")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Genetic and epigenetic mechanisms of epilepsy: a review
Authors Chen T, Giri M, Xia ZY, Subedi YN, Li Y
Received 18 May 2017
Accepted for publication 15 June 2017
Published 13 July 2017 Volume 2017:13 Pages 1841—1859
DOI https://doi.org/10.2147/NDT.S142032
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Tian Chen,1,* Mohan Giri,2,* Zhenyi Xia,3 Yadu Nanda Subedi,2 Yan Li1
1Department of Health Management Center, Chongqing Three Gorges Central Hospital, Chongqing, People’s Republic of China; 2National Center for Rheumatic Diseases, Ratopul, Gaushala, Kathmandu, Nepal; 3Department of Thoracic Surgery, Chongqing Three Gorges Central Hospital, Chongqing, People’s Republic of China
*These authors contributed equally to this work
Abstract: Epilepsy is a common episodic neurological disorder or condition characterized by recurrent epileptic seizures, and genetics seems to play a key role in its etiology. Early linkage studies have localized multiple loci that may harbor susceptibility genes to epilepsy, and mutational analyses have detected a number of mutations involved in both ion channel and nonion channel genes in patients with idiopathic epilepsy. Genome-wide studies of epilepsy have found copy number variants at 2q24.2-q24.3, 7q11.22, 15q11.2-q13.3, and 16p13.11-p13.2, some of which disrupt multiple genes, such as NRXN1, AUTS2, NLGN1, CNTNAP2, GRIN2A, PRRT2, NIPA2, and BMP5, implicated for neurodevelopmental disorders, including intellectual disability and autism. Unfortunately, only a few common genetic variants have been associated with epilepsy. Recent exome-sequencing studies have found some genetic mutations, most of which are located in nonion channel genes such as the LGI1, PRRT2, EFHC1, PRICKLE, RBFOX1, and DEPDC5 and in probands with rare forms of familial epilepsy, and some of these genes are involved with the neurodevelopment. Since epigenetics plays a role in neuronal function from embryogenesis and early brain development to tissue-specific gene expression, epigenetic regulation may contribute to the genetic mechanism of neurodevelopment through which a gene and the environment interacting with each other affect the development of epilepsy. This review focused on the analytic tools used to identify epilepsy and then provided a summary of recent linkage and association findings, indicating the existence of novel genes on several chromosomes for further understanding of the biology of epilepsy.
Keywords: epilepsy, genetics, neurodevelopment disorder, neurological disorder
Introduction
Epilepsy is a common chronic neurological disorders characterized by recurrent seizures due to excessive electrical discharges in a group of brain cells. Seizures are characterized by brief episodes of involuntary shaking that may involve a part or the entire body and sometimes are accompanied by loss of consciousness and loss of bowel or bladder control. While the classification of epilepsy is often evolving clinically, epilepsies tend to have two broad categories: focal and nonfocal epilepsy.1 Focal epilepsy is defined as a seizure originating within one cerebral hemisphere, whereas the nonfocal epilepsy is mostly a generalized epilepsy (GE). Nonfocal epilepsy such as absence seizure is characterized by a transitory alteration of consciousness, with or without other clinical signs, and each episode lasts between 3 and 30 seconds. Bimodal distribution for age of onset with the first peak at 6–7 years (childhood) and the second peak at ~12 years of age (juvenile) is a feature of typical absence seizure, but it is possible to have an absence seizure at any age. Absence seizures are broadly divided into typical and atypical types: typical absence seizure has a frequency >2.5 Hz and is characterized by transient impairment of consciousness accompanied by one or more other features such as staring, behavioral arrest, and eyelid fluttering. Atypical absence seizures have a frequency <2.5 Hz, have less abrupt onset and offset and variable alteration of consciousness, and tend to last longer than typical absence seizures.1–3 In addition, a smaller proportion of epilepsies are unclassifiable;2 ~60% of individuals with epilepsy are classified as focal.3
The development of epilepsy has a great variation in age and geographic distribution. Epilepsy affects ~1% of worldwide populations at the age of 20 years and 3% at the age of 75 years.4 According to the World Health Organization (WHO),5 ~80% of epilepsy cases occur in the developing countries and three-fourth of affected individuals do not get appropriate treatment. The onset of new cases occurs most frequently in infants and the elderly in the developed countries, whereas new cases are often seen in older children and young adults in the less developed world.6 Therefore, epilepsy may cause a different disease burden between the more developed and the less developed world and has different implications for the etiology.
Epilepsy is believed to have a genetic component. Berkovic et al7 revealed that environmental insults such as trauma, hypoxia, and vascular lesions may alter function of ion channels and result in altered transcription, thus causing acquired channelopathies. Both the inherited and acquired domains are important for epilepsy genetics. Twin studies suggest that the heritability of epilepsy is ~25%–70% and appears to increase with age. Studies have shown that the probandwise concordance rate is 0.37 in monozygotic twins, which is significantly higher than that in dizygotic twins.8 A recent study reported that the heritability is 32% for all epilepsy, 23% for focal epilepsy, and 36% for nonfocal epilepsy.9 While genetic linkage analyses have identified several loci that may harbor susceptibility loci to epilepsy. Recent advances in genomic technology have made it feasible to identify common genetic variants and to screen for copy number variants (CNVs) associated with epilepsies through genome-wide analysis or detect single-point mutations through exome sequencing or whole-genome sequencing. Epilepsy may result from infection and trauma or can also develop because of brain damage from other disorders such as brain tumor, alcoholism, Alzheimer’s disease, and stroke.10
Recent genetic research has led to the identification of a significant number of genes implicated in monogenic forms of epilepsy. However, other causative genes involved in a considerable proportion of familial epilepsies need to be identified to fully elucidate the etiology of epilepsy. Here, we provided a review on the genetics of epilepsy by discussing the genetic variants identified through a variety of approaches and epigenetic processes for further understanding of the biology of epilepsy.
Search strategy
The relevant studies were searched using key words in four acknowledged databases: Web of Science, Springer, Scopus, and MEDLINE. This review screened for studies conducted in the period from 1988 to April 2017 using the following key words: epilepsy, genetics, neurodevelopment disorder, and neurological disorder. A study was included if it matched the corresponding period from 1988 to April 2017. This review included randomized controlled trials (RCTs), observational studies, descriptive studies, web pages, study protocols, seminar papers, retrospective studies, reviews, books, book chapters, and abstracts on the research topic.
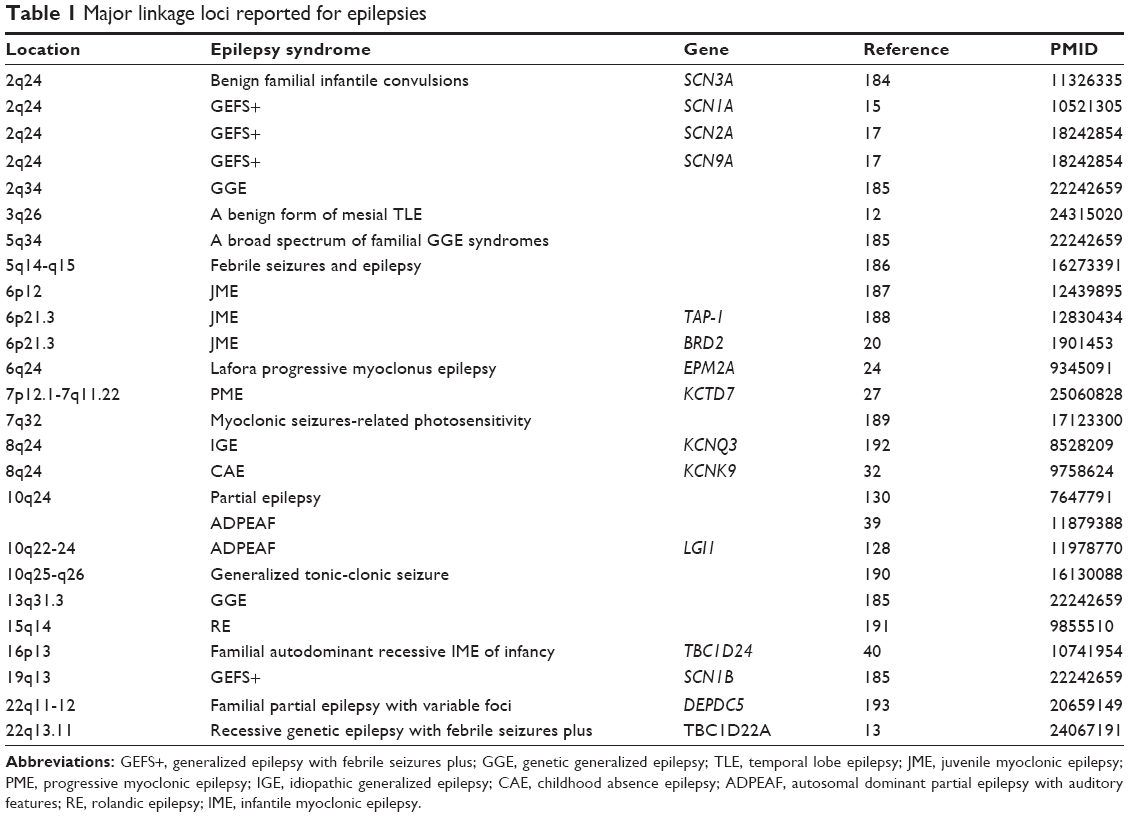
Genetic linkage loci
Genome-wide linkage scans and linkage analyses of some candidate regions have reported about 20 chromosome loci for epilepsy (Table 1). An acquired structural problem in the temporal lobe is a frequent feature of temporal lobe epilepsy (TLE). TLE consists of a heterogeneous group of seizure disorders, such as lateral familial TLE, mesial familial TLE, and familial partial epilepsy with variable foci.11,12 A single nucleotide polymorphism (SNP)-based linkage study of familial mesial TLE provided a suggestive linkage to chromosome 3q26.12 Belhedi et al13 demonstrated that chromosome 22q13.31 has been linked to recessive genetic generalized epilepsy with febrile seizures plus (GEFS+) in a Tunisian consanguineous family.
 | Table 1 Major linkage loci reported for epilepsies |
Chromosome 2q24
GEFS+ has been linked with mutations of three sodium channel genes and two gamma-aminobutyric acid (GABA)A receptor genes. The components of GEFS+ include febrile seizures persisting beyond childhood; afebrile, generalized, tonic-clonic, absence, myoclonic, atonic, uncomplicated febrile seizures; and temporal lobe seizures.14 The linkage locus 2q24 is localized with both GEFS+ and familial febrile seizures.15 In families with GEFS+, mutations in gene encoding ligand-gated GABAA receptor (GABAR) subunits such as GABRG2 and GABRD cause epilepsy by haploinsufficency.14,15 Two distinct regions at 2q24.2-q24.3, harboring possible SCN cluster genes and SLC4A10, respectively, are linked to idiopathic epilepsy.16 Interestingly, four of nine genes encoding the sodium channel pore-forming α subunits, SCN1A, SCN2A, SCN3A, and SCN9A, are located on the 2q24.3 locus.17 MASS1 was mapped to chromosome 5q14 and a loss-of-function mutation in MASS1 was associated with seizure phenotype, but MASS1 gene was not likely to be a contributing factor in most families with febrile seizures.14–17
Chromosome 6p21.3
Juvenile myoclonic epilepsy (JME) is one of the most common forms of idiopathic generalized epilepsy (IGE), and a locus for JME linked to human leukocyte antigen on chromosome 6p21.3 was originally termed EJM1 (OMIM 254770).18–20 Pal et al21 revealed that microsatellite marker for the bromodomain-containing 2 (BRD2) locus on chromosome 6p21.3 was associated with a highly significant linkage disequilibrium with several SNPs. They also found that along with two SNPs in the promoter region of BRD2, one haplotype (rs620202-rs516535-brd2-rs635688-rs2066741, G-C-6-T-T) was also associated with JME. BRD2 (RING3) is a putative nuclear transcriptional regulator from a family of genes that is expressed during development, suggesting that abnormalities in neural development may be a cause of common idiopathic epilepsy. Another candidate gene identified in the 6p21.3 locus is the transporter 1 ATP-binding cassette sub-family B (TAP-1), a gene encoding the transporter associated with antigen processing that may be a susceptibility factor to JME.22 CLCN2 encodes the voltage-gated chloride channel ClC-2, and it is regarded as a predisposing gene for IGE. CLCN2 mutation impairs GABAergic neurotransmission in the brain by altering chloride gradient across the cellular membrane.18–20 CACNB4 encodes a member of the beta subunit family of voltage-dependent calcium channel complex protein, and mutations in this gene have been associated with IGE juvenile episodic ataxia, type 5, and myoclonic epilepsy.18,19 JRK/JH8, which maps to 8q24, has been associated with some forms of IGE.18
The genetic mechanisms of BRD2 associated with epilepsy might be because 1) BRD2 haploinsufficiency is associated with a deficit in GABAergic neurons, which lead to an increased susceptibility to provoked seizure and 2) haploinsufficiency is associated with the basal ganglia path in structures, critical for control of seizure activity.23
Chromosome 6q24
Sainz et al24 reported an extended area of homozygosity on chromosome 6q23-25 in nine patients of Lafora progressive myoclonus epilepsy from four families with consanguineous marriages. Using positional cloning, a novel gene epilepsy progressive myoclonus type 2A (EPM2A) gene was identified.25 So far distinct mutations in EPM2A and EPM2B (or MALIN) have been discovered in patients with the Lafora progressive myoclonus epilepsy in over 200 independent families with the Lafora disease. Among these mutations, half of them are missense mutations and one-quarter are deletions.26 EPM2A encodes a protein with consensus amino acid sequence, indicative of a protein tyrosine phosphatase (PTP), whereas EPM2B encodes an ubiquitin ligase. Mutations in these regions show a critical impact on the functionality of laforin, which works together with malin as a complex to promote the degradation of malin targets through the ubiquitin-proteasome system.
