorder. The etiology of PHACEs and the pathophysiology of its associated vascular disease are undetermined. A potential cause may be a somatic mutation, affecting vascular development. Aplasia, hypoplasia, steno-occlusive arterial disease, an aberrant circle of Willis, and coexisting aortic arch anomalies lead to arterial ischemic stroke and moyamoya-like vasculopathy. Cerebral angiopathy characteristics include multiple congenital arterial lesions (eg, dysplasia-hypoplasia-aplasia, aneurysms, and arteries with an aberrant course) and progressive arterial lesions (eg, stenosis, occlusion, and MA).4,182,183
Congenital dwarfing syndromes comprise MOPDII (microcephalic osteodysplastic primordial dwarfism type II) and Seckel syndrome. MOPDII is an autosomal recessive disorder, which may be a cause of MMS. PCNT (21q22.3) encodes pericentrin, a centrosomal protein potentially involved in vascular homeostasis. Cerebral angiopathy characteristics involve aneurysms and MA. Seckel syndrome (microcephalic primordial dwarfism) is an MMS with autosomal recessive inheritance, encoded by ATR (3q23), RBBP8 (18q11.2), CENPJ (13q12.12-q12.13), CEP152 (15q21.1), CEP63 (3q22.2), NIN (14q22.1), and PCNT (21q22.3). Loss-of-perfusion reserve capacity in hypoperfused brain regions creates tortuous arterial collateral development that results in cerebral ischemia and stroke. Cerebral angiopathy characteristics frequently involve aneurysms and rarely MA.4,42,43
Alagille syndrome is an autosomal dominant disorder that may be a cause of MMS.4,25–27 Overall, 94% of cases are due to JAG1 loss-of-function mutations, 5%-7% are due to deletions incorporating JAG1. NOTCH2 receptor mutations cause less than 1% of cases. JAG1 (20p12.2) encodes a cell surface protein, protein jagged-1, a ligand for Notch receptors 1, 2, 3, and 4. NOTCH2 (1p12-p11) encodes the Notch2 transmembrane protein. Kamath et al31 suggests involvement of Jagged1 (JAG1) and the Notch signaling pathway in vascular development in reference to vascular anomalies in Alagille syndrome, indicating that the vascular anomalies evident in Alagille syndrome may be a direct consequence of a disruption in the Notch pathway. However, their study does not directly show the change of protein levels of JAG1 in MA.31 Cerebral arteries of patients with Alagille syndrome and intracranial hemorrhage show thin-walled vessels with myointimal hyperplasia. Cerebral angiopathy characteristics include unilateral or bilateral MA, other abnormalities of the distal ICA or MCA (stenosis, occlusion, and aneurysm), and basilar aneurysm.4,25–34
Noonan syndrome, an autosomal dominant disorder that may be a cause of MMS, is encoded by PTPN11 (12q24.13) in 50% of cases, SOS1 (2p22.1) in 10%–13% of cases, and RAF1 (3p25.2) in 3%–17% of cases. Rarely involved genes include KRAS (12p12.1), NRAS (1p13.2), BRAF (7q34), and MAP2K1 (15q22.31). Gain-of-function mutations in these genes activate the Ras pathway and enhance cellular proliferation.4,35 Gain-of-function mutations in SHP-2 phosphatase (PTPN11) associated with Noonan syndrome and various childhood leukemias enhance cell migration of endothelial cells, hematopoietic cells, and fibroblasts and in vivo angiogenesis in SHP-2 D61G knockin mice.86 Cerebral angiopathy characteristics include MA, AVMs, aneurysms, and hypoplasia of the posterior circulation (BA, PCA, superior cerebellar artery, PICA, anterior inferior cerebellar artery).4,35,184,185
Costello syndrome, an autosomal dominant disorder that may cause MMS, is encoded by HRAS (11p15.5) and, less often, KRAS (12p12.1). Gain-of-function mutations that affect p.Gly12 or p.Gly13 cause Ras pathway dysregulation, leading to hyperplasia and vascular remodeling.4,85 Cerebral angiopathy characteristics include MA.4
Molecular Biomarkers of Moyamoya Angiopathy
Vascular Smooth Muscle Enzymes
The serine protease inhibitor alpha-1 antitrypsin, encoded by Serpin family A member 1 (SERPINA1) (14q32.13), is an antiapoptotic factor in vascular SMCs. Increased alpha-1 antitrypsin serum levels, inflammation, and alterations of the alpha-1 antitrypsin gene lead to inhibition of elastase or collagenase, which causes proliferation of SMCs and synthesis of vascular matrix components elastin and collagen; these, in turn, lead to structural alterations of the vascular wall and intimal hyperplasia. Changes in alpha-1 antitrypsin serum levels may be indicative of MA progression, intracranial aneurysm formation, and fibromuscular dysplasia.89
MMP-3 (stromelysin-1) inhibits SMC migration. Decreased MMP-3 levels facilitate SMC migration and lead to intimal hyperplasia. The MMP3 6A allele and 6A/6A genotype are associated with an increased risk of MA.20,78 The MMP-3–1171 5A/6A (rs3025058) single nucleotide polymorphism may be associated with sporadic and familial MA in the Chinese Han population.5,79
The dominant type (GG vs GA+AA) of MMP-9 Q279R (rs17576) may be related to MA.6 MMP-9 (gelatinase B) overexpression leads to a decrease in angiostatin level, which degrades the vascular basement membrane and causes extracellular matrix remodeling. This remodeling increases the permeability of the blood-brain barrier, leading to intimal hyperplasia, cerebral ischemia, increased angiogenesis, collateral vessel formation (moyamoya vessels), and intracranial aneurysm formation and rupture.20,39,52,54,122 A paracrine effect on the MA vasculature causes increased MMP-9, leading to pathologic angiogenesis and microhemorrhages.5,39,122 Macrophage MMP-9 may be involved in capillary branching in angiogenic revascularization triggered by ischemia (experimental).121 Sonobe et al123 induced vascular hyperplasia by common carotid artery ligation in RNF213 knockout mice and their wild-type littermates. The results of this research group suggest that increased vascular expression of MMP-9 and subsequent vascular wall thinning in RNF213 knockout mice may reflect early distinctive changes in MA, consistent with the proposed constrictive remodeling theory, indicating that the RNF213 c.14576G>A polymorphism in MA patients may be a loss-of-function mutation.123 Increased serum MMP-9 in MA patients may be associated with spontaneous intracranial hemorrhage and cerebral hyperperfusion syndrome after direct surgical revascularization.15,20
MMP-12 degrades elastin and other extracellular matrix molecules. Increased interleukin (IL)-1β and monocyte chemoattractant protein-1 (MCP-1) levels increase MMP-12 expression in human peripheral blood monocytes and monocyte-derived macrophages, leading to macrophage and SMC migration.20
Tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2 inhibit SMC migration. The vascular injury repair model suggests an MMP/TIMP imbalance as a pathogenic mechanism of MA, which causes increased MMP-9 and decreased MMP-3, TIMP-1, and TIMP-2 levels, leading to SMC migration and proliferation and intimal hyperplasia. In 2010, Kang et al20 compared TIMP2 levels in the CSF between 20 patients and 9 controls. They showed that TIMP2 levels were significantly lower in patients with MA (p = 0.046). The G/C heterozygous genotype in the TIMP-2-418 G>C (rs8179090) promoter may be associated with MA. Park et al6 observed a significant difference exclusively in 11 familial MA patients, who were not necessarily unrelated individuals. Wang et al48 performed an association analysis in 96 MA patients and 96 controls and showed that PDGFRB, MMP-3, or TIMP-2 was not associated with MA in the Chinese Han population. Roder et al108 also failed to replicate the association in 40 MA patients and 68 controls in the Central European population. Paez et al186 also denied the association in the Japanese population. Bersano et al stated in their 2016 review article, “Again, conflicting results between studies did not allow any conclusions to be drawn about the significance of these associations.”187 MMP-2 (gelatinase A) is encoded by MMP2 (16q12.2). The MMP-2 −1575GA/-1306CC genotype may be a predisposing genetic factor for MA.6
The methylenetetrahydrofolate reductase (MTHFR) (1p36.22) 677C>T polymorphism is associated with ischemic stroke in children. Homozygous MTHFR 677C>T is associated with MA. Recessive MTHFR 677C>T and the C677T/A1298C compound genotype are associated with adult MA. The frequency of the CT/AA sequence of MTHFR 677/1298 is specifically increased in hemorrhagic MA. rs9651118 in MTHFR and rs117353193 in transcobalamin 2 (TCN2) (22q12.2) were associated with high-serum homocysteine in MA cases. Tissue enrichment analysis showed that the genes of associated loci were highly expressed in the immune system.6,37
Retinal dehydrogenase 2 (RALDH2) is encoded by ALDH1A2 (15q21.3). Epigenetic suppression of RALDH2 in pediatric MA is linked to dysfunction of endothelial colony-forming cells (ECFC), which may be reversed by retinoic acid.16,90
Endothelial NO is encoded by NOS3 (7q36.1). The NOS3 haplotype a-4b-G is associated with adult-onset MA. Age-specific cerebral angiopathy characteristics may include cerebral ischemia and hemorrhage.6
Growth Factors
VEGF is inversely correlated with the number of circulating endothelial progenitor cells.137 VEGF receptor (VEGFR)2 or kinase insert domain receptor (KDR) is considered an early marker of endothelial and hematopoietic progenitor cells.5 Increased VEGF and PDGF-BB plasma levels may contribute to collateral vessel formation and may be responsible for intracranial microhemorrhages.5,20 The VEGF*634G allele is associated with MA and defective collateral vessel formation.54 The CC genotype of VEGF*634 and decreased levels of VEGFR1 or flt-1 and VEGFR2 may correlate with synangiosis-induced, favorable postoperative collateral development.5,6,55 In 2010, Kang et al20 showed that increased plasma levels of VEGF and MCP-1 in MA patients might be involved in vascular progenitor cell recruitment and collateral vessel formation. Increased angiogenesis in the dura mater may lead to increased VEGF expression in the dura mater.20 Progressive ICA stenosis causes ischemia, leading to the induction of proinflammatory cascades and VEGF expression.16,54 VEGF immunoreactivity has been detected in ischemia.5 VEGF polymorphisms are associated with pediatric MA and poor collateral vessel formation.16 Park et al55 showed in a case-control study of 107 Korean MA patients and 243 healthy controls that the 2634CC genotype occurred less frequently in the pediatric MA group, whereas the KDR 2604C/1192A/1719T haplotype increased the risk of pediatric MA. Patients with the CC genotype of VEGF 2634 showed better collateral vessel formation after surgery, suggesting that the VEGF 2634G allele may be associated with pediatric MA and poor collateral vessel formation. In summary, Park et al55 suggested that VEGF and KDR polymorphisms may influence MA and the formation of synangiosis-induced collaterals after bypass surgery.55 Crosstalk between Notch and VEGF signaling is involved in maintaining physiological angiogenesis.56
Endoglin modulates cellular responses to TGF-β1 signaling. In the intimal MCA, HIF-1α and endoglin overexpression may lead to intimal hyperplasia.59,70,71
Aoyagi et al117 studied the response of vascular SMCs in MA after PDGF stimulus application and found an alteration in DNA synthesis and proliferation of SMCs compared to controls. In their 1993 follow-up study, Aoyagi et al188 showed that the diminished cellular response to PDGF is caused by a decreased number of PDGF receptors on SMCs of MA patients. Yamamoto et al63 demonstrated decreased reactivity of replication and migration after a PDGF (PDGF-BB and PDGF-AA) stimulus, suggesting that prolonged exposition of various cytokines caused by delayed repair processes after arterial wall injury due to a decreased PDGF reaction may cause intimal thickening in MA. Kang et al20 demonstrated an 18-fold increase in the PDGF-BB plasma level in MA patients compared to controls, suggesting a compensative mechanism to a systemically decreased amount of PDGFB receptors. The results by Roder et al108 on rs3828610, located in the promoter region of PDGFRB (5q32), may explain a decreased transcriptional activity and thus a decreased number of PDGFRB receptors.
