")
Back to Journals » Journal of Multidisciplinary Healthcare » Volume 15
Generalized Arterial Calcification of Infancy (GACI): Optimizing Care with a Multidisciplinary Approach
Authors Kawai K, Sato Y, Kawakami R, Sakamoto A, Cornelissen A, Mori M, Ghosh S, Kutys R, Virmani R, Finn AV
Received 31 December 2021
Accepted for publication 22 April 2022
Published 1 June 2022 Volume 2022:15 Pages 1261—1276
DOI https://doi.org/10.2147/JMDH.S251861
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Kenji Kawai,1,* Yu Sato,1,* Rika Kawakami,1 Atsushi Sakamoto,1 Anne Cornelissen,1 Masayuki Mori,1 Saikat Ghosh,1 Robert Kutys,1 Renu Virmani,1 Aloke V Finn1,2
1CVPath Institute, Gaithersburg, MD, USA; 2University of Maryland, School of Medicine, Baltimore, MD, USA
*These authors contributed equally to this work
Correspondence: Aloke V Finn, 19 Firstfield Road, Gaithersburg, MD, 20878, USA, Tel +301.208.3570, Fax +301.208.3745, Email [email protected]
Abstract: It is very unusual to see evidence of arterial calcification in infants and children, and when detected, genetic disorders of calcium metabolism should be suspected. Generalized arterial calcification of infancy (GACI) is a hereditary disease, which is characterized by severe arterial calcification of medium sized arteries, mostly involving the media with marked intimal proliferation and ectopic mineralization of the extravascular tissues. It is caused by inactivating variants in genes encoding either ENPP1, in a majority of cases (70– 75%), or ABCC6, in a minority (9– 10%). Despite similar histologic appearances between ENPP1 and ABCC6 deficiencies, including arterial calcification, organ calcification, and cardiovascular calcification, mortality is higher in subjects carrying the ENPP1 versus ABCC6 variants (40% vs 10%, respectively). Overall mortality in individuals with GACI is high (55%) before the age of 6 months, with 24.4% dying in utero or being stillborn. Rare cases show spontaneous regression with age, while others who survive into adulthood often manifest musculoskeletal complications (osteoarthritis and interosseous membrane ossification), enthesis mineralization, and cervical spine fusion. Despite recent advances in the understanding of the genetic mechanisms underlying this disease, there is still no ideal therapy for the resolution of vascular calcification in GACI. Although bisphosphonates with anti-calcification properties have been commonly used for the treatment of CAGI, their benefit is controversial, with favorable results reported at one year and questionable benefit with delayed initiation of treatment. Enzyme replacement therapy with administration of recombinant form of ENPP1 prevents calcification and mortality, improves hypertension and cardiac function, and prevents intimal proliferation and osteomalacia in mouse models of ENPP1 deficiency. Therefore, newer treatments targeting genes are on the horizon. In this article, we review up to date knowledge of the understanding of GACI, its clinical, pathologic, and etiologic understanding and treatment in support of more comprehensive care of GACI patients.
Keywords: generalized arterial calcification of infancy, ectonucleotide pyrophosphatase/phosphodiesterase 1, ATP binding cassette subfamily C member 6, vascular calcification
Introduction
Vascular calcification (VC) is the deposition of calcium and phosphate crystals in blood vessels, which is a common phenomenon in elderly patients with atherosclerosis. Cardiovascular calcification has also been associated with type 2 diabetes, chronic kidney disease, and renal dialysis in adults for many years.1 The association of VC and aortic valve calcification with increased mortality is well known.2–6 There are a few rare hereditary syndromes, such as generalized arterial calcification of infancy (GACI), pseudoxanthoma elasticum (PXE; OMIM no. 264800), and deficiency of CD73, which are also associated with arterial calcification. The disease GACI was first described in 1899 and there have been over 250 reported cases since. In 2003, the discovery of the mutation responsible for GACI was reported by Rutsch and collaborators as a loss of function mutation in the ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1, OMIM no. 173335) gene.7 ENPP1 is a transmembrane protein with a hydrolase extracellular domain, which hydrolyzes ATP into AMP and inorganic phosphate (PPi).7 A less severe form of generalized calcification involving calcification of joints and arteries (CALJA; OMIM no. 211800), an extremely rare Mendelian disorder of isolated calcification, characterized by late onset calcification of the arteries of the extremities and hand and foot joint capsules and caused by mutations on the NT5E gene, will also be briefly discussed.
GACI is characterized by calcification of arteries and intimal proliferation that leads to stenosis, especially involving the coronary arteries with cardiac complications. GACI patients also develop joint and parenchymal organ calcification involving renal, gastrointestinal, and pulmonary complications. The disease occurs in childhood and may also manifest prenatally, with approximately 55% mortality within the first 6 months of life.8 However, the disease after 6 months is not well characterized. While key molecular pathways leading to premature calcific artery disease have been identified, we still need a better understanding of hereditary diseases of calcification. In this review, we present the current knowledge and provide a comprehensive guide for the care of GACI, the most common form of genetic vascular calcification.
Etiology, Prevalence, and Prognosis
GACI is a rare autosomal recessive disease caused by loss-of-function gene mutations. In approximately 70–75% of the cases, the disease occurs from the loss of activity of ENPP1, while in 9% of cases it occurs from ATP-binding cassette, subfamily C member 6 (ABCC6, OMIM no. 614473), which is suspected to be involved in ATP export. This leads to continued calcium accumulation in the arterial wall, resulting in arterial calcification and narrowing due to intimal proliferation. The incidence of GACI is estimated to be 1 out of every 200,000 pregnancies, with the carrier rate being 1:200.9 The survival rate varies widely, but the mortality rate is high, reaching as high as 70% in cases of prenatal polyhydramnios/fetal hydrops, myocardial infarction, heart failure, and arrhythmias secondary to severe calcification. The major causes of death are myocardial infarction, congestive heart failure, persistent arterial hypertension, and multiple organ failure.8,10 Infants who survive the first few months have a significantly lower mortality.8 On the other hand, spontaneous regression of arterial and ectopic calcification has been observed in several cases with arterial calcifications documented in infancy.11,12 Bisphosphonate treatment has been used to reduce the mortality of these patients with GACI by 65%.8 However, a recent review contradicts their usefulness and is dependent on the combined use of Etidronate, the first-generation bisphosphonate (chemically stable derivatives of inorganic pyrophosphate (PPi)), which binds directly to hydroxyapatite with high affinity, resulting in inhibition of bone mineralization, while the second and third generation (are nitrogen-containing) cause osteoclast apoptosis and limit bone resorption.13–15
Diagnosis
About half of GACl cases (48%) are diagnosed in utero, and most of the other half (52%) are diagnosed at a median age of 3 months.16 However, no rigid consensus has been established for clinical diagnostic criteria for systemic arterial calcification in infancy. The reason is related to phenotypic overlap observed between GACI and PXE, which is discussed below.17,18 GACI is characterized by the onset of widespread arterial calcification in the first month of life, involving large and medium-sized vessels, resulting in signs and symptoms related to the cardiovascular system (including heart failure, respiratory distress, edema, cyanosis, hypertension, and/or cardiomegaly). Additional findings include skin and retinal manifestations typically also seen in PXE, including periarticular calcification, development of rickets after infancy, cervical spine fusion, and hearing loss. In some individual patients, the diagnosis of GACI may entail a combination of clinical manifestations, imaging, and histopathologic findings, as well as genetic testing. The cardiovascular phenotype of GACI disease is also very diverse and includes fetal distress, polyhydramnios, fetal edema, heart failure, hypertension, cardiomegaly, visceral effusion, dyspnea, cyanosis, and decreased peripheral pulses.16 Vascular calcification may be evident during prenatal ultrasound.19 Although calcification is detected as a subtle radiopaque area even on routine radiographic films, these findings are often overlooked, which has been reported as a pitfall and could result in delaying the diagnostic process.20,21 Therefore, whole-body computed tomography is favored as an imaging modality to assess the development of systemic calcification in GACI22 (Table 1).
