")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
Gene Expression Profile Analyses of the Skin Response of Balb/c-Nu Mice Model Injected by Staphylococcus aureus
Authors Zhang J , Wang C, An Q, Quan Q, Li M, Zhao D
Received 17 November 2021
Accepted for publication 20 January 2022
Published 15 February 2022 Volume 2022:15 Pages 217—235
DOI https://doi.org/10.2147/CCID.S348961
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Jiachan Zhang,1 Changtao Wang,1 Quan An,2 Qianghua Quan,2 Meng Li,2 Dan Zhao1
1Beijing Key Lab of Plant Resource Research and Development, College of chemistry and materials engineering, Beijing Technology and Business University, Beijing, 100048, People’s Republic of China; 2Yunnan Baiyao Group Co., Ltd., Kunming, 650000, People’s Republic of China
Correspondence: Changtao Wang, Beijing Key Lab of Plant Resource Research and Development, College of chemistry and materials engineering, Beijing Technology and Business University, Fucheng Road, Beijing, 100048, People’s Republic of China, Tel +8610-68984917, Email [email protected] Quan An, Yunnan Baiyao Group Co., Ltd., Kunming, 650000, People’s Republic of China, Email [email protected]
Background: Pathogenesis and persistence of many skin diseases are related to Staphylococcus aureus (S. aureus) colonization. S. aureus infection can cause varying degrees of changes in cell gene expression, resulting in complex changes in cell phenotype and finally changes in cell life activities.
Materials and Methods: The transcriptomes of healthy and Staphylococcus aureus (S. aureus)-infected murine skin tissues were analyzed. We identified 638 differentially expressed genes (DEGs) in the infected tissues compared to the control samples, of which 324 were upregulated and 314 were downregulated, following the criteria of P < 0.01 and |log2FC| > 3. The DEGs were functionally annotated by Gene Ontology (GO), KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway and the protein–protein interaction (PPI) network analyses.
Results: The upregulated DEGs were mainly enriched in GO terms, such as response to stimulus, immune system process and signal transduction, as well as in the complement and coagulation cascade pathway. Thus, S. aureus infection likely activates these pathways to limit the influx of neutrophils and prevent skin damage. Four clusters were identified in the PPI network, and the major hubs were mainly related to cell cycle and proliferation, and mostly downregulated. The expression levels of Nox4, Mmrn1, Mcm5, Msx1 and Fgf5 mRNAs were validated by qRT-PCR and found to be consistent with the RNA-Seq data, confirming a strong correlation between the two approaches.
Conclusion: The identified genes and pathways are potential drug targets for treating skin inflammation caused by S. aureus and should be investigated further.
Keywords: Staphylococcus aureus, inflammation, gene expression profile, transcriptome
Introduction
Staphylococcus aureus (S. aureus) is a conditional pathogen that forms part of normal intestinal flora of mammals but can infect other organs, especially the skin, nose and pharynx.1 Historically, S. aureus infections have been obtained within hospital environments, but the incidence or occurrence of S. aureus infections is increasing in community settings.2 Normally, S. aureus does not cause harm to human carriers; while, it can cause both invasive and noninvasive infections. Recurrent S. aureus skin infections have been correlated to atopic dermatitis (AD),3–5 and almost 80% of the individuals with AD show cutaneous S. aureus colonization.6–9 Based on its susceptibility to beta-lactams, S. aureus is commonly described as methicillin-susceptible S. aureus (MSSA) or methicillin-resistant S. aureus (MRSA).
A series of host responses will be occurred induced by both MSSA and MRSA. After S. aureus reaches the sub epidermal space, it is either locally controlled or spread in the dermis, resulting in soft tissue infection. These different processes depend not only on the virulence of S. aureus, but also on the defense ability of the host. Inflammation is the key pathological aspect of dermatological S. aureus infections, although the skin itself acts as an immune barrier, because of its unsuitable surface temperature and pH, existing antimicrobial peptides, and skin commensals (Staphylococcus epidermidis and their secreted products). However, if infection does exist, the body produces a series of pro- and anti-inflammatory mediators, chemotaxis, cell signaling, keratins, and TH1/TH17 cytokines to mediate neutrophil recruitment, and other acute and systemic immune response. Besides, S. aureus modulates the host environment through super-antigens that stimulate pro-inflammatory cytokine production by the keratinocytes, and disrupt the skin barrier. Polymorphonuclear neutrophils (PMNs) play a particularly important role in effectively eliminating S. aureus infections, and they contribute to the massive inflammation typically accompanying S. aureus infections.
The rapid evolution of whole transcriptome profiling using next-generation high-throughput RNA sequencing (RNA-Seq) has shed light on the complex mechanisms and pathways of skin infections.10 Investigators are beginning to understand some facets of the host response to S. aureus infection using high-throughput sequencing technology, especially the responses induced by MRSA strains, which are often resistant to multiple antibiotics.
Malachowa et al established a soft tissue model of infection caused by USA300, an epidemic community-associated methicillin-resistant S. aureus (CA-MRSA) strain in a rabbit to investigate host–pathogen interactions. Of the host genes tested, they found that transcripts encoding IL-8, IL1β, oncostatin M-like, CCR1, CXCR1 (IL8RA), CCL4 (MIP-1β) and CCL3 (MIP1α)-like proteins were up-regulated during S. aureus abscess formation.11 Brady et al established a murine SSTI model infected with S. aureus sub-type USA300 (MRSA) in ear epicutaneously and compared the infected and non-infected skin from challenged mice. Besides, comparisons of expression profiles of non-infected ears from challenged mice to ears of naïve mice were also discussed. In their study, the activation and inhibition pathways happen first locally, and lay behind systemically. However, they did not focus on the comparison of the infected skin to the normal skin from different individuals, which also contains a lot of information during the host response.12 In addition, transcriptomic profiling of healthy and inflamed skin biopsies has revealed significant differences in the expression levels of genes related to inflammation, type 2 immune response pathways, cornification and lipid biosynthesis.13,14
However, published information is still limited. The differences of host responses between MSSA and MRSA infections should be discussed. The understanding of the molecular mechanism induced by MSSA will be added in the existing knowledge. To this end, we established an intradermal S. aureus infection model in mice and compared the transcriptomes of the inflamed versus normal skin tissues using the RNA-Seq technology. The differentially expressed genes (DEGs) were functionally annotated by GO mRNA expression of some randomly selected genes was validated by qRT-PCR, and showed consistency with the RNA-Seq data.
Materials and Methods
Experimental Materials
S. aureus S1 strain was purchased from China General Microbiological Culture Collection Center (CGMCC Number: 1.4519) and identified as S. aureus (ATCC Number: 25923). Following overnight culture on a nutrient agar (NA) at 37°C, single colonies were picked and inoculated into a nutrient broth (NB). The inoculum was cultured overnight at 37°C with constant shaking at 200 rpm, followed by 2% subculture at 37°C for 2h to obtain mid-logarithmic phase bacteria. The broth culture was centrifuged, and the bacterial cell pellet was washed and resuspended in normal saline at the density of 3×107 CFU/100μL.15 The absorbance at 600 nm was measured to estimate the number of CFUs, which was verified by overnight culture on NA.
Establishment of Inflammation Model
Ten Balb/c-NU 6–8 weeks old male nude mice weighing ~20 g were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). They were housed in the animal facility under standard conditions of light and temperature and fed standard laboratory chow and water ad libitum. After being fed adaptively for a week, the mice were then randomly divided into the control and model groups. Intradermal S. aureus infection was established as previously described.16 The mice were injected subcutaneously into their right hind limb with 100μL S. aureus suspension (3×107 CFU), and the control mice received the same volume of sterile normal saline. The following day, the mice were anesthetized by intraperitoneal injection of tribromoethanol (98.5%; J & K Scientific Ltd) at the dose of 240 mg/kg BW. The inflamed and normal skin were peeled, and divided into two groups. Samples in one group were fixed with paraformaldehyde and embedded in paraffin. The tissue sections were stained with hematoxylin and eosin (H & E), and the thickness of stratum spinosum and stratum epidermis were measured using the image analysis software program Case Viewer. The other skin samples in another group were taken for RNA isolation.
RNA Isolation
Total RNA was extracted from the skin samples using TRIzol® Reagent (Sigma Technologies, St Louis, USA) according to the manufacturer’s instructions, and genomic DNA was removed with DNase I (TaKara Bio, Japan). RNA quality was determined using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara CA, USA) and quantified using NanoDrop 2000 (Thermo Scientific™ Technologies, Wilmington, DE, USA). Only high-quality RNA samples (OD260/280=1.8~2.2, OD260/230≥2.0, RIN≥6.5, 28S:18S≥1.0, >2μg) were used to construct the sequencing library (see supplementary material for technical details).
Screening for Differentially Expressed Genes (DEGs)
The critical information like platform, quality control, bioinformatics tools and the RNA sequencing data analysis (quality control, assembly, and mapping) were exhibited in detail in the supplementary file. Table A in the supplementary file showed the summary of trimming and read mapping results of the sequences generated from the skin samples with or without the injection of Staphylococcus aureus. A P-value and the fold change (FC) for each gene were calculated to denote its expression difference between libraries.
The DEGs between the control and inflamed skin samples were screened using P-value < 0.01 and |log2FC| > 3 as the thresholds. The DEGs were functionally annotated using Database for Annotation, Visualization and Integrated Discovery (DAVID ver 6.8, http://david.ncifcrf.gov/). The significant gene ontology (GO) terms for biological process (PB), molecular function (MF), and cellular component (CC) for the up- and down-regulated genes were identified on the basis of enrichment score >2.17 In addition, the significantly enriched pathways were identified from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database using the P < 0.05 as the criterion.