Hepatocyte growth factor (HGF) is involved in angiogenesis, intracranial aneurysm formation, tumorigenesis, and tissue regeneration.5,65 Cerebral ischemia and infection lead to upregulation of HGF and c-met protooncogene. This upregulation affects HGF binding to high-affinity c-met tyrosine kinase receptor and epithelial and endothelial cells via the c-met receptor, leading to activation of tyrosine kinase signaling. This signaling increase stimulates cell growth, vascular SMC migration, extracellular matrix invasion, and morphogenesis, causing intimal hyperplasia and occlusion of the carotid fork.5,66 Endothelial injury alters endothelial barrier properties, which may cause an accumulation of growth factors and cytokines, leading to occlusive changes.5,67 HGF has been detected in the ICA tunica media and intima.5,67 HGF-induced SMC migration suggests HGF involvement in neointima formation.5,63 Increased media HGF is associated with elevated CSF HGF, which is linked to increased angiogenesis, intimal proliferation, and collateral vessel formation (moyamoya vessels).67
bFGF is involved in vascular development and dose-dependent increases in angiogenesis or arteriogenesis.5,45 bFGF receptor overexpression causes local bFGF expression and release, leading to intimal thickening and angiogenesis.20,46 Increased CSF bFGF may correlate with favorable postoperative collateral development in indirect revascularization procedures.5,47 bFGF immunoreactivity has been detected in superficial temporal artery endothelial cells and SMCs.5
TGF-β is involved in vasculogenesis, angiogenesis, cell growth and differentiation, and extracellular matrix gene expression.5,109 TGF-β1 expression is upregulated in response to shear stress.5 TGF-β1 regulates the expression of IL-1, bFGF, PDGF, and TGF-β by human peripheral blood monocyte chemotaxis.5,110 TGF-β1 overexpression is correlated with increased production of extracellular matrix components (eg, procollagen, collagen, and proteoglycans) and accumulation of elastin synthase in SMCs of MA patients.5,111 Elastin synthesis and accumulation via the TGF-β pathway causes intimal and medial hyperplasia and collateral vessel formation.5,111,112 TGF-β increase in abnormal vascular growth inhibits HGF expression in vascular SMCs.67,113 MA in European patients may be associated with single polymorphisms in TGFB1 (rs1800471[C/G]) and PDGFRB (rs382861[A/C]), and with rs599839, a single nucleotide polymorphism located in the 3′ UTR region of PSRC1 (1p13.3).9,17,108 Liu et al114 demonstrated a negative correlation between TGFB1 polymorphisms rs1800471 and rs1800470 and Japanese MA patients.
Angiopoietins (ANGs) are secreted protein ligands of Tie receptor tyrosine kinases (Tie).24,56 ANG1 is encoded by ANGPT1 (8q23.1), ANG2 is encoded by ANGPT2 (8p23.1).24,69 ANG1 facilitates vascular network maturation, ANG2 initiates neovascularization.124 TIE1 (1p34.2) and TEK/TIE2 (9p21) are endothelial cell-specific type 1 transmembrane protein receptor tyrosine kinases.24 Tie1-Tie2 heterodimers attenuate signaling from Tie2 homodimers.24 ANG1 is an agonistic paracrine Tie2 ligand that stimulates Akt-dependent phosphorylation and nuclear exclusion of forkhead box protein O1 (FOXO1) transcription factor.87 FOXO1 inactivation decreases the expression of genes involved in endothelial destabilization, apoptosis, and growth control.87,88 ANG2 is an endothelial autocrine ligand that acts as a context-dependent weak agonist or antagonist of Tie2.87 ANG1-Tie2 signaling regulates proliferation, migration, adhesion, and endothelial cell survival; it also regulates endothelial cell–SMC communication in venous morphogenesis, angiogenesis, vessel maturation and stability, vascular development and maintenance during embryogenesis, inflammation, vascular permeability, and vascular leakage.56,125,126 ANG1-mediated Tie2 activation is linked to the maintenance of an endothelial barrier and a quiescent vasculature.126 ANG2 activation of Tie2 supports the stable enlargement of normal non-leaky vessels.127 ANG2-Tie2 binding is linked to vascular destabilization and sprouting during VEGF-induced neovascularization.124,128 The relative ratio of ANG1 versus ANG2 regulates the effect of ANG-VEGF on angiogenesis.128 Integrins regulate ANG-Tie signaling.125 ANG1 and ANG2 weakly bind integrins, with accentuated integrin effects in low Tie2 expression.24,69 Tie2 regulates vascular barrier function.24 Various ANG proteins may induce Tie1 phosphorylation, and Tie1 activation may be amplified through Tie2.189 Tie1 is involved in maintaining vascular endothelial cell integrity and angiogenic capillary growth, modulating the effects of ANG1 and ANG2 on Tie2 and regulating ANG1-Tie2 signaling.125 Tie1 deletion leads to compromised vascular integrity, hemorrhage, and embryo death.87 ANG2 acts as a Tie2 agonist in nonpathologic conditions, in the absence of ANG1, and at high levels. In inflammation and at low levels, ANG2 functions as a Tie2 antagonist, promoting vascular dysfunction.24,56,69,87,125,127 Blecharz et al analyzed ANG2 gene expression in MCA lesions obtained from MA patients and atherosclerotic cerebrovascular disease patients. Cerebral endothelial cells incubated with serum of the same patients in vitro were applied. In contrast to the sera of patients with atherosclerotic cerebrovascular disease, the sera of MA patients induced ANG2 overexpression and secretion, accompanied by the loss of endothelial integrity. The results obtained by the group may identify cerebral endothelial cells as a potential source of vessel-destabilizing factors, inducing plastic and unstable neovascularization and cerebrovascular disintegration in MA, mediated by the autocrine release of the pro-angiogenic cytokine ANG2, thereby elucidating a mechanism that may contribute to cerebrovascular hemorrhagic and ischemic events in MA.190 Yu et al191 showed that ANG2 may be involved in the pathophysiology of increased angiogenesis in adult hemorrhagic MA and may also be a prognostic factor of intracranial hemorrhage in adult MA patients. Infection is linked to Tie2 signaling disruption that is related to vascular leakage.24 The A20 binding inhibitor of nuclear factor-κB (NF-κB) activation-2 (ABIN-2) is a potential vascular protective protein that acts on Tie2 with antiapoptotic and anti-inflammatory effects on endothelial cells.126,129
Caveolin-1 is involved in endothelial progenitor cell recruitment, inhibits endothelial NOS (eNOS), is critical for VEGF-induced angiogenesis, and may be protective against ischemia/reperfusion injury.76,131–133 Some research groups showed an association of CAV1 with the RNF213 variant, whereas other groups showed the absence of such an association. Caveolin-1 may be decreased in MA patients with the RNF213 variant.16 Bang et al192 prospectively analyzed clinical data from 139 MA patients, 61 intracranial atherosclerotic stroke patients, and 68 healthy controls, comparing protein biomarkers for caveolae, genetic, angiogenesis, and endothelial dysfunction between MA patients and intracranial atherosclerotic stroke patients. The group used pathological analysis to evaluate whether a certain protein biomarker would mediate the association between genes and MA, suggesting that MA is a disorder of caveolae, potentially related to caveolin-1, involving the regulation of endothelial vesicular trafficking and signal transduction, but may not be related to dysregulation of circulating cytokines or endothelial dysfunction. Chung et al193 in a study with 77 consecutive MA patients and 17 patients with intracranial atherosclerotic stroke and no RNF213 mutation, who served as controls, showed that CAV1 downregulation induced apoptosis in SMCs, and suppressed angiogenesis in endothelial cells, suggesting that caveolin-1 may be involved in pathological negative arterial remodeling in adult MA, indicating that negative remodeling may be associated with MA symptomatology. In their 2019 review article, Mikami et al170 stated that caveolin-1 might be involved in MA and inflammation. They reported that caveolin-1 serum levels were shown to be decreased in MA and shown to be markedly decreased in MA patients with the RNF213 variant. They finally cite that caveolin-1 may be associated with angiogenesis in addition to an association between the caveolin-1/ERK and Wnt/β-catenin pathways. A decrease in caveolin-1 leads to an increase in eNOS and systemic NO,131 which alters VEGF-induced ERK activation in endothelial cells.132 This alteration increases proliferation and decreases stabilization, capillary tube formation, and endothelial cell migration.16,130,131 Increased endothelial caveolin-1 and VEGF result in decreased angiogenesis in mice.132 Aberrant angiogenesis in caveolin-1 gene-deficient mice may be restored by the ablation of eNOS. Caveolin-1 may regulate angiogenesis through multiple pathways, including NO-independent and NO-mediated mechanisms.131 Absent CAV1 gene expression, cerebral infarction volume, and ischemic injury increase in mice.130,133 Transient decreases in caveolin expression enhance progenitor cell engraftment.76
Circulating endothelial progenitor cells (EPCs) reflect mixed conditions of vascular occlusion and abnormal vasculogenesis.134,135 Decreased tube formation in EPCs may indicate defects in paracrine function.134,135,137 Increased levels of EPCs are associated with aberrant angiogenesis, arteriogenesis, and vascular hyperplasia and may contribute to neovascularization (moyamoya vessels) after cerebral ischemia and MA progression.16,134,135,137,139,140 With an inverse correlation between EPCs and VEGF, increased numbers of EPCs may not be entirely mediated by VEGF or granulocyte-macrophage–colony-stimulating factor.137 CD34+ cells, a subpopulation of EPCs, have been detected at increased levels with impaired function in the peripheral blood of adult MA patients compared to decreased CD34+ cells with defective angiogenic function in pediatric MA.2,16,77,136,139 An inadequate response to vascular progenitor cell recruitment may cause an early-onset severe form of pediatric MA.135 Detection of CD34+ cells and VEGFR2 in intimal thickening of supraclinoid ICAs from adult MA patients suggests that EPCs may participate in vascular stenosis.135,138 Decreased endothelial progenitor cell–colony-forming units may be associated with early-stage MA or MA progression. Increased endothelial progenitor cell–colony-forming units and outgrowth cells have been demonstrated after revascularization surgery.5,134,135 EPCs are associated with increased VEGF, granulocyte colony-stimulating factor, and SDF-1α.5
