")
Back to Journals » Biologics: Targets and Therapy » Volume 14
Gene, Cell and Antibody-Based Therapies for the Treatment of Age-Related Macular Degeneration
Received 24 June 2020
Accepted for publication 4 August 2020
Published 11 September 2020 Volume 2020:14 Pages 83—94
DOI https://doi.org/10.2147/BTT.S252581
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shein-Chung Chow
Engin Akyol, Andrew Lotery
Clinical Neurosciences Research Group, Faculty of Medicine, University of Southampton, Southampton General Hospital, Southampton SO16 6YD, UK
Correspondence: Andrew Lotery
Clinical Neurosciences Research Group, Faculty of Medicine, University of Southampton, Southampton General Hospital, Mailpoint 806, South Lab & Path Block, Tremona Road, Southampton SO16 6YD, UK
Tel +44 23 8120 5049
Email [email protected]
Abstract: Here we discuss antibody, cell and gene-based therapies that are currently available and under investigation for both wet and dry age-related macular degeneration (AMD). We initially discuss ocular anatomy, AMD modelling as well as the underlying pathophysiology of AMD. The antibody-based trials which have revolutionised the management of wet AMD are reviewed. The latest concepts in antibody therapy for wet AMD such as the port delivery systems, bispecific antibodies, designed ankyrin repeat protein (DARPINs) and brolucizumab are explored. Furthermore, the antibody-based trials targeting the complement pathway to reduce progression of geographic atrophy (GA) in dry AMD are discussed. Stem cell therapy and gene therapy are novel treatment modalities with no established clinical use in wet or dry AMD. Here, we discuss their efficacy so far in clinical trials. Their benefits and risk in the treatment of both wet and dry AMD are evaluated.
Keywords: choroidal neovascularisation, geographic atrophy, gene therapy, macular degeneration, stem cell therapy, monoclonal antibodies
Introduction
Age-related macular degeneration (AMD) is the leading cause of irreversible blindness in those over the age of 65 in the developed world.1 In the United Kingdom, the prevalence of advanced AMD in patients over 50 is approximately 2.4%.2 The global pooled prevalence (age range of 45–85 years) of early, late, and any AMD is, respectively, 8.01%, 0.37%, and 8.69%.3
AMD can be classified either as dry or wet AMD. Dry AMD is characterised by atrophic degeneration of the macula and gradual visual loss.4 The end stage of dry AMD is geographic atrophy (GA) and it accounts for circa 90% of total AMD (Figure 1). In contrast, wet AMD is responsible for rapid and substantial visual loss due to the development of choroidal neovascularisation (CNV) (Figure 1). These new vessels leak and cause exudation of blood and fluid into the macula. Wet AMD is subclassified as Type 1 “occult”/polypoidal choroidal vasculopathy, Type 2 “classic” and Type 3 “retinal angiomatous proliferation”. The angiogenesis and increased vascular permeability seen in wet AMD is driven partly by upregulation of vascular endothelial growth factor (VEGF).5 The emergence of monoclonal antibodies which inhibit VEGF have revolutionised the management of wet AMD and reduced the incidence of significant visual loss secondary to wet AMD.6–12 The most commonly used anti-VEGF therapies are Ranibuzimab (Lucentis), Bevacizumab (Avastin) used off label and Aflibercept (Eylea).13 There are currently no effective treatments for dry AMD although drugs targeting the complement pathway are under investigation.