Construction of Protein–Protein Interaction Network
The PPI network was constructed using Cytoscape ver. 3.5.1. Weighted PPIs of human proteins were downloaded from HPRD (Human Protein Reference Database, http://www.hprd.org/, Release 8).18,19 The DEGs were converted from mouse to human using the HCOP: Orthology Predictions Search in HGNC database tool (https://www.genenames.org/tools/hcop/).20–22
Quantitative RT-PCR
The RNA was reverse transcribed using the FastQuant RT Kit (with gDNase, TIANGEN Biotech, Beijing, China) in a 20µL reaction volume according to the manufacturer’s instructions. QRT-PCR for Nox4, Mmrn1, Mcm5, Msx1, Fgf5 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was performed using SuperReal PreMix Plus (SYBR Green, TIANGEN Biotech, Beijing, China) and gene-specific primers (Table S1) on AmpliTaq Gold (PerkinElmer, USA). All reactions were run in triplicates and GAPDH was used as the internal control. The cycling parameters were as follows: initial denaturation for 2 min at 95°C, followed by 40 cycles of 15s at 95°C, 20s at 57°C and 30s at 72°C, and a final extension at 95°C for 15s.
Data Availability Statement
All raw sequencing reads have been submitted to the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under the BioProject accession number PRJNA647241.
Statistics
Statistical analysis was conducted using GraphPad Prism software version 7.0. All data were presented as mean value where error bars indicated standard deviation (SD). Two-tailed Student’s test was used to compare two groups, and P < 0.05 was considered statistically significant.
Results
Histological Evaluation of the Skin
Localized S. aureus infection significantly altered the epidermal morphology. Innate immune recognition of S. aureus promotes proinflammatory signals, leading to neutrophil recruitment and abscess formation, which is a hallmark of S. aureus infection. As shown in Figure 1A and B, the surface of the infected skin turned grey and rough. Histological examination showed complete epidermis in the control group, with neatly arranged epidermal cells, and clear reticular ridges and dermal papillae. In addition, the healthy dermis had wavy fibrous tissues, intact and uniformly distributed hair follicles and sebaceous glands, and no infiltration of inflammatory cells (shown in Figure 1C). In contrast, the S. aureus-infected skin displayed significantly thickened granular and spinous layers, uneven epidermal thickness, discontinuity between the epidermis and dermis, aberrant hair follicles and sebaceous glands, and massive neutrophil infiltration into the dermal and subcutaneous tissues (shown in Figure 1D). The thickness of the stratum spinosum and epidermis were measured, and were significantly greater in the infected versus the control skin (P-value <0.001, shown in Figure 1E and F).
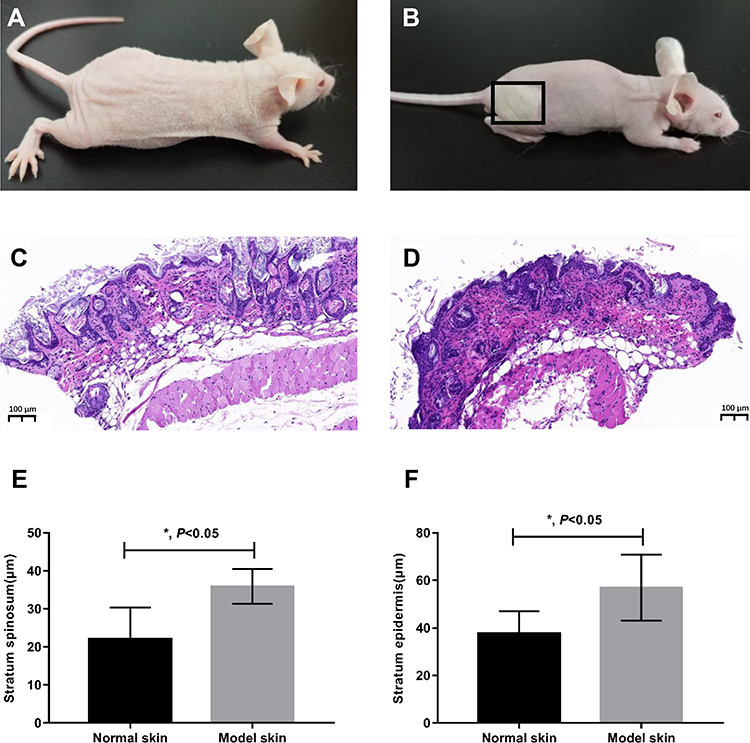 |
Figure 1 Histological evaluation of the skin (n = 5 mice for each group). (A and B), representative images of normal untreated control mouse (A) and the treated mouse with injection of S. aureus (B). (C and D), representative images of the hematoxylin-eosin staining performed to observe the morphological structure of skin in the control group (C) and treated group (model) (D). (E and F) were thicknesses quantifications of stratum spinosum and stratum epidermis from (C) and (D) (n=5). P-value from t-test. *P <0.05. Error bars indicate mean ± SD. |
Identification of DEGs
A total of 638 genes were identified in the inflamed skin samples relative to normal skin (|log2 fold change|> 3 and P-value <0.01), of which 324 were up-regulated and 314 were down-regulated (shown Figure S1). As shown in Table 1, 38 of the top 100 upregulated genes encode immune response-related proteins, of which five (C7, C5ar1, F13a1, C6 and Cfh) are involved in complement and coagulation cascades, six (Serpina3n, Saa2, Saa1, Saa3, Lbp and Cd163) are related to the acute-phase response, five (Saa1, Saa2, Saa3, Cxcl13 and Ccl6) regulate chemotaxis, and four (Oas1a, Oas1f, Oas1g and Tlr8) are involved in virus-specific immune response. Twenty-one genes with the highest increase in transcript levels encode proteins that have known functions in metabolism and cell proliferation, whereas 8 are functionally uncharacterized. In addition, 14 upregulated genes encode structural proteins of the dermis, including tenascin N (Tnn), osteomodulin (Omd), cartilage intermediate layer protein (Clip), myocilin (Myoc), and multimerin 1 (Mmrn1). Three metallopeptidase-encoding genes (Mmp8, Mmp11 and Lvrn) and seven genes encoding redox proteins also showed significant increase in their transcript levels.
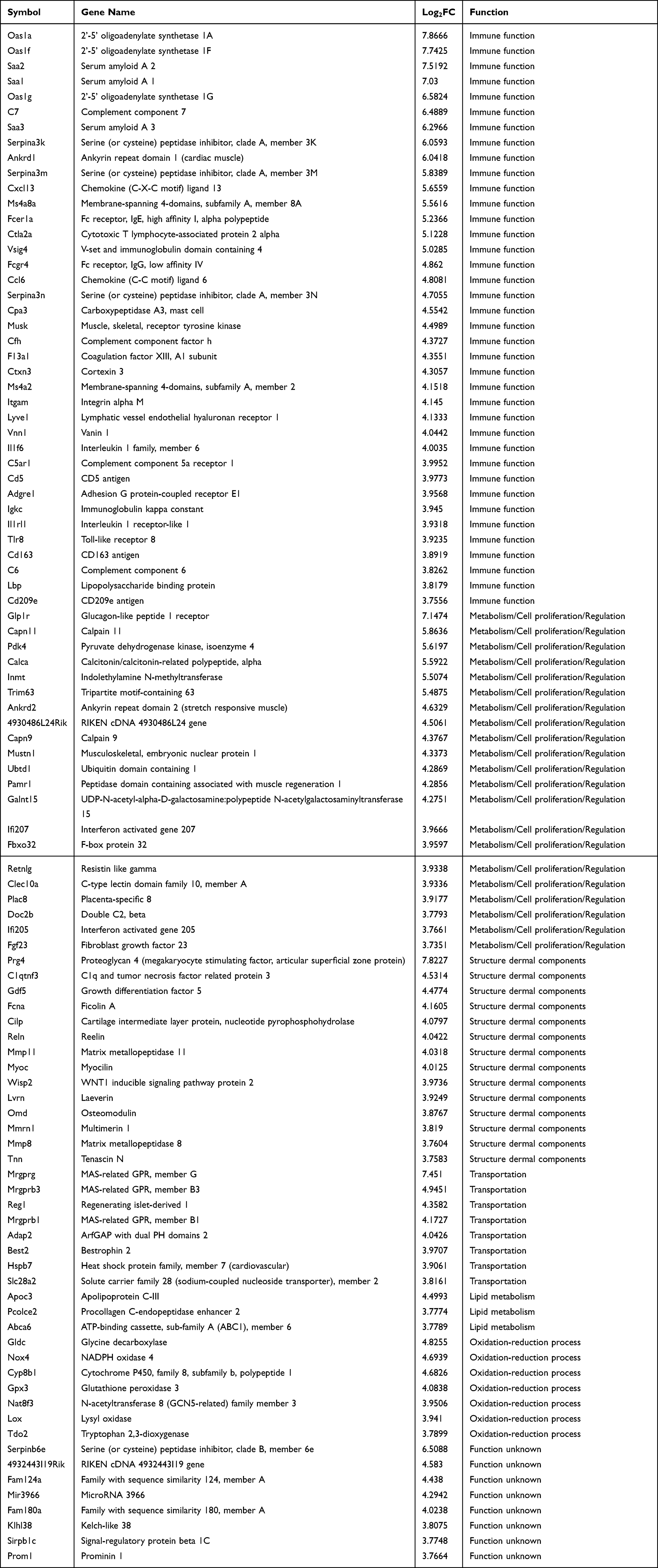 |
Table 1 Top Genes with Significantly Increased Transcript Levels |
The top 100 downregulated genes are listed in Table 2, of which 24 have unknown function. Only two genes encode immune response-related proteins, including PDZ binding kinase (Pbk) and transient receptor potential cation channel (Trpm1). Thirty-two genes regulate metabolism and cell proliferation, of which seven encode cell cycle proteins (Ccnb1, Cdk1, Ccnb2, Plk1, Cdc20, Cdc25c and Ccna2), and four encode negative regulators of cell proliferation (Hist1h2ag, Hist1h2ai, Nppc and Hist1h2an). The proteins encoded by Hist1h2ag and Hist1h2ai also play important roles in DNA repair. In addition, genes encoding DNA damage response proteins, including Exo1, Uhrf1, Ticrr, Kif22, Pclaf, Tgm6 and Pif1, were also significantly downregulated by S. aureus infection. Furthermore, 16 genes encode dermal proteins, of which 14 genes encode proteins related to keratin or keratin filament. Other major downregulated genes were those encoding proteins involved lipid metabolism (5) and transportation (10).