Vascular smooth muscle progenitor cells (SPC) isolated from the peripheral blood of MA patients demonstrate decreased PDGF receptor-α, major histocompatibility complex, and calponin expression levels, suggesting a defective cell maturation process.16,135,141 Increased VEGF causes failure of SPC outgrowth.135,141 SPCs of MA patients show irregular tube formation and differential expression of over 200 genes, resulting in reduced CD31 expression.2,141 Gene ontology analysis of upregulated transcripts identified ephrin receptor signaling, and analysis of downregulated transcripts identified cell adhesion, cell migration regulation, innate immune response, enzyme-linked receptor protein signaling pathways, and vessel development.135,141
Transcription Factors
Hypoxia-inducible factor (HIF)-1α is involved in tissue oxygen homeostasis and helps regulate VEGF, PDGF-BB, TGF, and inducible NO synthase (NOS).59,70,71 HIF is a sequence-specific, DNA-binding, heterodimeric transcription factor that binds to hypoxia-response elements and transcriptionally activates genes linked to angiogenesis (eg, VEGF, PDGFB), erythropoiesis (eg, erythropoietin), extracellular matrix turnover, and apoptosis.71 Hypoxia may regulate transcription factors including HIF and NF-κB in innate and adaptive immune cells.194 HIF-1 and NF-k B may be involved in the inflammatory response.170,194 The PI3K/Akt signaling pathway may regulate HIF-1 expression.170,195 Mikami et al hypothesized that signal transduction pathways may be activated through RNF213, triggered by inflammatory cytokines present due to infection or autoimmune diseases.170 Takagi et al59 aimed to clarify the pathogenesis of MA through immunohistochemical analyses of human MCA samples. The research group compared 12 MCA terminal segments (M4) samples obtained at the time of superficial temporal artery (STA)-MCA bypass in MA patients with those obtained from patients with other causes of MCA or ICA occlusion. The group reported that the MCA samples from MA patients showed a thicker intima, higher staining for HIF-1α in the intima and endothelium, and higher staining for endoglin in the endothelium compared with the controls. TGF-β3 staining was co-localized with staining of HIF-1α and endoglin, predominantly in the endothelium.59
Adhesion Molecules
Vascular cell adhesion molecule 1 (VCAM-1),5,16,20 intercellular adhesion molecule 1 (ICAM-1),5,16,20 and E-selectin5,16,20 activate leukocytes, causing sticking and rolling of leukocytes along the endothelium before transmigration through the endothelial cell barrier, leading to vascular inflammation.5 E-selectin is a molecule on the endothelial surface that binds to β2-integrin Mac-1 (CD11b/CD18) on neutrophils. β2-integrin Mac-1 mediates firm adherence of neutrophils to the vessel wall by binding to its endothelial ligand, ICAM-1. E-selectin is involved in endothelial progenitor cell recruitment and angiogenesis.16 Increased expression of VCAM-1, ICAM-1, and E-selectin in the CSF in pediatric MA poststroke or with subarachnoid hemorrhage activates leukocytes and extravascular migration, leading to inflammation and neovascularization.16 Isolated increases in CSF VCAM, ICAM, and E-selectin in pediatric patients with MMS may be associated with immunologic activation.5,90,196 Lee et al90 compared the gene expression profiles of ECFCs isolated from pediatric MA patients and normal controls. They concluded that the expression of RALDH2 was epigenetically suppressed in ECFCs in pediatric MA patients, which may be critical in their functional impairment. Soriano et al196 studied CSF and serum samples obtained from 20 MMS patients and 20 patients with congenital spinal deformities. The research group demonstrated increased CSF levels of soluble endothelial adhesion molecules, suggesting that MMS patients may have ongoing central nervous system inflammation and impairment of the blood-brain barrier. These soluble adhesion molecules may be used as indicators of that inflammatory process.
Cellular retinoic acid-binding protein 1 (CRABP1) decreases retinoid activity, which increases growth factor expression that stimulates SMC migration and proliferation, causing neointimal hyperplasia.91 Increased CSF CRABP1 may be associated with bilateral MA in adults and a postoperative decrease in basal collaterals.16,92 Roder et al108 demonstrated that the two SNPs (rs2280367 and rs3813573) genotyped in the promoter region of CRABP1 (15q25.1) did not show a significant association with MA.
Inflammation/Coagulation
MCP-1 upregulation enhances migration of bone marrow stromal cells (experimental).20
IL-1β stimulation of SMCs causes increased expression of prostaglandin E2 and cyclooxygenase-2, endothelial cell and SMC proliferation, macrophage activation, increased vascular permeability, and endothelial dysfunction; these events increase vasodilation and pial hyperemia.5,20 Absent IL-1 and MCP-1 gene expression reduce ischemic injury in mice.39
IL-6 is involved in the activation of intracellular signal transduction pathways, including the Janus kinase (JAK)-signal transducer and activator of transcription protein (STAT) signaling pathway.61–63 VEGF and IL-6 receptor binding are involved in JAK-STAT signaling pathway activation, which amplifies IL-6 signal transduction and enhances MMP-9, VEGF, and VEGFR2 activation and vascular SMC proliferation.60,62 Cerebral oxygen-glucose deprivation upregulates IL-6 messenger RNA that increases transcription of angiogenesis-associated genes.61 Increased angiogenesis and extracellular matrix breakdown, initiated in part by IL-6, are associated with vascular instability and AVM rupture.60,62 IL-6-mediated endothelial blood-brain barrier disturbances may be decreased by IL-6 receptor inhibition in cerebral microvascular endothelial cells (experimental).64 IL-6 inhibits migration of moyamoya SMCs.63 Kang et al20 showed that IL-6 plasma levels were not different between MA patients and controls.
Perinuclear antineutrophil cytoplasmic antibodies (p-ANCA) are associated with inflammation, arterial stenosis, and ICA occlusion.106 Yanagawa et al106 describe a case showing bilateral ICA stenosis, moyamoya vessels, and seropositivity of rheumatoid factor and MPO-ANCA, suggesting that immunological factors, inflammation, and vasculitis may be involved in the obstruction and/or stenosis of the ICA.
D-dimer is a potential prognostic biomarker for childhood arterial ischemic stroke and is predictive of stroke recurrence risk and adverse neurologic outcomes.142
Roder et al17 analyzed 40 DNA samples of MA patients and 68 healthy controls from Central Europe and demonstrated a strong association between MA and rs599839, located on 1p13.3 in the 3′ UTR region of PSRC1, in a Central European cohort. This SNP was described as being associated with coronary artery disease in a European cohort. Stromal cell-derived factor 1 alpha (SDF-1α) is also known as C-X-C motif chemokine 12 (CXCL12a). Ischemia creates HIF-1-induced expression of platelet-derived SDF-1α, SDF-1α plasma levels increase, and SDF-1α interacts with caveolae-mediated endocytosed C-X-C chemokine receptor type 4 (CXCR4) (fusin or CD184). These interactions cause SDF-1α/CXCR4 axis-mediated CD34+ EPC mobilization, migration from the bone marrow to the peripheral blood, and peripheral homing, which are associated with increased arteriogenesis and vasculogenesis.76,77 Increased SDF-1α is associated with rapid MA progression.77
Fujimura et al143 showed that sCD163 serum levels of MA patients were significantly increased compared to controls. CXCL5 serum levels of MA patients were significantly increased compared to controls. The research group observed no differences in both sCD163 and CXCL5 serum levels between each genotype of the RNF213 polymorphism, variant or wild-type, between MA patients. The group indicated that MA patients might show an increase in autoimmunity, further elucidating the pathogenesis of MA by the use of CD163+ M2-polarized macrophages. Phi et al144 showed that peripheral blood-derived ECFCs of MA patients are critical to the pathogenesis of MA and the phenomenon of impaired tube formation in vitro, respectively. The C-C motif chemokine ligand 5 (CCL5) may mediate these interactions. In MA patients, defective ECFCs may direct aberrant recruitment of smooth muscle progenitor cells (SPCs) to critical vascular locations through the action of CCL5.
Immune-Related Factors/Autoantibodies
Protein S100-A4, encoded by S100A4 (1q21.3), is a marker in the media of vascular SMCs. Hypothetically, it may be linked to the migration of S100A4-positive SMCs into the intima through a broken internal elastic lamina, with potential loss of the contractile phenotype by an endothelial-to-mesenchymal transition. This hypothesized transition may create a proliferation of S100A4-positive SMCs that may cause luminal stenosis with intimal thickening, leading to compensatory small-vessel proliferation. Immunoglobulin G may deposit in the internal elastic lamina and disrupt it.18 MA may be considered a hyperplastic vascular myopathy, promoting migration and proliferation of SMCs and matrix production.18,19
Ogawa et al145 showed that the sera of 32 MA patients, who were tested for anti-endothelial cell antibodies through enzyme-linked immunoassays and flow cytometric analysis, contained an increased incidence of anti–α-fodrin autoantibodies, providing further insight into arterial occlusion mechanisms in MA.