Chromosome 7p12.1-7q11.22
A homozygous region on chromosome 7p12.1-7q11.22 has been identified for progressive myoclonic epilepsy (PME). Multiple mutations (eg, p.Tyr276Cys and p.R94W) are detected for PME in potassium channel tetramerization domain-containing 7 (KCTD7).27–29 KCTD7 is a member of the KCTD gene family. It is predominantly expressed in the brain to hyperpolarize cell membrane and reduce the excitability of transfected neurons.27 The effect on the plasma membrane resting potential is possibly mediated by KCTD7 in interaction with Cullin-3, an ubiquitin-ligase component.30
Chromosome 8q24
Chromosome 8q24 has been found to be associated with IGE, familial adult myoclonic epilepsy,31 and childhood absence epilepsy (CAE).32 A mutation in Twik-like acid-sensitive K+ channel (KCNK9) was revealed in a genetic model of absence epilepsy.33 KCNK9 encodes K2P9.1, a protein that contains multiple transmembrane regions and two pore-forming P domains and functions as a pH-dependent potassium channel. A standard karyotype analysis had detected a 2.3 Mb duplication of chromosome 8q24.3 associated with severe mental retardation and epilepsy.34 Unfortunately, previous mutational screening of 8q24.3 in epilepsy failed to find any association with variants in JRK/JH8 or ARC, genes that have been implicated in other neuropsychiatric disorders such as schizophrenia and bipolar.35,36
Chromosome 10q24
An initial linkage evidence was detected in a 10 cM interval on 10q in a large pedigree with 10 individuals affected with partial epilepsy,37 which was replicated in a follow-up study of a four-generation family with nine individuals affected with temporal partial epilepsy.38 Another follow-up linkage study in the four multiplex families with autosomal dominant partial epilepsy with auditory features (ADPEAF) also confirmed this linkage locus.39 Mutation in the LGI1 gene/Epitempin on 10q24 may cause autosomal dominant lateral temporal epilepsy (ADLTE).
Chromosome 16p13.3
A genome-wide linkage scan in a large Italian pedigree segregating recessive idiopathic myoclonic epilepsy mapped a linkage locus on chromosome 16p13. Haplotype analysis defined the critical region within a 3.4-cM interval between D16S3024 and D16S423.40 Mutations including two compound heterozygous missense mutations (D147H and A509V) in TBC1D24 were identified for familial infantile myoclonic epilepsy.41–44 The gene mutations together with its predominant expression in critical epileptogenic brain areas highlighted that TBC1D24 plays a fundamental role in the regulation of neuronal network excitability. In situ hybridization analysis revealed that TBC1D24 is mainly expressed in the cerebral cortex and the hippocampus. TBC1D24 modulates the small GTPase ADP-ribosylation factor 6 (ARF6), which has been implicated for the modulation of brain excitability and regulates neuronal migration and maturation through modulation of the ARF6-dependent pathway.44
CNVs
CNVs are alterations in the DNA that results in the cell having an abnormal variation in the number of copies of one or more sections of the DNA. CNVs are caused by structural rearrangements of the genome, such as deletions, duplications, inversions, and translocations; they are causes of some nervous system disorders.45 Methods for the direct detection of CNVs across the whole genome have become effective instruments for identifying genetic risk factors of human diseases.46 CNVs can be private and recurrent, normally rare but highly penetrant for a disease. While the private CNVs can occur anywhere across the genome, the recurrent CNVs occur at genomic hot spots that can increase the risk of a range of disorders. One CNV often disrupts multiple genes and therefore may cause multiple diseases or a syndrome.
CNVs vary according to phenotype and have been reported in up to 28% of patients with epilepsy.47 Genome-wide analysis of 517 individuals with epilepsy and 2,493 controls suggests that 8.9% of patients carry one and more rare CNVs that were not present in controls. Of these CNVs, 2.9% of patients have deletions at loci 15q11.2, 15q13.3, or 16q13.11, which are genomic hotspots previously associated with intellectual disability, autism, and schizophrenia.48 Similar deletions were confirmed in several independent samples.49–52 Other studies53–56 have also found recurrent distal deletion at 7q11.22, maternally derived duplication at 15q11.2-q13.3, duplication of 16p11.2, and duplication of 16p13 in patients with epilepsy (Table 2). In patients with infantile epilepsy, duplication at 2q24.2-q24.3 was found. In a recent study, microduplication at 6p12.1 and microdeletion at 7q32.3, predicting BMP5 and PODXL, respectively, were found in multiple patients with epilepsy or epilepsy plus mental retardation or with JME.57
 | Table 2 Summary of CNVs identified for epilepsy |
In patients with IGEs, recurrent microdeletions were found more frequent at 15q11.2.49 The 15q11.2 microdeletion disrupted non-imprinted genes in Prader–Willi syndrome (PWS)/Angelman syndrome (AS) 2 (NIPA2) in patients with CAE58 and schizophrenia59 in two independent studies in the Han Chinese populations. This region contained four highly conserved and non-imprinted genes, NIPA1, NIPA2, CYFIP1, and TUBGCP5. A study of 52 patients carrying this CNV found that 15q11.2 was prevalent in neurodevelopment disorders such as developmental delay (68.3%), speech impairment (85.4%), and psychological issues (63.4%) that included attention-deficit hyperactivity disorder (ADHD), autistic spectrum disorder (ASD), or obsessive-compulsive disorder (OCD), and 18.7% of patients were noted with seizures and 17.3% with associated congenital heart diseases.60
Several other deletions that disrupt multiple genes are also detected for neurodevelopmental disorders, such as autism, schizophrenia, mental retardation, and epilepsy. In the 16p13.11 deletion, the gene nuclear distribution protein nudE homolog 1 (NDE1) is suspected as the leading disease-causing gene at 16p13.11.51 A novel 16p13.2 microdeletion, disrupting ionotropic glutamate N-methyl-D-aspartate (NMDA) receptor subunit 2A (GRIN2A) and proline-rich transmembrane protein 2 (PRRT2), is detected in patients with rolandic epilepsy (RE).61 A 20-kb microdeletion was found in two sisters with auditory focal seizure at 200 kb away from exon 1 of NRXN1, and in the same family, a 50.5-kb microdeletion within the neuroligin 1 (NLGN1) gene at 3q26.31 was co-segregated with both lateral temporal epilepsy and with the NRXN1 deletion simultaneously.62 Deletions in AUTS2 and CNTNAP2 implicated for other neurodevelopmental disorders such as autism may also cause epilepsy.48,63 Kim et al64 revealed that patients with 22q11.2DS tend to have a higher risk of epilepsy.
Chromosome translocations may cause epilepsy. A de novo balanced translocation t(6;22) (p21.32;q11.21) has been detected in patients with epilepsy with myoclonic absences (EMA) and intellectual disability.65 The breakpoint, between exon 5 and exon 6 of SYNGAP1, is found to truncate the NMDA receptor-associated gene SYNGAP1, encoding the synaptic Ras GTPase activating protein 1. The protein is a major component of the postsynaptic density, exclusively expressed in the brain, predominantly in the cortex, hippocampus, and olfactory bulb, and it is activated using NMDA receptor activator.66 Mutations in SYNGAP1 may result in nonsyndromic intellectual disability.65 Dysfunction of SYNGAP1 contributes to the development of generalized EMA. Another translocation involving chromosome 6p21, t(4;6) (q35;p21), is reported in two patients with IGE within a nuclear family.67 A de novo duplication, found in a single patient with epilepsy and development delay, is likely linked to genes (GABBR1, BRD2, and GRM4) that are involved with brain function and synaptic transmission.68
Common genetic variants
Genome-wide association studies have successfully identified numerous susceptibility genes for epilepsy. Using two cohorts of case–control samples from Hong Kong and Taiwan, a study showed that two SNPs rs2292096 and rs6660197 located at CAMSAP1L1 on 1q32.1 were associated with epilepsy at a genome-wide significance.69 Aside from rs702416 in KCND2, the rs1980416 at DSCAM and rs13020314 at ERBB4 were also associated with epilepsy. ERBB4 encodes ErbB4, a member of the type I receptor tyrosine kinase subfamily that promotes synapse formation of GABA-containing interneurons in the hippocampus. Two SNPs rs9390745 and rs4840200 at GRIK2 (encoding GluR6, glutamate ionotropic receptor, kainate 2) on 6q21 also showed strong association signals in the discovery sample. Meta-analysis70 of 12 genome-wide association studies, including nine cohorts of European ancestry populations, two African-Americans, and one Chinese from Hong Kong and Taiwan, suggested that common variants at SCN1A and PCDH7 are associated with epilepsies. Several other variants at GOLIM4, GABRA2, VRK2/FANCL, and MMP8 are likely associated with different forms of epilepsy, although not genome-wide significant.