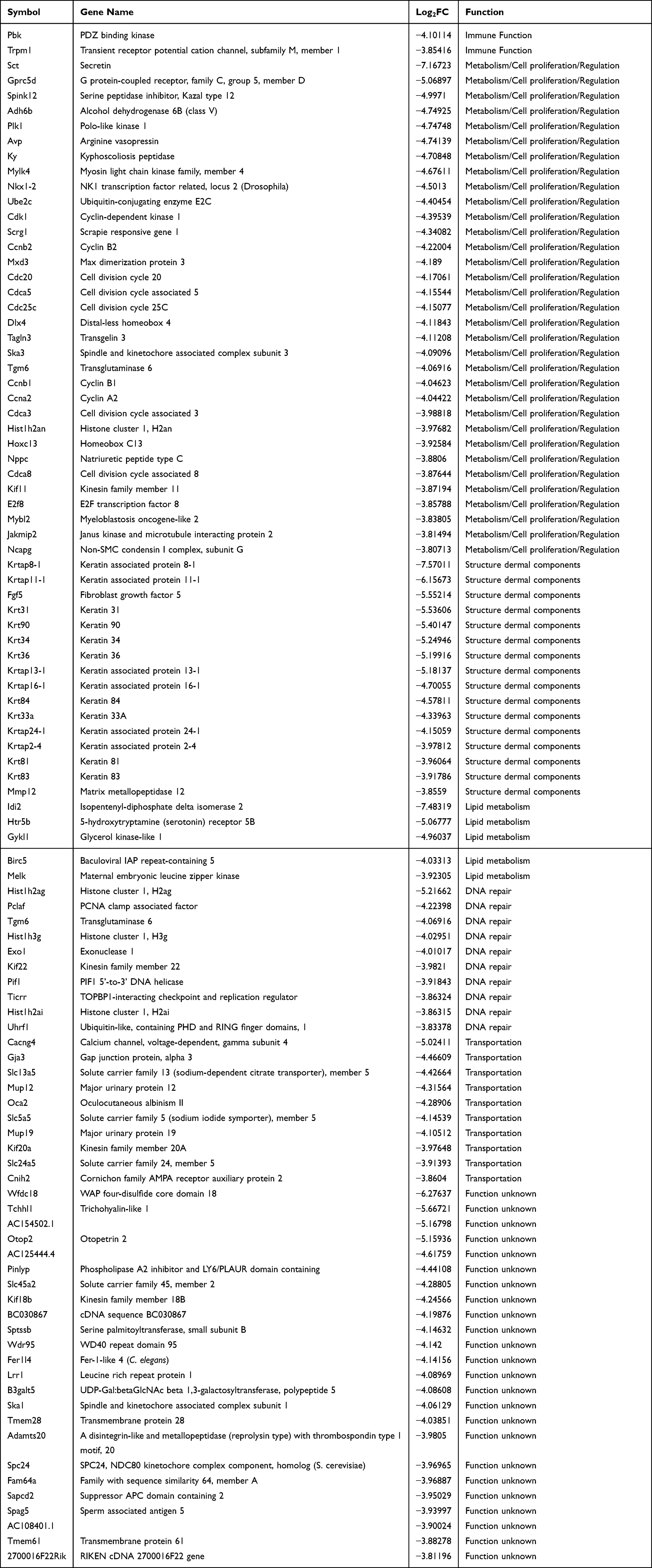 |
Table 2 Top Genes with Significantly Decreased Transcript Levels |
GO and KEGG Pathway Analysis
The top 20 GO terms of up- and down-regulated DEGs are shown in Figure 2. The top 5 GO terms for the up-regulated genes were regulation of response to stimulus, extracellular space, defense response, regulation of response to external stimulus and immune system process (shown in Figure 2A), and that for the down-regulated DEGs were keratin filament, chromosome segregation, mitotic cell cycle process, cell cycle process and intermediate filament (shown in Figure 2B). On the other hand, the most enriched KEGG pathways of the up-regulated genes were complement and coagulation cascades and Staphylococcus aureus infection (shown in Figure 2C), and that of the down-regulated genes were cell cycle and oocyte meiosis (shown in Figure 2D). GO clustering enrichment analysis further showed that the up-regulated DEGs were mainly involved in acute-phase response (enrichment score ES = 3.06, gene count GC = 8), chemotaxis (ES = 2.86, GC = 18), negative regulation of inflammation factor production (ES = 2.12, GC=6) and lipid metabolism (ES = 2, GC = 10) (shown in Figure 3A). The GO term of signal transduction was enriched in most up-regulated genes (39). As shown in Figure 3B, the down-regulated DEGs were associated with cell cycle (ES = 39.15, GC = 70), chromosome segregation (ES = 20.58, GC = 59), structural molecule (ES = 9.10, GC = 22), microtubule-based movement (ES = 8.72, GC = 32), melanin biosynthetic process (ES = 4.42, GC = 7), and DNA metabolism (ES = 2.21, GC = 8). The detailed information is presented in Tables S2 and S3.
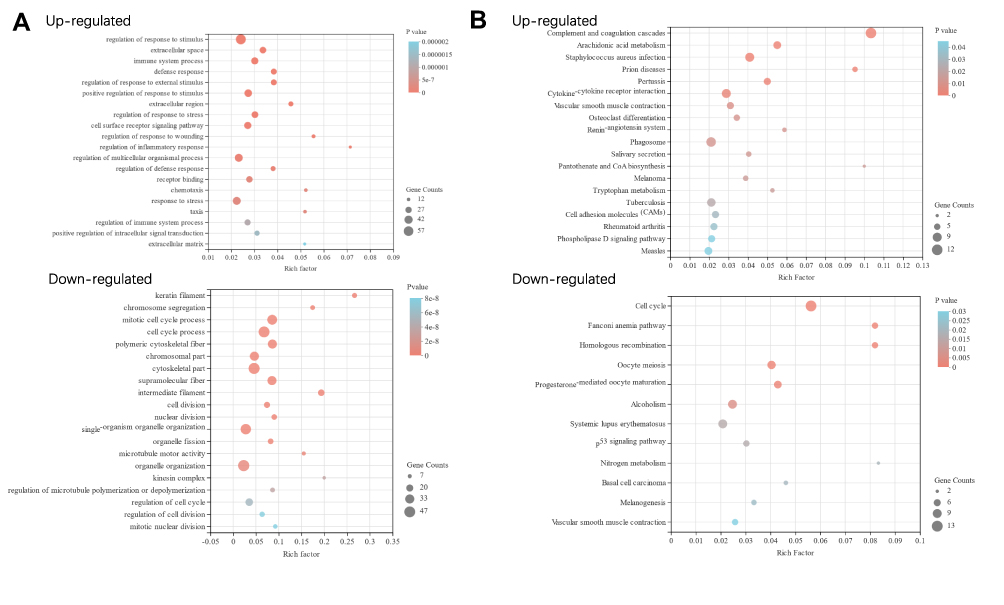 |
Figure 2 The top 20 GO terms (A) and KEGG pathways (B) of different expressed genes enrichment in RNA-sequencing (P-value ≤ 0.05). Gene Counts: number of target genes in each GO term or KEGG pathway. Rich factor: the ratio of the number of target genes divided by the number of all the genes in each GO term or KEGG pathway. The size of pot indicated the gene counts, and the color reflected the different P-value. |
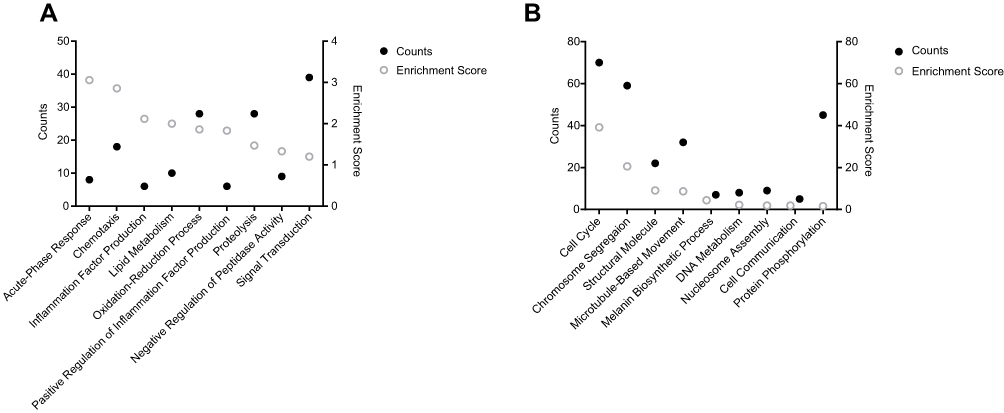 |
Figure 3 GO clustering results of DEGs with a criterion of Enrichment Score > 1. (A) Up-regulated. (B) Down-regulated. Counts represent the gene number enriched in each term. The Enrichment score was obtained according to the built-in program in DAVID, which ranks the biological significance of gene groups based on overall P-value of all enriched annotation terms. The higher the enrichment score is, the more important the term is. |
PPI Network of DEGs and Core Genes in the PPI Network
The PPI network contained 1498 nodes (DEGs) and 6701 edges (interactions between the DEGs). Using the twofold median value of node degree in this network as a cutoff point,23 1104 nodes were identified as hubs. The hub interaction network was constructed (Figure S2), and topological features, such as “Average Shortest Path Length”, “Closeness Centrality”, “Betweenness Centrality”, “Neighborhood Connectivity”, and “Degree” were calculated for each hub (shown in Table S4). Clustering analysis further revealed 11 groups based on the physiological processes (shown in Figure S3), and the hub genes in each cluster were mainly associated with cell growth and death, cellular community, cell cycle, organismal systems, immune system, infectious diseases, signal transduction and apoptosis (shown in Table 3). A total of 295 major hubs (degree higher than 2-fold of all hubs; median value = 6) were screened and 4 clusters were built (shown in Figure 4). As shown in Table 4, the top 5 pathways in Cluster 1 were cell cycle (P- value = 1.17E-30), p53 signaling pathway (P-value = 1.76E-12), DNA replication (P-value = 7.53E-09), viral carcinogenesis (P-value = 3.20E-07), and homologous recombination (P-value = 2.31E-06), that in Cluster 2 were ErbB signaling pathway (P-value = 1.57E-18), Natural killer cell mediated cytotoxicity (P-value = 4.44E-17), PI3K-Akt signaling pathway (P-value = 2.55E-16), pathways in cancer (P-value = 8.16E-16) and proteoglycans in cancer (P-value = 3.22E-15), in Cluster 3 were pathways in cancer (P-value = 1.29E-27), chronic myeloid leukemia (P-value = 1.52E-21), hepatitis B (P-value = 1.13E-20), pancreatic cancer (P-value = 1.40E-19), and prostate cancer (P-value = 1.57E-19), and in Cluster 4 were oocyte meiosis (P-value = 0.005), legionellosis (P-value = 0.05), and salmonella infection (P-value = 0.08). Furthermore, the top 4 high-degree hub nodes in each cluster included CDK1, MCM10, CD5, PDGFRA, BRCA1, LEF1, AURKA, BUB1 etc. (shown in Table 5), and were the most down-regulated genes.