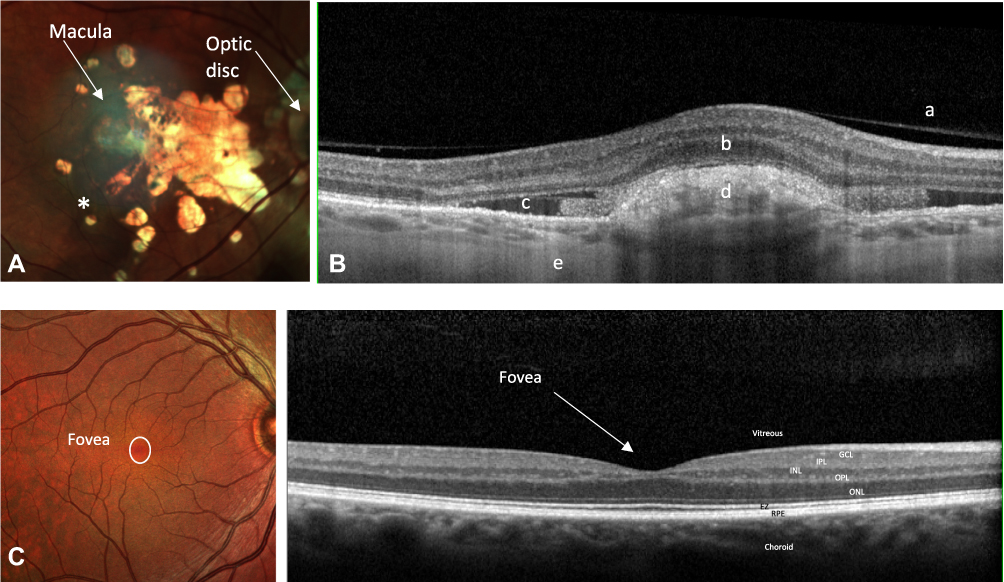 |
Figure 1 (A) A right colour fundus photograph demonstrating geographic atrophy in a patient with advanced dry AMD. There is atrophy of the neurosensory retina allowing visualization of the underlying choroid and scleral layers, seen as yellow white in this image. Increased pigmentation is also noted (*on diagram). (B) A right optical coherence tomography (OCT) through the macula demonstrating CNV and subretinal fluid in a patient with advanced wet AMD. The CNV is inferred by the presence of sub-retinal fluid and sub-retinal hyperreflective material. (C) A right colour fundus photograph and an OCT through the macula of a patient with a healthy retina demonstrating the fovea. |
In this review, we will discuss the aetiology of AMD and how this is directing the development of novel therapies. We will describe current and potential future antibody, gene and cell-based therapies for AMD.
Neurosensory Retina and the Retinal Pigment Epithelium (RPE)
The neurosensory retina and RPE are derived from the neuroectoderm. The neurosensory retina and RPE are surrounded externally by Bruch’s membrane and internally by the vitreous. The tissue is composed of a combination of neural cells, glial cells, vascular endothelium, pericytes and microglia. This complexity makes replacement of tissues by cell-based therapies challenging. It is damage to the photoreceptors particularly the cones that cause significant daytime visual loss in AMD patients.14,15 Loss of rod photoreceptors is also common as they are the predominant photoreceptor in the macula and patients are often aware of reduced scotopic vision early in the disease.16 They often have a central visual scotoma when dark adapted, eg, getting up in the middle of the night. Cone cells located in the macula and at the fovea are responsible for photopic, ie, central, fine colour vision. The fovea is a depression in the inner retinal surface, about 1.5mm wide. Within the fovea is a region of 0.5mm diameter called the foveal avascular zone: an area without any blood vessels. The fovea is arranged anatomically in such a way that the light arriving to the central cones are undisturbed as the overlying retinal layers are displaced to the side of the fovea (Figure 1).
The RPE is a single layer of cuboidal/columnar epithelial cells surrounding the neurosensory retina externally. The RPE is essential for the normal functioning of the neural retina. The tight junctions between the RPE cells are the site of the outer blood retinal barrier highlighting its importance in maintaining ocular integrity.
The functions of the RPE include storage of metabolites and vitamin A as well as the recycling and production of the photopigments. RPE cells surround and support photoreceptors. The RPE cells provide nutrients to the photoreceptors and phagocytose the photoreceptor discs.17 Therefore, malfunction of RPE cells as in AMD will rapidly result in accumulation of cellular debris beneath the RPE and in loss of photoreceptors. Replacement of the RPE is a key strategy emerging cell-based therapies for AMD.