Candidate gene approach elucidated several common variants or mutational variants associated with epilepsy. Consistent genetic associations with epilepsy have been observed in carboxypeptidase A6 (CPA6) on 8q13.2 and the calcium homeostasis modulator 1 (CALHM1) on 10q24.33.71–73 CPA6 protein plays a role in the activation of peptides that have neuroprotective effects or in the inactivation of peptides that cause neuronal stimulation. Mutations of G267R and A270V in CPA6 are linked to familial TLE. These mutations cause a decreased level of CPA6 protein in the extracellular matrix. The reduction in CPA6 activity may lead to epilepsy by an imbalance of excitation and inhibition.72,74 CALHM1 encodes calcium channel, interferes with calcium homeostasis and increases amyloid beta (Aβ) levels. CALHM1 rs2986017 might be associated with an increased risk for Alzheimer’s disease in multiple populations.75–77 However, there are multiple inconsistent replications.78 In an analysis of five SNPs in CALHM1, an SNP rs11191692 was associated with TLE after correction for multiple testing, and a haplotype analysis of five SNPs provided a support that this association was independent of apolipoprotein E (APOE) epsilon 4.79 APOE, a gene widely associated with neurological disorders and some cardiovascular traits, was also associated with TLE. A meta-analysis of seven published studies showed that carrying of APOE4 allele tends to have an early age at the onset of TLE.80
Rare mutational variants
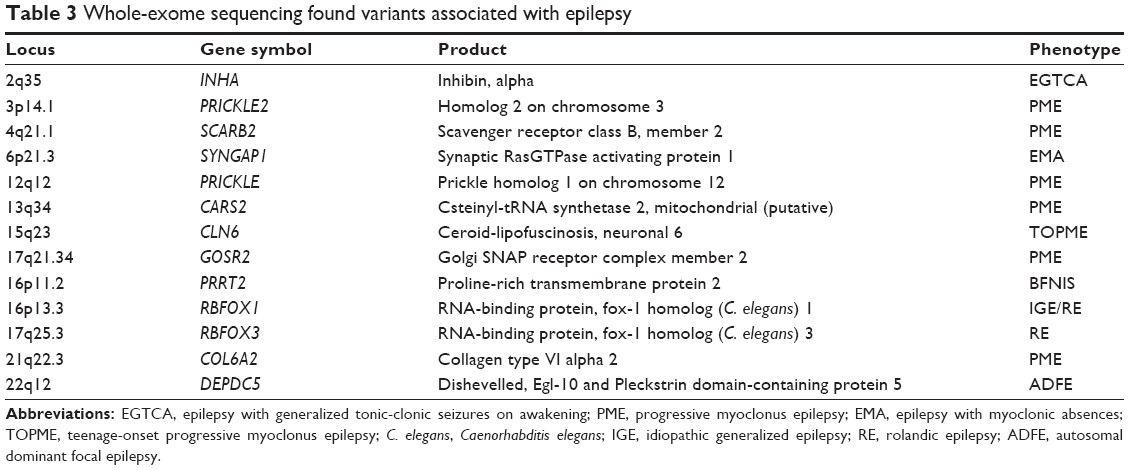
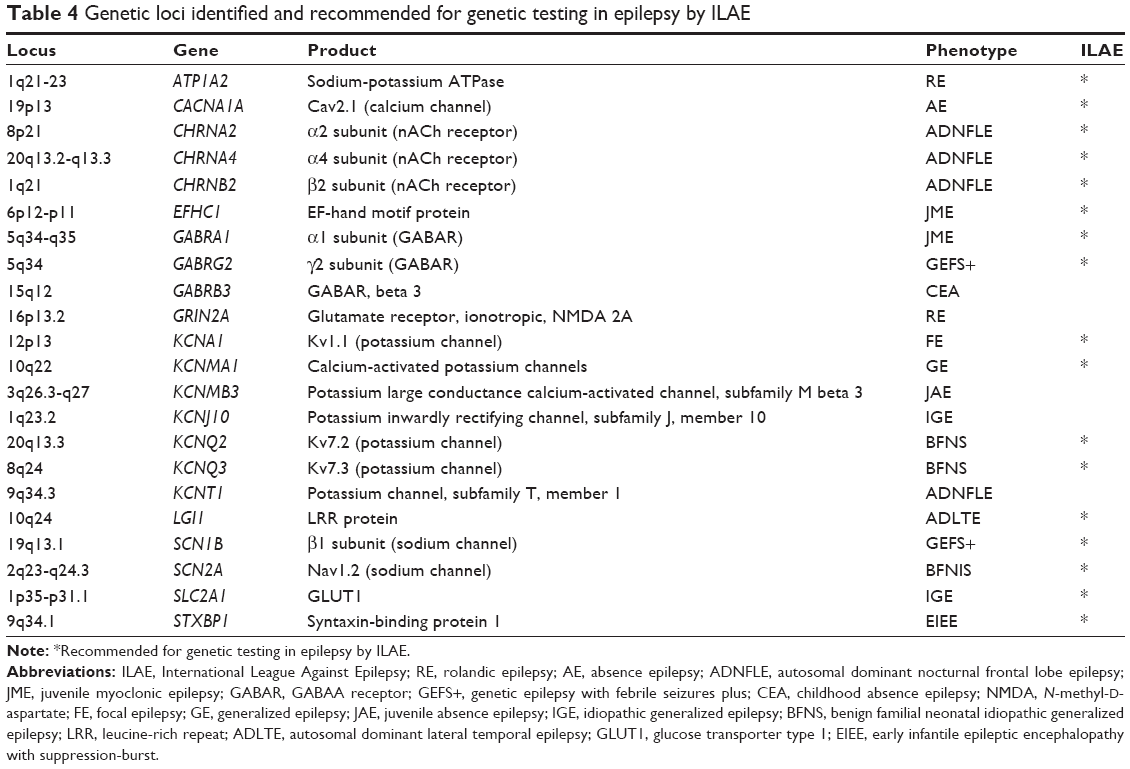
Exome sequencing is a technique for sequencing all the protein-coding genes in the genome. It is an effective approach to study rare Mendelian diseases. Exome-sequencing analysis has opened an avenue to discover genetic variants for epileptogenesis across the whole genome and to determine the physiologic effects of mutations in epilepsy-associated genes.81 Because of genetic heterogeneity or population stratification, however, exome sequencing is likely difficult in detecting rare variants associated with complex disorders in a population-based sample such as unrelated cases and controls. For example, through exome sequencing of 237 channel genes in cases and controls, Klassen et al82 found little evidence or biological rationale for an SNP load effect in ion channelopathy. Another study using exome sequencing followed by genotyping in a larger sample failed to identify single rare variants of large effect in IGE.83 Whole-genome sequencing has revealed a de novo SCN8A mutation and de novo mutations in HCN1.81–83 Other examples of recent gene discoveries are listed in Table 3. Exome-sequencing analysis of family samples has detected >200 rare variants in ~20 genes in patients with epilepsies, of which some have been recommended by the International League Against Epilepsy (ILAE) as likely causal variants for idiopathic epilepsy (Table 4). While most of these variants are identified in individual families with a small sample size, only a small portion (<3%) of these variants is found in the 1000 Genomes Project database or NHLBI Exome Variant Server (EVS). This suggests that using exome-sequencing approach to identify rare variants in the family with individuals affected with epilepsy is robust.84 These rare variants are mostly located in genes encoding for ion channels, including voltage-gated sodium, potassium, calcium, chloride channels, and receptors such as ligand-gated GABAA, glutamate, and acetylcholine receptors.
 | Table 3 Whole-exome sequencing found variants associated with epilepsy |
 | Table 4 Genetic loci identified and recommended for genetic testing in epilepsy by ILAE |
Ion channel gene mutations
Ion channel is a protein macromolecule spanning a biological membrane that allows ions to pass from one side of the membrane to the other.85 There are two types of ion channel: voltage gated and leakage. A voltage-gated ion channel can be opened or closed at some circumstances. The opening of voltage-gated ion channel can be archived by either a voltage change across the membrane or a binding of a chemical substance such as a ligand to a receptor in or near the channel (chemically activated or ligand-gated channel).
Voltage-gated sodium channels
Voltage-gated sodium channels play an essential role in the initiation and propagation of action potentials in neurons and other electrically excitable cell.86 They include nine α subunits named Nav1.1–Nav1.9 that are encoded by the gene from SCN1A–SCN11A, respectively, in which the SCN6/7A gene is part of the Nax subfamily, and four β subunits named Nav β1–Nav β4 that are encoded by SCN1B–SCN4B, respectively. In the mammalian brain, normally voltage-gated sodium channels consist of an α subunit that forms the ion conduction pore and one to two β subunits, but only the α subunit is required for function.87 Genetic epilepsy syndromes with a wide range of severity may result from voltage-gated sodium channel mutation. Benign neonatal infantile seizure is caused by mutations in NaV1.2 channels, and patients may experience episodes of discoordination and continuous muscle movement (myokymia) between attacks of ataxia.86,87
Mutations in the genes encoding for α subunits play an important role in the development of epilepsy. Following up the linkage locus 2q24, sequencing of SCN1A demonstrated that individuals affected with epilepsy are heterozygous for missense mutations (T875M and R1648H).88 Direct sequencing of SCN1A revealed a novel heterozygous mutation (c.5383G>A) in one Chinese family with GEFS+.89 Two novel mutations (p.V1612I and p.C1756G) in SCN1A were identified in Indonesian patients with severe myoclonic epilepsy in infancy.90
So far, >800 mutations in genes encoding neuronal Nav channels, including SCN1A and SCN2A, have been described in patients with epilepsy.91 Many of these mutations are sporadic and cause loss of function, which demonstrates haploinsufficiency of SCN1A.92 In SCN2A, a heterozygous de novo nonsense mutation R102X is found with intractable epilepsy plus mental decline.93 The de novo missense mutation (p.Ala263Val) in SCN2A, which leads to a pronounced gain of function, is found with neonatal-onset seizures and variable episodes of ataxia.94 Only a few mutations,17 such as a charge-neutralizing mutation K345Q and four novel missense variants (R357Q, D766N, E1111K, and M1323V), are found in SCN3A with focal epilepsy in children.91
In addition, a mutation in SCN1B predicting a deletion of five amino acids was found in a family with both febrile seizure and early onset absence epilepsy.95 The genes SCN1A, SCN1B, and SCN2A have been recommended by the ILAE for genetic testing in epilepsy.
Voltage-gated potassium channels
Potassium channels are a diverse family of membrane proteins that are present in both excitable and non-excitable cells found in all living organisms. Members of this channel family play critical roles in cellular signaling processes regulating neurotransmitter release, heart rate, insulin secretion, neuronal excitability, smooth muscle contraction, and cell volume.96 Based on the primary amino acid sequence of the pore-containing subunit, potassium channels are generally classified into: 1) six transmembranes with one-pore channel (voltage-gated potassium channels); 2) two transmembranes with one-pore channel (inward rectifier K+ channels); 3) the calcium-activated potassium channels; and 4) four transmembranes with two-pore channels.
The voltage-gated potassium channels (Kv) include α and β subunits. While α subunits form the actual conductance pore, β subunits are auxiliary proteins that associate with α subunits. There are about 40 known human voltage-gated potassium channel α subunits that can be grouped into 12 classes, labeled as Kvα1–12 or Kv1–12.97 The grouping is according to function and then subgrouped according to the Kv sequence homology classification scheme. The most common class includes the Shaker-related potassium channels Kv1.1–Kv1.7, which is encoded by KCNA1–KCNA7. Human ether-a-go-go-related K+ channels Kv11.1 or hERG, Kv11.2, and Kv11.3 are encoded by KCNH2, KCNH6 and KCNH7, and KCNQ channels, respectively. Kv7.1–7.5 is encoded by KCNQ1–KCNQ5.98 In addition, the known human calcium-activated (Kca) channels are grouped into Kca 2/3 (Kca2.1, Kca2.3, Kca2.3, and Kca3.1, encoded by KCNN1–KCNN4, respectively) and Kca1/4/5 (Kca1.1, Kca4.1, Kca4.2, and Kca5.1, encoded by KCNMA1, KCNT1, KCNT2, and KCNU1, respectively).99 The inward rectifier K+ channels consist of seven subfamilies denoted as Kir1–Kir7.
Mutations in multiple genes in the voltage-gated potassium channels are detected in epilepsy. They include mutations in KCNA1 in families with autosomal dominant episodic ataxia type 1 (EA1) and epilepsy.100 KCNA1 mutations have been associated with phenotypic variability and may increase vagus nerve activity sufficiently to cause sudden unexplained death in epilepsy.97–100 A mutation in KCNMA1 has been identified in a family with generalized epilepsy and paroxysmal dyskinesia (GEPD), and multiple mutations in KCNQ2 and KCNQ3 were associated with benign familial neonatal convulsion or epilepsy.101,102 KCNMA1 encodes the a subunit of the large conductance, voltage-gated, and calcium-activated K+ channel The four loci KCNA1, KCNMA1, KCNQ2, and KCNQ3 are recommended for genetic testing in epilepsy by the ILAE.
Mutations in several other genes such as KCNH1, KCND2, and KCTD7 encoding for potassium channels are implicated for epilepsy. Damaging de novo mutations in KCNH1 (Kv10.1) were found in six individuals with Temple–Baraitser syndrome (TBS), in which two mothers of children with TBS had epilepsy.103 Whole-exome sequencing of a family with identical twins affected with autism and severe intractable seizures found the de novo mutation p.Val404Met in KCND2.104 A homozygous missense mutation (p.R94W) in a highly conserved segment of exon 2 of KCTD7 was found in two independent families with PME.29 Recently, whole-exome sequencing of a family with three daughters affected with PME and ataxia revealed an additional mutation p.Tyr276Cys in KCTD7.27 The gene KCNJ10 encoding a member of the inward rectifier-type potassium channel family has been implicated in multiple neurological disorders, including epilepsy. A common missense variation (Arg271Cys) in KCNJ10 is associated with susceptibility to both focal and generalized epilepsies as well as common IGE syndromes.105 Heterozygous missense mutations in KCNJ10 may cause epilepsy, ataxia, sensorineural deafness, and tubulopathy (EAST).106,107 KCNJ10 is characterized by having a greater tendency to allow potassium to flow into a cell. It may affect the potassium-buffering action of glial cells in the brain.