Molecules listed in this paragraph were identified by Sigdel et al21 via an analysis of 165 significantly elevated autoantibodies in the sera of a multiethnic group of MA patients. Amyloid-beta A4 precursor protein (APP), encoded by APP (21q21.3), is associated with an increased risk of hemorrhagic stroke, Alzheimer disease pathogenesis, and DNA damage response/repair.21,115 Cerebral amyloid angiopathy in APP transgenic mice leads to loss of vascular SMCs, intracranial aneurysms, vessel obliteration, and vasculitis. Extracellular deposition of neuron-derived β-amyloid in the vessel wall disrupts the vessel wall and causes parenchymal hemorrhage.115 Catenin beta-1 is encoded by CTNNB1 (3p22.1). The Wnt/β-catenin pathway is associated with angiogenesis, reduced microvessel density, and decreased expression of VEGF and SDF-1.21 EGF-like repeat and discoidin I-like domain-containing protein 3 (EDIL3), encoded by EDIL3 (5q14.3), is involved in angiogenesis and vessel wall remodeling and development.21 EDIL3 mediates adhesion, migration, and proliferation of vascular SMCs via interaction with integrin-receptor αvβ3 and may regulate vascular morphogenesis or remodeling during embryonic development.49,50 Receptor tyrosine kinase-like orphan receptor 1 (ROR1), encoded by ROR1 (1p31.3), may be involved in cell migration and cytoskeleton remodeling, potentially explaining the function of ROR1 in neuron development.21,22 Adult MA is associated with neuronal impairment.23 COP9 signalosome complex subunit 1 (GPS1), encoded by GPS1 (17q25.3), is a component of the COP9 signalosome involved in DNA damage response/repair, which could be compromised in MA.21 Stimulated by retinoic acid 13 (STRA13), also known as centromere protein X (CENPX) or MHF2, is encoded by CENPX (17q25.3). STRA13 is the DNA-binding component of the Fanconi anemia core protein complex, which is a component of the kinetochore. The Fanconi anemia complex is involved in DNA damage response/repair. Fanconi anemia is a disease associated with MMS.21
Biomarker Candidate Proteins
Proteome screening of the CSF and the blood serum in MA patients using mass spectrometry has identified several specific proteins involved in the arteriopathy.2
Rüggeberg et al146 performed a CSF proteome analysis in 29 White MA patients and 10 arteriosclerosis patients as controls. The research group demonstrated a novel post-translational modification of human transthyretin. Gamma-carboxylation of the Glu-42 (Gla-42), Gla-42 transthyretin, was demonstrated by two-dimensional electrophoresis and subsequent peptide sequencing with ESI-MS/MS. The research group reports that Gla-42 or similar post-translational modifications have not been shown in Asian MA patients so far. Therefore, the pathophysiological relevance of Gla-42 transthyretin in MA patients remains speculative.
Koh et al147 demonstrated 22 differently-expressed serum proteomes of 6 MA patients and 6 normal controls by matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry and electrospray ionization quadruple time-of-flight mass spectrometry. Complement C1 inhibitor protein was upregulated (7.23 ± 0.10141-fold) in MA patients, and apolipoprotein C-III was the lowest expressed protein (0.066 ± 0.05231). Koh et al147 suggested that complement C1 inhibitor protein overexpression in MA may be related to progressive obstruction of the distal ICA and protection against ischemia. Masuda et al160 demonstrated SMC proliferation in occlusive lesions in major intracranial arteries in MA. Koh et al147 suggested that apolipoprotein C-III downregulation may be biologically protective, reducing the formation of intracranial occlusive lesions.
Molecules listed in this paragraph were identified by Araki et al38 in a pilot study of CSF proteomics via an analysis of CSF samples of 20 MA patients and 12 control patients using surface-enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF-MS). Several CSF proteins with molecular weights corresponding to the 4473-Da peptide are potential biomarkers of vasculogenesis.38 These proteins include oxyntomodulin (P01275), urocortin-2 (Q96RP3), beta-defensin 133 (Q30KQ1), antibacterial protein LL-37 (P49913), liver-expressed antimicrobial peptide 2 (Q969E1), and proenkephalin-A (143–183) (P01210). Aberrant vasculogenesis may be involved in MA.38
Maruwaka et al2,149 performed a CSF proteome analysis in 20 MA patients and 17 controls by SELDI-TOF-MS. The research group detected the 4473-Da peptide, a possible prognostic biomarker for antihypoxic response and intracranial inflammation, which is associated with postoperative angiogenesis and favorable postoperative collateral development when found in the CSF of pediatric MA patients.2,149
Hamauchi et al197 analyzed induced pluripotent stem cell-derived endothelial cells from the peripheral blood of 3 MA patients carrying the RNF213 R4810K variant and 3 healthy controls, suggesting that downregulation of ECM receptor-related genes may be related to impaired angiogenic activity in endothelial cells derived from induced pluripotent stem cells of MA patients. The upregulation of splicing regulation-related proteins may suggest differences in splicing patterns between endothelial cells of MA patients and controls.
Kashiwazaki et al148 performed a CSF proteome study in 10 MA patients and 4 controls, using comparative 2-dimensional gel electrophoresis (2D-PAGE) and mass spectrometry. According to the proteome analysis technique, this study demonstrates that the CSF of MA includes 3 downregulated proteins (apolipoprotein-E, apoE precursor, and apolipoprotein-J) and 2 upregulated proteins (haptoglobin and alpha-1-B-glycoprotein). Downregulation of apolipoprotein-E and apolipoprotein-J suggests an involvement of the lipid metabolism in MA pathogenesis. Downregulated apolipoprotein-E may be involved in neurovascular unit dysfunction and neuronal vulnerability against cerebral ischemia. Downregulated apolipoprotein-J may induce apoptosis of endothelial cells. Haptoglobin overexpression may indicate angiogenesis and/or inflammation in MA. The role of alpha-1-B-glycoprotein in the central nervous system remains to be elucidated.
Implementation and Future Considerations
Future research on MA should carefully consider past studies and current concepts.150 Continued research into MA pathophysiology and associated signaling pathways may identify new treatment strategies, therapeutic applications, and mechanism-tailored interventions that may halt MA progression.4,5,187
Ideally, biomarkers should assist in the diagnosis, prognosis, and treatment of MA.2 Directing future research toward the identification of biomarkers that comprise an MA-specific fingerprint compared to more nonspecific markers of general physiological processes, such as angiogenesis or ischemia, will be essential.2 Determining the relationship between any given measurable biomarker and relevant clinical endpoints is essential.2,198
Biomarkers may improve therapeutic efficacy. Studies of cell function may help to predict the capacity for therapeutic angiogenesis. These data could be used to decide between direct or indirect bypass. Measuring circulating peptides may provide additional data to more accurately predict the response to surgery. Changes in biomarker levels may suggest novel therapeutics, such as growth factors, that may modulate surgical collateral growth.2,20 Nonsurgical approaches may include the application of trophic factors or chemicals increasing angiogenesis,199 anti-cancer drugs decreasing SMC proliferation,73 retinoids,90,91 increasing caveolin-1 levels,200 and stem-cell therapy compensating for impaired endothelial or smooth muscle progenitor cells.16,199 Development of cost-effective, noninvasive screening tests based on biomarkers may revolutionize MA patient care.2 Accurate and timely diagnosis of MA may profoundly improve outcomes for MA patients. Neurologic status at the time of treatment critically influences long-term outcomes.2,8,201,202 Continued monitoring of at-risk patients with MMS, early referral to neurosurgeons once the arteriopathy becomes evident, considering surgical revascularization in asymptomatic MA patients,201 and long-term neurological and radiological follow-up along with a separate management guideline is advocated to prevent ischemic and hemorrhagic strokes in asymptomatic MA patients.203 Clinical features, prognosis, outcomes, and risk factors causing ischemic and hemorrhagic stroke in asymptomatic MA patients may be ascertained by prospective multicenter studies, eg, the Japanese AMORE study.204 Biomarker development may also aid in more accurately predicting patients who would benefit from surgery. Data enabling surgeons to prospectively identify the increased probability of posterior circulation stroke or higher risk of contralateral progression in unilateral MA is already employed in clinical practice.2,201,205,206 STA-to-MCA anastomosis may decrease the level of endothelial progenitor cells, decrease the formation of moyamoya vessels, halt MA progression, and alleviate headache in ischemic MA by potentially improving perfusion pressure and cerebral circulation.12,140 Compared with drug treatment, STA–MCA bypass surgery may significantly lower the risk of postoperative rebleeding and ischemia.140
In vivo studies and animal models of MA are warranted.135 Identification of MA-associated genes may allow the development of moyamoya animal models, essential for preclinical trials. Novel sequencing technologies, such as next-generation sequencing, and high-throughput technologies, such as genome-wide association studies, may accelerate the identification of new familial MA and monogenic MMS.4,187 Refining subgroups of MA by mutational genetic analysis, the definition of the function of relevant genes, and evaluation of potential epigenetic factors that serve as important modulators of the MA phenotype are advocated. Identifying protein function from mutated MA-associated genes is a promising area of research. Cell-based biomarkers may provide the best combination of genomic, proteomic, and functional markers of MA.2 The study of perivascular cells or pericytes, cells involved in post-injury tissue regeneration, may be a promising approach to elucidate the mechanisms leading to the formation of aberrant collaterals in MA.5 Research into the pathophysiological association between MA and inflammation is advocated.158 Studying levels of circulating biomarkers in relation to the disease stage is another promising path for future research.16 Establishing separate management guidelines for unilateral and bilateral MA is warranted.151
Clear articulation of strengths and weaknesses of new diagnostic and prognostic capabilities provided by research may avoid unrealistic expectations. Generating consensus on how best to utilize new data provided by biomarkers will be important.2 The relative rarity of MA necessitates collaboration among centers in validating novel candidate proteins. Shared data, multicenter prospective longitudinal, large-cohort, case-control studies, family-based analyses, comparative trials, and close collaboration between the clinical, laboratory, and basic research settings will be essential for generating meaningful results.2,5,16,21,84,198
Ethics
To the best of our knowledge, this manuscript meets all updated current ethics statements.