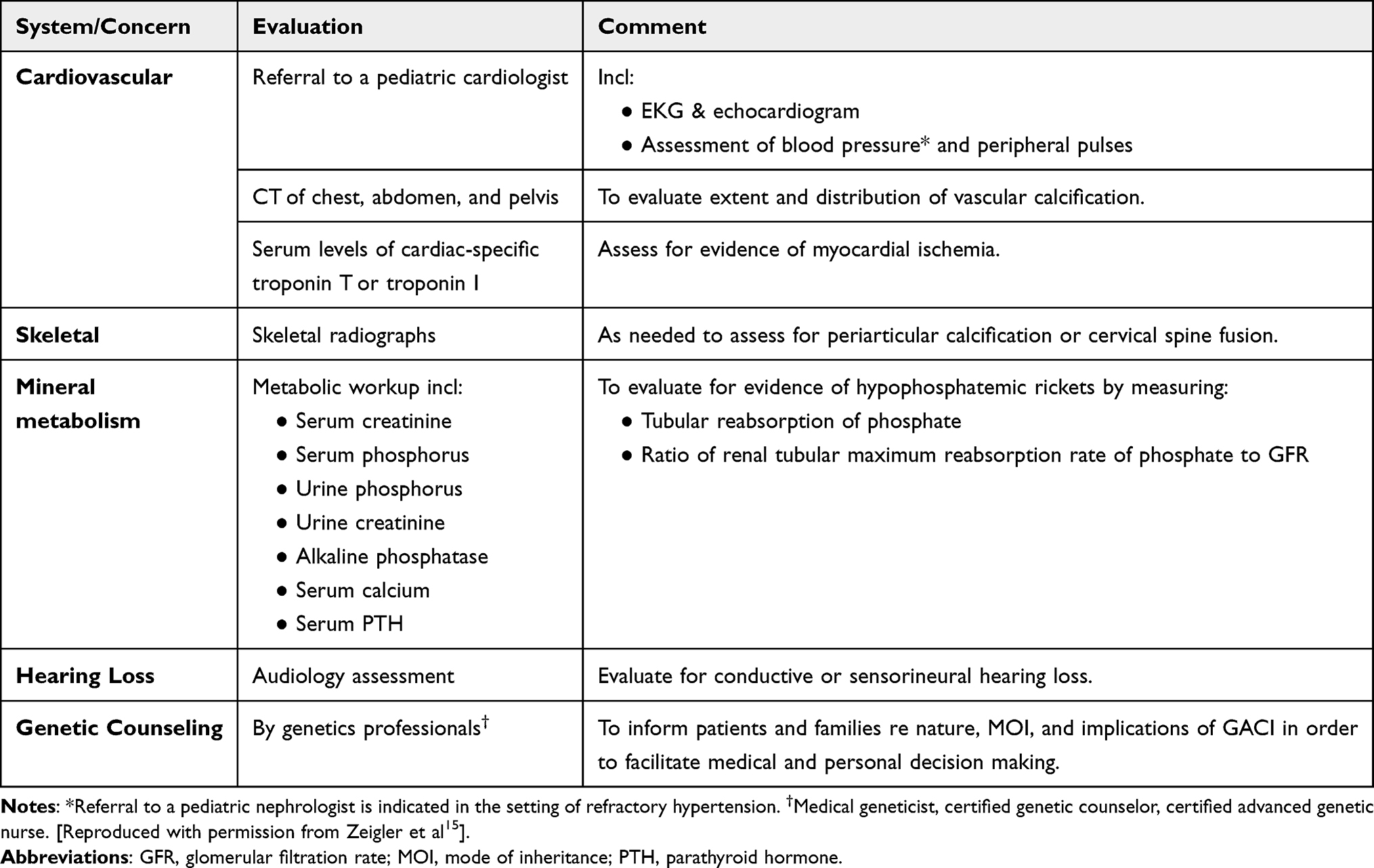 |
Table 1 Recommended Evaluations Following Initial Diagnosis in Individuals with GACI |
Genetic Background
GACI is a genetic disorder caused by biallelic inactivating variants in the ENPP1 gene in about 70% of cases, with more than 40 different mutations have been identified in about 200 cases.8,23 However, in about 10% of GACI patients, a different mutation has also been seen in the ABCC6 gene.23 GACI (OMIM no. 208000, OMIM no. 603234) is associated with mutations in either the ENPP1 or ABCC6 genes respectively, and deactivating mutations in these genes results in decreased inorganic pyrophosphate (PPi). Originating from these two gene mutations, the phenotype of GACI is classified into two types (see below). Most patients with GACI eventually develop hypophosphatemic rickets24 (Table 2).
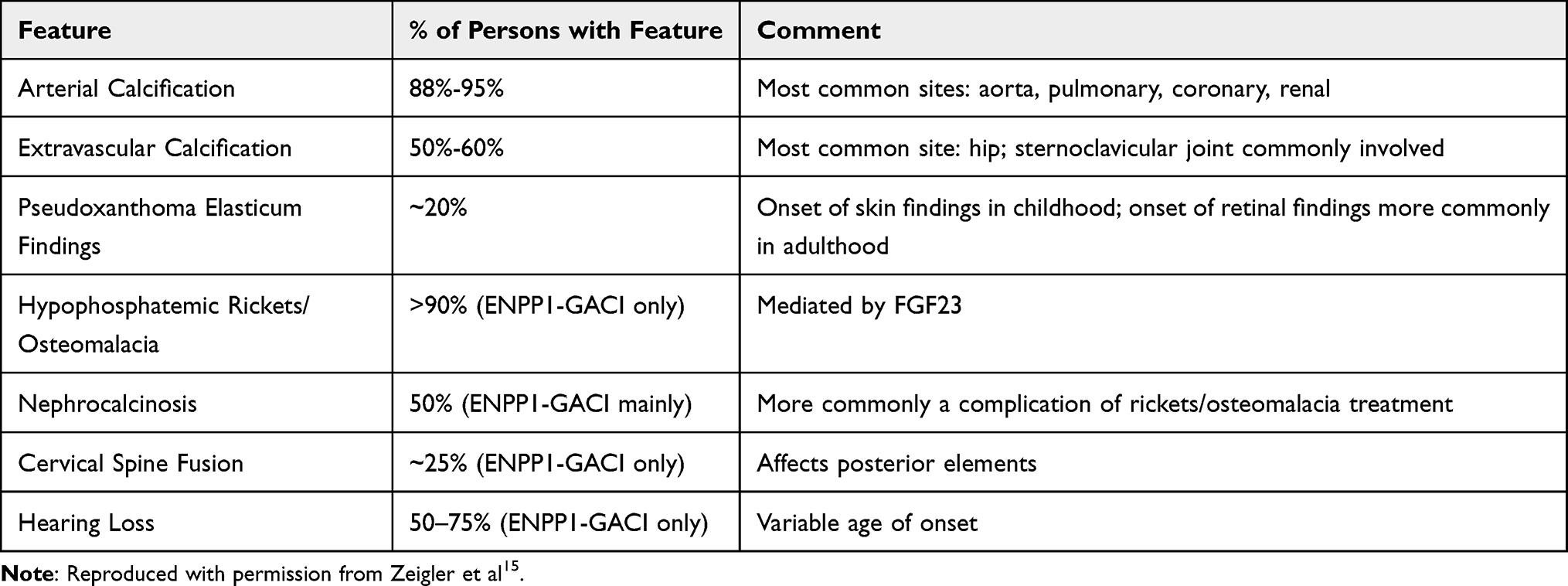 |
Table 2 GACI: Frequency of Select Features |
ENPP1 Gene
The GACI gene was identified in 2003 as due to a loss-of-function mutation in the ENPP1 gene.7 ENPP1 is a type II extracellular membrane-bound glycoprotein that is expressed in various cell types, including osteoblasts, osteoclasts, chondrocytes, and vascular smooth muscle cells.25–29 About 70% of GACI cases are caused by biallelic mutations in the ENPP1 gene, with more than 40 different mutations identified.8 ENPP1 is an extracellular enzyme that hydrolyzes extracellular ATP into AMP and PPi.7 Extracellular PPi is an important inhibitor of calcification in vivo and is hydrolyzed by the tissue cell surface enzyme, nonspecific alkaline phosphatase (TNAP), to produce inorganic phosphate (Pi) from PPi. Since the essence of vascular calcification is the deposition of calcium phosphate in the vessel wall, the balance between PPi and Pi is key to inhibiting vascular calcification.30 Therefore, loss-of-function mutations in ENPP1 lead to a decrease in PPi and an imbalance in the PPi/Pi ratio, thus promoting the formation of calcification in blood vessels and soft tissues.28,31 It is known that mouse models with genetically disrupted ENNP1 expression exhibit high levels of ectopic calcification, leading to cardiovascular disease and joint hyperostosis.32 TNAP activity is elevated in patients with GACI, resulting in increased Pi levels and accelerated calcification formation (Figure 1).33 Patients with GACI caused by ENPP1 who survive the first 6 months of life are at risk of developing bone deformities, hypophosphatemia, hyperphosphaturia, and elevated alkaline phosphatase, with all the manifestations of autosomal recessive hypophosphatemic rickets type 2 (ARHR2, OMIM no. 613312).