Pathophysiology
AMD is a complex multifactorial disease and the underlying pathophysiology is not fully understood. Drusen formation is the hallmark of AMD. Drusen are accumulations of yellow, Periodic acid-Schiff (PAS)-positive, amorphic extracellular material deposited between the RPE and the inner collagenous zone of Bruch’s membrane. The size, shape and morphology of drusen are used to classify the stages of AMD. Recent advances in imaging technologies have enabled identification of other imaging biomarkers of AMD such as reticular pseudodrusen (subretinal deposits) that develop and progress independent of drusen.18 OCT imaging indicates that there are 38 different types of drusen that may co-exist in a patient and their significance in AMD progression is unknown.19 RPE cell debris entrapped between the RPE monolayer and Bruch’s membrane is thought to serve as a chronic inflammatory stimulus allowing the deposition of drusen. Thereby it is proposed that inflammation potentiates biogenesis of drusen.20 The prevention of inflammation is targeted in complement pathway inhibiting therapy as discussed below.
One of the main environmental risk factors involved in AMD formation is smoking. A recent systematic review demonstrated a 2–3 fold increase in risk of AMD in current smokers compared with never-smokers.21 An interesting observation has been the reduction in complement factor H (CFH) levels in smokers which may explain the increased risk of AMD in smokers.22
The importance of genetic factors in the development of AMD has been highlighted through numerous studies. It is the first complex disease for which a genome-wide association study (GWAS) was successful with the discovery of a strong association with polymorphisms in the CFH gene. It is known that during the course of the disease the alternative complement pathway is activated.23 Genetic variants at chromosome 1q31 encompassing the CFH and CFH related genes (CFHR1-5) are major determinants of AMD susceptibility.24–26 The risk of developing AMD is 4.6 times greater in heterozygous and 7.4 times greater in homozygous compared to wild type for a CFH risk allele (a tyrosine-histidine polymorphism at amino acid 402).26 CFH acts to suppress the complement pathway. Therefore, in the presence of an abnormal CFH the complement pathway is activated resulting in an inflammatory environment. It is this inflammatory environment which is thought to initiate and causes progression in AMD.
A recent paper has shown that Factor H-Related Protein 4 (FHR-4) plays a prominent role in AMD pathogenesis.27 The study showed that systemic FHR-4 levels are elevated in AMD patients and FHR-4 accumulates in the choriocapillaris, Bruch’s membrane and drusen. This is a new potential therapeutic target in future trials.