Acknowledgments
The authors thank Samantha Soto, Rogena Lake, and Laura Repak for manuscript preparation; Mary Ann Clifft, Paula Higginson, and Lynda Orescanin for editing; Cassandra Todd, Cindy Giljames, and Kristen Larson for illustrations; all in Neuroscience Publications at Barrow Neurological Institute; and Nicole Galvan at St. Joseph’s Hospital and Medical Center for help with the literature search.
Funding
The authors have not received a fee from any organization to write this review.
Disclosure
The authors report no potential conflict of interest relevant to this article.
References
1. Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. 2009;360(12):1226–1237. doi:10.1056/NEJMra0804622
2. Smith ER. Moyamoya Biomarkers. J Korean Neurosurg Soc. 2015;57(6):415–421.
3. Roach ES, Golomb MR, Adams R, et al. Management of stroke in infants and children: a scientific statement from a Special Writing Group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. Stroke. 2008;39(9):2644–2691.
4. Guey S, Tournier-Lasserve E, Herve D, Kossorotoff M. Moyamoya disease and syndromes: from genetics to clinical management. Appl Clin Genet. 2015;8:49–68.
5. Bedini G, Blecharz KG, Nava S, et al. Vasculogenic and angiogenic pathways in moyamoya disease. Curr Med Chem. 2016;23(4):315–345.
6. Park YS. Single nucleotide polymorphism in patients with moyamoya disease. J Korean Neurosurg Soc. 2015;57(6):422–427.
7. Kuroda S, Houkin K. Moyamoya disease: current concepts and future perspectives. Lancet Neurol. 2008;7(11):1056–1066.
8. Scott RM, Smith JL, Robertson RL, Madsen JR, Soriano SG, Rockoff MA. Long-term outcome in children with moyamoya syndrome after cranial revascularization by pial synangiosis. J Neurosurg. 2004;100(2Suppl Pediatrics):142–149.
9. Hishikawa T, Sugiu K, Date I. Moyamoyadisease: a review of clinical research. Acta Med Okayama. 2016;70(4):229–236.
10. Hishikawa T, Tokunaga K, Sugiu K, Date I. Clinical and radiographic features of moyamoya disease in patients with both cerebral ischaemia and haemorrhage. Br J Neurosurg. 2013;27(2):198–201. doi:10.3109/02688697.2012.717983
11. Kang S, Liu X, Zhang D, et al. Natural course of moyamoya disease in patients with prior hemorrhagic stroke. Stroke. 2019;50(5):1060–1066.
12. Okada Y, Kawamata T, Kawashima A, Yamaguchi K, Ono Y, Hori T. The efficacy of superficial temporal artery-middle cerebral artery anastomosis in patients with moyamoya disease complaining of severe headache. J Neurosurg. 2012;116(3):672–679.
13. Takeuchi K, Shimizu K. Hypoplasia of the bilateral internal carotid arteries. Brain Nerve. 1957;9:37–43.
14. Suzuki J, Takaku A. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch Neurol. 1969;20(3):288–299.
15. Fujimura M, Sonobe S, Nishijima Y, et al. Genetics and biomarkers of moyamoya disease: significance of RNF213 as a susceptibility gene. J Stroke. 2014;16(2):65–72.
16. Bang OY, Fujimura M, Kim SK. The pathophysiology of moyamoya disease: an update. J Stroke. 2016;18(1):12–20.
17. Roder C, Peters V, Kasuya H, et al. Common genetic polymorphisms in moyamoya and atherosclerotic disease in Europeans. Childs Nerv Syst. 2011;27(2):245–252.
18. Lin R, Xie Z, Zhang J, et al. Clinical and immunopathological features of moyamoya disease. PLoS One. 2012;7(4):e36386.
19. Milewicz DM, Kwartler CS, Papke CL, Regalado ES, Cao J, Reid AJ. Genetic variants promoting smooth muscle cell proliferation can result in diffuse and diverse vascular diseases: evidence for a hyperplastic vasculomyopathy. Genet Med. 2010;12(4):196–203.
20. Kang HS, Kim JH, Phi JH, et al. Plasma matrix metalloproteinases, cytokines and angiogenic factors in moyamoya disease. J Neurol Neurosurg Psychiatry. 2010;81(6):673–678.
21. Sigdel TK, Shoemaker LD, Chen R, et al. Immune response profiling identifies autoantibodies specific to moyamoya patients. Orphanet J Rare Dis. 2013;8:45.
22. Tseng HC, Kao HW, Ho MR, et al. Cytoskeleton network and cellular migration modulated by nuclear-localized receptor tyrosine kinase ROR1. Anticancer Res. 2011;31(12):4239–4249.
23. Harada M, Miyoshi H, Uno M, et al. Neuronal impairment of adult moyamoya disease detected by quantified proton MRS and comparison with cerebral perfusion by SPECT with tc-99m HM-PAO: a trial of clinical quantification of metabolites. J Magn Reson Imaging. 1999;10(2):124–129.
24. Milam KE, Parikh SM. The angiopoietin-Tie2 signaling axis in the vascular leakage of systemic inflammation. Tissue Barriers. 2015;3(1–2):e957508.
25. Kamath BM, Bauer RC, Loomes KM, et al. NOTCH2 mutations in Alagille syndrome. J Med Genet. 2012;49(2):138–144.
26. Oda T, Elkahloun AG, Pike BL, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16(3):235–242.
27. Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16(3):243–251.
28. Connor SE, Hewes D, Ball C, Jarosz JM. Alagille syndrome associated with angiographic moyamoya. Childs Nerv Syst. 2002;18(3–4):186–190.
29. Emerick KM, Krantz ID, Kamath BM, et al. Intracranial vascular abnormalities in patients with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2005;41(1):99–107.
30. Gaba RC, Shah RP, Muskovitz AA, Guzman G, Michals EA. Synchronous moyamoya syndrome and ruptured cerebral aneurysm in Alagille syndrome. J Clin Neurosci. 2008;15(12):1395–1398.
31. Kamath BM, Spinner NB, Emerick KM, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109(11):1354–1358.
32. Rocha R, Soro I, Leitao A, Silva ML, Leao M. Moyamoya vascular pattern in Alagille syndrome. Pediatr Neurol. 2012;47(2):125–128.
33. Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012;20(3):251–257.
34. Woolfenden AR, Albers GW, Steinberg GK, Hahn JS, Johnston DC, Farrell K. Moyamoya syndrome in children with Alagille syndrome: additional evidence of a vasculopathy. Pediatrics. 1999;103(2):505–508.
35. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381(9863):333–342.
36. Shoemaker LD, Clark MJ, Patwardhan A, et al. Disease variant landscape of a large multiethnic population of moyamoya patients by exome sequencing. G3 (Bethesda). 2015;6(1):41–49.
37. Duan L, Wei L, Tian Y, et al. Susceptibility loci for moyamoya disease revealed by a genome-wide association study. Stroke. 2018;49(1):11–18.
38. Araki Y, Yoshikawa K, Okamoto S, Sumitomo M, Maruwaka M, Wakabayashi T. Identification of novel biomarker candidates by proteomic analysis of cerebrospinal fluid from patients with moyamoya disease using SELDI-TOF-MS. BMC Neurol. 2010;10:112.
39. Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4(5):399–415.
40. Ikeda H, Sasaki T, Yoshimoto T, Fukui M, Arinami T. Mapping of a familial moyamoya disease gene to chromosome 3p24.2-p26. Am J Hum Genet. 1999;64(2):533–537.
41. Bader HL, Systemic HT. VHL gene functions and the VHL disease. FEBS Lett. 2012;586(11):1562–1569.
42. Codd PJ, Scott RM, Smith ER. Seckel syndrome and moyamoya. J Neurosurg Pediatr. 2009;3(4):320–324.
43. Willems M, Genevieve D, Borck G, et al. Molecular analysis of pericentrin gene (PCNT) in a series of 24 Seckel/microcephalic osteodysplastic primordial dwarfism type II (MOPD II) families. J Med Genet. 2010;47(12):797–802.
44. Herve D, Philippi A, Belbouab R, et al. Loss of alpha1beta1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am J Hum Genet. 2014;94(3):385–394.
45. Baffour R, Berman J, Garb JL, Rhee SW, Kaufman J, Friedmann P. Enhanced angiogenesis and growth of collaterals by in vivo administration of recombinant basic fibroblast growth factor in a rabbit model of acute lower limb ischemia: dose-response effect of basic fibroblast growth factor. J Vasc Surg. 1992;16(2):181–191.
46. Takagi Y, Kikuta K, Nozaki K, Hashimoto N. Histological features of middle cerebral arteries from patients treated for moyamoya disease. Neurol Med Chir (Tokyo). 2007;47(1):1–4.
47. Malek AM, Connors S, Robertson RL, Folkman J, Scott RM. Elevation of cerebrospinal fluid levels of basic fibroblast growth factor in moyamoya and central nervous system disorders. Pediatr Neurosurg. 1997;27(4):182–189.
48. Wang X, Zhang Z, Liu W, et al. Impacts and interactions of PDGFRB, MMP-3, TIMP-2, and RNF213 polymorphisms on the risk of moyamoya disease in Han Chinese human subjects. Gene. 2013;526(2):437–442.
49. Hidai C, Zupancic T, Penta K, et al. Cloning and characterization of developmental endothelial locus-1: an embryonic endothelial cell protein that binds the alphavbeta3 integrin receptor. Genes Dev. 1998;12(1):21–33.
50. Rezaee M, Penta K, Quertermous T. Del1 mediates VSMC adhesion, migration, and proliferation through interaction with integrin alpha(v)beta(3). Am J Physiol Heart Circ Physiol. 2002;282(5):H1924–H1932.
51. Inoue TK, Ikezaki K, Sasazuki T, Matsushima T, Fukui M. Linkage analysis of moyamoya disease on chromosome 6. J Child Neurol. 2000;15(3):179–182.
52. Weinberg DG, Arnaout OM, Rahme RJ, Aoun SG, Batjer HH, Bendok BR. Moyamoya disease: a review of histopathology, biochemistry, and genetics. Neurosurg Focus. 2011;30(6):E20.
53. Roder C, Nayak NR, Khan N, Tatagiba M, Inoue I, Krischek B. Genetics of moyamoya disease. J Hum Genet. 2010;55(11):711–716.
54. Young AM, Karri SK, Ogilvy CS, Zhao N. Is there a role for treating inflammation in moyamoya disease? A review of histopathology, genetics, and signaling cascades. Front Neurol. 2013;4:105.