Genes in the sodium-activated and calcium-activated potassium channels are implicated for epilepsy. A missense heterozygous de novo mutation p.Phe932Ile in KCNT1 is identified for severe epilepsy.108 Several mutations (M896I, R398Q, Y796H, and R928C) have been recently detected in severe cases of autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE).109 The gene mutations may increase the sensitivity of potassium channel to cytosolic sodium concentrations, which account for increased excitability. ADNFLE/NFLE is characterized by clusters of brief stereotyped seizures and generally occurs during light sleep. Sometimes it is preceded by psychiatric symptoms and occurs mostly during non-rapid eye movement sleep.109 KCNT1 gene encodes a sodium-activated potassium channel subunit, which is thought to function in ion conductance and developmental signaling pathways. In addition, a genetic locus for familial febrile seizures and epilepsy has been localized on chromosome 3q26.2-q26.33 and a single base pair deletion (delA750) in exon 4 of KCNMB3 at this locus was found.110 KCNMB3 encodes for calcium-activated potassium channel subunit beta-3 and alters or truncates the terminal 21 amino acids of the three subunits. KCNMB3 delA750 mutation confers susceptibility to common IGE syndrome.111
Voltage-gated calcium channels
Calcium is a very important signaling molecule that mediates muscle contraction, neurotransmission, and gene transcription.112 The voltage-gated calcium channels are key transducers of membrane potential changes into intracellular Ca2+ transients that initiate many physiological events. The channels are multi-subunit proteins comprising a large pore-forming α1 subunit and four other types of subunits, including α2, δ, an intracellular β, and transmembrane glycoprotein γ. The latter four types are auxiliary subunits and may not be directly involved with conducting calcium or voltage gating.112 According to amino acid sequence, in mammals, the principal α1 subunits have ten members grouped into three subfamilies, including Cav1s, Cav2s, and Cav3s. The Cav1s comprises Cav1.1, Cav1.2, Cav1.3, and Cav1.4 that are encoded by CACNA1S, CACNA1C, CACNA1D, and CACNA1F, respectively; the Cav2s includes Cav2.1, Cav2.2, and Cav2.3 that are encoded by CACNA1A, CACNA1B, and CACNA1E, respectively; and the Cav3s includes Cav3.1, Cav3.2, and Cav3.3 that are encoded by CACNA1G, CACNA1H, and CACNA1I, respectively. While Cav1s and Cav2s are high-voltage activated members, the Cav3s is a low-voltage activated member. A mutation in the Cav2.1 channel has been linked to both generalized and complex partial seizures and severe neurological disorders, including ataxias and congenital migraine.112,113 Mutations in multiple genes encoding for the calcium channels are implicated for epilepsy. A proband with complex phenotype comprising primary GE is heterozygous for an exon 36 mutation (C5733T) in CACNA1A.113 Mutations in CACNA1A have a strong link with familial hemiplegic migraine, episodic ataxia type 2, and spinocerebellar ataxia type 6.114 Family based studies have also found some common genetic variants in CACNA1A associated with IGE.115,116 In addition, two mutations (R482X and C104F) in CACNB4 are detected in epilepsy.117 In screening for mutations in 38 SCN1A-mutation-positive probands with myoclonic epilepsy in infancy (SMEI), a missense mutation (R468Q) in CACNB4 was detected in one proband.118
Nicotinic acetylcholine receptors (nAChRs)
nAChRs are cholinergic receptors that form ligand-gated ion channels in the plasma membranes of certain neurons on the presynaptic and postsynaptic sides of the neuromuscular junction. As ionotropic receptors, nAChRs are directly linked to ion channels. In all, 17 different genes encode for ACh receptor subunits, including ten α, four β, and γ, δ, and ε subunits. The α subunits are principal, and others are complementary.
Several genes encoding for nAChRs are implicated for some rare forms of epilepsy. A missense mutation in CHRNA4 that replaces serine with phenylalanine at codon 248 was found in all 21 affected family members with ADNFLE, but not in 333 healthy control subjects.119 Two mutations (V287L and V287M) in CHRNB2 were also detected in patients with ADNFLE.120 Mutations in CHRNA2 were detected in an Italian family with nocturnal frontal lobe epilepsy (NFLE), but they were not found in 47 families affected with ADNFLE.121 These three genes have been recommended for genetic testing in epilepsy by the ILAE.
GABARs
The GABARs are one of the two classes of the GABA receptors that respond to the neurotransmitter GABA. GABARs, known as ionotropic receptors, are ligand-gated ion channels that mediate most fast synaptic inhibition in brain. Mutation in GABAR subunit expression and function may result in epileptogenesis. There are 19 genes that encode a number of different GABAR subunits, including α1–6, β1–3, and γ1–3, and several other subunits. Mutations in GABRA1 and GABRG2 have been recommended by the ILAE for genetic testing in epilepsy. Recently, mutation R46W in the GABAR GABRA6 was detected in patients with CAE, and it was located in a region homologous to a GABRG2 missense mutation, R82Q, that was associated with both CAE and febrile seizures in human beings.122 Mutations (P11S, S15F, and G32R) in the GABAR β3 subunit gene (GABRB3) are found in multiple families with CAE.123 GABRB3 is located at the human chromosome 15q11-13 complex locus, which contains two GABARs (GABRA5 and GABRG3). PWS and AS are unique clinical disorders that may arise from deletion of chromosome 15q11-q13 during paternal or maternal gametogenesis, respectively.124 GABRB3 is regulated by epigenetic modulation and highly expressed in embryonic brain, where repressor-element-1-silencing transcription factor (REST) regulates neuronal genes. Reduced expression of GABRB3 and UBE3A, both of which are within a close genomic region, is observed in each of three neurological syndromes (Rett syndrome, AS, and autism) as well as mental retardation and epilepsy. Mutations in GABRB3 are detected in epilepsy. Individuals with epilepsy tend to have more severe symptom when a deficiency of UBE3A is also present.125 GABRB3 is a major inhibitory neurotransmitter of the mammalian nervous system.
Glutamate receptors
Glutamate (glutamic acid) is the main excitatory neurotransmitter in the mammalian central nervous system. It acts at a number of receptors, which are defined by the agonists that activate them. The receptors are classified as ligand-gated ion channels (ionotropic receptors) and G-protein-coupled (metabotropic) receptors.85 Metabotropic receptors consist of Class I (mGluR1, 5), Class II (mGluR2, 3), and Class III (mGluR4, 6, 7, 8) receptors. Ionotropic receptors consist of AMPA (GluR1 to GluR4), kainate (GluR5 to GluR6), NMDA (GluN1, GluN2A to GluN2D, and GluN3A to GluN3B) and Glu δ receptors. The NMDA receptors have a high permeability for Ca2+ ion and a high affinity for glutamate. One of the major functions of glutamate receptors appears to modulate synaptic plasticity, a property of the brain that is believed to be vital for memory and learning. An increase in the number of ionotropic glutamate receptors on a postsynaptic cell may lead to long-term potentiation of that cell and a decrease may lead to long-term depression. Metabotropic glutamate receptors trigger postsynaptic protein synthesis, which modulates synaptic plasticity through second messenger systems.
The GRIN2A on 16p13.2 locus may have an important implication for diagnostic testing. In a target gene capture of 18 epilepsy-associated genes in 519 probands with epileptic encephalopathies (EE) with diverse epilepsy syndromes, three different genetic mutations (c1005-1C>T, p.Thr531Met, and Phe139Ilefs*15) in GRIN2A, a NMDA subunit, were found in four probands and were segregated with their families, and all four probands presented with epilepsy–aphasia syndromes (EAS), accounting for 9% (4/44) of EE cases with EAS. However, the mutations were found neither in the rest of 475 EE cases nor in 6,500 control exomes.126 The mutations in GRIN2A could be used for diagnosis testing for EE with EAS. A chromosomal translocation t(16;17) (p13.2;q11.2) involving GRIN2A was found in a family with epilepsy and neurodevelopmental defects. A novel de novo missense mutation in GRIN2A (c.2449A>G, p.Met817Val) is the likely cause of refractory epilepsy and global developmental delay.127
Nonion channel gene mutations
In addition to the mutations in the ion channel genes, recently exome-sequencing study also showed that mutations in nonion channel genes are involved with the development of epilepsy. Some mutations such as LGI1, PRRT2, EFHC1, PRICKLE, RBFOX1, and DEPDC5 are found in individuals with a clear inheritance of epilepsy or with a specific phenotype (Table 3).
LGI1
The LGI1 gene is located at a linkage region on chromosome 10q24. Mutations in LGI1 are more prevalent in ADLTE, ADPEAF, or sporadic epilepsy.128 More than two dozens of mutations have been detected with LGI1 in patients with ADLTE.129 LGI1 mutations were found in seven out 14 families with ADPEAF and are believed as a common cause of epilepsy.130 The LGI1 encodes a member of the secreted leucine-rich repeat (LRR) superfamily and shares homology with members of the SLIT protein family.
Two distinct molecular mechanisms may underlie ADLTE. The one recognizes LGI1 as a novel subunit of the presynaptic Kv1 potassium channel implicated in the regulation of channel inactivation, while the other suggests that LGI1 acts as a ligand that selectively binds to the postsynaptic receptor ADAM22, thereby regulating the glutamate–AMPA neurotransmission.131 Both mechanisms imply that LGI1 mutations result in an alteration of synaptic currents.
PRRT2
PRRT2 encodes a transmembrane protein containing a proline-rich domain in its N-terminal half and is located at 16p11.2 microduplications implicated for epilepsies.61 A whole-exome sequencing analysis of eight Han Chinese families with histories of paroxysmal kinesigenic dyskinesia (PKD) detected c.514_517delTCTG (p.Ser172Argfs*3) and c.972delA (p.Val325Serfs*12) each in one individual family and c.649dupC (p.Arg217Profs*8) in six families.132 The truncating mutation c.649dupC in PRRT2 is a hotspot mutation resulting in benign familial infantile epilepsy (BFIE) or infantile convulsions with paroxysmal choreoathetosis (ICCA) regardless of the ethnic background. A Japanese study found heterozygous truncated mutation c.649dupC in 15 of 26 individuals with benign infantile epilepsy (52.1%), and all three families of ICCA harbored the same mutation (100%).133
The PRRT2 mutation appears to affect a spectrum of neurological disorders and is common to multiple racial groups, including Chinese, Japanese, and European ancestry populations, as well as African American family.134,135 The mutation c.649_650insC was found in five of six families affected with ICCA syndrome and in 14 of 17 families affected with BFIE,136 and a majority of the sample was Australian with Western heritage. A mutation c.649delC is detected in a family with four people affected with different phenotypes of febrile convulsion, epileptic seizures, PKD, or a headache. However, in another study of 24 child probands with BFIE, convulsions with mild gastroenteritis, and benign early infantile epilepsy in Japanese families, the insertion mutation c.649_650insC was found in five families.137 Wang et al138 identified c.649-650insC mutation in all patients of two Han Chinese families with benign familial infantile seizure.