Another significant genetic locus responsible for conferring risk of AMD is the High‐Temperature Requirement Protein A1 (HTRA1) locus which is a serine protease secreted by a number of tissues including RPE. It is located on chromosome 10q26 (Table 1).28 The mechanism by which the HTRA1/ARMS2 (age-related maculopathy susceptibility 2) allele causes AMD is not clear. A recent large genome-wide association study of AMD identified very rare coding variants (frequency <0.1%) in CFH, CFI, and TIMP3 suggesting causal roles for these genes, as did a splice variant in SLC16A8 (Table 2). Furthermore, they identified the first signal specific to wet AMD near Matrix Metalloproteinase (MMP-9).29
 |
Table 1 Showing the Odds Ratio for AMD of ARMS2 and HTRA1 Genes |
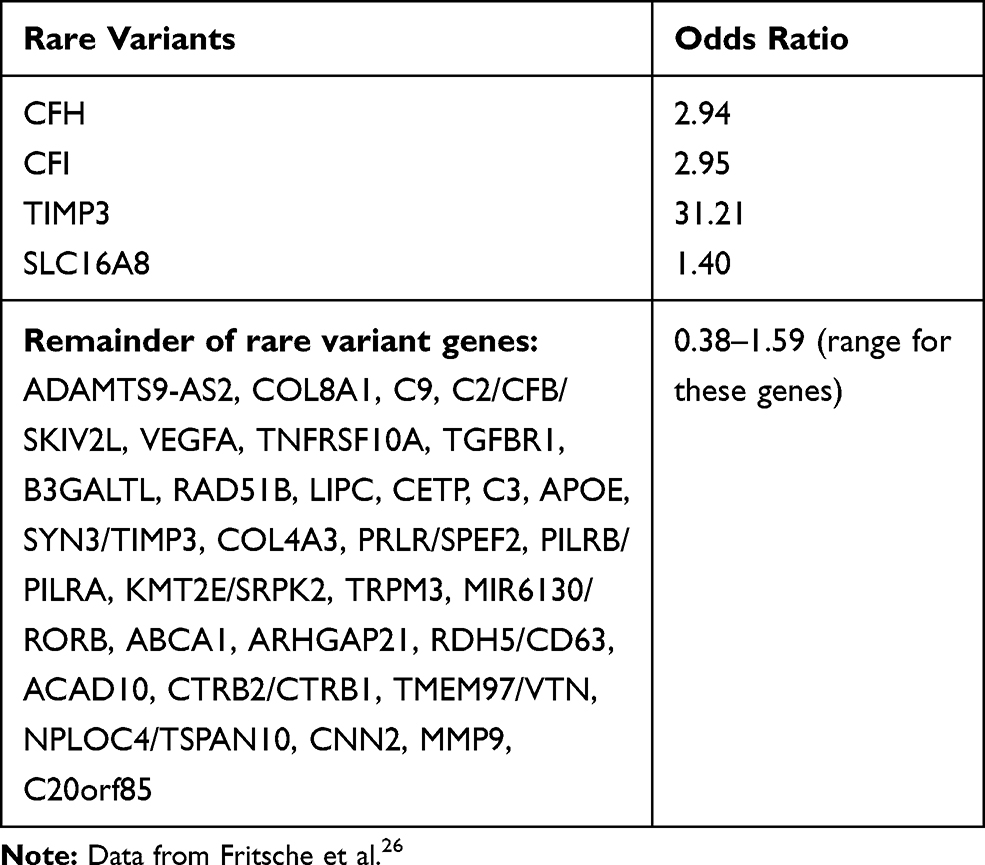 |
Table 2 Showing the Odds Ratio for AMD of 34 Rare Variants. The First 4 Genes Have a Significant Rare Variant Burden |
It is essential to understand the molecular and genetic basis of AMD in order to be able to target treatment effectively.
Modelling of AMD and Its Challenges
We will briefly discuss modelling of both dry and wet AMD. As of yet no single model captures all histological features of AMD. Accurate models can assist in the development of new treatment therapies but there are many challenges in modelling AMD. One of the main challenges is the fact that AMD is a complex multifactorial disease involving genetic and environmental factors. Secondly, anatomical differences between the human retina and other species lead to other difficulties with modelling. For example, mice and rats lack an anatomical macula meaning it is impossible to make direct comparisons and inferences. In contrast, non-human primates have a macula. However, they are costly, difficult to manipulate genetically and have a slow course of disease progression in comparison to mice and rats. In addition, it is considered best practice not to use non-human primates for research.
Cellular models of AMD can be derived from patient-specific induced pluripotent stem (iPS) cells. In order to model AMD the retinal homeostatic unit consisting of the neurosensory retina, the RPE, Bruch’s membrane and the endothelial cells that line the choriocapillaris need to be formed. However, there are numerous challenges with this the most obvious being the oversimplification of a complex disease process which takes years to develop.
Antibody Therapy for Macular Degeneration
Until the introduction of anti-VEGF antibody therapy for wet AMD the main treatment modality for patients with neovascular AMD was either laser photocoagulation or photodynamic therapy (PDT). Neither of these approaches resulted in significant visual gain for patients. The main target of antibody therapies has been VEGF which is responsible for driving the angiogenesis seen in CNV. Blockage of VEGF with monoclonal antibodies leads to regression of CNV (Table 3). We will discuss antibody therapies aimed at treating both CNV in wet AMD and GA in dry AMD.