 |
Table 3 Main GO and Pathway Enrichment Analysis Relating to All Hubs |
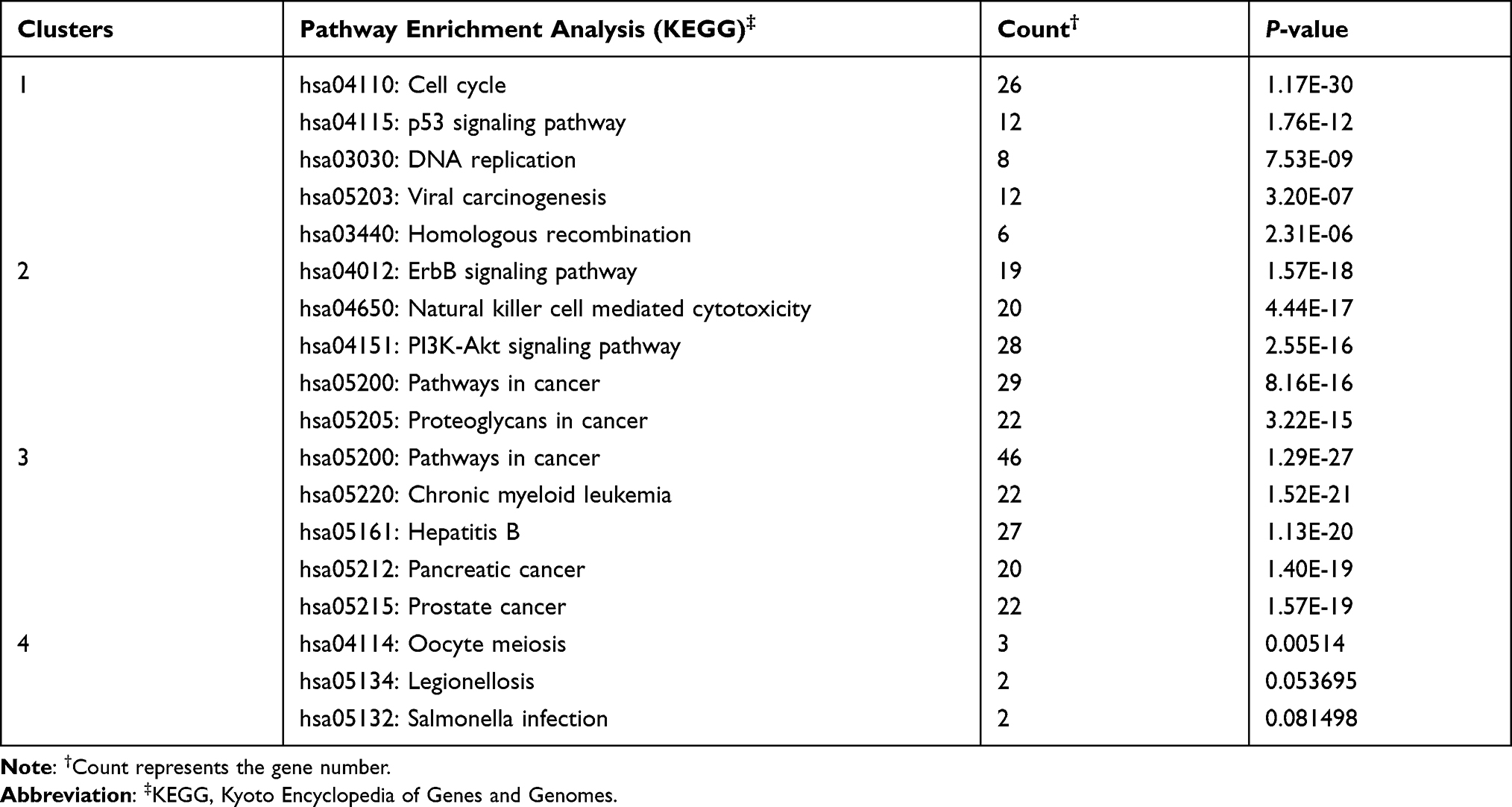 |
Table 4 Pathway Enrichment Analysis of PPI Clusters Related to Major Hubs (Top 5) |
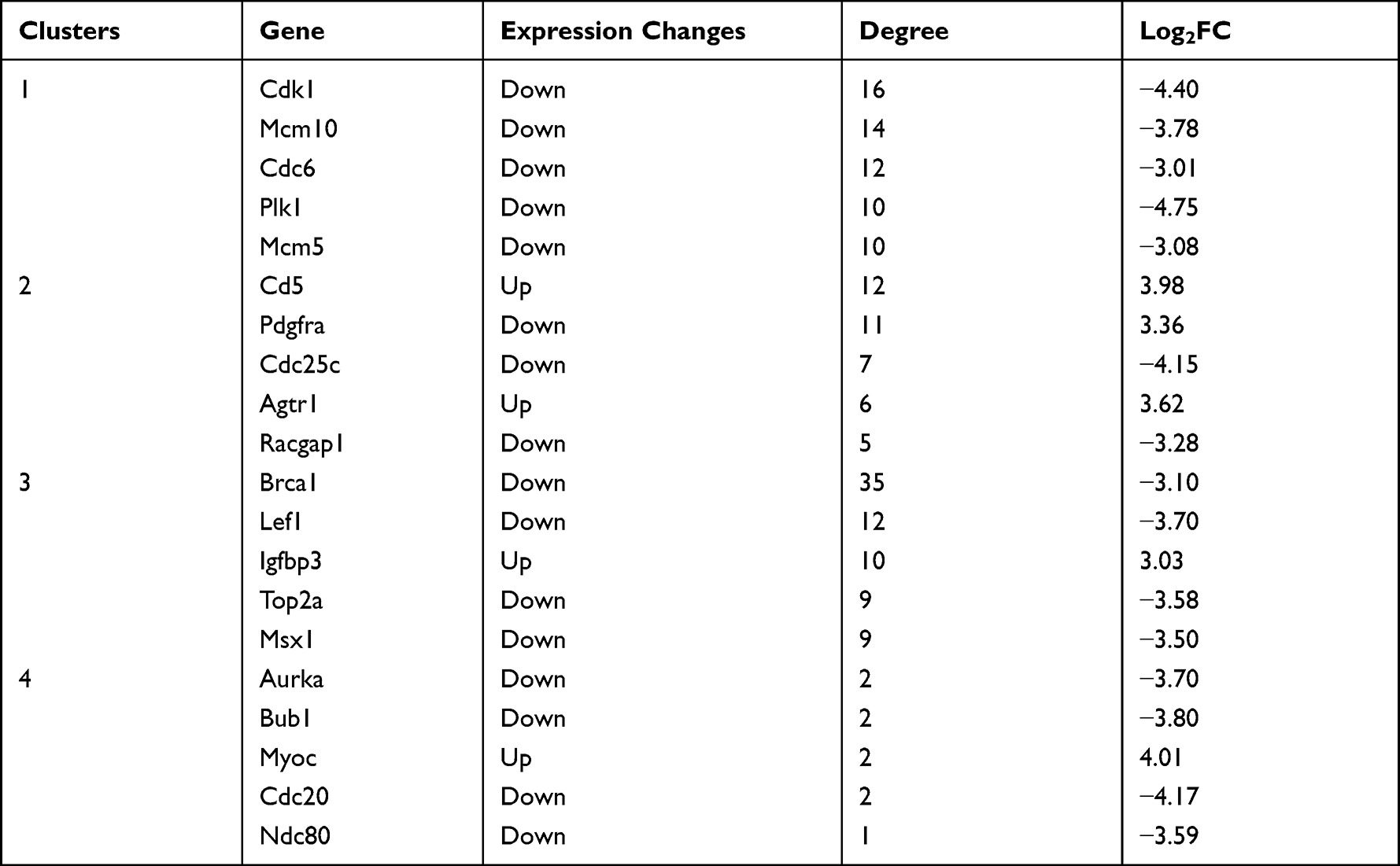 |
Table 5 The Top 5 DEGs with Higher Degrees in the Clusters Related to Major Hubs |
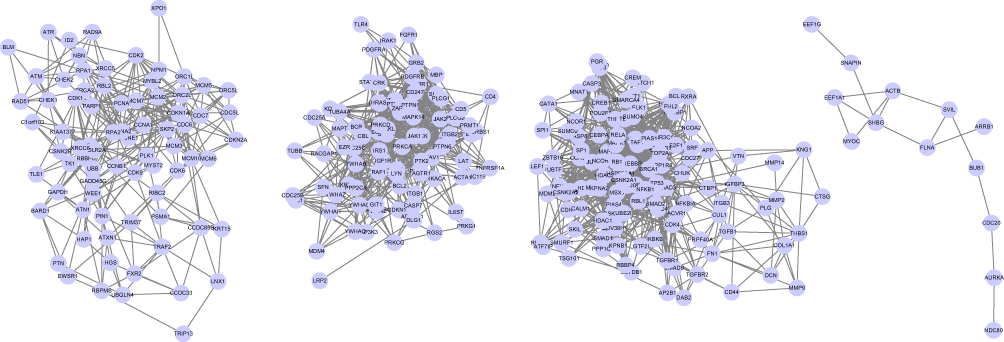 |
Figure 4 The top clusters of model with relatively high degrees selected from the protein–protein interaction network. The hubs were selected following the criteria of degree higher than 2-fold of all hubs, and median value=6. The clusters were built using Cytoscape ver. 3.5.1. |
qRT-PCR Confirmation of Differential mRNA Expression
Expression of Nox4, Mmrn1, Mcm5, Msx1, and Fgf5 was further validated by qRT-PCR and were coincident with RNA-Seq data (shown in Figure 5). Thus, both approaches showed a strong correlation.