Studies in mice suggest that PRRT2 is predominantly expressed in brain and spinal cord in embryonic and postnatal stages. A recent study suggested that mutant PRRT2 may affect glutamate signaling and glutamate receptor activity through interaction with SNAP25, resulting in the increase of glutamate release and subsequent neuronal hyperexcitability.139
EFHC1
EFHC1 is located at 6p12-p11, and it encodes an EF-hand-containing calcium-binding protein, which plays a key role in calcium homeostasis. Five missense mutations in EFHC1 were found in six unrelated families affected with JME, one of the most frequent forms of IGE, but none of these mutations were present in ~400 controls.140 In all, 9% of consecutive JME cases from Mexico and Honduras clinics and 3% of clinic patients from Japan carry mutations in Myoclonin1/EFHC1.141 EFHC1 is shown to regulate cell division and neuronal migration during cortical development; and disruption of its functions may lead to JME.142 A recent study showed that EFHC1 may be involved in the regulation of Wnt signaling that plays a role in human neuronal development and may affect the development of epilepsy.143
Prickle homolog 1 (PRICKLE1)
PRICKLE1 encodes a nuclear receptor that may be a negative regulator of the Wnt/beta-catenin signaling pathway. A homozygous mutation in human PRICKLE1 identified in three pedigrees causes an autosomal recessive progressive myoclonus epilepsy–ataxia syndrome.144 The possible mechanisms are likely through disrupted motor neuron migration and neurite extension and affected neuronal positioning. PRICKLE1 is expressed in brain regions implicated for epilepsy and ataxia in mice and human beings. Moreover, prickle-mediated calcium signaling might underlie prickle function in the nervous system, reduce calcium activation, and dysregulate calcium currents.145
RNA-binding protein, fox-1 homolog 1 (RBFOX1) and RNA-binding protein, fox-1 homolog 3 (RBFOX3)
Mutations in RBFOX1 on 16p13.3 and RBFOX3 on 17q25.3 are detected in patients with epilepsy. In addition to two hemizygous deletions, a 365 kb deletion affecting two untranslated 59-terminal exons of RBFOX1 and a 43 kb deletion spanning exon 3 of RBFOX3 were identified. An exome-sequencing analysis of 242 patients with RE revealed novel frameshift mutation (p.A233Vfs*74) and a hexanucleotide deletion (p.A299_A300del) in RBFOX1 and a novel nonsense mutation (p.Y287*) in RBFOX3.146 Moreover, five exon-disrupting RBFOX1 deletions ranging from 68 to 896 kb affecting the untranslated 5′-terminal RBFOX1 exons were detected in 1,408 patients with IGE, but not in 2,256 controls.147
The possible mechanism is that knockdown of RBFOX1 transcripts in human neurons changes the alternative splicing pattern, and the expression of primarily neuronal genes involved in synapse formation and function that lead to an increased susceptibility for seizure events.148 RBFOX and PTBP1 proteins interacting together regulate the alternative splicing of microexons (<51 nucleotides) in human brain transcripts.149 This may have important implications because RBFOX proteins are well known to play crucial roles in both brain development and function. Aberrant splicing induced by RBFOX dysregulation is associated with a variety of brain-related disorders, including autism, mental retardation, and epilepsy.150,151
DEPDC5
DEPDC5 is located on chromosome 22q12.2-q12.3, and it encodes a member of the IML1 family of proteins involved in G-protein signaling pathways. Mutations in DEPDC5 are identified in 10 of 82 families with autosomal dominant familial with focal epilepsy with variable foci (FFEVF), suggesting a common cause for FFEVF.152 Whole-exome sequencing analysis revealed a splice acceptor mutation (c.2355-2A>G) and three nonsense mutations (p.Arg487*, p.Arg1087*, and p.Trp1369*) in DEPDC5 in probands of 30 European families with ADNFLE.153 In addition to a frameshift mutation in DEPDC5 identified in a family with FFEVF, five additional mutations in DEPDC5 are found in 15 families affected with a broad spectrum of focal epilepsies.154 The mutations in DEPDC5 are associated with both lesional and nonlesional epilepsies even within the same family.155 At the clinical level, however, 78% of the patients with DEPDC5 mutations are drug resistant.153
DEPDC5 may have implications for a broad spectrum of familial focal epilepsies. Previous studies have linked ion channel subunit genes (eg, CHRNA4, CHRNA2, CHRNB2, and KCNT1) to ADNFLE and neuronal-secreted protein (LGI1-encoding epitempin) to ADPEAF. The mTORC1-repressor gene DEPDC5 has recently been reported in multiple inherited focal epilepsies such as ADNFLE, FTLE, and FFEVF.156 This suggests that DEPDC5 may have broad implications for epilepsy syndrome.157 DEPDC5 negatively regulates the mammalian target of rapamycin (mTOR) pathway, which regulates cell growth by sensing the availability of nutrients.
Mutations in multiple other genes (eg, CARS2, CLN6, COL6A2, and GOSR2) are reported in patients with PME. A homozygous mutation (c.655G>A) in CARS2 is confirmed in the family with PME.158 This mutation is located at the last nucleotide of exon 6, results in a removal of exon 6, and leads to an in-frame deletion of 28 amino acids in a conserved sequence motif of the protein involved in the stabilization of the acceptor end hairpin of tRNA. A whole exome-sequencing combined with homozygosity mapping identified mutations in CLN6 associated with the teenage-onset PME.159 The p.Asp215Asn mutation in COL6A2 is identified in a consanguineous family with PME.160 Mutations (c.430G>T, p.Gly144Trp) in GOSR2, a Golgi vesicle transport gene encoding a vesicle docking proteins, are identified in patients with PME.161
Lastly, a novel homozygous nonsense mutation in exon 11(c.1270C>T) changing arginine to a premature stop codon (p.R424X) in SCARB2 is found for epilepsy.162 A loss-of-function mutation in SCARB2 has been identified in families with the action myoclonus–renal failure syndrome, which is an autosomal recessive form of PME. This gene encodes a lysosomal integral membrane protein of 478 amino acids (IMP2), which is important for lysosomal mannose-6-phosphate-independent trafficking of β-glucocerebrosidase.
Epigenetics of epilepsy
While numerous single mutations have been identified for epilepsy, they account for little of the heritability. Most of these mutations, especially those in the ion channel genes, are rare or not even found in the genomic database.84 The heritability of epilepsy is estimated to be ~0.30, which is moderate and lower than most of the neurodevelopmental and neuropsychiatric disorders such as autism. Environmental factors alone or interacting with genetic variants may play some roles in the development of epilepsy. Epigenetic regulation is another mechanism through which environmental factors affect the development of epilepsy.
Epigenetics refers to “heritable” changes in gene expression or phenotype that do not involve changes to the underlying DNA sequence. However, sometimes researchers also seek to describe dynamic alterations in the transcriptional potential of a cell, which may not be heritable. Common epigenetic mechanisms include DNA methylation, post-translational histone modification, and non-coding RNA. These epigenetic processes determine not only gene expression or silencing but also where, when, and how gene are expressed and then which proteins are transcribed.163 The epigenetic process is influenced by age; physical environments such as nutrients, smoking, drugs, and chemicals; and even psychosocial events such as stressful life events.
Epigenetics plays a role in neuronal function from embryogenesis and early brain development to tissue-specific gene expression. Evidence has shown that micro-RNAs, one type of non-coding RNAs, modulate the translation of target genes in early stages of brain development and are critical to dendritogenesis, synapse formation, and maturation.164 Therefore, dysregulation of epigenetics would have a significant role in neuropsychiatric disorders, including stroke and epilepsy, as well as other diseases such as cancer, autoimmune diseases, and diabetes.165 Because epigenetic changes are reversible and such pathological epigenetic modifications are readily curable through pharmacological interventions, the epigenetic machinery may provide a new platform for therapeutic approaches against these diseases.166
DNA methylation
Aberrant DNA methylation may contribute to the pathophysiology of epilepsy and other neurodevelopmental, neuropsychiatric, and neurodegenerative disorders. Using biopsy brain specimens from 16 adult patients with temporal lope epilepsy (TLE) and autopsy brain samples from three age-matched controls, a study found hypermethylation at the reelin promoter in the dentate gyrus of TLE patients.167 The aberrant DNA methylation may cause dispersion of granule cell and the deficiency in the reelin expression that are found in epilepsy. A genome-wide DNA methylation analysis of hippocampus in mice to define the pattern of status epilepticus and epileptic tolerance also showed that >300 genes showed altered DNA methylation, with 90% of the promoters of these genes undergoing hypomethylation.168 The DNA methylation has been implicated in genome stability, silencing transposable element, and repressing gene expression that affects brain development, leading to multiple neurological disorders.169 DNA methyltransferase (DNMT) enzymes catalyze DNA methylation and results in the formation of 5-methylcytosyine, primarily at CpG dinucleotide-containing regulatory sequences. The role of DNA methylation in the pathogenesis of epileptic syndromes, including TLE, PWS, and AS, has already been elucidated by some studies.167–169
Histone modification
Histone acetylation and phosphorylation are two most common processes of histone modification.170 While histone hyperacetylation indicates an increase in gene activity, its hypoacetylation marks gene repression. The hyper- and hypo- methylation are controlled by a dynamic interplay of histone acetyltransferase (HAT) and histone deacetylase (HDAC). A study has shown that acetylation of histone H4 in rat hippocampal CA3 neurons is reduced at the promoter of glutamate receptor 2 (GluR2, encoded by GRIA2) but increased at brain-derived neurotrophic factor (BDNF) promoter P2 as soon as 3 hours after induction of status epilepticus by pilocarpine.171 In examining the effect of kainate-induced status epilepticus on two different histone modifications on histone H3 phosphorylation at serine 10 and histone H4 acetylation, a study demonstrated that histone modifications induced by kainate are involved not only in immediate early genes (IEGs) expression but also in the development of epilepsy.172 Mutations in the histone demethylase KDM5C are linked to X-linked mental retardation and epilepsy.170–172 Electroconvulsive seizures, produced by repetitive electronic stimulation, may lead to increased H4 acetylation at c-fos and BDNF gene promoters while decreasing at the CREB gene promoter in rat hippocampus.173 CREB is a transcription factor that modulates the expression of the GABARs in the hippocampus and plays an important role in the epileptogenic process.
Non-coding RNA
MicroRNA is a class of small non-coding RNA that plays a key role in epigenetic regulation by controlling the translation and/or stability of mRNAs, mainly via sequence-specific binding within the 3′ untranslated region of mRNA transcripts. In human beings, there are ~1,600 miRNAs, which regulate more than half of protein coding genes, and each miRNA can regulate multiple proteins.174 Because the brain expresses several unique miRNAs such as miR-34a that control dendritic morphology, ion channel level, neuronal migration, and glial function microRNA may have an important implication for neuropsychiatric disorders.175 For example, miR-34a affected bipolar risk through directly impacting the ANK3 and CACNB3 expressions,176 and upregulation of miR-34a is associated with the risk of epilepsy.177 miRNAs regulates neural differentiation, maintenance, and plasticity. Fragile X mental retardation protein (FMRP) results in epilepsy associated with Fragile X syndrome by glutaminergic and GABAergic synaptic dysfunctions at the mRNA and protein levels.175–177
In summarizing three independent epigenome-wide analyses of microRNA expression, Hwang et al164 showed that miR-132 was consistently upregulated in the hippocampal CA3 in the rat animal model after status epilepticus. Five microRNAs, including miR-24, miR-29a, miR-99a, miR134, and miR375, are shown upregulated in at least two of three studies. However, no downregulated microRNA was consistently found across three studies. Because microRNAs tend to regulate multiple proteins and play a role in neurodevelopment, they may hold promising for novel therapeutic development.
Conclusion and future implication
Epilepsy is fundamentally a disorder of neuronal excitability, and its etiology is elusive. Genetic studies have indicated a number of single-point mutations in genes that involve voltage-gated sodium channels (SCN1A, SCN1B, SCN2A, and SCN9A), potassium channels (KCNA1, KCNMA1, KCNMB3, LCNJ10, KCNQ2, KCNQ3, and KCNT1), calcium channels (CACNA1A), ligand-gated GABARs (GBAEA, GBARG2, and GABRB3), glutamate receptors (GRIN2A), and acetylcholine receptors (CHRNA2, CHRNA4, and CHRNB2). All of these are molecular mediators of excitability, and excitability molecules are predominant cellular components affected in epilepsies.178 However, some of the genetic mutations identified in the ion channel genes are very rare and even hardly found in a normal population.