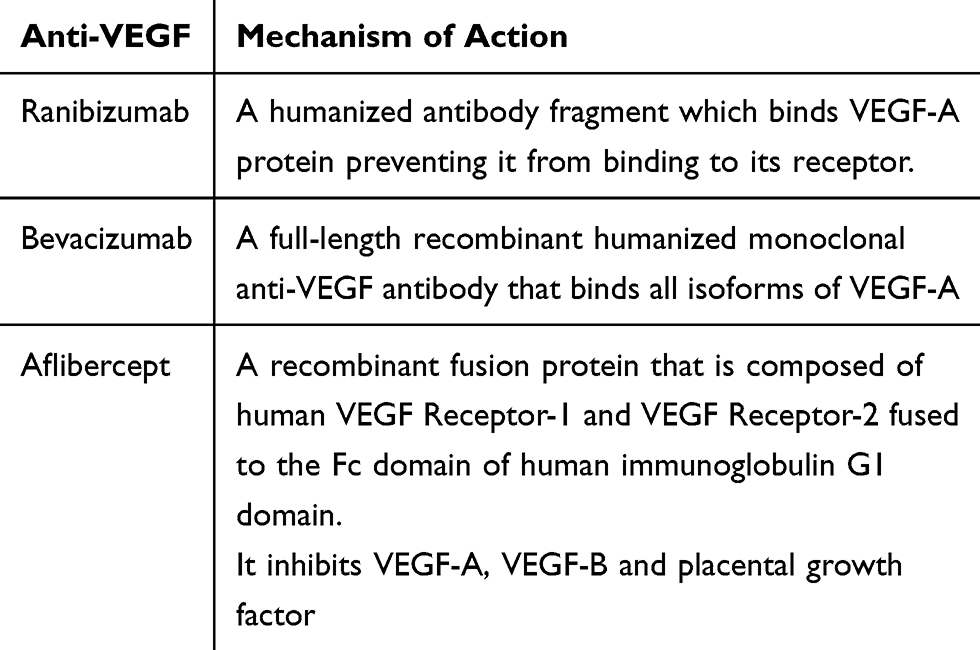 |
Table 3 Showing the Different Types of Anti-VEGF and Their Mechanisms of Action |
Dry AMD Antibody Therapy
There are no current effective antibody therapies for GA. However, antibodies targeting the complement pathway have been and currently are in clinical trials.
Lampalizumab is a fragment antigen-binding segment of a humanised monoclonal antibody. It selectively binds to and inhibits complement factor D. It was thought that lampalizumab may reduce the rate of enlargement of GA in dry AMD as it had already passed Phase I and II clinical trials. However, a recent Phase III double-masked, randomized, sham-controlled clinical trial involving 1881 participants unfortunately did not show a reduction in GA progression versus sham.30
Prior to this, another complement pathway inhibitor Eculizumab was trialled in the COMPLETE study to assess the progression of GA in dry AMD.31 Eculizumab functions as a complement component C5 inhibitor. Patients were randomized 2:1 to receive intravenous eculizumab or placebo over 6 months. Although systemic complement inhibition with eculizumab was well tolerated the treatment did not decrease the growth rate of GA significantly. Another C5 inhibitor LFG316 a fully human antibody failed to halt GA progression in a Phase II study.32 Currently, a Phase IIb trial using an intravitreal C5 inhibitor (Zimura) is being used to assess GA progression.33
In another study, a humanized monoclonal anti-amyloid-β antibody therapy (GSK933776) delivered via the intravenous route was trialled for the treatment of GA in dry AMD.34 This was a prospective, randomized, multicentre Phase II clinical trial. Their results showed that intravenous amyloid-β inhibition with GSK933776 did not slow the rate of GA enlargement compared with placebo.
On a more positive note, a Phase II clinical trial named the FILLY trial showed that the intravitreal administration of APL-2, a complement C3 inhibitor slowed the rate of GA atrophy35 and it is now progressing in to Phase III studies. Furthermore, combined antibody therapies are now being trialled. A Phase I trial is being conducted using a combination of LFG316 (C5 inhibitor) and CLG561 (Properdin inhibitor).36 The results are awaited.
Antibody Therapy for Wet AMD
The first Phase III trial looking at the effects of ranibizumab in wet AMD was the MARINA trial.11 In this multicentre, double-blind, sham-controlled study, AMD patients with either minimally classic or occult CNV were randomly assigned to receive 24 monthly intravitreal injections of ranibizumab (0.3mg or 0.5mg) or sham injections. At 12 months, 94.5% of the group given 0.3mg of ranibizumab and 94.6% of those given 0.5 mg lost fewer than 15 letters, as compared with 62.2% of patients receiving sham injections. Visual acuity (VA) improved by 15 or more letters in 24.8% of the 0.3 mg group and 33.8% of the 0.5 mg group, compared with 5.0% of the sham-injection group. Additionally, there were low rates of adverse events.