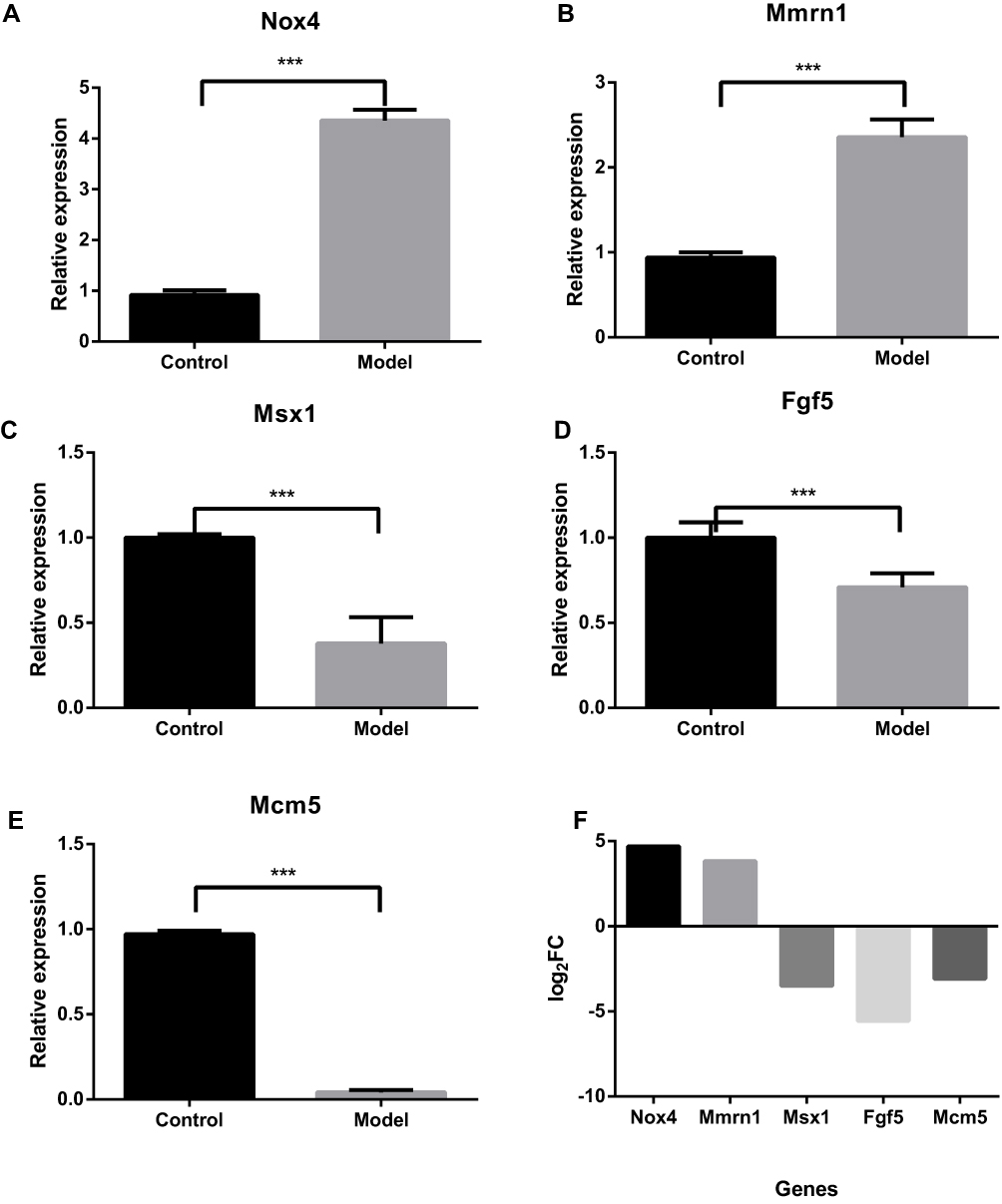 |
Figure 5 Relative expression of each selected gene validated by qRT-PCR using GAPDH as the housekeeping gene. The expression of each gene relative to GAPDH was calculated using a modified method of delta Ct(2−ΔCt). Data represent the mean ± SD of triplicate experiments. ***P value <0.001. (A) The relative expression of Nox4. (B) The relative expression of Mmrn1. (C) The relative expression of Msx1. (D) The relative expression of Fgf5. (E) The relative expression of Mcm5. (F) Log2FC values of the selected genes from the RNA-Seq data. |
Discussion
The Gram-positive bacterium S. aureus is the most common cause of skin infections in humans,15 although the molecular mechanisms underlying its pathological process are largely unknown. In this study, we established a mouse model of intradermal S. aureus infection, and observed the cardinal signs of localized inflammation, including epidermal thickening and massive infiltration of neutrophils in the dermis and epidermis. Miller et al summarized the host defense against S. aureus cutaneous infections and pointed again that one hallmark of S. aureus infections is the formation of neutrophil abscess, which is necessary for bacterial clearance.24 The recruitment of neutrophils to a site of S. aureus infection in the skin involves two steps, one is the innate immune process, recognition pathogen-associated molecular patterns of invading microorganisms, and the other is the subsequent initiation of pro-inflammatory immune responses, such as the secretion or production of cytokines, chemokines and adhesion molecules which promote neutrophil rolling, adhesion and diapedesis.24,25 While the inflammatory response combats pathogen invasion, uncontrolled infiltration of granulocytes can cause extensive tissue damage and epidermal dysfunction.26–28 In fact, several regulatory mechanisms limit the influx of neutrophils and prevent collateral damage due to a persistent pro-inflammatory state.29
To reveal the gene expression changes behind the above observations we carried out a transcriptome analysis. A total of 638 DEGs were identified in the study, of which 324 and 314 genes were, respectively, up- and down-regulated in the inflamed skin relative to the normal skin. The up-regulated DEGs were mainly enriched in GO terms like response to stimulus, immune system process and signal transduction, and pathways related to complement and coagulation cascades and S. aureus infection. Furthermore, 38 genes with the highest increase in transcript levels encoded immune response-related proteins like cytokines and chemokines, and the complement and coagulation cascades were enriched in most upregulated genes. Consistent with our findings, Brady et al12 reported 39 highly upregulated genes associated with the immune system in a mouse model of S. aureus soft tissue infection. A major part of these 39 genes (17 genes) encoding proteins included cytokines, chemokines, and chemokine-like proteins, as well as Cxcr1. Three genes encoding cytolytic granule proteins were included according to Brady’s study.12 While, we did not find related genes encoding cytolytic granule proteins in our study.
The complement system is a protein hydrolysis cascade that opsonizes pathogens and augments the effect of antibodies and phagocytic cells.30 Thirteen complement-related genes were differentially expressed in the inflamed skin, of which C7, C5ar1, F13a1, C6 and Cfh were highly upregulated. In addition, the top 4 enriched GO clusters following S. aureus infection were acute-phase response, chemotaxis, negative regulation of inflammation factor production and lipid metabolism.
Pathogenic infection also triggers the release of pro-inflammatory cytokines, chemokines and adhesion molecules by keratinocytes and the resident skin immune cells (such as Langerhans cells and γδ T cells in the epidermis, and dendritic cells, macrophages, fibroblasts, mast cells, B and T cells, plasma cells and natural killer cells in the dermis).25,28,29,31 These molecules promote the neutrophils recruitment which plays an important role in bacterial clearance.