The genome-wide analysis also shows that CNVs and single-point mutations in nonion channel genes affect epilepsy. While epilepsy shares some common genetic lesions such as microdeletions at 15q11.2, some of them disrupt a few credible candidate genes such as NRXN1, AUTS2, NLGN1, CNTNAP2, GRIN2A, PRRT2, NIPA2, BMP5, and PODXL that have been implicated in other neurodevelopmental disorders, including intellectual disability, autism, and schizophrenia.179 Recent exome-sequencing study of families with epilepsy revealed that mutations in some genes such as LGI1, PRRT2, EFHC1, SYNGAP1, PRICKLE1, RBFOX1, RBFOX3, and DEPDC5 may account for a certain proportion of familial epilepsy cases. Some mutations such as c.649_650insC in PRRT2 are present in a majority of families with probands of PKD, BFIE, and ICCA syndrome. Some of these genes strongly suggest that neurodevelopment was involved in the development of epilepsy.
While genetic study allows us to understand the etiology of epilepsy, it is likely that genetic variants only directly contribute to a proportion of the liability of epilepsy. Genome-wide association studies have only identified a few common variants associated with epilepsy through genome-wide association study. Given that the heritability of epilepsy is just moderate, environmental factors may contribute to the etiology of epilepsy.180 So far, preeminent genetic variants that have been found in epilepsies are single-point mutations (de novo or inherited). The epigenetic mechanism may play a key role in interpreting how environmental factors cause the phenotype or phenotypic variations and how the DNA mutational variants may contribute to phenotypic traits.7,181
Epigenetic silencing may cause the loss of function of genes and predisposes to a genetic mutation. Genes controlling the cell cycle and DNA repair, such as RB, BRCA1/2, and PTEN, have all been reported to be hypermethylated or mutated/deleted in cancer. Promoter hypermethylation is the predominant mechanism for the loss of function of some genes.182 Evidence has shown that altered epigenetics promote genome instability and formation of genetic mutations.183 CpG DNA methylation may facilitate a C to T conversion, the most frequent point mutation. Translocation events and inversions are also influenced by histone modifications, DNA methylation, and ncRNA.182 A recent study indicates that environmentally induced epigenetic transgenerational inheritance of disease and phenotypic variation can promote genetic mutations (ie, CNVs) in later generations.182
The future study of epilepsy should focus on integrating whole-genome sequencing variants and environmental factors into epigenetic change to understand the potential mechanism of epilepsy. Given that epigenetic regulation is tissue specific and may be susceptible to confounding factors, a careful design using systems biology approach that unifies genetics, environmental factors, epigenetic regulation, and gene expression will be required to understand the epigenetic mechanism of epilepsy. Family based samples allow us to effectively control for the potential genetic heterogeneity or population stratification that arises from the case–control study. The epigenetic study may also facilitate biomarker discovery and the potential development of novel therapeutics for epilepsy.
Disclosure
The authors report no conflicts of interest in this work.
References
Korff CM, Scheffer IE. Epilepsy classification: a cycle of evolution and revolution. Curr Opin Neurol. 2013;26(2):163–167. | ||
Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51(4):676–685. | ||
Banerjee PN, Filippi D, Hauser WA. The descriptive epidemiology of epilepsy – a review. Epilepsy Res. 2009;85(1):31–45. | ||
Browne TR, Holmes GL. Handbook of Epilepsy. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008. | ||
WHO [webpage on the Internet]. WHO Fact Sheet on Epilepsy [updated February 2017]. Available from: http://www.who.int/mediacentre/factsheets/fs999/en/. Accessed April 3, 2017. | ||
Newton CR, Garcia HH. Epilepsy in poor regions of the world. Lancet. 2012;380(9848):1193–1201. | ||
Berkovic SF, Mulley JC, Scheffer IE, Petrou S. Human epilepsies: interaction of genetic and acquired factors. Trends Neurosci. 2006;29(7):391–397. | ||
Miller LL, Pellock JM, DeLorenzo RJ, Meyer JM, Corey LA. Univariate genetic analyses of epilepsy and seizures in a population-based twin study: the Virginia Twin Registry. Genet Epidemiol. 1998;15(1):33–49. | ||
Speed D, O’Brien TJ, Palotie A, et al. Describing the genetic architecture of epilepsy through heritability analysis. Brain. 2014;137(10):2680–2689. | ||
Institute of Medicine (US) Committee on the Public Health Dimensions of the Epilepsies; England MJ, Liverman CT, et al, editors. Epidemiology and prevention. Epilepsy Across the Spectrum: Promoting Health and Understanding. (Chap. 3). Washington, DC: National Academies Press (US); 2012. | ||
Hwang SK, Hirose S. Genetics of temporal lobe epilepsy. Brain Dev. 2012;34(8):609–616. | ||
Fanciulli M, Di Bonaventura C, Egeo G, et al. Suggestive linkage of familial mesial temporal lobe epilepsy to chromosome 3q26. Epilepsy Res. 2014;108(2):232–240. | ||
Belhedi N, Bena F, Mrabet A, et al. A new locus on chromosome 22q13.31 linked to recessive genetic epilepsy with febrile seizures plus (GEFS+) in a Tunisian consanguineous family. BMC Genet. 2013;14:93. | ||
Kullmann DM. Genetics of epilepsy. J Neurol Neurosurg Psychiatry. 2002;73(suppl II):ii32–ii35. | ||
Moulard B, Guipponi M, Chaigne D, Mouthon D, Buresi C, Malafosse A. Identification of a new locus for generalized epilepsy with febrile seizures plus (GEFS+) on chromosome 2q24-q33. Am J Hum Genet. 1999;65(5):1396–1400. | ||
Krepischi AC, Knijnenburg J, Bertola DR, et al. Two distinct regions in 2q24.2-q24.3 associated with idiopathic epilepsy. Epilepsia. 2010;51(12):2457–2460. | ||
Holland KD, Kearney JA, Glauser TA, et al. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci Lett. 2008;433(1):65–70. | ||
Pinto D. Dissecting the Genetic Basis of Idiopathic Epilepsies [PhD thesis] Utrecht University. 2006. ISBN 90-393-4191-5. | ||
Greenberg DA, Delgado-Escueta AV, Widelitz H, et al. Juvenile myoclonic epilepsy (JME) may be linked to the BF and HLA loci on human chromosome 6. Am J Med Genet. 1988;31(1):185–192. | ||
Weissbecker KA, Durner M, Janz D, Scaramelli A, Sparkes RS, Spence MA. Confirmation of linkage between juvenile myoclonic epilepsy locus and the HLA region of chromosome 6. Am J Med Genet. 1991;38(1):32–36. | ||
Pal DK, Evgrafov OV, Tabares P, Zhang F, Durner M, Greenberg DA. BRD2 (RING3) is a probable major susceptibility gene for common juvenile myoclonic epilepsy. Am J Hum Genet. 2003;73(2):261–270. | ||
Layouni S, Buresi C, Thomas P, Malafosse A, Dogui M. BRD2 and TAP-1 genes and juvenile myoclonic epilepsy. Neurol Sci. 2010;31(1):53–56. | ||
Velíšek L, Shang E, Velíšková J, et al. GABAergic neuron deficit as an idiopathic generalized epilepsy mechanism: the role of BRD2 haploinsufficiency in juvenile myoclonic epilepsy. PLoS One. 2011;6(8):e23656. | ||
Sainz J, Minassian BA, Serratosa JM. Lafora progressive myoclonus epilepsy: narrowing the chromosome 6q24 locus by recombinations and homozygosities. Am J Hum Genet. 1997;61(5):1205–1209. | ||
Minassian BA, Lee JR, Herbrick JA, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20(2):171–174. | ||
Singh S, Ganesh S. Lafora progressive myoclonus epilepsy: a meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum Mutat. 2009;30(5):715–723. | ||
Farhan SM, Murphy LM, Robinson JF, et al. Linkage analysis and exome sequencing identify a novel mutation in KCTD7 in patients with progressive myoclonus epilepsy with ataxia. Epilepsia. 2014;55(9):e106–e111. | ||
Kousi M, Anttila V, Schulz A, et al. Novel mutations consolidate KCTD7 as a progressive myoclonus epilepsy gene. J Med Genet. 2012;49(6):391–399. | ||
Krabichler B, Rostasy K, Baumann M, et al. Novel mutation in potassium channel related gene KCTD7 and progressive myoclonic epilepsy. Ann Hum Genet. 2012;76(4):326–331. | ||
Azizieh R, Orduz D, Van Bogaert P, et al. Progressive myoclonic epilepsy-associated gene KCTD7 is a regulator of potassium conductance in neurons. Mol Neurobiol. 2011;44(1):111–121. | ||
Plaster NM, Uyama E, Uchino M, et al. Genetic localization of the familial adult myoclonic epilepsy (FAME) gene to chromosome 8q24. Neurology. 1999;53(6):1180–1183. | ||
Fong GC, Shah PU, Gee MN, et al. Childhood absence epilepsy with tonic-clonic seizures and electroencephalogram 3-4-Hz spike and multispike-slow wave complexes: linkage to chromosome 8q24. Am J Hum Genet. 1998;63(4):1117–1129. | ||
Holter J, Carter D, Leresche N, Crunelli V, Vincent P. A TASK3 channel (KCNK9) mutation in a genetic model of absence epilepsy. J Mol Neurosci. 2005;25(1):37–51. | ||
Bonaglia MC, Giorda R, Tenconi R, et al. A 2.3 Mb duplication of chromosome 8q24.3 associated with severe mental retardation and epilepsy detected by standard karyotype. Eur J Hum Genet. 2005;13(5):586–591. | ||
Haug K, Kremerskothen J, Hallmann K, et al. Mutation screening of the chromosome 8q24.3-human activity-regulated cytoskeleton-associated gene (ARC) in idiopathic generalized epilepsy. Mol Cell Probes. 2000;14(4):255–260. | ||
Morita R, Miyazaki E, Shah PU, Castroviejo IP, Delgado-Escueta AV, Yamakawa K. Exclusion of the JRK/JH8 gene as a candidate for human childhood absence epilepsy mapped on 8q24. Epilepsy Res. 1999;37(2):151–158. | ||
Ottman R, Risch N, Hauser WA, et al. Localization of a gene for partial epilepsy to chromosome 10q. Nat Genet. 1995;10(1):56–60. | ||
Mautner VF, Lindenau M, Gottesleben A, Goetze G, Kluwe L. Supporting evidence of a gene for partial epilepsy on 10q. Neurogenetics. 2000;3(1):31–34. | ||
Winawer MR, Martinelli Boneschi F, Barker-Cummings C, et al. Four new families with autosomal dominant partial epilepsy with auditory features: clinical description and linkage to chromosome 10q24. Epilepsia. 2002;43(1):60–67. | ||
Zara F, Gennaro E, Stabile M, et al. Mapping of a locus for a familial autosomal recessive idiopathic myoclonic epilepsy of infancy to chromosome 16p13. Am J Hum Genet. 2000;66(5):1552–1557. | ||
Afawi Z, Mandelstam S, Korczyn AD, et al. TBC1D24 mutation associated with focal epilepsy, cognitive impairment and a distinctive cerebro-cerebellar malformation. Epilepsy Res. 2013;105(1–2):240–244. | ||
Corbett MA, Bahlo M, Jolly L, et al. A focal epilepsy and intellectual disability syndrome is due to a mutation in TBC1D24. Am J Hum Genet. 2010;87(3):371–375. | ||
Falace A, Buhler E, Fadda M, et al. TBC1D24 regulates neuronal migration and maturation through modulation of the ARF6-dependent pathway. Proc Natl Acad Sci U S A. 2014;111(6):2337–2342. | ||
Falace A, Filipello F, La Padula V, et al. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am J Hum Genet. 2010;87(3):365–370. | ||
Lee JA, Lupski JR. Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron. 2006;52(1):103–121. | ||
Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19(9):1586–1592. | ||
Mullen SA, Carvill GL, Bellows S, et al. Copy number variants are frequent in genetic generalized epilepsy with intellectual disability. Neurology. 2013;81(17):1507–1514. | ||