In a separate trial called ANCHOR, ranibizumab was compared with PDT with verteporfin in the treatment of predominantly classic neovascular AMD.6 This multicentre, double-blind study randomly assigned patients in a 1:1:1 ratio to receive monthly intravitreal injections of ranibizumab (0.3 mg or 0.5 mg) plus sham verteporfin therapy or monthly sham injections plus active verteporfin therapy. Of the 423 patients enrolled, 94.3% of those given 0.3 mg of ranibizumab and 96.4% of those given 0.5 mg lost fewer than 15 letters, as compared with 64.3% of those in the verteporfin group. VA improved by 15 letters or more in 35.7% of the 0.3mg group and 40.3% of the 0.5mg group, compared with 5.6% of the verteporfin group. Mean VA increased by 8.5 letters in the 0.3mg group and 11.3 letters in the 0.5mg group, compared with a decrease of 9.5 letters in the verteporfin group. The authors concluded that ranibizumab was superior to verteporfin in the treatment of predominantly classic neovascular AMD, with low rates of serious ocular adverse events.
Two years later the PIER study evaluated the efficacy and safety of ranibizumab administered monthly for 3 months and then quarterly in patients with subfoveal CNV secondary to AMD.10 In this Phase IIIb, sham injection-controlled trial patients were randomized 1:1:1 to 0.3mg ranibizumab, 0.5mg ranibizumab, or sham treatment groups. They found that ranibizumab administered monthly for three months and then quarterly provided significant VA benefit to patients with AMD-related subfoveal CNV and was well tolerated.
Bevacizumab was initially licensed for use in the treatment of colonic tumours. The ABC trial12 evaluated the efficacy and safety of intravitreal bevacizumab injections for the treatment of wet AMD. Patients received intravitreal bevacizumab or standard treatment available at the start of the trial. In the bevacizumab group, 32% of patients gained 15 or more letters from baseline VA compared with 3% in the standard care group. In addition, the proportion of patients who lost fewer than 15 letters of visual acuity from baseline was significantly greater among those receiving bevacizumab treatment 91% versus 67% in standard care group. They concluded bevacizumab 1.25mg intravitreal injections given as part of a six-weekly variable retreatment regimen is superior to standard care, with low rates of serious ocular adverse events.
Comparison between bevacizumab and ranibizumab was conducted later in the IVAN7 and CATT8 trials. In the CATT trial patients were assigned to 4 treatment groups defined by drug (ranibizumab or bevacizumab) and dosing regimen (monthly or as needed). At 1 year, patients initially assigned to monthly treatment were reassigned randomly to monthly or as-needed treatment, without changing the drug assignment. Among patients following the same regimen for 2 years, mean gain in VA was similar for both drugs. Furthermore, mean gain was greater for monthly than for as-needed treatment. The authors concluded that ranibizumab and bevacizumab had similar effects on VA over a 2-year period.
The IVAN trial was a multicentre, non-inferiority randomised trial. Adults with active, previously untreated wet AMD and a best corrected VA (BCVA) of at least 25 letters were recruited. Participants were randomly assigned (1:1:1:1) to intravitreal injections of ranibizumab or bevacizumab in continuous (every month) or discontinuous (PRN) regimens. For BCVA, bevacizumab was neither non-inferior nor inferior to ranibizumab. Discontinuous treatment was neither non-inferior nor inferior to continuous treatment. The authors concluded that ranibizumab and bevacizumab have similar efficacy.
Finally, aflibercept was investigated as an antibody therapy for wet AMD in the VIEW 1 and VIEW 2 studies.9 Both Phase III studies compared monthly and every 2-monthly dosing of intravitreal aflibercept injection with monthly ranibizumab. Patients in the study had active, subfoveal CNV secondary to AMD. They concluded that intravitreal aflibercept dosed monthly or every 2 months after 3 initial monthly doses produced similar efficacy and safety outcomes as monthly ranibizumab.
Studies such as the PrONTO study3737 demonstrated that anti-VEGF treatment can be given as required depending on the OCT scan of the macula. The results showed that using an OCT-guided variable-dosing regimen with intravitreal ranibizumab resulted in VA outcomes comparable with the outcomes from the Phase III clinical studies, but with fewer intravitreal injections.
The HORIZON study38 evaluated the long-term safety and efficacy of multiple intravitreal ranibizumab injections. It showed that multiple ranibizumab injections were well tolerated for ≥4 years, ie, this indicated that patients may need treatment for many years with these antibody therapies.