55. Park YS, Jeon YJ, Kim HS, et al. The role of VEGF and KDR polymorphisms in moyamoya disease and collateral revascularization. PLoS One. 2012;7(10):e47158.
56. Hansen TM, Singh H, Tahir TA, Brindle NP. Effects of angiopoietins-1 and −2 on the receptor tyrosine kinase Tie2 are differentially regulated at the endothelial cell surface. Cell Signal. 2010;22(3):527–532.
57. Han H, Pyo CW, Yoo DS, Huh PW, Cho KS, Kim DS. Associations of moyamoya patients with HLA class I and class II alleles in the Korean population. J Korean Med Sci. 2003;18(6):876–880.
58. Hong SH, Wang KC, Kim SK, Cho BK, Park MH. Association of HLA-DR and -DQ genes with familial moyamoya disease in Koreans. J Korean Neurosurg Soc. 2009;46(6):558–563.
59. Takagi Y, Kikuta K, Nozaki K, et al. Expression of hypoxia-inducing factor-1 alpha and endoglin in intimal hyperplasia of the middle cerebral artery of patients with moyamoya disease. Neurosurgery. 2007;60(2):338–345.
60. Mouchtouris N, Jabbour PM, Starke RM, et al. Biology of cerebral arteriovenous malformations with a focus on inflammation. J Cereb Blood Flow Metab. 2015;35(2):167–175.
61. Gertz K, Kronenberg G, Kalin RE, et al. Essential role of interleukin-6 in post-stroke angiogenesis. Brain. 2012;135(Pt 6):1964–1980.
62. Hoffmann CJ, Harms U, Rex A, et al. Vascular signal transducer and activator of transcription-3 promotes angiogenesis and neuroplasticity long-term after stroke. Circulation. 2015;131(20):1772–1782.
63. Yamamoto M, Aoyagi M, Fukai N, Matsushima Y, Yamamoto K. Differences in cellular responses to mitogens in arterial smooth muscle cells derived from patients with moyamoya disease. Stroke. 1998;29(6):1188–1193.
64. Blecharz-Lang KG, Wagner J, Fries A, et al. Interleukin 6-mediated endothelial barrier disturbances can be attenuated by blockade of the IL6 receptor expressed in brain microvascular endothelial cells. Transl Stroke Res. 2018;9(6):631–642.
65. Pena-Silva RA, Chalouhi N, Wegman-Points L, et al. Novel role for endogenous hepatocyte growth factor in the pathogenesis of intracranial aneurysms. Hypertension. 2015;65(3):587–593.
66. Nakamura T, Mizuno S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86(6):588–610.
67. Nanba R, Kuroda S, Ishikawa T, Houkin K, Iwasaki Y. Increased expression of hepatocyte growth factor in cerebrospinal fluid and intracranial artery in moyamoya disease. Stroke. 2004;35(12):2837–2842.
68. Sakurai K, Horiuchi Y, Ikeda H, et al. A novel susceptibility locus for moyamoya disease on chromosome 8q23. J Hum Genet. 2004;49(5):278–281.
69. Felcht M, Luck R, Schering A, et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J Clin Invest. 2012;122(6):1991–2005.
70. Jin KL, Mao XO, Nagayama T, Goldsmith PC, Greenberg DA. Induction of vascular endothelial growth factor and hypoxia-inducible factor-1alpha by global ischemia in rat brain. Neuroscience. 2000;99(3):577–585.
71. Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol. 2007;2:145–173.
72. Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18(8):509–516.
73. Guo DC, Papke CL, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84(5):617–627.
74. Munot P, Crow YJ, Ganesan V. Paediatric stroke: genetic insights into disease mechanisms and treatment targets. Lancet Neurol. 2011;10(3):264–274.
75. Munot P, Saunders DE, Milewicz DM, et al. A novel distinctive cerebrovascular phenotype is associated with heterozygous Arg179 ACTA2 mutations. Brain. 2012;135(Pt 8):2506–2514.
76. Sbaa E, Dewever J, Martinive P, et al. Caveolin plays a central role in endothelial progenitor cell mobilization and homing in SDF-1-driven postischemic vasculogenesis. Circ Res. 2006;98(9):1219–1227.
77. Ni G, Liu W, Huang X, et al. Increased levels of circulating SDF-1alpha and CD34+ CXCR4+ cells in patients with moyamoya disease. Eur J Neurol. 2011;18(11):1304–1309.
78. Ma J, You C. Association between matrix metalloproteinase-3 gene polymorphism and moyamoya disease. J Clin Neurosci. 2015;22(3):479–482.
79. Li H, Zhang ZS, Liu W, et al. Association of a functional polymorphism in the MMP-3 gene with moyamoya disease in the Chinese Han population. Cerebrovasc Dis. 2010;30(6):618–625.
80. Adam SS, Key NS, Greenberg CS. D-dimer antigen: current concepts and future prospects. Blood. 2009;113(13):2878–2887.
81. Dobson SR, Holden KR, Nietert PJ, et al. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood. 2002;99(9):3144–3150.
82. Smith ER, McClain CD, Heeney M, Scott RM. Pial synangiosis in patients with moyamoya syndrome and sickle cell anemia: perioperative management and surgical outcome. Neurosurg Focus. 2009;26(4):E10.
83. Switzer JA, Hess DC, Nichols FT, Adams RJ. Pathophysiology and treatment of stroke in sickle-cell disease: present and future. Lancet Neurol. 2006;5(6):501–512.
84. Smith ER, Scott RM. Spontaneous occlusion of the circle of Willis in children: pediatric moyamoya summary with proposed evidence-based practice guidelines. A review. J Neurosurg Pediatr. 2012;9(4):353–360.
85. Schuhmacher AJ, Guerra C, Sauzeau V, Canamero M, Bustelo XR, Barbacid M. A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J Clin Invest. 2008;118(6):2169–2179.
86. Wang S, Yu WM, Zhang W, McCrae KR, Neel BG, Qu CK. Noonan syndrome/leukemia-associated gain-of-function mutations in SHP-2 phosphatase (PTPN11) enhance cell migration and angiogenesis. J Biol Chem. 2009;284(2):913–920.
87. Korhonen EA, Lampinen A, Giri H, et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J Clin Invest. 2016;126(9):3495–3510.
88. Wilhelm K, Happel K, Eelen G, et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529(7585):216–220.
89. Amano T, Inoha S, Wu CM, Matsushima T, Ikezaki K. Serum alpha1-antitrypsin level and phenotype associated with familial moyamoya disease. Childs Nerv Syst. 2003;19(9):655–658.
90. Lee JY, Moon YJ, Lee HO, et al. Deregulation of retinaldehyde dehydrogenase 2 leads to defective angiogenic function of endothelial colony-forming cells in pediatric moyamoya disease. Arterioscler Thromb Vasc Biol. 2015;35(7):1670–1677.
91. Kim SK, Yoo JI, Cho BK, et al. Elevation of CRABP-I in the cerebrospinal fluid of patients with moyamoya disease. Stroke. 2003;34(12):2835–2841.
92. Jeon JS, Ahn JH, Moon YJ, et al. Expression of cellular retinoic acid-binding protein-I (CRABP-I) in the cerebrospinal fluid of adult onset moyamoya disease and its association with clinical presentation and postoperative haemodynamic change. J Neurol Neurosurg Psychiatry. 2014;85(7):726–731.
93. Yamauchi T, Tada M, Houkin K, et al. Linkage of familial moyamoya disease (spontaneous occlusion of the circle of Willis) to chromosome 17q25. Stroke. 2000;31(4):930–935.
94. Xu J, Ismat FA, Wang T, Yang J, Epstein JA. NF1 regulates a Ras-dependent vascular smooth muscle proliferative injury response. Circulation. 2007;116(19):2148–2156.
95. Hamilton SJ, Friedman JM. Insights into the pathogenesis of neurofibromatosis 1 vasculopathy. Clin Genet. 2000;58(5):341–344.
96. Nanba R, Tada M, Kuroda S, Houkin K, Iwasaki Y. Sequence analysis and bioinformatics analysis of chromosome 17q25 in familial moyamoya disease. Childs Nerv Syst. 2005;21(1):62–68.
97. Mineharu Y, Liu W, Inoue K, et al. Autosomal dominant moyamoya disease maps to chromosome 17q25.3. Neurology. 2008;70(24 Pt 2):2357–2363.
98. Liu W, Hashikata H, Inoue K, et al. A rare Asian founder polymorphism of Raptor may explain the high prevalence of moyamoya disease among East Asians and its low prevalence among Caucasians. Environ Health Prev Med. 2010;15(2):94–104.
99. Kamada F, Aoki Y, Narisawa A, et al. A genome-wide association study identifies RNF213 as the first moyamoya disease gene. J Hum Genet. 2011;56(1):34–40.
100. Morito D, Nishikawa K, Hoseki J, et al. Moyamoya disease-associated protein mysterin/RNF213 is a novel AAA+ ATPase, which dynamically changes its oligomeric state. Sci Rep. 2015;4(1):4442. doi:10.1038/srep04442
101. Liu W, Morito D, Takashima S, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS One. 2011;6(7):e22542.
102. Miyatake S, Miyake N, Touho H, et al. Homozygous c.14576G>A variant of RNF213 predicts early-onset and severe form of moyamoya disease. Neurology. 2012;78(11):803–810.
103. Miyatake S, Touho H, Miyake N, et al. Sibling cases of moyamoya disease having homozygous and heterozygous c.14576G>A variant in RNF213 showed varying clinical course and severity. J Hum Genet. 2012;57(12):804–806.
104. Mineharu Y, Takagi Y, Takahashi JC, et al. Rapid progression of unilateral moyamoya disease in a patient with a family history and an RNF213 risk variant. Cerebrovasc Dis. 2013;36(2):155–157.
105. Itokawa K, Fukui M, Yamamoto T, Tamura N, Ishihara S, Araki N. Dural arteriovenous fistula as a possible cause of Tolosa-Hunt syndrome: a case report. J Neurol. 2010;257(5):846–847.
106. Yanagawa Y, Sugiura T, Suzuki K, Okada Y. Moyamoya disease associated with positive findings for rheumatoid factor and myeloperoxidase-anti-neutrophil cytoplasmic antibody. West Indian Med J. 2007;56(3):282–284.