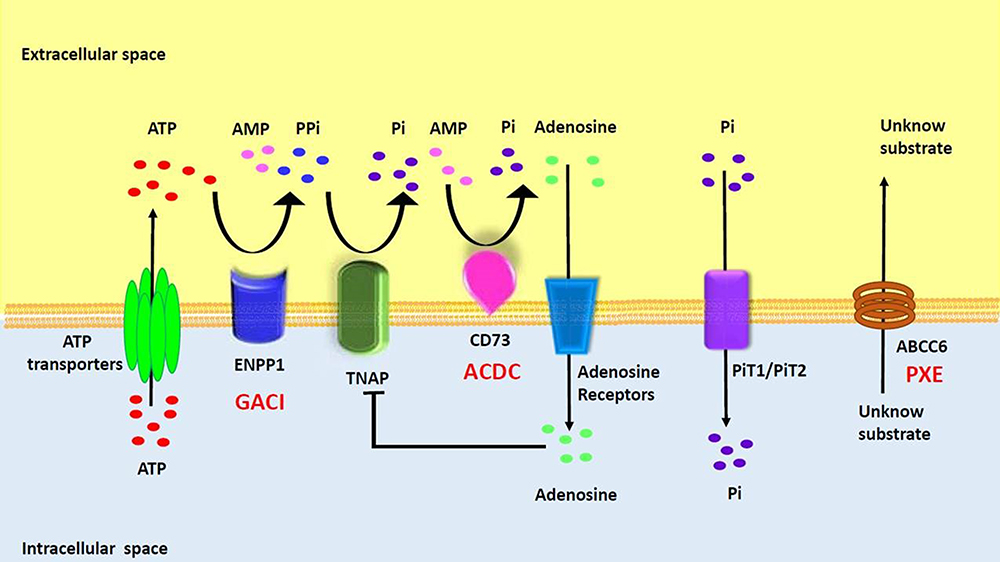 |
Figure 1 Overview of Rare Monogenic Diseases with Vascular Calcification Due to Mutation of the Purine Metabolism Pathway. ATP gains access via ATP transporters into extracellular space, where it is hydrolyzed by ENPP1 into AMP and PPi. AMP is then hydrolyzed by CD73 into adenosine and Pi while PPi is hydrolyzed by TNAP into inorganic phosphate. PiT1/2 (Na+-dependent Pi cotransporter) mediates cellular Pi uptake and transport into the cell. PPi is a potent inhibitor of calcification. Adenosine can interact with adenosine receptors and suppress TNAP expression, as well as have various effects based on specific receptors. The specific substrate for ABCC6 has yet to be identified. ENPP1, CD73, and ABCC6 deficiencies drive the molecular pathophysiology of several monogenic diseases associated with arterial calcification (GACI, ACDC, and PXE). Reproduced with permission from Cudrici CD et al.33 |
As mentioned earlier, a less severe form of generalized calcification involving CALJA (OMIM no. 211800) also exists and is an extremely rare Mendelian disorder of isolated calcification that is characterized by late onset calcification of the arteries of the extremities and hand and foot joint capsules. It is caused by mutations on the NT5E gene. AMP is hydrolyzed by CD73, which is encoded by the 5’-nucleotidase ecto (NT5E) gene, producing Pi and adenosine (Figure 1). NT5E mutations cause the loss of CD73, resulting in the occurrence of CALJA. This autosomal recessive disease, also called arterial calcification due to the deficiency of CD73 (ACDC), is characterized by calcification of the hand and foot joints and severe medial arterial and periarterial calcification, including aneurysmal dilation that primarily affects the arteries of the lower extremities. Coronary calcification has not been reported and none of the patients have any significant atherosclerosis, impaired renal function, hypertension, dyslipidemia, or diabetes. GACI is deficient in both adenosine and its precursor AMP, whereas in CALJA there is only reduced extracellular adenosine levels and enhanced TNAP activation, which degrades PPi to Pi and promotes tissue calcification. Therefore, patients with CALJA have adult onset of calcification of hand and foot capsule joints and severe calcification of aneurysmally dilated arteries of the lower extremities (eg, iliac, femoral, tibial), but not of the carotid, aorta or coronary arteries.34–36
ABCC6 Gene
GACI can also be caused by homozygous mutations in the ABCC6 gene (also known as ABCC6 deficiency), which directs the production of a protein called multidrug resistance-related protein 6 (MRP6, also known as ABCC6 protein). This mutation has been identified in 10% of GACI patients. MRP6 protein belongs to a group of proteins that transport molecules across the cell membrane, but little is known about the substances transported by MRP6. Jansen et al reported that loss of ABCC6 reduces extracellular ATP, the main substrate required for the generation of extracellular PPi.37 Several studies have also suggested that MRP6 promotes the release of the molecule adenosine triphosphate (ATP) into the extracellular space through an unknown mechanism.38 ATP is then immediately broken down into adenosine monophosphate (AMP) and pyrophosphate, and pyrophosphate inhibits the deposition of minerals such as calcium (calcification).39 MRP6 is found primarily in the liver and kidneys, but also in small amounts in other tissues such as skin, stomach, blood vessels, and eyes.40,41 Therefore, patients with GACI due to deficiency of ABCC6 may develop symptoms similar to PXE, an autosomal recessive disorder also caused by mutations in the ABCC6 gene, as they grow older.
Clinical Spectrum of ENPP1 and ABCC6 Deficiency Syndromes
GACI is a multiorgan disease with a complex phenotype, with genotypic and phenotypic diversity observed between GACI and several other diseases, such as PXE. The phenotypes caused by ENPP1 and ABCC6 deficiencies may overlap, while also varying in severity (Table 3).42 GACI and PXE have overlapping genotypes and phenotypes and have a common pathway of diffuse vascular calcification, however, they ultimately differ in disease severity. PXE is an autosomal recessive disorder that causes calcium and mineral deposition in elastic fibers, leading to degeneration and ectopic calcification of the skin, retina, and cardiovascular systems. PXE often occurs in adolescence and is usually diagnosed by the early twenties. Patients with PXE have arterial disease consisting of fragmentation of elastic laminae, mineralization of elastic fibers, and diffuse wall thickening. Although PXE was thought to be a distinct disease from GACI, which is primarily associated with ENPP1, it was determined that mutations of the ENPP1 gene also may cause PXE in a small number of patients.23,38,43,44 An enormous number of different ABCC6 mutations have been described for PXE but no clear correlation with genotype and phenotype has been found. Furthermore, even the same ABCC6 mutation may lead to a serious progression, resulting in death from myocardial infarction in early infancy in GACI, while the PXE phenotype leads to a relatively mild progression. In 2010, Boulanger et al reported the case with two brothers born to unrelated parents, where the older brother developed uncomplicated PXE in adolescence while the younger brother died at 15 months of age from myocardial infarction. The autopsy showed calcification of the endocardium and extensive calcification of coronary arteries, medium-sized arteries, and aorta, leading to a diagnosis of GACI. Genetic analysis was performed, but no tissue was available from the younger brother. The elder brother had two heterozygous missense mutations of ABCC6. Each gene was inherited from one of his heterozygous asymptomatic parents. No ENPP1 mutation was found in the living family members.45 At present, however, it is still not completely known how mutations in the different genes ENPP1 and ABCC6 can lead to similar pathophysiological consequences. GACI and PXE do not simply represent two distinct disorders but rather represent a spectrum of differences in ectopic calcification.
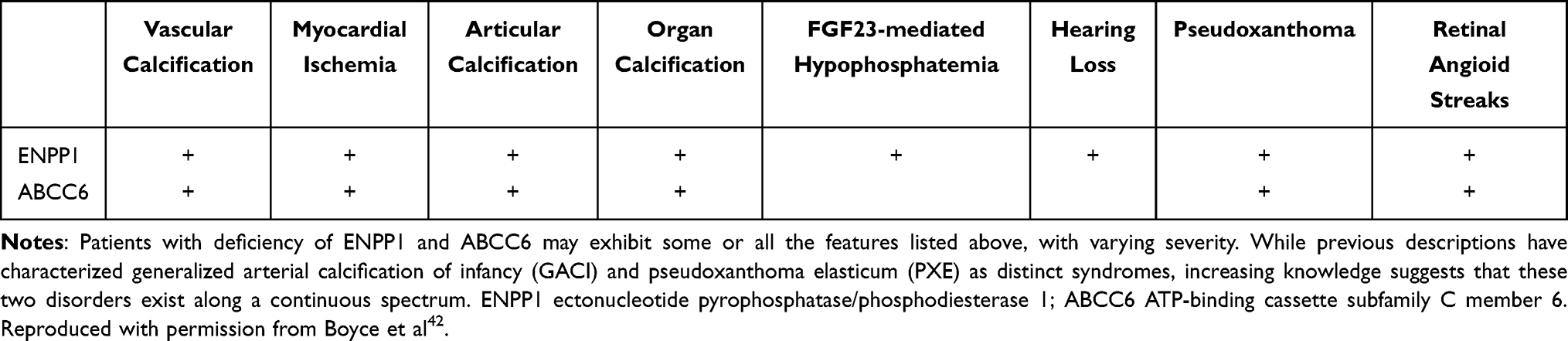 |
Table 3 Clinical Spectrum of ENPP1 and ABCC6 Deficiency Syndromes |
Pathophysiology
The main pathogenesis of GACI is vessel calcification, which is considered to be the most serious cause of morbidity and mortality in GACI cases. The histological characteristics include severe calcification, with the fragmentation of elastic fibers of the medium and large caliber arteries, calcification of the internal elastic layer, and intimal proliferation of smooth muscle cells. It is noted that severe arterial calcification, with severe arterial stenosis, can occur in utero (earliest calcification seen at 18 weeks) or in the postnatal weeks.16 Thus, in cases surviving beyond infancy, the vascular involvements may affect a wide variety of other organs, like the kidneys, liver, and spleen. Overall, males and females are equally affected and many different ethnic groups have had GACI cases reported.
Vascular Calcification
In the past, vascular calcification was thought to be a passive phenomenon. While it is well accepted today that vascular calcification is highly regulated, involving a variety of molecular signalling pathways, it remains incompletely understood. Calcification of the viscera is divided into two major forms; 1) dystrophic calcification observed in injured or necrotic tissue with normal serum calcium levels, occurring in soft tissues or organs that usually do not calcify, eg, the vascular system, valves, heart, kidney, lungs, brain, etc. and 2) metastatic calcification occurs in tissues that originally are normal but calcify in the presence of hypercalcemia, which is related to calcium imbalance, such as hypervitaminosis D or hyperparathyroidism.46 The calcific deposits are composed of amorphous calcium phosphate or crystals of hydroxyapatite, and may also consist of calcium oxalates, octacalcium phosphate, and other calcium salts. Various mechanisms of vascular calcification have been proposed, and calcification of the arterial wall can be generally classified into three types.47 The three main types of calcification are inflammatory, which is mainly atherosclerotic in nature, metabolic, which manifests as medial arterial calcification, also known as Monckeberg medial sclerosis, and is often associated with but not limited to diabetes mellitus, chronic kidney disease, and aging, and genetic causes such as GACI, PXE, and ACAD. It should be noted that these classifications overlap for each category, especially inflammatory and metabolic (Category 1 and 2). Diabetic patients frequently have both hyperlipidemia and chronic kidney disease (CKD), thus, both intimal and medial calcification may be present in the same arterial lesion. GACI is classified as category 3, on the basis of the genetic factors, and is characterized as calcification developing in the media of the artery.