An international, retrospective, observational study monitored the real-life use and effectiveness of ranibizumab injections in patients with neovascular AMD.39 It showed that real world as required treatment was less successful compared to clinical trials such as the PrONTO study. This seemed to be because in real-world situations patients could not access treatment as quickly as in randomised clinical trials. Countries with less bureaucratic hurdles and less delay in delivering treatment obtained the best outcomes for their patients.
Current Developments in Antibody Therapy
The goal for the future is fewer injections for the same or better visual outcomes as current therapy. This will reduce the treatment burden for patients who are typically older and less mobile. Currently, there are several therapeutic options that are being investigated – designed ankyrin repeat proteins (DARPINs), brolucizumab, use of a port delivery systems (PDS) and bispecific antibodies.
The PDS is a system used with ranibizumab. It is a device implanted into the wall of the eye. Here it slowly releases ranibizumab into the vitreous. The port can be re-filled in clinic visits and is simple to do so. The expectation is that the interval between port injections will be significantly longer that repeat intravitreal injections. A Phase II randomised clinical trial showed the PDS was generally well tolerated and the PDS 100-mg/mL arm of the trial showed visual and anatomic outcomes comparable with monthly intravitreal ranibizumab 0.5mg injections.40
Bifunctional antibodies are now being developed such faricimab which targets both Angiopoietin-2 and VEGF-A. It is being trialled for both diabetic macula oedema and AMD. In wet AMD IBI302, a novel bispecific decoy receptor fusion protein designed with both a VEGF inhibition and complement inhibition domain has been trialled. A dose escalation study of intravitreal IBI302 in patients with wet AMD is underway.41 This dual targeting of 2 different pathways may mean patients will need a reduced number of intravitreal injections and possibly better VA outcomes. Furthermore, it can potentially help in the treatment of patients who are refractory to anti-VEGF treatment.
Brolucizumab is a humanized single-chain antibody fragment inhibitor of VEGF-A. Two recent Phase III trials HAWK and HARRIER compared brolucizumab with aflibercept.42 These trials showed brolucizumab was noninferior to aflibercept in visual function at week 48, and >50% of brolucizumab 6mg-treated eyes were maintained on 12 weekly dosing intervals through week 48. This reduced the injection burden for the patients. Anatomic outcomes also favoured brolucizumab over aflibercept and overall safety was similar to aflibercept in the study. Though recently there have been post marketing reports of some cases of retinal vasculitis and retinal artery occlusion using this drug.43
Finally, intravitreal abicipar pegol a novel DARPin blocks all isoforms of VEGF-A. In a Phase III study abicipar 8 and 12 weekly were both noninferior to ranibizumab 4 weekly in terms of vision stability. However, intraocular inflammation was more frequent with abicipar.44 It has not been granted a license by the Food and Drug Administration (FDA) due to the levels of intraocular inflammation reported. Subsequently, Allergan has withdrawn application filings with both the European Medicines Agency (EMA) and the Japanese Regulatory Agency (PMDA) for abicipar pegol.
Gene Therapy for Macular Degeneration
The aim of gene therapy is to replace a protein that is either non-functional or deficient in a particular tissue. The replacement of this protein should halt or reverse the disease process. The eye is an excellent organ in which to investigate gene therapy for several different reasons. As a relatively small organ, a small amount of clinical grade genetic vector is needed which helps with clinical manufacture. The eye has relative immune privilege which limits immunological response to implanted genetic material. It has a built-in control, namely the contra-lateral eye. As a transparent organ, gene therapy can be delivered precisely to the target area. Furthermore, changes induced by gene therapy can be viewed through direct clinical examination and imaging such as OCT.
Viral Vectors
Viral vectors are the transport mechanisms for the delivery of genetic material. Examples of vectors used in gene delivery are adenoviral vectors, helper-dependent adenoviral vectors, gamma retrovirus vectors and lentiviral vectors. The choice of viral vector depends on several different factors including safety, cloning capacity and tissue tropism of the vector. The most commonly used viral vectors in ocular gene therapy are adeno-associated viral vectors. This is because they have a good safety profile and do not cause significant intra-ocular inflammation.