107. Joutel A, Andreux F, Gaulis S, et al. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest. 2000;105(5):597–605.
108. Roder C, Peters V, Kasuya H, et al. Polymorphisms in TGFB1 and PDGFRB are associated with moyamoya disease in European patients. Acta Neurochir (Wien). 2010;152(12):2153–2160.
109. Pepper MS. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997;8(1):21–43.
110. McCartney-Francis N, Mizel D, Wong H, Wahl L, Wahl S. TGF-beta regulates production of growth factors and TGF-beta by human peripheral blood monocytes. Growth Factors. 1990;4(1):27–35.
111. Yamamoto M, Aoyagi M, Tajima S, et al. Increase in elastin gene expression and protein synthesis in arterial smooth muscle cells derived from patients with Moyamoya disease. Stroke. 1997;28(9):1733–1738.
112. Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630.
113. Nakamura Y, Morishita R, Higaki J, et al. Expression of local hepatocyte growth factor system in vascular tissues. Biochem Biophys Res Commun. 1995;215(2):483–488.
114. Liu C, Roder C, Schulte C, et al. Analysis of TGFB1 in European and Japanese moyamoya disease patients. Eur J Med Genet. 2012;55(10):531–534.
115. Winkler DT, Bondolfi L, Herzig MC, et al. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J Neurosci. 2001;21(5):1619–1627.
116. Leno C, Mateo I, Cid C, Berciano J, Sedano C. Autoimmunity in Down’s syndrome: another possible mechanism of moyamoya disease. Stroke. 1998;29(4):868–869.
117. Aoyagi M, Fukai N, Sakamoto H, et al. Altered cellular responses to serum mitogens, including platelet-derived growth factor, in cultured smooth muscle cells derived from arteries of patients with moyamoya disease. J Cell Physiol. 1991;147(2):191–198.
118. Lim M, Cheshier S, Steinberg GK. New vessel formation in the central nervous system during tumor growth, vascular malformations, and moyamoya. Curr Neurovasc Res. 2006;3(3):237–245.
119. Herve D, Touraine P, Verloes A, et al. A hereditary moyamoya syndrome with multisystemic manifestations. Neurology. 2010;75(3):259–264.
120. Meyer S, Zanardo L, Kaminski WE, et al. Elastosis perforans serpiginosa-like pseudoxanthoma elasticum in a child with severe Moya Moya disease. Br J Dermatol. 2005;153(2):431–434.
121. Johnson C, Sung HJ, Lessner SM, Fini ME, Galis ZS. Matrix metalloproteinase-9 is required for adequate angiogenic revascularization of ischemic tissues: potential role in capillary branching. Circ Res. 2004;94(2):262–268.
122. Fujimura M, Watanabe M, Narisawa A, Shimizu H, Tominaga T. Increased expression of serum Matrix Metalloproteinase-9 in patients with moyamoya disease. Surg Neurol. 2009;72(5):476–480.
123. Sonobe S, Fujimura M, Niizuma K, et al. Increased vascular MMP-9 in mice lacking RNF213: moyamoya disease susceptibility gene. Neuroreport. 2014;25(18):1442–1446.
124. Asahara T, Chen D, Takahashi T, et al. Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ Res. 1998;83(3):233–240.
125. Eklund L, Saharinen P. Angiopoietin signaling in the vasculature. Exp Cell Res. 2013;319(9):1271–1280.
126. Fagiani E, Christofori G. Angiopoietins in angiogenesis. Cancer Lett. 2013;328(1):18–26.
127. Kim M, Allen B, Korhonen EA, et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J Clin Invest. 2016;126(9):3511–3525.
128. Scheufler KM, Drevs J, van Velthoven V, et al. Implications of vascular endothelial growth factor, sFlt-1, and sTie-2 in plasma, serum and cerebrospinal fluid during cerebral ischemia in man. J Cereb Blood Flow Metab. 2003;23(1):99–110.
129. Tadros A, Hughes DP, Dunmore BJ, Brindle NP. ABIN-2 protects endothelial cells from death and has a role in the antiapoptotic effect of angiopoietin-1. Blood. 2003;102(13):4407–4409.
130. Jasmin JF, Malhotra S, Singh Dhallu M, Mercier I, Rosenbaum DM, Lisanti MP. Caveolin-1 deficiency increases cerebral ischemic injury. Circ Res. 2007;100(5):721–729.
131. Morais C, Ebrahem Q, Anand-Apte B, Parat MO. Altered angiogenesis in caveolin-1 gene-deficient mice is restored by ablation of endothelial nitric oxide synthase. Am J Pathol. 2012;180(4):1702–1714.
132. Sonveaux P, Martinive P, DeWever J, et al. Caveolin-1 expression is critical for vascular endothelial growth factor-induced ischemic hindlimb collateralization and nitric oxide-mediated angiogenesis. Circ Res. 2004;95(2):154–161.
133. Xu L, Guo R, Xie Y, Ma M, Ye R, Liu X. Caveolae: molecular insights and therapeutic targets for stroke. Expert Opin Ther Targets. 2015;19(5):633–650.
134. Jung KH, Chu K, Lee ST, et al. Circulating endothelial progenitor cells as a pathogenetic marker of moyamoya disease. J Cereb Blood Flow Metab. 2008;28(11):1795–1803.
135. Kang HS, Wang KC, Kim SK. Circulating vascular progenitor cells in moyamoya disease. J Korean Neurosurg Soc. 2015;57(6):428–431.
136. Kim JH, Jung JH, Phi JH, et al. Decreased level and defective function of circulating endothelial progenitor cells in children with moyamoya disease. J Neurosci Res. 2010;88(3):510–518.
137. Rafat N, Beck G, Pena-Tapia PG, Schmiedek P, Vajkoczy P. Increased levels of circulating endothelial progenitor cells in patients with moyamoya disease. Stroke. 2009;40(2):432–438.
138. Sugiyama T, Kuroda S, Nakayama N, Tanaka S, Houkin K. Bone marrow-derived endothelial progenitor cells participate in the initiation of moyamoya disease. Neurol Med Chir (Tokyo). 2011;51(11):767–773.
139. Yoshihara T, Taguchi A, Matsuyama T, et al. Increase in circulating CD34-positive cells in patients with angiographic evidence of moyamoya-like vessels. J Cereb Blood Flow Metab. 2008;28(6):1086–1089.
140. Zhang W, Wang X, Wang Z, Shang J, Wang H, Yin G. Effectiveness of superficial temporal artery-to-middle cerebral artery anastomosis in treating moyamoya disease by reducing endothelial progenitor cells. World Neurosurg. 2016;93:365–370.
141. Kang HS, Moon YJ, Kim YY, et al. Smooth-muscle progenitor cells isolated from patients with moyamoya disease: novel experimental cell model. J Neurosurg. 2014;120(2):415–425.
142. Bernard TJ, Fenton LZ, Apkon SD, et al. Biomarkers of hypercoagulability and inflammation in childhood-onset arterial ischemic stroke. J Pediatr. 2010;156(4):651–656.
143. Fujimura M, Fujimura T, Kakizaki A, et al. Increased serum production of soluble CD163 and CXCL5 in patients with moyamoya disease: involvement of intrinsic immune reaction in its pathogenesis. Brain Res. 2018;1679:39–44.
144. Phi JH, Suzuki N, Moon YJ, et al. Chemokine ligand 5 (CCL5) derived from endothelial colony-forming cells (ECFCs) mediates recruitment of smooth muscle progenitor cells (SPCs) toward critical vascular locations in moyamoya disease. PLoS One. 2017;12(1):e0169714.
145. Ogawa K, Nagahiro S, Arakaki R, Ishimaru N, Kobayashi M, Hayashi Y. Anti-alpha-fodrin autoantibodies in moyamoya disease. Stroke. 2003;34(12):e244–e246.
146. Ruggeberg S, Horn P, Li X, Vajkoczy P, Franz T. Detection of a gamma-carboxy-glutamate as novel post-translational modification of human transthyretin. Protein Pept Lett. 2008;15(1):43–46.
147. Koh EJ, Kim HN, Ma TZ, Choi HY, Kwak YG. Comparative analysis of serum proteomes of moyamoya disease and normal controls. J Korean Neurosurg Soc. 2010;48(1):8–13.
148. Kashiwazaki D, Uchino H, Kuroda S. Downregulation of apolipoprotein-E and apolipoprotein-J in moyamoya disease - a proteome analysis of cerebrospinal fluid. J Stroke Cerebrovasc Dis. 2017;26(12):2981–2987.
149. Maruwaka M, Yoshikawa K, Okamoto S, et al. Biomarker research for moyamoya disease in cerebrospinal fluid using surface-enhanced laser desorption/ionization time-of-flight mass spectrometry. J Stroke Cerebrovasc Dis. 2015;24(1):104–111.
150. Houkin K, Ito M, Sugiyama T, et al. Review of past research and current concepts on the etiology of moyamoya disease. Neurol Med Chir (Tokyo). 2012;52(5):267–277.
151. Chen JB, Liu Y, Zhou LX, Sun H, He M, You C. Increased prevalence of autoimmune disease in patients with unilateral compared with bilateral moyamoya disease. J Neurosurg. 2016;124(5):1215–1220.
152. Suzuki J, Kodama N. Moyamoya disease–a review. Stroke. 1983;14(1):104–109.
153. Hishikawa T, Tokunaga K, Sugiu K, Date I. Assessment of the difference in posterior circulation involvement between pediatric and adult patients with moyamoya disease. J Neurosurg. 2013;119(4):961–965.
154. Fujimura M, Tominaga T. Current status of revascularization surgery for moyamoya disease: special consideration for its ‘internal carotid-external carotid (IC-EC) conversion’ as the physiological reorganization system. Tohoku J Exp Med. 2015;236(1):45–53.
155. Bower RS, Mallory GW, Nwojo M, Kudva YC, Flemming KD, Meyer FB. Moyamoya disease in a primarily white, midwestern US population: increased prevalence of autoimmune disease. Stroke. 2013;44(7):1997–1999.
156. Chen JB, Liu Y, Zhou LX, Sun H, He M, You C. Prevalence of autoimmune disease in moyamoya disease patients in Western Chinese population. J Neurol Sci. 2015;351(1–2):184–186.
157. Suzuki S, Mitsuyama T, Horiba A, Fukushima S, Hashimoto N, Kawamata T. Moyamoya disease complicated by Graves’ disease and type 2 diabetes mellitus: report of two cases. Clin Neurol Neurosurg. 2011;113(4):325–329.