In addition, several cytokines that regulate keratinocyte proliferation and differentiation are often dysregulated during inflammation.32 The chemokines in particular play an important part in the recruitment, activation and differentiation of inflammatory cells.33 For instance, Boeckmans et al detected increased expression of inflammatory cytokines such as CCL2, CCL5, CCL7, CCL8, CXCL5, CXCL8, IL1a, IL6 and IL11 in a human skin stem cell-derived model of non-alcoholic steatohepatitis (NASH).34 Consistent with this, 38 immune response-related genes (cytokines and chemokines, and the complement and coagulation cascades) above mentioned were upregulated in the S. aureus-infected skin, Cxcl13, Cxcl5, Ccl6 and Ccl9, for instance. There still existed other genes associated with neutrophil recruitment showing an increase in the expression. Several adhesion molecules, such as cell adhesion molecule3 (Cadm3), selectins (Sell and Selp) and adhesion molecule interacting with CXADR antigen 1(Amica1), playing an important part in neutrophil rolling, adhesion and diapedesis, increased in the inflamed skin.
The production and secretion of pro-inflammatory mediators depends on the activation of sensors like the toll-like receptors (TLRs), interleukin-1 receptor (IL-1R) that recognize pathogen-associated molecular patterns (PAMPs), and trigger downstream signaling pathways. The TLR or IL-1R pathway is associated with the sensitivity of various tissues and organs to S. aureus infection. The defects in these pathways can result in an increased susceptibility to the infections.35,36 The TLR or IL-1R family members signal through myeloid differentiation primary response protein 88 (MYD88), which subsequently interacts with IL-1R-associated kinase 4 (IRAK4) to activate TNFR-associated factor 6(TRAF6).24 In this study, both TLR and IL-1R family members were detected at high transcript levels. The TLR pathway members, such as toll-like receptor 4(Tlr4), toll-like receptor 7(Tlr7), toll-like receptor 8(Tlr8), cathepsin K (Ctsk), and lipopolysaccharide binding protein (Lbp), were up-regulated in the inflamed skin. It has been shown that TLRs are activated by recognizing S. aureus lipopeptides and lipoteichoic acid.37 These genes subsequently participate in the regulation of interleukin biosynthetic process, together with the IL-1R family members like Il1r2 and Il1rl1.
Miller et al reported that the activation of IL-1R is due to the stimulation of IL-1α and IL-1β.24 They found that different establishment of model results in differences in immune response. Both IL-1α and IL-1β contribute to host defense during a superficial S. aureus skin infection, whereas IL-1β has a predominant role during a deeper S. aureus skin infection.38 In our study, the subcutaneous model was established. Meanwhile, we did not detect the increase of IL-1β, which might be due to the differences in mice and the strains we used between the literature38 and our study.
Inflammasomes are molecular platforms activated in response to cell infection or stress that trigger the maturation of proinflammatory cytokines to participate in innate immune defense.39 Miller et al reported that inflammasome-mediated production of IL-1β is required for neutrophil recruitment against S. aureus in vivo.40 There are several types of inflammasome, such as NLRP1, NLRP3, NLRC4, and AIM2. In our study, we detected the gene expressions participating in the inflammasome complex assembly, although these genes were not included as DEGs by the subjective criteria used. The increased expression of Nlrp3 (NLR family, pyrin domain containing 3, Log2FC = 1.57, P-Adjust=0.001), Gsdmd (gasdermin D, Log2FC = 1.30, P-Adjust=5.01E-14), and Cd36 (CD36 antigen, Log2FC = 1.59, P-Adjust=1.48E-21) were detected, which all participated in assembly of NLRP3 inflammasome, and the secretion of IL-1β. Nlrc4 (NLR family, CARD domain containing 4, Log2FC = 1.15, P-Adjust=0.021) participating in the IL-1β secretion, also played an important part in the forming of NLRC4 inflammasome complex. In the study, Casp12 (caspase 12, Log2FC = 1.58, P-Adjust=3.59E-20) was increased, taking part in three types of inflammasome complex (NLRP3, AIM2 and NLRC4).
People believed that TH17 cells have a role in host defense against S. aureus at cutaneous site of infection.24,41 TH17 cells produce IL-17, IL-17F, and IL-22, thereby inducing a massive tissue reaction owing to the broad distribution of the IL-17 and IL-22 receptors.41 In the study, genes participating in IL-17 signaling pathway were accelerated, including lipocalin 2(Lcn2), chemokine (C-X-C motif) ligand 5(Cxcl5), S100 calcium binding protein A8 (calgranulin A) (S100a8), and S100 calcium binding protein A9 (calgranulin B) (S100a8). Inflammatory responses involving IL-17 probably contribute to arthritis, asthma, skin immune reactions and autoimmune disorders. Meanwhile, interleukin 17 receptor A (Il17ra), chemokine (C-C motif) ligand 7 (Ccl7), chemokine (C-C motif) ligand 12 (Ccl12) were also detected in a relatively high level, though they were not included as DEGs by the subjective criteria used. The activation of IL-17 receptors (IL-17RA-IL-17RC) on epidermal keratinocytes results in the production of antimicrobial peptides that have bacteriostatic and bactericidal activity against S. aureus.24 Antimicrobial peptides such as β-defensins and cathelicidins are induced production subsequently to come into play bacteriostatic or bactericidal activity against S. aureus.25 In the study, two genes participating in cathelicidins related process were changed in the expression. Neutrophilic granule protein (Ngp) was accelerated. Kininogen 2 (Kng2) was also increased, which plays a role in peptide secretion. While, cathelicidin antimicrobial peptide (Camp) decreased in the study due to the different period of inflammatory process.
In addition, several growth factors promote the accumulation and proliferation of inflammatory cells in infected tissues, and the fibroblast growth factor genes Fgf14 and Fgf23 were activated in the infected skin. Fgf14 is the receptor of TNF-related weak inducer of apoptosis (TWEAK), which triggers the release of pro-inflammatory factors like IL-6, IL-8, MMP-1, IFNγ-inducible protein 10 and prostaglandin E2 from fibroblasts and synoviocytes.42 Fgf23 is a direct stimulator of systemic inflammation, and promotes the secretion of IL-6 and C-reactive protein by liver cells.43 Prostaglandins (PG), including PG2, prostaglandin D2 (PGD2), prostaglandin F2α (PGF2α) and prostacyclin (PGI2), play a key role in the development of acute inflammation. Genes encoding for PGD2 synthase and PGI2 synthase were up-regulated in the model group, which is consistent with the high levels of PGD2 and PGI2 observed in inflamed tissues.44 The PGF receptor, a member of the prostanoid receptor subfamily, was also upregulated in the infected skin, which may be related to increased function of PGF2α.
The aryl hydrocarbon receptor (AhR) plays an important role in B cell maturation, and activation of macrophages, dendritic cells and neutrophils following lipopolysaccharide challenge or influenza virus infection. It is known to increase the expression levels of cytochrome P450 (Cyp), which is a key regulator of xenobiotic metabolism and the generation of intracellular reactive oxygen species (ROS).45 The S. aureus-infected tissues showed high transcript levels of AhR, and the Cyp2b23, Cyp2e1, Cyp4a10, Cyp4b1 and Cyp8b1 genes. ROS act as second messengers in redox-sensitive signal transduction, inflammation and apoptosis.46 Furthermore, several antioxidants maintain intracellular redox homeostasis by neutralizing end products of oxidation, and mitigate tissue damage due to oxidative stress. Genes encoding glutathione peroxidases (Gpx3 and Gpx7), glutathione S-transferases (Gsta3, Gstp2, Gstt1 and Gstt2) and other antioxidants (for eg, Nox4, Gpx3 and Lox) were significantly upregulated in the model group.
Consistent with the morphological changes seen in the epidermis following S. aureus infection, the expression levels of genes encoding extracellular matrix proteins were significantly altered. Fourteen of the most upregulated genes encode proteins related to the dermis structure. For instance, Cilp encodes cartilage intermediate layer protein that prevents fibrosis during pressure overload-induced fibrotic remodeling.47 In addition, Mmrn1 is involved in platelet adhesion and can bind to multiple proteins, which is related to the occurrence and development of many diseases.48 Four genes (Mmp2, Mmp8, Mmp11 and Lvrn) encoding different MMPs were significantly upregulated in the model group. The MMPs are a family of structurally related matrix-degrading enzymes that accelerate tissue disintegration during various pathological processes, including inflammation.49 They are normally expressed at low levels, and rapidly induced by various extracellular stimuli including growth factors, cytokines, oncoproteins, and UV and infrared radiations.49 MMP8 or collagenase-2 (neutrophil collagenase) can cleave collagens, elastins, fibronectins, gelatins and laminins in the extracellular matrix (ECM). MMP2 or gelatinase A, degrades denatured collagens or gelatins, stromelysins or proteoglycanases. Consistent with the high transcript levels of MMPs in the inflamed tissues, ECM components including the keratin family, keratin associated proteins, and cytoskeletal proteins were also downregulated in the model group.