Autopsy studies have shown that the location of calcified arteries in GACI varies depending on the time of onset of the disease.16 The most frequently calcified artery in early-onset GACI is the hepatic artery (81%), followed by the aorta (80%), pulmonary artery (67%), the coronary artery (53%), and the renal artery (39%). The most commonly calcified artery in late-onset GACI is the coronary artery (88%), followed by the renal artery (55%), the pulmonary artery (49%), the aorta (36%), the adrenal artery (34%), the splenic artery (31%), the pancreatic artery (28%), and the mesenteric artery (26%). In the cohort of long-term survivors of GACI, the most common locations of arterial calcification are in the aorta, renal, mesenteric, coronary, iliac, and pulmonary arteries.9 There are also reports of cerebral artery involvement in a few individuals, with low frequency, which are associated with seizures, cerebral infarction, and recurrent transient ischemic attacks due to cerebral vascular insufficiency, also included in the symptoms of GACI.48–51 Calcification of the peripheral arteries also leads to a reduction in peripheral pulse pressure, resulting in gangrene of the distal extremities.52,53 It has been noted that development in the pulmonary artery may result in drug-resistant pulmonary hypertension.54,55 On the other hand, arterial stenosis with intimal thickening has been observed in individuals with no evidence of progressive calcification.9,23,56,57 These arterial calcifications or intimal proliferations explain the high frequency of recurrent pregnancy loss.9
Histological Characteristics of Arterial Calcification in GACI
We have seen 2 cases of GACI in the last 15 years at CVPath, and one was reported showing diffuse coronary arterial calcification of infancy.58 This case is from an 8-month-old boy who presented with cardiogenic shock and had a myocardial infarction in the anterolateral and septal wall of the left ventricle. Coronary angiography revealed severe multivessel disease with diffuse stenosis of the left anterior descending artery. The patient’s older sister had died of sudden cardiac arrest at 3-months of age. Genetic testing revealed a mutation of the ENPP1 gene confirming the diagnosis of GACI. The patient died after a short period of a relatively stable course. At autopsy, the coronary arteries showed both early and late phase histological features of calcified lesions, especially involving the distal left anterior descending (LAD). The LAD, left diagonal, left circumference, left obtuse marginal, and right coronary arteries showed severe intimal proliferation with marked stenosis and calcification (Figures 2 and 3A–C). Note there were a few macrophages and scattered T lymphocytes around the regions of calcification, but overall foamy macrophages were not seen (Figure 3D–F). In intermediate lesions, there was destruction of elastin fibers, inflammatory cell infiltration, and a thickened layer of calcification with neovascularization. Elastin fiber regeneration was also observed near the luminal surface (Figure 3G–I). In arteries with severe stenosis, there were greater calcium deposits, not only along the elastin fibers but extending into the intima and media, with an accompanying decrease in SMCs and focal infiltration by T cells and macrophages (Figure 3J–L). Further expansion of calcium deposition was also observed in the intima and media, with marked SMC proliferation and matrix deposition, which resulted in severe luminal narrowing. The progression of calcification in GACI begins with the deposition of calcium in the elastic lamina and expands into the surrounding intima and media. At the border of the calcified area in the initial lesion, SMCs are abundant, and as the calcified deposits expand and invade the lumen as the lesion progresses, chronic inflammatory cells, mainly composed of T lymphocytes and macrophages, infiltrate (Figure 3M–O). Similar early calcification and SMC-rich neointima were seen in other vascular beds, such as the aorta and its major branches (Figure 4).
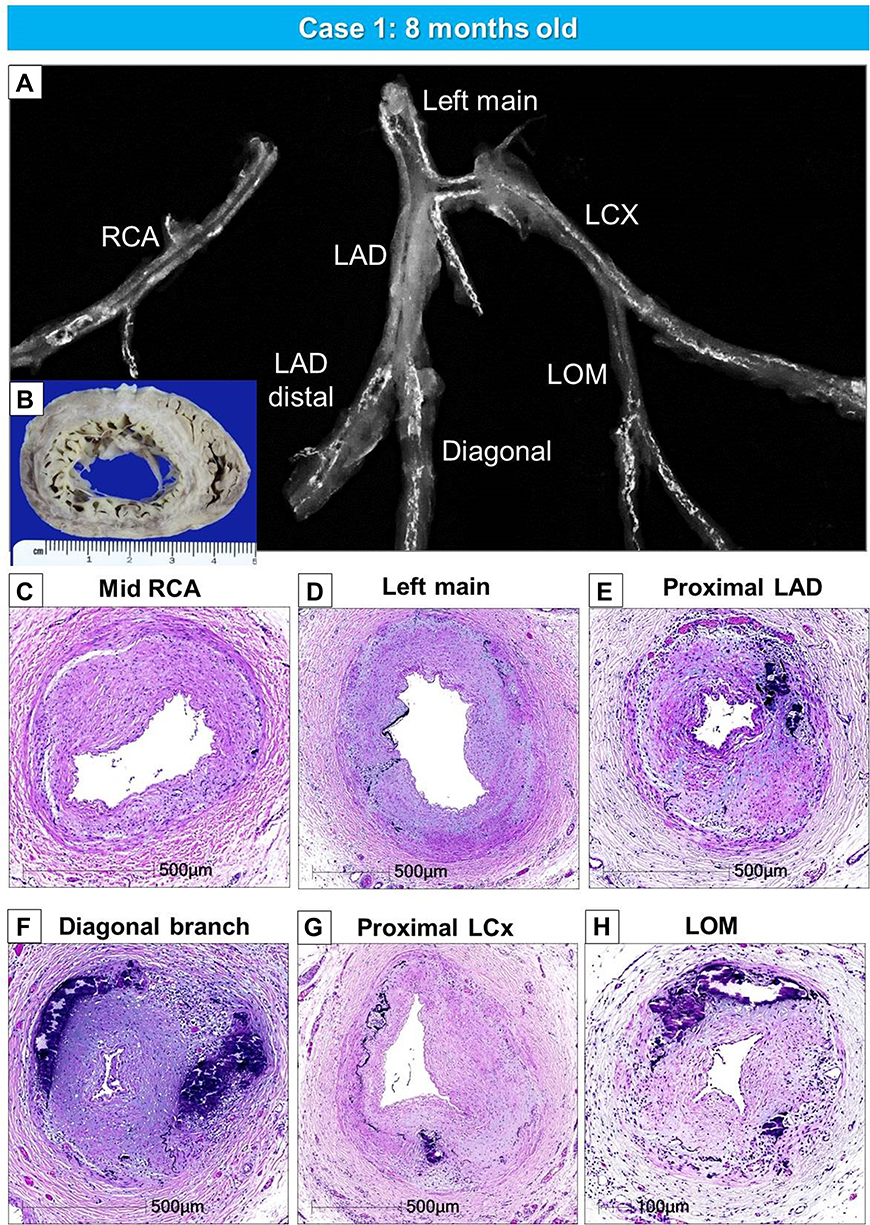 |
Figure 2 Gross, radiographic, and histologic images of the heart and coronary artery from a 8-months-old male with GACI. Radiographs of excised coronary arteries (CA) (A) demonstrate focal calcification of all four major branches [left main, left anterior descending (LAD), left circumflex (LCX), and right CA (RCA)] and smaller branches. (B) Mid-left and right ventricular slices of the heart show dilatation of the LV and subendocardial circumferential healed myocardial infarction, along with transmural scarring of the posterolateral wall of LV. (C–H). Hematoxylin and eosin-stained sections of the major coronary arteries and branch vessels demonstrating moderate to severe luminal narrowing with or without medial calcification (A, B, E–H) are reproduced with permission from Federici et al58. Abbreviation: LOM, left obtuse marginal. |
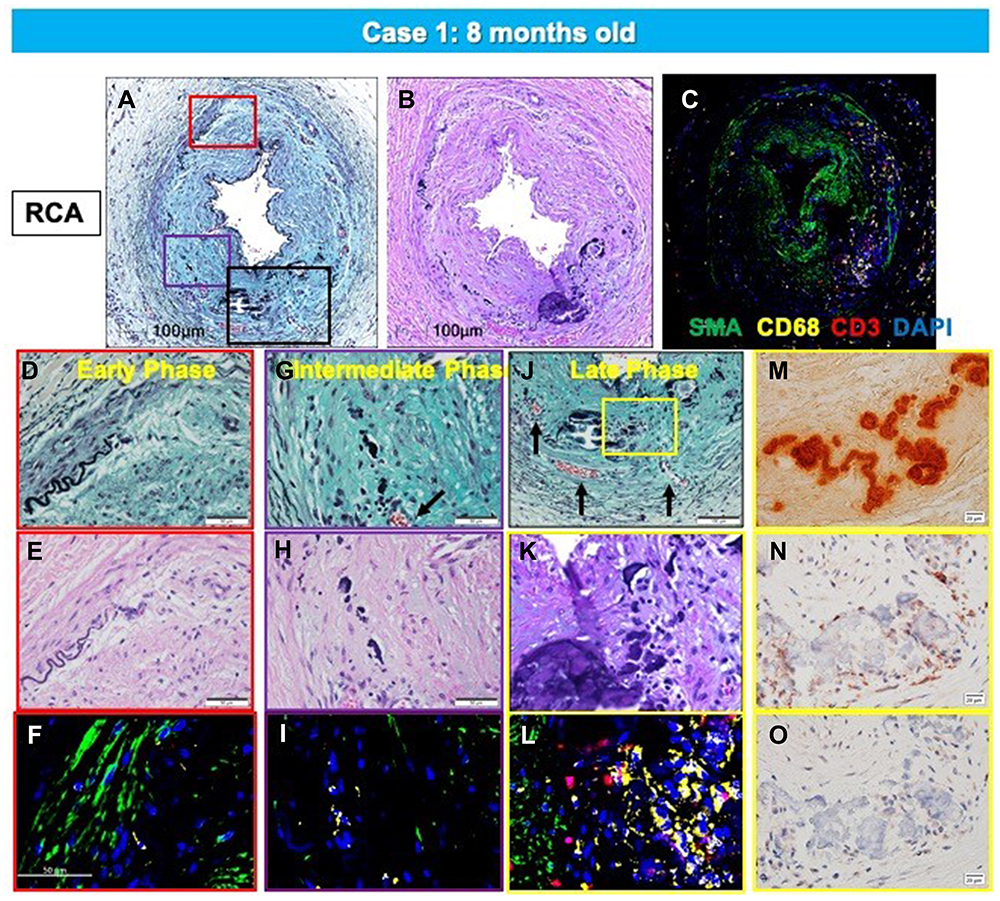 |
Figure 3 Representative histologic sections of the right coronary artery, from the same case shown in Figure 2. Top row: low-power images of RCA stained with Movat Pentachrome (A), hematoxylin and eosin (B), and triple immunofluorescence for SMC and inflammatory markers [macrophage (CD68) and T cells (CD3)], and α-actin (SMA) (C). Early- (D–F), intermediate- (G–I), and late-phase (J–O) lesion morphologies are shown. There is moderate luminal narrowing with SMC-rich neointima. Regions of interest shown at higher magnification are within the colored boxed areas on the low-magnification image (A). Areas highlighted by the red box (D–F) show early calcification with degradation of the IEL and relative absence of inflammation. The purple-boxed area (G–I) represents fragmentation and focal thickening of elastic fibers with calcification, along with early inflammation (macrophages, yellow in panel (I) and neoangiogenesis (black arrow). A more advanced late phase (black in panel (A) and yellow boxes in panel (J) shows more prominent calcification (M, alizarin red) and neoangiogenesis (black arrows in panel (J) with inflammatory infiltrate (C and L) consisting mostly of macrophages and T cells together with a loss of adjacent SMCs. These areas were also typically positive for the calcification markers, osteoprotegerin(N) and receptor activator of the nuclear κB (RANK) (O). (A–O) are reproduced with permission from Federici et al.58 |