158. Mejia-Munne JC, Ellis JA, Feldstein NA, Meyers PM, Connolly ES. Moyamoya and inflammation. World Neurosurg. 2017;100:575–578.
159. Takagi Y, Hermanto Y, Takahashi JC, et al. Histopathological characteristics of distal middle cerebral artery in adult and pediatric patients with moyamoya disease. Neurol Med Chir (Tokyo). 2016;56(6):345–349.
160. Masuda J, Ogata J, Yutani C. Smooth muscle cell proliferation and localization of macrophages and T cells in the occlusive intracranial major arteries in moyamoya disease. Stroke. 1993;24(12):1960–1967.
161. Chalouhi N, Ali MS, Jabbour PM, et al. Biology of intracranial aneurysms: role of inflammation. J Cereb Blood Flow Metab. 2012;32(9):1659–1676.
162. Yu X, Li Z. MicroRNAs regulate vascular smooth muscle cell functions in atherosclerosis (review). Int J Mol Med. 2014;34(4):923–933.
163. Oka K, Yamashita M, Sadoshima S, Tanaka K. Cerebral haemorrhage in moyamoya disease at autopsy. Virchows Arch a Pathol Anat Histol. 1981;392(3):247–261.
164. Czabanka M, Pena-Tapia P, Schubert GA, Woitzik J, Vajkoczy P, Schmiedek P. Characterization of cortical microvascularization in adult moyamoya disease. Stroke. 2008;39(6):1703–1709.
165. Baltsavias G, Khan N, Valavanis A. The collateral circulation in pediatric moyamoya disease. Childs Nerv Syst. 2015;31(3):389–398.
166. Kim SJ, Son TO, Kim KH, et al. Neovascularization precedes occlusion in moyamoya disease: angiographic findings in 172 pediatric patients. Eur Neurol. 2014;72(5–6):299–305.
167. Hou K, Ji T, Guo Y, Xu K, Yu J. The coexistence of persistent primitive trigeminal artery, moyamoya disease, and multiple intracranial aneurysms: a case report and literature review. World Neurosurg. 2019;23.
168. Nishino K, Ito Y, Sorimachi T, Shimbo J, Fujii Y. Sturge-Weber syndrome associated with arteriovenous malformation in a patient presenting with progressive brain edema and cyst formation. J Neurosurg Pediatr. 2010;5(5):529–534.
169. Wu Z, Jiang H, Zhang L, et al. Molecular analysis of RNF213 gene for moyamoya disease in the Chinese Han population. PLoS One. 2012;7(10):e48179.
170. Mikami T, Suzuki H, Komatsu K, Mikuni N. Influence of inflammatory disease on the pathophysiology of moyamoya disease and quasi-moyamoya disease. Neurol Med Chir (Tokyo). 2019;59(10):361–370.
171. Choi EH, Lee H, Chung JW, et al. Ring finger protein 213 variant and plaque characteristics, vascular remodeling, and hemodynamics in patients with intracranial atherosclerotic stroke: a high-resolution magnetic resonance imaging and hemodynamic study. J Am Heart Assoc. 2019;8(20):e011996.
172. Zhu B, Liu X, Zhen X, et al. RNF213 gene polymorphism rs9916351 and rs8074015 significantly associated with moyamoya disease in Chinese population. Ann Transl Med. 2020;8(14):851.
173. Cecchi AC, Guo D, Ren Z, et al. RNF213 rare variants in an ethnically diverse population with moyamoya disease. Stroke. 2014;45(11):3200–3207.
174. Zhang Q, Liu Y, Zhang D, et al. RNF213 as the major susceptibility gene for Chinese patients with moyamoya disease and its clinical relevance. J Neurosurg. 2017;126(4):1106–1113.
175. Hitomi T, Habu T, Kobayashi H, et al. Downregulation of Securin by the variant RNF213 R4810K (rs112735431, G>A) reduces angiogenic activity of induced pluripotent stem cell-derived vascular endothelial cells from moyamoya patients. Biochem Biophys Res Commun. 2013;438(1):13–19.
176. Hitomi T, Habu T, Kobayashi H, et al. The moyamoya disease susceptibility variant RNF213 R4810K (rs112735431) induces genomic instability by mitotic abnormality. Biochem Biophys Res Commun. 2013;439(4):419–426.
177. Sonobe S, Fujimura M, Niizuma K, et al. Temporal profile of the vascular anatomy evaluated by 9.4-T magnetic resonance angiography and histopathological analysis in mice lacking RNF213: a susceptibility gene for moyamoya disease. Brain Res. 2014;1552:64–71.
178. Kanoke A, Fujimura M, Niizuma K, et al. Temporal profile of the vascular anatomy evaluated by 9.4-tesla magnetic resonance angiography and histological analysis in mice with the R4859K mutation of RNF213, the susceptibility gene for moyamoya disease. Brain Res. 2015;1624:497–505.
179. Kanoke A, Fujimura M, Niizuma K, et al. Temporal profile of magnetic resonance angiography and decreased ratio of regulatory T cells after immunological adjuvant administration to mice lacking RNF213, a susceptibility gene for moyamoya disease. Brain Res. 2016;1642:1–9.
180. Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39(12):1488–1493.
181. Milewicz DM, Ostergaard JR, Ala-Kokko LM, et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A. 2010;152A(10):2437–2443.
182. Heyer GL, Dowling MM, Licht DJ, et al. The cerebral vasculopathy of PHACES syndrome. Stroke. 2008;39(2):308–316.
183. Siegel DH, Tefft KA, Kelly T, et al. Stroke in children with posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities (PHACE) syndrome: a systematic review of the literature. Stroke. 2012;43(6):1672–1674.
184. Ganesan V, Kirkham FJ. Noonan syndrome and moyamoya. Pediatr Neurol. 1997;16(3):256–258.
185. Hinnant CA. Noonan syndrome associated with thromboembolic brain infarcts and posterior circulation abnormalities. Am J Med Genet. 1995;56(2):241–244.
186. Paez MT, Yamamoto T. Single nucleotide polymorphisms of tissue inhibitor of metalloproteinase genes in familial moyamoya disease. Neurosurgery. 2007;60(3):E582.
187. Bersano A, Guey S, Bedini G, et al. Research progresses in understanding the pathophysiology of moyamoya disease. Cerebrovasc Dis. 2016;41(3–4):105–118.
188. Aoyagi M, Fukai N, Matsushima Y, Yamamoto M, Yamamoto K. Kinetics of 125I-PDGF binding and down-regulation of PDGF receptor in arterial smooth muscle cells derived from patients with moyamoya disease. J Cell Physiol. 1993;154(2):281–288.
189. Saharinen P, Kerkela K, Ekman N, et al. Multiple angiopoietin recombinant proteins activate the Tie1 receptor tyrosine kinase and promote its interaction with Tie2. J Cell Biol. 2005;169(2):239–243.
190. Blecharz KG, Frey D, Schenkel T, et al. Autocrine release of angiopoietin-2 mediates cerebrovascular disintegration in moyamoya disease. J Cereb Blood Flow Metab. 2016;5.
191. Yu J, Huang K, Pan J, Shen J, Zhan R. Significance of serum angiopoietin-2 in patients with hemorrhage in adult-onset moyamoya disease. Biomed Res Int. 2020;2020:8209313.
192. Bang OY, Chung JW, Kim SJ, et al. Caveolin-1, Ring finger protein 213, and endothelial function in moyamoya disease. Int J Stroke. 2016;11(9):999–1008.
193. Chung JW, Kim DH, Oh MJ, et al. Cav-1 (caveolin-1) and arterial remodeling in adult moyamoya disease. Stroke. 2018;49(11):2597–2604.
194. Taylor CT, Doherty G, Fallon PG, Cummins EP. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J Clin Invest. 2016;126(10):3716–3724.
195. Zhang Z, Yao L, Yang J, Wang Z, Du G. PI3K/Akt and HIF1 signaling pathway in hypoxiaischemia (Review). Mol Med Rep. 2018;18(4):3547–3554.
196. Soriano SG, Cowan DB, Proctor MR, Scott RM. Levels of soluble adhesion molecules are elevated in the cerebrospinal fluid of children with moyamoya syndrome. Neurosurgery. 2002;50(3):544–549.
197. Hamauchi S, Shichinohe H, Uchino H, et al. Cellular functions and gene and protein expression profiles in endothelial cells derived from moyamoya disease-specific iPS cells. PLoS One. 2016;11(9):e0163561.
198. Strimbu K, Tavel JA. What are biomarkers? Curr Opin HIV AIDS. Nov. 2010;5(6):463–466.
199. Bang OY, Goyal M, Liebeskind DS. Collateral circulation in ischemic stroke: assessment tools and therapeutic strategies. Stroke. 2015;46(11):3302–3309.
200. Navarro G, Borroto-Escuela DO, Fuxe K, Franco R. Potential of caveolae in the therapy of cardiovascular and neurological diseases. Front Physiol. 2014;5:370.
201. Lin N, Baird L, Koss M, et al. Discovery of asymptomatic moyamoya arteriopathy in pediatric syndromic populations: radiographic and clinical progression. Neurosurg Focus. 2011;31(6):E6.
202. Koss M, Scott RM, Irons MB, Smith ER, Ullrich NJ. Moyamoya syndrome associated with neurofibromatosis Type 1: perioperative and long-term outcome after surgical revascularization. J Neurosurg Pediatr. 2013;11(4):417–425.
203. Kuroda S, Hashimoto N, Yoshimoto T, Iwasaki Y. Research Committee on Moyamoya Disease in Japan. Radiological findings, clinical course, and outcome in asymptomatic moyamoya disease: results of multicenter survey in Japan. Stroke. 2007;38(5):1430–1435.
204. Kuroda S, Group AS. Asymptomatic moyamoya disease: literature review and ongoing AMORE study. Neurol Med Chir (Tokyo). 2015;55(3):194–198.
205. Lee JY, Kim SK, Cheon JE, et al. Posterior cerebral artery involvement in moyamoya disease: initial infarction and angle between PCA and basilar artery. Childs Nerv Syst. 2013;29(12):2263–2269.
206. Smith ER, Scott RM. Progression of disease in unilateral moyamoya syndrome. Neurosurg Focus. 2008;24(2):E17.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.