S. aureus infection also increased transcript levels of filaggrin (Flg) and the filaggrin family member 2 (Flg2). Flg overexpression is associated with the disruption of keratin and other cytoskeletal elements, loss of desmosomal proteins from intercellular junctions, and cell cycle arrest.50 Tight junction proteins maintain the integrity of the epidermal barrier and prevent transepidermal water loss (TEWL).51,52 We found that genes encoding small proline-rich protein 2D (Sprr2d) and Keratin 78 (Krt78) were increased in the model group. Likewise, Brady et al found that Sprr2a1, Sprr2j-ps, Sprr3, Sprr2k and Krt6b, which have known functions in epidermal formation, were upregulated in S. aureus-infected tissues.30 Angiopoietins 1–4 (Angpt1-4) are growth factors belonging to the angiopoietin/TIE (tyrosine kinase with Ig and EGF homology domains) family that are involved in angiogenesis.53 In our study, we observed that Angpt1, Angpt2 and angiopoietin-like (Angptl) 1, 2, 4 and 7 transcripts were upregulated following S. aureus infection. Both Angpts and Angptls are activated by inflammatory mediators and other stimuli,54 and exacerbate inflammation-related tissue damage by increasing endothelial permeability and pericyte shedding from the basement membrane. These changes induce vascular leakage and promote immune or/and trans-endothelial migration of cancer cells.54
The down-regulated genes were mainly linked to GO terms like cell cycle, cell division and cytoskeletal parts, and the cell cycle (mmu:04110), oocyte meiosis (mmu:04114) and homologous recombination (mmu:03440) pathways. Two genes encoding immunity-related proteins were downregulated, of which Pbk promotes biosynthesis of pro-inflammatory factors like TNF-α, IL-1 and cyclooxygenase-2 via the p38 MAPK pathway.55 Most downregulated genes were enriched in cell cycle and chromosome segregation GO clusters, and 32 of the top 100 downregulated genes participate in the processes of metabolism and cell proliferation. These results indicate that genes related to cell growth and death are repressed by inflammation. The GO term structural molecule was the third most enriched among these DEGs, and related to the increased expression of MMPs. Fourteen of the most downregulated genes are related to keratin or keratin filaments, which modulate hair formation.30 Furthermore, keratinocytes also regulate the immune barrier function of the skin by secreting cytokines and chemokines. However, since we used the Balb/c nude mice, altered gene expression related to hair formation did not translate to any phenotypic changes. In addition, skin inflammation in the model group was accompanied by reduced expression of the junction protein claudin, which is consistent with the findings reported before.51,52
There are four enriched clusters in the PPI network, and most hub genes were down-regulated and mainly related to cell proliferation. CDK1, CD5, IGFBP3 and CDC20 were identified as the major hub proteins. CDK1 is a member of the cyclin-dependent kinase that regulates cell cycle progression. In the present study, Cdks (including Cdk1, Cdk2 and Cdk4), cyclins (Cyclin A2, Cyclin B1 and Cyclin B2) and their downstream transcription factors (E2f1, E2f2, and E2f3) were down-regulated, which can lead to cell cycle arrest. Other proliferation-related genes including Cdc20, Ccna2, Cdc6, Bub1, Bub1b, Ccnb1 and Ccnb2 were also downregulated. This is consistent with the observation that eukaryotic cells respond to DNA damage by inducing cell cycle arrest and DNA repair. However, in a previous study using USA300 strain-injected mice, cell cycle-related genes were not among the top 50 DEGs. In their study, genes related to immune function, signal transduction and epidermal formation account for a large proportion.12 Perhaps, it is because the experimental designs of the two studies are quite different, such as the selection of animals, the type of Staphylococcus aureus, the conditions of model establishment, and the way of obtaining samples, etc. On the other hand, it also shows the complexity of the organisms, for there are differences in occurrence time in the biological processes directly and indirectly related to the inflammatory process. As for our study, CD5 was up-regulated. It is reported that CD5 can be considered an immune checkpoint inhibitor used to potentiate anti-tumoral T cell responses or promote immunity against infections and modulate autoimmune reactions. Increased CD5 expression has been shown in anergized T cells after DC antigen targeting, whereas CD5 downregulation allows cytotoxic T cells to overcome tumor evasion and promotes cell death of autoreactive T cells and B-CLL cells.56 Furthermore, the pathogen-recognition receptors CD209d and CD209e that mediate pathogen endocytosis and lysosomal degradation57 were also upregulated. This is expected since the increased expression of pathogenic antigens is a major determinant of the immune system’s efficacy. The insulin-like growth factor binding protein (IGFBP) is a family of 6 proteins that bind to IGF and regulate insulin signaling. IGFBP3 induces apoptosis of retinal endothelial cells by stimulating PKA.58 While the Igfbp3 gene was up-regulated in the S. aureus-infected skin, Pka transcript levels were unaffected, indicating that protein IGFBP3 likely does not interact with PKA in the inflamed skin. IGFBP also regulates EMT,59 which is an essential cellular program in embryogenesis, tumor metastasis and wound healing,60 and is triggered by growth factors, inflammatory cytokines, chemokines, transcription factors and other proteins.61 These results further confirm that cell proliferation is inhibited by S. aureus-induced inflammation.
Based on its susceptibility to beta-lactams, S. aureus is commonly described as methicillin-susceptible S. aureus (MSSA) or methicillin-resistant S. aureus (MRSA). Moran et al found that S. aureus USA300 subtype (MRSA) is the cause of most community-acquired skin infections.62 More importantly, the impact of MSSA on infection still needs to be fully addressed. Although, we used the ATCC 25923 strain (one kind of MSSA) to induce model in the study. The transcriptomic changes in genes controlling the immune system, cell proliferation and dermal structure were more or less similar with that observed in USA300-induced soft tissue infection.12 The results were fully discussed above.
There are still limitations in our research. It would be better if we considered the genes related to S. aureus infection reported publicly when performing gene expression verification by qRT-PCR. Uninfected skin tissues from the same challenged mice should be added in the research to evaluate the local response to infection. Besides, inflammation is a process of change and development, and our research pays more attention to the genetic changes in the period of acute inflammation.
Nevertheless, our results still provide some theoretical support in the study of the pathogenesis of S. aureus and the screening of anti-inflammatory or anti- S. aureus drugs.
Statement of Ethics
The Guide for the Care and Use of Laboratory Animals (National Institutes of Health, USA) was closely adhered to in the formulation of all animal experimental procedures. The animal experimental procedures described below had the permission of College of Chemistry and materials engineering, Beijing Technology and Business University, Ethics Committee.
Acknowledgments
The work was supported by Yunnan Science and technology project (2018ZF005).
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
The work is supported by Yunnan Science and technology project (2018ZF005).
Disclosure
The authors have no conflicts of interest to declare.
References
1. Kaspar U, Kriegeskorte A, Schubert T, et al. The culturome of the human nose habitats reveals individual bacterial fingerprint patterns. Environ Microbiol. 2016;18:2130–2142. doi:10.1111/1462-2920.12891
2. Tong SY, Davis JS, Eichenberger E, et al. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. 2015;28:603–661. doi:10.1128/CMR.00134-14
3. Henriksen L, Simonsen J, Haerskjold A, et al. Incidence rates of atopic dermatitis, asthma, and allergic rhinoconjunctivitis in Danish and Swedish children. J Allergy Clin Immun. 2015;136:360–366.e2. doi:10.1016/j.jaci.2015.02.003
4. Carroll CL, Balkrishnan R, Feldman SR, et al. The burden of atopic dermatitis: impact on the patient, family, and society. Pediatr Dermatol. 2005;22(3):192–199. doi:10.1111/j.1525-1470.2005.22303.x
5. Cai SCS, Chen H, Koh WP, et al. Filaggrin mutations are associated with recurrent skin infection in Singaporean Chinese patients with atopic dermatitis. Brit J Dermatol. 2012;166:200–203. doi:10.1111/j.1365-2133.2011.10541.x
6. Zollner TM, Wichelhaus TA, Hartung A, et al. Colonization with superantigen-producing Staphylococcus aureus is associated with increased severity of atopic dermatitis. Clin Exp Allergy. 2000;30:994–1000. doi:10.1046/j.1365-2222.2000.00848.x
7. Joanna W, Hanna T, Dorota J, et al. Skin and nasal vestibule colonisation by Staphylococcus aureus, and its susceptibility to drugs in atopic dermatitis patients. Ann Agr Env Med. 2018;25:334–337. doi:10.26444/aaem/85589
8. Izabela B, Maciej J, Marta B, et al. Decolonization of Staphylococcus aureus in patients with atopic dermatitis: a reason for increasing resistance to antibiotics? Adv Dermatol Allergol. 2017;34:553–560. doi:10.5114/ada.2017.72461
9. Salava A, Lauerma A. Role of the skin microbiome in atopic dermatitis. Clin Transl Allerg. 2014;4:33. doi:10.1186/2045-7022-4-33