 |
Figure 4 Representative histologic sections of the aorta and major branches with varying degrees of IEL fragmentation and calcification. Low-power images of the thoracic aorta (A), abdominal aorta (B), left subclavian artery (C), left common carotid artery (D), celiac artery (E), superior mesenteric artery (F), renal artery (G), and left common iliac artery (H) showing varying degrees of IEL fragmentation and calcificationLeft images are stained with hematoxylin and eosin stain and right images are stained with von Kossa stain. (A–H) are reproduced with permission from Federici et al.58 |
The second case is from a 20-day-old female infant, who died from severe coronary stenosis with calcification involving all four major coronary arteries (Figure 5A–M). These calcified lesions were not seen in the arteries within the myocardium (Figure 5O–Q). Radiography showed diffuse calcification of the pulmonary artery, with varying degrees of luminal narrowing (Figure 6A–F). Arterial calcification was also observed in the thoracic aorta and histologic examination showed bone formation at sites of severe calcification (Figure 6G–L). This GACI case illustrates the importance of intimal proliferation along with severe calcification of the coronary as a cause of early mortality in newborn infants. In addition, this case also showed bone metaplasia in the calcified lesions in the proximal coronary arteries and aorta (Figures 5C, K and 6K). Bone metaplasia has been observed in areas of calcification in muscular and elastic arteries, in patients with atherosclerosis, diabetes mellitus, and chronic kidney disease (CKD), with and without dialysis.59 The mechanism likely involve elevated levels of the phosphate-regulating hormone fibroblast growth factor-23 (FGF-23), which increases phosphorylation of Erk1/2 (one of the major subfamilies of mitogen activated protein kinases and regulate cell differentiation) and promotes phosphorus-induced transformation of vascular smooth muscle cells to osteoblasts, with Klotho expression in smooth muscle cells being the key determinant.60,61 Given that Enpp1 knockout mice show abnormal calcium/phosphate homeostasis and elevated blood levels of FGF-23,62 increasing serum FGF-23 levels by mutations in ENPP1 may regulate the vascular calcification phenotype through the transformation of smooth muscle cells.
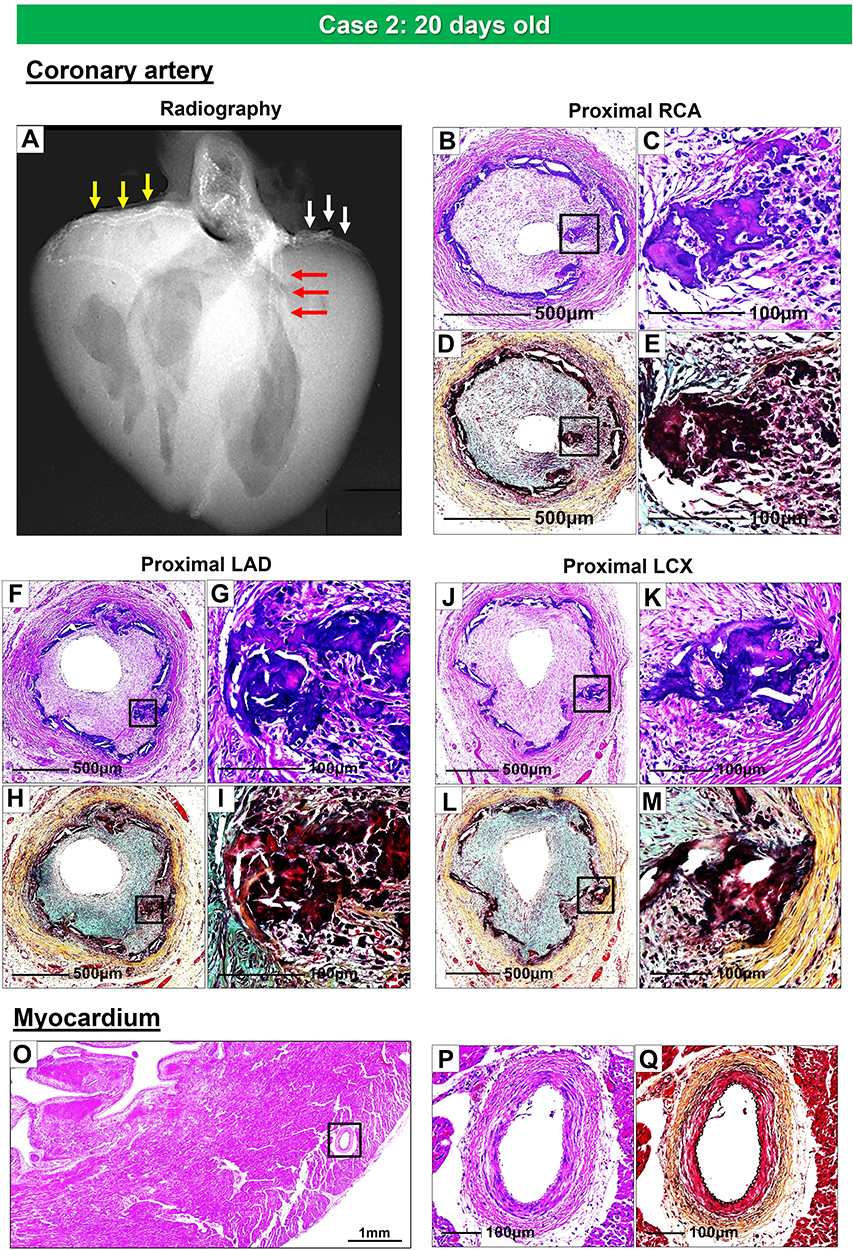 |
Figure 5 Radiography and histologic images of the coronary arteries and myocardium from a 20-days-old female infant with GACI. Radiograph of the heart shows calcification of the major coronary arteries: RCA (yellow arrows), LAD (red arrows), and LCX (white arrows) (A). Proximal RCA (B–E), proximal LAD (F–I), and proximal LCX (J–M) show moderate to severe luminal narrowing with circumferential calcification of IEL. Images B, C, F, G, J, and K of coronary arteries are stained with hematoxylin and eosin and Movat Pentachrome D, E, H, I, L, and M, stains, respectively. The boxed areas in the left panels, B, D, F, H, J, and L, are shown at high power in the right panels, C, E, G, I, K, and M. Panels C and K show bone metaplasia within calcified areas. Histologic section of the left ventricular wall showing normal histology of the intramyocardial coronary arteries [boxed area in (O); note an absence of calcification and intimal proliferation (P and Q)]. (O and P) are stained by hematoxylin and eosin; (Q) is stained by Movat pentachrome stain. Abbreviations: RCA, right coronary artery; LAD, left anterior descending; LCX, left circumflex. |
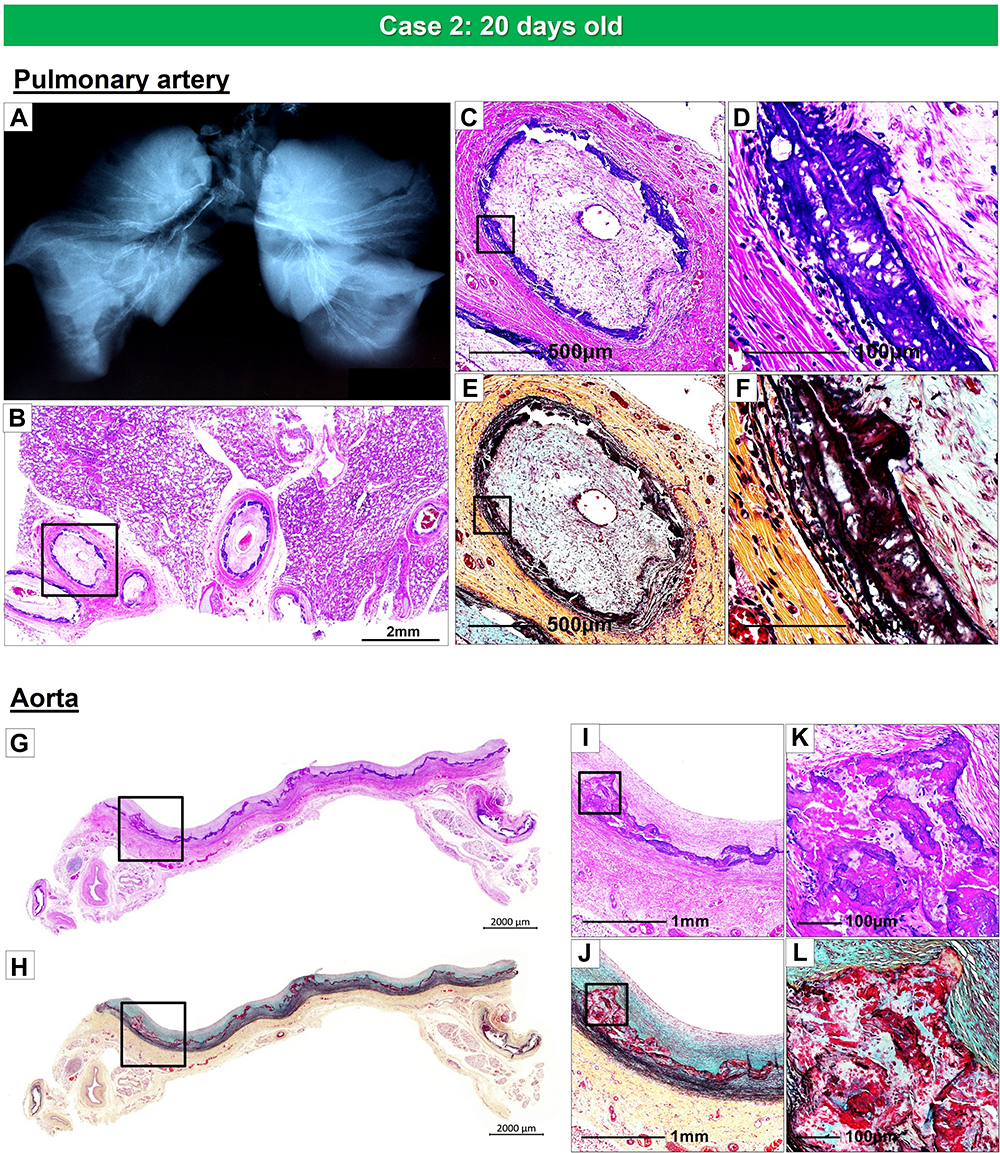 |
Figure 6 Radiography of the lungs and histology of pulmonary artery and thoracic aorta, from the same case as Figure 5. Radiography of right and left lungs demonstrates diffuse calcification of pulmonary arteries (A). (B). Histologic section of the right lung showing diffuse narrowing of multiple branches of the pulmonary arteries (hematoxylin and eosin). The boxed area in panel B is shown at higher magnification, demonstrating severe luminal narrowing from intimal proliferation and circumferential calcification (C–F). Images (C and D) are stained with hematoxylin and eosin and (E and F) are stained with Movat Pentachrome. A section of the thoracic aorta showing circumferential calcification (G and H). (I and J) are higher-power magnifications from the boxed areas in (G and H), showing diffuse calcification of the media. (K and L) show an area of bone metaplasia within the lesions of calcification. The upper and lower panels are stained with hematoxylin and eosin (G, I, and K) and Movat Pentachrome (H, J, and L). |
Extravascular Calcification
Ectopic calcification occurs in 94% of patients with GACI and is most frequent in heart valves, followed by kidneys. Joint calcification has been reported in approximately 30–50% of infants with GACI, most often in the shoulders, hips, ankles, wrists, and sternoclavicular joint, and is reported to occur in both early and late cases of GACI.42 Other locations of extravascular calcification include the earlobe,23,63,64 myocardium, and cerebral parenchyma.9 Calcification was also found in cervical fusion, tendons, and ligaments in contact with bone and was associated with local musculoskeletal pain. Calcification of the Achilles tendon may result in spontaneous rupture in adulthood.42 There is no difference in the frequency or location of ectopic calcification in ENPP1 mutants compared to ABCC6 mutants, although the relationship between the presence of ectopic calcification and pathological features in GACI has not been fully elucidated.65