Mefford HC, Muhle H, Ostertag P, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6(5):e1000962. | ||
de Kovel CG, Trucks H, Helbig I, et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133(1):23–32. | ||
Heinzen EL, Radtke RA, Urban TJ, et al. Rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am J Hum Genet. 2010;86(5):707–718. | ||
Liu JY, Kasperavičiūtė D, Martinian L, Thom M, Sisodiya SM. Neuropathology of 16p13.11 deletion in epilepsy. PLoS One. 2012;7(4):e34813. | ||
Miteff CI, Smith RL, Bain NL, Subramanian G, Brown JE, Kamien B. 16p13.11 microdeletion in a patient with hemiconvulsion-hemiplegia-epilepsy syndrome: a case report. J Child Neurol. 2015;30(1):83–86. | ||
Gurrieri F, Battaglia A, Torrisi L, et al. Pervasive developmental disorder and epilepsy due to maternally derived duplication of 15q11-q13. Neurology. 1999;52(8):1694–1697. | ||
Stewart LR, Hall AL, Kang SH, Shaw CA, Beaudet AL. High frequency of known copy number abnormalities and maternal duplication 15q11-q13 in patients with combined schizophrenia and epilepsy. BMC Med Genet. 2011;12:154. | ||
Bassuk AG, Geraghty E, Wu S, et al. Deletions of 16p11.2 and 19p13.2 in a family with intellectual disability and generalized epilepsy. Am J Med Genet A. 2013;161A(7):1722–1725. | ||
Reinthaler EM, Lal D, Lebon S, et al. 16p11.2 600 kb duplications confer risk for typical and atypical Rolandic epilepsy. Hum Mol Genet. 2014;23(22):6069–6080. | ||
Naseer MI, Faheem M, Chaudhary AG, et al. Genome wide analysis of novel copy number variations duplications/deletions of different epileptic patients in Saudi Arabia. BMC Genomics. 2015;16(suppl 1):S10. | ||
Jiang Y, Zhang Y, Zhang P, et al. NIPA2 located in 15q11.2 is mutated in patients with childhood absence epilepsy. Hum Genet. 2012;131(7):1217–1224. | ||
Zhao Q, Li T, Zhao X, et al. Rare CNVs and tag SNPs at 15q11.2 are associated with schizophrenia in the Han Chinese population. Schizophr Bull. 2013;39(3):712–719. | ||
Vanlerberghe C, Petit F, Malan V, et al. 15q11.2 microdeletion (BP1-BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: a series of 52 patients. Eur J Med Genet. 2015;58(3):140–147. | ||
Dimassi S, Labalme A, Lesca G, et al. A subset of genomic alterations detected in rolandic epilepsies contains candidate or known epilepsy genes including GRIN2A and PRRT2. Epilepsia. 2014;55(2):370–378. | ||
Fanciulli M, Pasini E, Malacrida S, et al. Copy number variations and susceptibility to lateral temporal epilepsy: a study of 21 pedigrees. Epilepsia. 2014;55(10):1651–1658. | ||
Poot M, van der Smagt JJ, Brilstra EH, Bourgeron T. Disentangling the myriad genomics of complex disorders, specifically focusing on autism, epilepsy, and schizophrenia. Cytogenet Genome Res. 2011;135(3–4):228–240. | ||
Kim EH, Yum MS, Lee BH, et al. Epilepsy and other neuropsychiatric manifestations in children and adolescents with 22q11.2 deletion syndrome. J Clin Neurol. 2016;12(1):85–92. | ||
Klitten LL, Møller RS, Nikanorova M, Silahtaroglu A, Hjalgrim H, Tommerup N. A balanced translocation disrupts SYNGAP1 in a patient with intellectual disability, speech impairment, and epilepsy with myoclonic absences (EMA). Epilepsia. 2011;52(12):e190–e193. | ||
Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron. 1998;20(5):895–904. | ||
Sáez-Hernández L, Peral B, Sanz R, et al. Characterization of a 6p21 translocation breakpoint in a family with idiopathic generalized epilepsy. Epilepsy Res. 2003;56(2–3):155–163. | ||
Andrieux J, Richebourg S, Duban-Bedu B, et al. Characterization by array-CGH of an interstitial de novo tandem 6p21.2p22.1 duplication in a boy with epilepsy and developmental delay. Eur J Med Genet. 2008;51(4):373–381. | ||
Guo Y, Baum LW, Sham PC, et al. Two-stage genome-wide association study identifies variants in CAMSAP1L1 as susceptibility loci for epilepsy in Chinese. Hum Mol Genet. 2012;21(5):1184–1189. | ||
International League Against Epilepsy Consortium on Complex Epilepsies. Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol. 2014;13(9):893–903. | ||
Lyons PJ, Callaway MB, Fricker LD. Characterization of carboxypeptidase A6, an extracellular matrix peptidase. J Biol Chem. 2008;283(11):7054–7063. | ||
Salzmann A, Guipponi M, Lyons PJ, et al. Carboxypeptidase A6 gene (CPA6) mutations in a recessive familial form of febrile seizures and temporal lobe epilepsy and in sporadic temporal lobe epilepsy. Hum Mutat. 2012;33(1):124–135. | ||
Yalçin O, Baykan B, Ağan K, et al. An association analysis at 2q36 reveals a new candidate susceptibility gene for juvenile absence epilepsy and/or absence seizures associated with generalized tonic-clonic seizures. Epilepsia. 2011;52(5):975–983. | ||
Sapio MR, Salzmann A, Vessaz M, et al. Naturally occurring carboxypeptidase A6 mutations: effect on enzyme function and association with epilepsy. J Biol Chem. 2012;287(51):42900–42909. | ||
Cui PJ, Zheng L, Cao L, et al. CALHM1 P86L polymorphism is a risk factor for Alzheimer’s disease in the Chinese population. J Alzheimers Dis. 2010;19(1):31–35. | ||
Dreses-Werringloer U, Lambert JC, Vingtdeux V, et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell. 2008;133(7):1149–1161. | ||
Boada M, Antúnez C, López-Arrieta J, et al. CALHM1 P86L polymorphism is associated with late-onset Alzheimer’s disease in a recessive model. J Alzheimers Dis. 2010;20(1):247–251. | ||
Tao QQ, Sun YM, Liu ZJ, et al. Lack of association between CALHM1 p.P86L variation and Alzheimer’s disease in the Han Chinese population. Neurobiol Aging. 2014;35(8):.e13–.e14. | ||
Lv RJ, He JS, Fu YH, et al. A polymorphism in CALHM1 is associated with temporal lobe epilepsy. Epilepsy Behav. 2011;20(4):681–685. | ||
Kauffman MA, Consalvo D, Moron DG, Lereis VP, Kochen S. ApoE epsilon4 genotype and the age at onset of temporal lobe epilepsy: a case-control study and meta-analysis. Epilepsy Res. 2010;90(3):234–239. | ||
Rossignol E, Kobow K, Simonato M, et al. WONOEP appraisal: new genetic approaches to study epilepsy. Epilepsia. 2014;55(8):1170–1186. | ||
Klassen T, Davis C, Goldman A, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145(7):1036–1048. | ||
Heinzen EL, Depondt C, Cavalleri GL, et al. Exome sequencing followed by large-scale genotyping fails to identify single rare variants of large effect in idiopathic generalized epilepsy. Am J Hum Genet. 2012;91(2):293–302. | ||
Cherepanova NS, Leslie E, Ferguson PJ, Bamshad MJ, Bassuk AG. Presence of epilepsy-associated variants in large exome databases. J Neurogenet. 2013;27(1–2):1–4. | ||
Barker RA, Barasi S, Neal MJ. Neuroscience at a Glance. Malden, MA: Blackwell Publishing; 2008:3. | ||
Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biol. 2003;4(3):207. | ||
Marban E, Yamagishi T, Tomaselli GF. Structure and function of voltage-gated sodium channels. J Physiol. 1998;508(3):647–657. | ||
Escayg A, MacDonald BT, Meisler MH, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24(4):343–345. | ||
Li N, Zhang J, Guo JF, Yan XX, Xia K, Tang BS. Novel mutation of SCN1A in familial generalized epilepsy with febrile seizures plus. Neurosci Lett. 2010;480(3):211–214. | ||
Herini ES, Gunadi F, van Kempen MJ, et al. Novel SCN1A mutations in Indonesian patients with severe myoclonic epilepsy in infancy. Pediatr Int. 2010;52(2):234–239. | ||
Vanoye CG, Gurnett CA, Holland KD, George AL Jr, Kearney JA. Novel SCN3A variants associated with focal epilepsy in children. Neurobiol Dis. 2014;62:313–322. | ||
Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115(8):2010–2017. | ||
Kamiya K, Kaneda M, Sugawara T, et al. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci. 2004;24(11):2690–2698. | ||
Liao Y, Anttonen AK, Liukkonen E, et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology. 2010;75(16):1454–1458. | ||
Audenaert D, Claes L, Ceulemans B, Löfgren A, Van Broeckhoven C, De Jonghe P. A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy. Neurology. 2003;61(6):854–856. | ||
Rang HP, Dale MM, Ritter JM, Gardner P. Pharmacology. 4th ed. New York, NY: Churchill Livingstone; 2001. | ||
Gutman GA, Chandy KG, Grissmer S, et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57(4):473–508. | ||
Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156(8):1185–1195. | ||
Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev. 2005;57(4):463–472. | ||
Spauschus A, Eunson L, Hanna MG, Kullmann DM. Functional characterization of a novel mutation in KCNA1 in episodic ataxia type 1 associated with epilepsy. Ann N Y Acad Sci. 1999;868:442–446. | ||
Borgatti R, Zucca C, Cavallini A, et al. A novel mutation in KCNQ2 associated with BFNC, drug resistant epilepsy, and mental retardation. Neurology. 2004;63(1):57–65. | ||
Soldovieri MV, Boutry-Kryza N, Milh M, et al. Novel KCNQ2 and KCNQ3 mutations in a large cohort of families with benign neonatal epilepsy: first evidence for an altered channel regulation by syntaxin-1A. Hum Mutat. 2014;35(3):356–367. | ||
Simons C, Rash LD, Crawford J, et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet. 2015;47(1):73–77. | ||
Lee H, Lin MC, Kornblum HI, Papazian DM, Nelson SF. Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum Mol Genet. 2014;23(13):3481–3489. | ||
Lenzen KP, Heils A, Lorenz S, et al. Supportive evidence for an allelic association of the human KCNJ10 potassium channel gene with idiopathic generalized epilepsy. Epilepsy Res. 2005;63(2–3):113–118. | ||
Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360(19):1960–1970. | ||
Reichold M, Zdebik AA, Lieberer E, et al. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci U S A. 2010;107(32):14490–14495. | ||
Vanderver A, Simons C, Schmidt JL, et al. Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr Neurol. 2014;50(1):112–114. | ||
Heron SE, Smith KR, Bahlo M, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 2012;44(11):1188–1190. | ||
Dai XH, Chen WW, Wang X, et al. A novel genetic locus for familial febrile seizures and epilepsy on chromosome 3q26.2-q26.33. Hum Genet. 2008;124(4):423–429. | ||
Lorenz S, Heils A, Kasper JM, Sander T. Allelic association of a truncation mutation of the KCNMB3 gene with idiopathic generalized epilepsy. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(1):10–13. | ||
Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3(8):a003947. | ||
Jouvenceau A, Eunson LH, Spauschus A, et al. Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet. 2001;358(9284):801–807. | ||
Kors EE, Melberg A, Vanmolkot KR. Childhood epilepsy, familial hemiplegic migraine, cerebellar ataxia, and a new CACNA1A mutation. Neurology. 2004;63(6):1136–1137. | ||
Chioza B, Osei-Lah A, Nashef L, et al. Haplotype and linkage disequilibrium analysis to characterise a region in the calcium channel gene CACNA1A associated with idiopathic generalised epilepsy. Eur J Hum Genet. 2002;10(12):857–864. | ||
Chioza B, Wilkie H, Nashef L, et al. Association between the alpha(1a) calcium channel gene CACNA1A and idiopathic generalized epilepsy. Neurology. 2001;56(9):1245–1246. | ||
Escayg A, De Waard M, Lee DD, et al. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66(5):1531–1539. | ||
Ohmori I, Ouchida M, Miki T, et al. A CACNB4 mutation shows that altered Ca(v)2.1 function may be a genetic modifier of severe myoclonic epilepsy in infancy. Neurobiol Dis. 2008;32(3):349–354. | ||