10. Suárez-Fariñas M, Ungar B, da Rosa JC, et al. RNA sequencing atopic dermatitis transcriptome profiling provides insights into novel disease mechanisms with potential therapeutic implications. J Allergy Clin Immun. 2015;135:1218–1227. doi:10.1016/j.jaci.2015.03.003
11. Malachowa N, Kobayashi SD, Sturdevant DE, Scott DP, DeLeo FR. Insights into the Staphylococcus aureus-host interface: global changes in host and pathogen gene expression in a rabbit skin infection model. PLoS One. 2015;10(2):e0117713. doi:10.1371/journal.pone.0117713
12. Brady RA, Bruno VM, Burns DL, Subbian S. RNA-seq analysis of the host response to Staphylococcus aureus skin and soft tissue infection in a mouse model. PLoS One. 2015;10:e0124877. doi:10.1371/journal.pone.0124877
13. Kõks S, Keermann M, Reimann E, et al. Psoriasis-Specific RNA isoforms identified by RNA-seq analysis of 173,446 Transcripts. Front Med (Lausanne). 2016;3:46. doi:10.3389/fmed.2016.00046
14. Baek JO, Lee JR, Roh JY, et al. Oral tolerance modulates the skin transcriptome in mice with induced atopic dermatitis. Allergy. 2018;73:962–966. doi:10.1111/all.13367
15. Liu H, Archer NK, Dillen CA, et al. Staphylococcus aureus epicutaneous exposure drives skin inflammation via IL-36-MT cell responses. Cell Host Microbe. 2017;22:653–666.e5. doi:10.1016/j.chom.2017.10.006
16. Cho JS, Guo Y, Ramos RI, et al. Neutrophil-derived IL-1beta is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS Pathog. 2012;8:e1003047. doi:10.1371/journal.ppat.1003047
17. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi:10.1093/nar/gkn923
18. Joseph JA, Peri S, Krishna US, et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13:2363–2371. doi:10.1101/gr.1680803
19. Keshava Prasad TS, Goel R, Kandasamy K, et al. Human protein reference database–2009 update. Nucleic Acids Res. 2009;37:767–772. doi:10.1093/nar/gkn892
20. Wright MW, Eyre TA, Lush MJ, et al. HCOP: the HGNC comparison of orthology predictions search tool. Mamm Genome. 2005;16:827–828. doi:10.1007/s00335-005-0103-2
21. Eyre TA, Wright MW, Lush MJ, et al. HCOP: a searchable database of human orthology predictions. Brief Bioinform. 2007;8:2–5. doi:10.1093/bib/bbl030
22. Gray KA, Daugherty LC, Gordon SM, et al. Genenames.org: the HGNC resources in 2013. Nucleic Acids Res. 2013;41:545–552. doi:10.1093/nar/gks1066
23. Li S, Zhang ZQ, Wu LJ, et al. Understanding ZHENG in traditional Chinese medicine in the context of neuro-endocrine-immune network. IET Syst Biol. 2007;1:51–60. doi:10.1049/iet-syb:20060032
24. Miller LS, Cho JS. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol. 2018;11(8):505–518. doi:10.1038/nri3010
25. Molne L, Verdrengh M, Tarkowski A, Clements JD. Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infect Immun. 2000;68:6162–6167. doi:10.1128/IAI.68.11.6162-6167.2000
26. Baron ED, Oyetakin-White P, Suggs A. Effect of botanicals on inflammation and skin aging: analyzing the evidence. Inflamm Allergy Drug Targets. 2014;13:168–176. doi:10.2174/1871528113666140526163052
27. Wang Y, Wen X, Hao D, et al. Insights into autophagy machinery in cells related to skin diseases and strategies for therapeutic modulation. Biomed Pharmacother. 2019;113:108775. doi:10.1016/j.biopha.2019.108775
28. Varma SR, Sivaprakasam TO, Arumugam I, et al. In vitro anti-inflammatory and skin protective properties of Virgin coconut oil. J Tradit Complem Med. 2019;9:5–14. doi:10.1016/j.jtcme.2017.06.012
29. Sugimoto MA, Vago JP, Perretti M, et al. Mediators of the resolution of the inflammatory response. Trends Immunol. 2019;40:212–227. doi:10.1016/j.it.2019.01.007
30. Jin X, Ma Q, Sun Z, et al. Airborne fine particles induce hematological effects through regulating the crosstalk of the kallikrein-kinin, complement, and coagulation systems. Environ Sci Technol. 2019;53:2840–2851. doi:10.1021/acs.est.8b05817
31. Jung SH, Kim KA, Sohn SW, et al. Cytokine levels of the aqueous humour in central serous chorioretinopathy. Clin Exp Optom. 2014;97:264–269. doi:10.1111/cxo.12125
32. Ye L, Mauro TM, Dang E, et al. Topical applications of an emollient reduce circulating pro-inflammatory cytokine levels in chronically aged humans: a pilot clinical study. J Eur Acad Dermatol. 2019;33:2197–2201. doi:10.1111/jdv.15540
33. Kuo PT, Zeng Z, Salim N, et al. The Role of CXCR3 and its chemokine ligands in skin disease and cancer. Front Med. 2018;5:271. doi:10.3389/fmed.2018.00271
34. Boeckmans J, Buyl K, Natale A, et al. Elafibranor restricts lipogenic and inflammatory responses in a human skin stem cell-derived model of NASH. Pharmacol Res. 2019;144:377–389. doi:10.1016/j.phrs.2019.04.016
35. Picard C, Puel A, Bonnet M, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076–2079. doi:10.1126/science.1081902
36. von Bernuth H, Picard C, Jin Z, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321:691–696. doi:10.1126/science.1158298
37. Kawai T, Akira S. The Role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi:10.1038/ni.1863
38. Cho JS, Zussman J, Donegan NP, et al. Noninvasive in vivo imaging to evaluate immune responses and antimicrobial therapy against Staphylococcus aureus and USA300 MRSA skin infections. J Invest Dermatol. 2011;131:907–915. doi:10.1038/jid.2010.417
39. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi:10.1016/j.cell.2010.01.040
40. Miller LS, Pietras EM, Uricchio LH, et al. Inflammasome-mediated production of IL-1β is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol. 2007;179:6933–6942. doi:10.4049/jimmunol.179.10.6933
41. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi:10.1146/annurev.immunol.021908.132710
42. Chicheportiche Y, Chicheportiche R, Sizing I, et al. Proinflammatory activity of TWEAK on human dermal fibroblasts and synoviocytes: blocking and enhancing effects of anti-TWEAK monoclonal antibodies. Arthritis Res. 2002;4:126–133. doi:10.1186/ar388
43. Singh S, Grabner A, Yanucil C, et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016;90:985–996. doi:10.1016/j.kint.2016.05.019
44. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi:10.1161/ATVBAHA.110.207449
45. Costa C, Catania S, De Pasquale R, et al. Exposure of human skin to benzo[a]pyrene: role of CYP1A1 and aryl hydrocarbon receptor in oxidative stress generation. Toxicol. 2010;271:83–86. doi:10.1016/j.tox.2010.02.014
46. Fuchs J, Zollner TM, Kaufmann R, et al. Redox-modulated pathways in inflammatory skin diseases. Free Radical Biol Med. 2001;30:337–353. doi:10.1016/S0891-5849(00)00482-2
47. Zhang CL, Zhao Q, Liang H, et al. Cartilage intermediate layer protein-1 alleviates pressure overload-induced cardiac fibrosis via interfering TGF-β1 signaling. J Mol Cell Cardiol. 2018;116:135–144. doi:10.1016/j.yjmcc.2018.02.006
48. Laszlo GS, Alonzo TA, Gudgeon CJ, et al. Multimerin-1 (MMRN1) as novel adverse marker in pediatric acute myeloid leukemia: a report from the children’s oncology group. Clin Cancer Res. 2015;21:3187–3195. doi:10.1158/1078-0432.CCR-14-2684
49. Kang SM, Han S, Oh JH, et al. A synthetic peptide blocking TRPV1 activation inhibits UV-induced skin responses. J Dermatol Sci. 2017;88:126–133. doi:10.1016/j.jdermsci.2017.05.009
50. Presland RB, Kuechle MK, Lewis SP, et al. Regulated expression of human filaggrin in keratinocytes results in cytoskeletal disruption, loss of cell-cell adhesion, and cell cycle arrest. Exp Cell Res. 2001;270:199–213. doi:10.1006/excr.2001.5348
51. Watson AJ. Claudins and barrier dysfunction in intestinal inflammation: cause or consequence? Gut. 2015;64:1501–1502. doi:10.1136/gutjnl-2014-309110
52. Turksen K, Troy TC. Barriers built on claudins. J Cell Sci. 2004;117:2435–2447. doi:10.1242/jcs.01235
53. Michalska-Jakubus M, Cutolo M, Smith V, et al. Imbalanced serum levels of Ang1, Ang2 and VEGF in systemic sclerosis: integrated effects on microvascular reactivity. Microvasc Res. 2019;125:103881. doi:10.1016/j.mvr.2019.103881
54. Akwii RG, Sajib MS, Zahra FT, et al. Role of angiopoietin-2 in vascular physiology and pathophysiology. Cells. 2019;8:471. doi:10.3390/cells8050471
55. Vermeulen L, Vanden Berghe W, Beck IM, et al. The versatile role of MSKs in transcriptional regulation. Trends Biochem Sci. 2009;34:311–318. doi:10.1016/j.tibs.2009.02.007
56. Burgueo‐Bucio E, Mier‐Aguilar CA, Gloria S. The multiple faces of CD5. J Leukoc Biol. 2019;105:1–14. doi:10.1002/JLB.MR0618-226R
57. Akahori H, Karmali V, Polavarapu R, et al. CD163 interacts with TWEAK to regulate tissue regeneration after ischaemic injury. Nat Commun. 2015;6:7792. doi:10.1038/ncomms8792
58. Liu L, Patel P, Steinle JJ. PKA regulates HMGB1 through activation of IGFBP-3 and SIRT1 in human retinal endothelial cells cultured in high glucose. Inflamm Res. 2018;67:1013–1019. doi:10.1007/s00011-018-1196-x
59. Xiao W, Tang H, Wu M, et al. Ozone oil promotes wound healing by increasing the migration of fibroblasts via PI3K/Akt/mTOR signaling pathway. Biosci Rep. 2017;37:BSR20170658. doi:10.1042/BSR20170658
60. Yin SY, Peng AP, Huang LT, et al. The phytochemical shikonin stimulates Epithelial-Mesenchymal Transition (EMT) in skin wound healing. Evid Based Compl Altern Med. 2013;2013:262796. doi:10.1155/2013/262796
61. Baek SH, Ko JH, Lee JH, et al. Ginkgolic acid inhibits invasion and migration and TGF-beta-induced EMT of lung cancer cells through PI3K/Akt/mTOR inactivation. J Cell Physiol. 2017;232:346–354. doi:10.1002/jcp.25426
62. Moran GJ, Krishnadasan A, Gorwitz RJ, et al. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355:666–674. doi:10.1056/NEJMoa055356
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.