Treatment
Currently, there is no curative treatment for GACI. Since ectopic arterial calcification can manifest in any systemic organ, individualized treatment is needed for each organ dysfunction. Furthermore, these systemic diseases need to be followed by a comprehensive medical team that involves multiple specialties for their management. Since there is a high mortality rate, both prenatally and in the first few months of life, associated with diffuse arterial calcification due to low PPi levels, it is recommended that therapeutic intervention be initiated as early as possible.65
Bisphosphonates are the most widely used drugs for patients with GACI, with varying degrees of success. Bisphosphonates consist of a core nonhydrolyzable carbon P-C-P motif that creates a more stable (less hydrolyzable) PPi analog. There are many different generations of bisphosphonates, which can be divided into nitrogen-containing and non-nitrogen-containing compounds. The non-nitrogen-containing bisphosphonate, disodium etidronate, the first generation, binds to hydroxyapatite, resulting in inhibition of bone mineralization. The subsequent generations of nitrogen-containing bisphosphonates, like disodium pamidronate, inhibit the action of the enzymes mevalonate pathway, with more potent antiresorptive properties. Both types of bisphosphonates induce apoptosis in osteoclasts through the interference with proteins that regulate bone metabolism. Rutsch et al reported a retrospective observational analysis of 55 patients with systemic arterial calcification in infancy and showed that treatment with bisphosphonates was effective in the regression of calcification and was associated with an improvement in mortality.8 On the other hand, a more recent report has shown that bisphosphonate therapy does not improve the rate of survival based on the analysis of matched starting times. The efficacy of bisphosphonate therapy for GACI is still controversial and further investigation is needed to determine their usefulness.65
A recombinant ENPP1-Fc protein is one of the therapies under investigation and has been demonstrated to prevent aortic calcification and myocardial infarction in a mouse model of ENPP1 deficiency.66 Subsequently, new evidence for the role of ENPP1 in vascular smooth muscle cells (VSMCs) has also been published, which showed that soluble recombinant human (rh) ENPP1-Fc not only reduces ectopic calcification by increasing levels of extracellular PPi, but also cleaves extracellular ATP to prevent AMP and adenosine anti-proliferative signaling and inhibits the proliferation of VSMCs.34 In a mouse model of GACI that demonstrates ectopic arterial calcification, oral administration of pyrophosphate significantly suppressed calcification. The effect of PPi was robust, with reduction ranging from 75% to 88% of calcium burden.67 Also, oral PPi administration to GACI pregnant mice was found to inhibit calcification in their offspring. These results suggest that PPi as a therapeutic intervention may be effective in man to inhibit prenatal GACI. While there is no evidence to date of the efficacy of oral PPi in patients with GACI, its safety has been confirmed by the US Food and Drug Administration when administered orally in humans. In GACI, another treatment option is heart transplantation, which has been reported in an 18-month-old child with severe myocardial infarction and end-stage heart failure due to diffuse coronary artery calcification. The patient was followed for 2 years and has thus far thrived without recurrence of calcification.68
Summary and Perspective
This review summarizes previous and current knowledge of the epidemiology, pathogenesis, and treatment of GACI to provide comprehensive management in clinical practice. Vascular calcification and intimal proliferation are the major causes of high mortality and morbidity in GACI, which causes myocardial infarction and heart failure in infants. GACI is an autosomal recessive disorder associated with genetic mutations of two genes, ENNP1 and ABCC6, which are the underlying cause of vascular calcification in infancy, with mortality due to coronary calcification and intimal proliferation. Hereditary vascular calcification occurring in adulthood from the deficiency of CD73 is known an CALJA and is related to NT5E genetic mutation. CALJA less severe than GACI and involves the arteries of the lower extremity. Although the mechanisms of these genetic mutations are becoming clearer, the substrates of the ABCC6 transporter remain unknown, and the mechanisms of vascular and soft tissue calcification caused by mutations in ABCC6 in patients with GACI and PXE are not fully elucidated. Further studies are needed to determine the mechanism by which these genetic mutations influence phenotypic manifestations of the disease. Early generation bisphosphonates have been used as treatment to reduce calcification in patients with GACI, however their efficacy remains controversial. Novel therapeutic agents, rhENPP1-Fc proteins, not only inhibit calcification but also affect intimal proliferation, the cause of severe stenosis in coronary arteries. Dedinski et al in mouse model administered oral PPi in water during gestation, which lead to suppressed ectopic calcification in offspring, suggesting that PPi could be a possible therapeutic option in humans, as PPi levels have been shown to increase from oral administration of PPi in water.67 Further research is needed to improve the ability to comprehensively treat patients with GACI.