Steinlein OK, Mulley JC, Propping P, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11(2):201–203. | ||
Phillips HA, Favre I, Kirkpatrick M, et al. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet. 2001;68(1):225–231. | ||
Gu W, Bertrand D, Steinlein OK. A major role of the nicotinic acetylcholine receptor gene CHRNA2 in autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) is unlikely. Neurosci Lett. 2007;422(1):74–76. | ||
Hernandez CC, Gurba KN, Hu N, Macdonald RL. The GABRA6 mutation, R46W, associated with childhood absence epilepsy, alters 6β22 and 6β2 GABA(A) receptor channel gating and expression. J Physiol. 2011;589(pt 23):5857–5878. | ||
Tanaka M, Olsen RW, Medina MT, et al. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am J Hum Genet. 2008;82(6):1249–1261. | ||
Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–175. | ||
Tanaka M, DeLorey TM, Delgado-Escueta A, Olsen RW. GABRB3, epilepsy, and neurodevelopment. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. SourceJasper’s Basic Mechanisms of the Epilepsies [Internet]. 4th ed. Bethesda, MD: National Center for Biotechnology Information (US); 2012. | ||
Carvill GL, Regan BM, Yendle SC, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet. 2013;45(9):1073–1076. | ||
Venkateswaran S, Myers KA, Smith AC, et al. Whole-exome sequencing in an individual with severe global developmental delay and intractable epilepsy identifies a novel, de novo GRIN2A mutation. Epilepsia. 2014;55(7):e75–e79. | ||
Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet. 2002;11(9):1119–1128. | ||
Ohkawa T, Fukata Y, Yamasaki M, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci. 2013;33(46):18161–18174. | ||
Ottman R, Winawer MR, Kalachikov S, et al. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology. 2004;62(7):1120–1126. | ||
Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313(5794):1792–1795. | ||
Chen WJ, Lin Y, Xiong ZQ, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet. 2011;43(12):1252–1255. | ||
Ishii A, Yasumoto S, Ihara Y, et al. Genetic analysis of PRRT2 for benign infantile epilepsy, infantile convulsions with choreoathetosis syndrome, and benign convulsions with mild gastroenteritis. Brain Dev. 2013;35(6):524–530. | ||
Hedera P, Xiao J, Puschmann A, Momčilović D, Wu SW, LeDoux MS. Novel PRRT2 mutation in an African-American family with paroxysmal kinesigenic dyskinesia. BMC Neurol. 2012;12:93. | ||
Wood H. Genetics: expanding the spectrum of neurological disorders associated with PRRT2 mutations. Nat Rev Neurol. 2012;8(12):657. | ||
Heron SE, Grinton BE, Kivity S, et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am J Hum Genet. 2012;90(1):152–160. | ||
Okumura A, Shimojima K, Kubota T, et al. PRRT2 mutation in Japanese children with benign infantile epilepsy. Brain Dev. 2013;35(7):641–646. | ||
Wang JL, Mao X, Hu ZM, et al. Mutation analysis of PRRT2 in two Chinese BFIS families and nomenclature of PRRT2 related paroxysmal diseases. Neurosci Lett. 2013;552:40–45. | ||
Li M, Niu F, Zhu X, et al. PRRT2 mutant leads to dysfunction of glutamate signaling. Int J Mol Sci. 2015;16(5):9134–9151. | ||
Suzuki T, Delgado-Escueta AV, Aguan K, et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004;36(8):842–849. | ||
Medina MT, Suzuki T, Alonso ME, et al. Novel mutations in Myoclonin1/EFHC1 in sporadic and familial juvenile myoclonic epilepsy. Neurology. 2008;70(22):2137–2144. | ||
de Nijs L, Léon C, Nguyen L, et al. EFHC1 interacts with microtubules to regulate cell division and cortical development. Nat Neurosci. 2009;12(10):1266–1274. | ||
Zhao Y, Shi J, Winey M, Klymkowsky MW. Identifying domains of EFHC1 involved in ciliary localization, ciliogenesis, and the regulation of Wnt signaling. Dev Biol. 2016;411(2):257–265. | ||
Bassuk AG, Wallace RH, Buhr A, et al. A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am J Hum Genet. 2008;83(5):572–581. | ||
Tao H, Manak JR, Sowers L, et al. Mutations in prickle orthologs cause seizures in flies, mice, and humans. Am J Hum Genet. 2011;88(2):138–149. | ||
Lal D, Reinthaler EM, Altmüller J, et al. RBFOX1 and RBFOX3 mutations in rolandic epilepsy. PLoS One. 2013;8(9):e73323. | ||
Lal D, Trucks H, Møller RS, et al; EMINet Consortium; EPICURE Consortium. Rare exonic deletions of the RBFOX1 gene increase risk of idiopathic generalized epilepsy. Epilepsia. 2013;54(2):265–271. | ||
Fogel BL, Wexler E, Wahnich A, et al. RBFOX1 regulates both splicing and transcriptional networks in human neuronal development. Hum Mol Genet. 2012;21(19):4171–4186. | ||
Li YI, Sanchez-Pulido L, Haerty W, Ponting CP. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015;25(1):1–13. | ||
Gehman LT, Stoilov P, Maguire J, et al. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat Genet. 2011;43(7):706–711. | ||
Weyn-Vanhentenryck SM, Mele A, Yan Q, et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014;6(6):1139–1152. | ||
Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet. 2013;45(5):546–551. | ||
Picard F, Makrythanasis P, Navarro V, et al. DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology. 2014;82(23):2101–2106. | ||
Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet. 2013;45(5):552–555. | ||
Scheffer IE, Heron SE, Regan BM, et al. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol. 2014;75(5):782–787. | ||
Baulac S. Genetics advances in autosomal dominant focal epilepsies: focus on DEPDC5. Prog Brain Res. 2014;213:123–139. | ||
Poduri A. DEPDC5 does it all: shared genetics for diverse epilepsy syndromes. Ann Neurol. 2014;75(5):631–633. | ||
Hallmann K, Zsurka G, Moskau-Hartmann S, et al. A homozygous splice-site mutation in CARS2 is associated with progressive myoclonic epilepsy. Neurology. 2014;83(23):2183–2187. | ||
Arsov T, Smith KR, Damiano J, et al. Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6. Am J Hum Genet. 2011;88(5):566–573. | ||
Karkheiran S, Krebs CE, Makarov V, et al. Identification of COL6A2 mutations in progressive myoclonus epilepsy syndrome. Hum Genet. 2013;132(3):275–283. | ||
Corbett MA, Schwake M, Bahlo M, et al. A mutation in the Golgi Qb-SNARE Gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am J Hum Genet. 2011;88(5):657–663. | ||
Berkovic SF, Dibbens LM, Oshlack A, et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet. 2008;82(3):673–684. | ||
Pulido Fontes L, Quesada Jimenez P, Mendioroz Iriarte M. Epigenetics and epilepsy. Neurologia. 2015;30(2):111–118. | ||
Hwang JY, Aromolaran KA, Zukin RS. Epigenetic mechanisms in stroke and epilepsy. Neuropsychopharmacology. 2013;38(1):167–182. | ||
Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463. | ||
Gräff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev. 2011;91(2):603–649. | ||
Kobow K, Jeske I, Hildebrandt M, et al. Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J Neuropathol Exp Neurol. 2009;68(4):356–364. | ||
Miller-Delaney SF, Das S, Sano T, et al. Differential DNA methylation patterns define status epilepticus and epileptic tolerance. J Neurosci. 2012;32(5):1577–1588. | ||
Kobow K, Blümcke I. The emerging role of DNA methylation in epileptogenesis. Epilepsia. 2012;53(suppl 9):11–20. | ||
Henshall DC, Kobow K. Epigenetics and epilepsy. Cold Spring Harb Perspect Med. 2015;5(12):a022731. | ||
Huang Y, Doherty JJ, Dingledine R. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci. 2002;22(19):8422–8428. | ||
Sng JC, Taniura H, Yoneda Y. Histone modifications in kainate-induced status epilepticus. Eur J Neurosci. 2006;23(5):1269–1282. | ||
Tsankova NM, Kumar A, Nestler EJ. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci. 2004;24(24):5603–5610. | ||
Jimenez-Mateos EM, Henshall DC. Epilepsy and microRNA. Neuroscience. 2013;238:218–229. | ||
Mollinari C, Racaniello M, Berry A, et al. miR-34a regulates cell proliferation, morphology and function of newborn neurons resulting in improved behavioural outcomes. Cell Death Dis. 2015;6:e1622. | ||
Bavamian S, Mellios N, Lalonde J, et al. Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol Psychiatry. 2015;20(5):573–584. | ||
Hu K, Xie YY, Zhang C, et al. MicroRNA expression profile of the hippocampus in a rat model of temporal lobe epilepsy and miR-34a-targeted neuroprotection against hippocampal neurone cell apoptosis post-status epilepticus. BMC Neurosci. 2012;13:115. | ||
Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. 2007;4(1):18–61. | ||
Bozzi Y, Casarosa S, Caleo M. Epilepsy as a neurodevelopmental disorder. Front Psychiatry. 2012;3:19. | ||
Sawyer NT, Escayg A. Stress and epilepsy: multiple models, multiple outcomes. J Clin Neurophysiol. 2010;27(6):445–452. | ||
Hartman JL 4th, Garvik B, Hartwell L. Principles for the buffering of genetic variation. Science. 2001;291(5506):1001–1004. | ||
Skinner MK. Environmental epigenetics and a unified theory of the molecular aspects of evolution: a neo-lamarckian concept that facilitates neo-Darwinian evolution. Genome Biol Evol. 2015;7(5):1296–1302. | ||
Feinberg AP. The epigenetics of cancer etiology. Semin Cancer Biol. 2004;14(6):427–432. | ||
Malacarne M, Gennaro E, Madia F, et al. Benign familial infantile convulsions: mapping of a novel locus on chromosome 2q24 and evidence for genetic heterogeneity. Am J Hum Genet. 2001;68(6):1521–1526. | ||
EPICURE Consortium, Leu C, de Kovel CG, et al. Genome-wide linkage meta-analysis identifies susceptibility loci at 2q34 and 13q31.3 for genetic generalized epilepsies. Epilepsia. 2012;53(2):308–318. | ||
Deprez L, Claes LR, Claeys KG, et al. Genome-wide linkage of febrile seizures and epilepsy to the FEB4 locus at 5q14.3-q23.1 and no MASS1 mutation. Hum Genet. 2006;118(5):618–625. | ||
Bai D, Alonso ME, Medina MT, et al. Juvenile myoclonic epilepsy: linkage to chromosome 6p12 in Mexico families. Am J Med Genet. 2002;113(3):268–274. | ||
Layouni S, Chouchane L, Malafosse A, Dogui M. Dimorphism of TAP-1 gene in Caucasian with juvenile myoclonic epilepsy and in Tunisian with idiopathic generalized epilepsies. Int J Immunogenet. 2010;37(2):117–123. | ||
Pinto D, Kasteleijn-Nolst Trenité DG, Cordell HJ, et al. Explorative two-locus linkage analysis suggests a multiplicative interaction between the 7q32 and 16p13 myoclonic seizures-related photosensitivity loci. Genet Epidemiol. 2007;31(1):42–50. | ||
Puranam RS, Jain S, Kleindienst AM, et al. A locus for generalized tonic-clonic seizure susceptibility maps to chromosome 10q25-q26. Ann Neurol. 2005;58(3):449–458. | ||
Neubauer BA, Fiedler B, Himmelein B, et al. Centrotemporal spikes in families with rolandic epilepsy: linkage to chromosome 15q14. Neurology. 1998;51(6):1608–1612. | ||
Zara F, Bianchi A, Avanzini G, et al. Mapping of genes predisposing to idiopathic generalized epilepsy. Hum Mol Genet. 1995;4(7):1201–1207. | ||
Morales-Corraliza J, Gómez-Garre P, Sanz R, Díaz-Otero F, Gutiérrez-Delicado E, Serratosa JM. Familial partial epilepsy with variable foci: a new family with suggestion of linkage to chromosome 22q12. Epilepsia. 2010;51(9):1910–1914. | ||
Goeggel Simonetti B, Rieubland C, Courage C, et al. Duplication of the sodium channel gene cluster on 2q24 in children with early onset epilepsy. Epilepsia. 2012;53(12):2128–2134. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.