Acknowledgements
CVPath Institute received Grant/Research/Clinical Trial Support from NIH-HL141425, Leducq Foundation Grant, 4C Medical, 4Tech, Abbott Vascular, Ablative Solutions, Absorption Systems, Advanced NanoTherapies, Aerwave Medical, Alivas, Amgen, Asahi Medical, Aurios Medical, Avantec Vascular, BD, Biosensors, Biotronik, Biotyx Medical, Bolt Medical, Boston Scientific, Canon USA, Cardiac Implants, Cardiawave, CardioMech, Cardionomic, Celonova, Cerus EndoVascular, Chansu Vascular Technologies, Children's National Medical Center, Concept Medical, Cook Medical, Cooper Health, Coramaze Technologies GmbH, CRL/AccelLab, Croivalve, CSI, Dexcom, Edwards Lifesciences, Elucid Bioimaging, eLum Technologies, Emboline, Endotronix, Envision, Filterlex, Imperative Care, Innovalve, Innovative Cardiovascular Solutions, Intact Vascular, Interface Biologics, Intershunt Technologies, Invatin Technologies, Lahav CRO, Limflow, L&J Biosciences, Lutonix, Lyra Therapeutics, Mayo Clinic, Maywell, MD Start, MedAlliance, Medanex, Medtronic, Mercator, Microport, Microvention, Neovasc, Nephronyx, Nova Vascular, Nyra Medical, Occultech, Olympus, Ohio Health, OrbusNeich, Ossio, Phenox, Pi-Cardia, Polares Medical, Polyvascular, Profusa, ProKidney LLC, Protembis, Pulse Biosciences, Qool Therapeutics, Recombinetics, Recor Medical, Regencor, Renata Medical, Restore Medical, Ripple Therapeutics, Rush University, Sanofi, Shockwave, Sahajanand Medical Technologies, SoundPipe, Spartan Micro, Spectrawave, Surmodics, Terumo Corporation, The Jacobs Institute, Transmural Systems, Transverse Medical, TruLeaf Medical, UCSF, UPMC, Vascudyne, Vesper, Vetex Medical, Whiteswell, WL Gore, Xeltis
Disclosure
Dr. Aloke V. Finn received consultant fees/honoraria from Abbott Vascular, Amgen, Biosensors, Boston Scientific, Celonova, Cook Medical, CSI, Lutonix Bard, Sinomed, and Terumo Corporation. Dr. Renu Virmani is a consultant of Abbott Vascular, Boston Scientific, Celonova, OrbusNeich Medical, Terumo Corporation, W. L. Gore, Edwards Lifesciences, Cook Medical, CSI, ReCor Medical, SinoMedical Sciences Technology, Surmodics, Bard BD and a scientific Advisory Board Member of Medtronic and Xeltis. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or any other conflict with the subject matter or materials discussed.
References
1. Lanzer P, Boehm M, Sorribas V, et al. Medial vascular calcification revisited: review and perspectives. Eur Heart J. 2014;35(23):1515–1525. doi:10.1093/eurheartj/ehu163
2. Carr JJ, Jacobs DR
3. Detrano R, Guerci AD, Carr JJ, et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. 2008;358(13):1336–1345. doi:10.1056/NEJMoa072100
4. Erbel R, Möhlenkamp S, Moebus S, et al. Coronary risk stratification, discrimination, and reclassification improvement based on quantification of subclinical coronary atherosclerosis: the Heinz Nixdorf Recall study. J Am Coll Cardiol. 2010;56(17):1397–1406. doi:10.1016/j.jacc.2010.06.030
5. Taylor AJ, Bindeman J, Feuerstein I, Cao F, Brazaitis M, O’Malley PG. Coronary calcium independently predicts incident premature coronary heart disease over measured cardiovascular risk factors: mean three-year outcomes in the Prospective Army Coronary Calcium (PACC) project. J Am Coll Cardiol. 2005;46(5):807–814. doi:10.1016/j.jacc.2005.05.049
6. Pradelli D, Faden G, Mureddu G, et al. Impact of aortic or mitral valve sclerosis and calcification on cardiovascular events and mortality: a meta-analysis. Int J Cardiol. 2013;170(2):e51–e55. doi:10.1016/j.ijcard.2013.10.081
7. Rutsch F, Ruf N, Vaingankar S, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34:379–381. doi:10.1038/ng1221
8. Rutsch F, Boyer P, Nitschke Y, et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet. 2008;1:133–140. doi:10.1161/CIRCGENETICS.108.797704
9. Ferreira CR, Hackbarth ME, Ziegler SG, et al. Prospective phenotyping of long-term survivors of generalized arterial calcification of infancy (GACI). Genet Med. 2021;23(2):396–407. doi:10.1038/s41436-020-00983-0
10. Moran JJ. Idiopathic arterial calcification of infancy: a clinicopathologic study. Pathol Annu. 1975;10:393–417.
11. Ciana G, Trappan A, Bembi B, et al. Generalized arterial calcification of infancy: two siblings with prolonged survival. Eur J Pediatr. 2006;165(4):258–263. doi:10.1007/s00431-005-0035-6
12. Sholler GF, Yu JS, Bale PM, Hawker RE, Celermajer JM, Kozlowski K. Generalized arterial calcification of infancy: three case reports, including spontaneous regression with long-term survival. J Pediatr. 1984;105(2):257–260. doi:10.1016/S0022-3476(84)80123-7
13. Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83(9):1032–1045. doi:10.4065/83.9.1032
14. Ralph D, van de Wetering K, Uitto J, Li Q. Inorganic pyrophosphate deficiency syndromes and potential treatments for pathologic tissue calcification. Am J Pathol. 2022;192(5):762–770. doi:10.1016/j.ajpath.2022.01.012
15. Ziegler SG, Gahl WA, Ferreira CR, et al. Generalized arterial calcification of infancy. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews is a Registered Trademark of the University of Washington, Seattle. Seattle WA: University of Washington, Seattle; 1993.
16. Chong CR, Hutchins GM. Idiopathic infantile arterial calcification: the spectrum of clinical presentations. Pediatr Dev Pathol. 2008;11(5):405–415. doi:10.2350/07-06-0297.1
17. Devriese M, Legrand A, Courtois MC, Jeunemaitre X, Albuisson J. Pseudoxanthoma elasticum with prominent arterial calcifications evoking CD73 deficiency. Vasc Med. 2019;24:461–464. doi:10.1177/1358863X19853360
18. Nitschke Y, Rutsch F. Generalized arterial calcification of infancy and pseudoxanthoma elasticum: two sides of the same coin. Front Genet. 2012;3:302. doi:10.3389/fgene.2012.00302
19. Spear R, Mack LA, Benedetti TJ, Cole RE. Idiopathic infantile arterial calcification. J Ultrasound Med. 1990;9:473–476. doi:10.7863/jum.1990.9.8.473
20. Stuart G, Wren C, Bain H. Idiopathic infantile arterial calcification in two siblings: failure of treatment with diphosphonate. Br Heart J. 1990;64:156–159. doi:10.1136/hrt.64.2.156
21. Brunod I, Tosello B, Hassid S, Gire C, Thomachot L, Panuel M. Generalized arterial calcification of infancy with a novel ENPP1 mutation: a case report. BMC Pediatr. 2018;18(1):217. doi:10.1186/s12887-018-1198-4
22. Greenberg SB, Gibson J. New findings in idiopathic arterial calcification of infancy detected by MDCT. AJR Am J Roentgenol. 2005;185(2):530–532. doi:10.2214/ajr.185.2.01850530
23. Nitschke Y, Baujat G, Botschen U, et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012;90(1):25–39. doi:10.1016/j.ajhg.2011.11.020
24. Lorenz-Depiereux B, Schnabel D, Tiosano D, Häusler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet. 2010;86:267–272. doi:10.1016/j.ajhg.2010.01.006
25. Goding JW, Terkeltaub R, Maurice M, Deterre P, Sali A, Belli SI. Ecto-phosphodiesterase/pyrophosphatase of lymphocytes and non-lymphoid cells: structure and function of the PC-1 family. Immunol Rev. 1998;161(1):11–26. doi:10.1111/j.1600-065X.1998.tb01568.x
26. Hajjawi MO, MacRae VE, Huesa C, et al. Mineralisation of collagen rich soft tissues and osteocyte lacunae in Enpp1(-/-) mice. Bone. 2014;69:139–147. doi:10.1016/j.bone.2014.09.016
27. Harahap AR, Goding JW. Distribution of the murine plasma cell antigen PC-1 in non-lymphoid tissues. J Immunol. 1988;141:2317–2320.
28. Hessle L, Johnson KA, Anderson HC, et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci U S A. 2002;99(14):9445–9449. doi:10.1073/pnas.142063399
29. Prosdocimo DA, Douglas DC, Romani AM, O’Neill WC, Dubyak GR. Autocrine ATP release coupled to extracellular pyrophosphate accumulation in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2009;296(4):C828–C839. doi:10.1152/ajpcell.00619.2008
30. Persy VP, McKee MD. Prevention of vascular calcification: is pyrophosphate therapy a solution? Kidney Int. 2011;79(5):490–493. doi:10.1038/ki.2010.478
31. Krug HE, Mahowald ML, Halverson PB, Sallis JD, Cheung HS. Phosphocitrate prevents disease progression in murine progressive ankylosis. Arthritis Rheum. 1993;36(11):1603–1611. doi:10.1002/art.1780361116
32. Maulding ND, Kavanagh D, Zimmerman K, et al. Genetic pathways disrupted by ENPP1 deficiency provide insight into mechanisms of osteoporosis, osteomalacia, and paradoxical mineralization. Bone. 2021;142:115656. doi:10.1016/j.bone.2020.115656
33. Cudrici CD, Ferrante EA, Boehm M. Chapter 3 - Basic molecular mechanism of vascular calcification. In: Finn AV, editor. Coronary Calcium. Academic Press; 2019:47–82.
34. Nitschke Y, Yan Y, Buers I, Kintziger K, Askew K, Rutsch F. ENPP1-Fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp Mol Med. 2018;50(10):1–12. doi:10.1038/s12276-018-0163-5
35. St Hilaire C, Ziegler SG, Markello TC, et al. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364(5):432–442. doi:10.1056/NEJMoa0912923
36. de Nijs T, Albuisson J, Ockeloen CW, et al. Isolated arterial calcifications of the lower extremities: a clue for NT5E mutation. Int J Cardiol. 2016;212:248–250. doi:10.1016/j.ijcard.2016.03.068
37. Jansen RS, Küçükosmanoglu A, de Haas M, et al. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc Natl Acad Sci U S A. 2013;110(50):20206–20211. doi:10.1073/pnas.1319582110
38. Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J. Pseudoxanthoma elasticum: mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci U S A. 2000;97(11):6001–6006. doi:10.1073/pnas.100041297
39. Jansen RS, Duijst S, Mahakena S, et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler Thromb Vasc Biol. 2014;34(9):1985–1989. doi:10.1161/ATVBAHA.114.304017
40. Matsuzaki Y, Nakano A, Jiang QJ, Pulkkinen L, Uitto J. Tissue-specific expression of the ABCC6 gene. J Invest Dermatol. 2005;125(5):900–905. doi:10.1111/j.0022-202X.2005.23897.x
41. Van Gils M, Nollet L, Verly E, Deianova N, Vanakker OM. Cellular signaling in pseudoxanthoma elasticum: an update. Cell Signal. 2019;55:119–129. doi:10.1016/j.cellsig.2018.12.009
42. Boyce AM, Gafni RI, Ferreira CR. Generalized arterial calcification of infancy: new insights, controversies, and approach to management. Curr Osteoporos Rep. 2020;18(3):232–241. doi:10.1007/s11914-020-00577-4
43. Bergen AA, Plomp AS, Schuurman EJ, et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet. 2000;25(2):228–231. doi:10.1038/76109
44. Le Saux O, Urban Z, Tschuch C, et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet. 2000;25(2):223–227. doi:10.1038/76102
45. Le Boulanger G, Labrèze C, Croué A, et al. An unusual severe vascular case of pseudoxanthoma elasticum presenting as generalized arterial calcification of infancy. Am J Med Genet A. 2010;152A(1):118–123. doi:10.1002/ajmg.a.33162
46. Senba M, Kawai K, Mori N. Pathogenesis of metastatic calcification and acute pancreatitis in adult T-cell leukemia under hypercalcemic state. Leuk Res Treatment. 2012;2012:128617. doi:10.1155/2012/128617
47. Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. 2014;34(4):715–723. doi:10.1161/ATVBAHA.113.302070
48. Glatz AC, Pawel BR, Hsu DT, Weinberg P, Chrisant MR. Idiopathic infantile arterial calcification: two case reports, a review of the literature and a role for cardiac transplantation. Pediatr Transplant. 2006;10(2):225–233. doi:10.1111/j.1399-3046.2005.00414.x
49. van der Sluis IM, Boot AM, Vernooij M, Meradji M, Kroon AA. Idiopathic infantile arterial calcification: clinical presentation, therapy and long-term follow-up. Eur J Pediatr. 2006;165(9):590–593. doi:10.1007/s00431-006-0146-8
50. Van Dyck M, Proesmans W, Van Hollebeke E, Marchal G, Moerman P. Idiopathic infantile arterial calcification with cardiac, renal and central nervous system involvement. Eur J Pediatr. 1989;148(4):374–377. doi:10.1007/BF00444138
51. Thomas P, Chandra M, Kahn E, McVicar M, Naidich J, LaCorte M. Idiopathic arterial calcification of infancy: a case with prolonged survival. Pediatr Nephrol. 1990;4(3):233–235. doi:10.1007/BF00857661
52. Witzleben CL. Idiopathic infantile arterial calcification–a misnomer? Am J Cardiol. 1970;26:305–309. doi:10.1016/0002-9149(70)90798-8
53. Lussier-Lazaroff J, Fletcher BD. Idiopathic infantile arterial calcification: roentgen diagnosis of a rare cause of coronary artery occlusion. Pediatr Radiol. 1973;1(4):224–228. doi:10.1007/BF00972856
54. Farquhar J, Makhseed N, Sargent M, Taylor G, Osiovich H. Idiopathic infantile arterial calcification and persistent pulmonary hypertension. Am J Perinatol. 2005;22(03):121–125. doi:10.1055/s-2005-863787
55. Shaireen H, Howlett A, Amin H, Yusuf K, Kamaluddeen M, Lodha A. The mystery of persistent pulmonary hypertension: an idiopathic infantile arterial calcification. BMC Pediatr. 2013;13(1):107. doi:10.1186/1471-2431-13-107
56. Marrott PK, Newcombe KD, Becroft DM, Friedlander DH. Idiopathic infantile arterial calcification with survival to adult life. Pediatr Cardiol. 1984;5:119–122. doi:10.1007/BF02424963
57. Thiaville A, Smets A, Clercx A, Perlmutter N. Idiopathic infantile arterial calcification: a surviving patient with renal artery stenosis. Pediatr Radiol. 1994;24(7):506–508. doi:10.1007/BF02015014
58. Federici D, Torii S, Ciuffreda M, et al. Coronary pathology of inherited generalized arterial calcification of infancy: a case report. Cardiovasc Pathol. 2018;36:15–19. doi:10.1016/j.carpath.2018.05.001
59. Mathew RO, Bangalore S, Lavelle MP, et al. Diagnosis and management of atherosclerotic cardiovascular disease in chronic kidney disease: a review. Kidney Int. 2017;91(4):797–807. doi:10.1016/j.kint.2016.09.049
60. Desjardins L, Liabeuf S, Renard C, et al. FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos Int. 2012;23(7):2017–2025. doi:10.1007/s00198-011-1838-0
61. Jimbo R, Kawakami-Mori F, Mu S, et al. Fibroblast growth factor 23 accelerates phosphate-induced vascular calcification in the absence of Klotho deficiency. Kidney Int. 2014;85(5):1103–1111. doi:10.1038/ki.2013.332
62. Mackenzie NC, Zhu D, Milne EM, et al. Altered bone development and an increase in FGF-23 expression in Enpp1(-/-) mice. PLoS One. 2012;7(7):e32177. doi:10.1371/journal.pone.0032177
63. Vera J, Lucaya J, Garcia Conesa JA, Aso C, Balaguer A. Idiopathic infantile arterial calcification: unusual features. Pediatr Radiol. 1990;20(8):585–587. doi:10.1007/BF02129060
64. Brachet C, Mansbach AL, Clerckx A, Deltenre P, Heinrichs C. Hearing loss is part of the clinical picture of ENPP1 loss of function mutation. Horm Res Paediatr. 2014;81(1):63–66. doi:10.1159/000354661
65. Ferreira CR, Kintzinger K, Hackbarth ME, et al. Ectopic calcification and hypophosphatemic rickets: natural history of ENPP1 and ABCC6 deficiencies. J Bone Miner Res. 2021;36(11):2193–2202. doi:10.1002/jbmr.4418
66. Albright RA, Stabach P, Cao W, et al. ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat Commun. 2015;6(1):10006. doi:10.1038/ncomms10006
67. Dedinszki D, Szeri F, Kozák E, et al. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol Med. 2017;9(11):1463–1470. doi:10.15252/emmm.201707532
68. Giovannoni I, Callea F, Travaglini L, et al. Heart transplant and 2-year follow up in a child with generalized arterial calcification of infancy. Eur J Pediatr. 2014;173(12):1735–1740. doi:10.1007/s00431-014-2447-7
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.