There are several ways of delivering ocular gene therapy. One way is to perform a pars plana vitrectomy, retinotomy and the delivery of the genetic material in a viral vector into the subretinal space. Alternatively, the vector is injected directly into the vitreous cavity. A newer method of delivery now being considered is via the choroid. The surgical approach will be determined by the cells which the physician is trying to transduce. Currently, sub-retinal gene therapy is needed to deliver a genetic vector to the RPE or photoreceptors. Although it is hoped modification of the capsid coat of viral vectors may allow in the future sufficient tropism to allow intravitreal injections to permit transduction of RPE and photoreceptors.
The potential of gene therapy in treating retinal disease has been demonstrated in a Phase III clinical trial in Lebers Congenital Amaurosis.45 In this open-label, randomised, controlled trial, patients received bilateral, subretinal injection of 1·5 × 1011 vector genomes of voretigene neparvovec. The results showed gene replacement improved functional vision in RPE65-mediated inherited retinal dystrophy previously medically untreatable. Voretigene neparvovec has now been approved for clinical use in both the USA and the European Union.
In this review, we will focus on the progress of gene therapy in AMD. Significant issues remain with gene therapy such as achieving a sufficient dosage of the required protein in the retina/vitreous and the spread of the proteins produced from the sub-retinal blebs that are formed. Common adverse events include intraocular inflammation, ie, retinitis, uveitis, vasculitis and choroiditis, infection (endophthalmitis), floaters, retinal problems including retinal detachment and failure of delivering the genetic material to the right place.
Dry AMD Gene Therapy
In contrast to wet AMD, there is currently no treatment for dry AMD. However, activation of the complement pathway is strongly associated with AMD formation and progression. Therefore, inhibition of the complement pathway is being assessed as a potential treatment for dry AMD.
A Phase I, dose-escalating, safety and tolerability study of a single intravitreal injection of AAVCAGsCD59 in patients with advanced AMD with GA is being conducted.46 This soluble recombinant version of naturally occurring CD59 is designed and intended to protect retinal cells that are responsible for central vision by inhibiting the formation of the membrane attack complex, the terminal step of complement-mediated cell lysis. The role of the viral vector is to induce normal retinal cells to increase the expression of a soluble form of CD59. The primary end point is evaluation of safety, and one of the secondary end points is change in area of GA with treatment. The study is now complete, and the full results are awaited.
A multi-centre Phase II trial evaluating intravitreal AAVCAGsCD59 compared to sham injection for the treatment of advanced dry AMD with GA is due to start in June 2020.47 This is the next phase of the above study. It aims to recruit 132 to patients with advanced dry AMD and evaluate the outcome 2 years following intervention.
Finally, given the importance of complement factor I in the regulation of the complement pathway and its significance in AMD progression, the FOCUS study group have started a Phase I/II multicentre study evaluating the safety, dose response and efficacy of GT005.48 CFI will be delivered via a recombinant non-replicating adeno-associated viral vector. The results are awaited.
Wet AMD Gene Therapy
The gene therapy target for wet AMD is primarily VEGF; however, other anti-angiogenic targets include pigment epithelium-derived factor (PEDF), endostatin and angiostatin. VEGF is responsible for driving neovascularisation via proliferation of vascular endothelial cells, increased vessel permeability, migration and survival of endothelial cells.49 The formation of leaky choroidal vessels that penetrate the subretinal space and ultimately form a scar is the main reason why patients lose vision in wet AMD. Intraocular delivery of VEGF antagonists via gene therapy could potentially reduce the need for regular intravitreal injection of anti-VEGF antibodies.
A Phase I clinical trial which progressed to Phase IIa trial investigated the effects of rAAV.sFLT-1 in the treatment of wet AMD with a single subretinal injection.50,51 sFLT-1 is a naturally occurring VEGF inhibitor that is a soluble variant of the full-length membrane-bound VEGFR-1 protein.52 In the Phase I trial patients were randomized into 3 groups low dose, high dose and control. The study found that rAAV.sFLT-1 was safe and well tolerated. Therefore, gene therapy with rAAV.sFLT-1 progressed to a Phase IIa trial. In this study, ocular side-effects were mainly procedure related and self-resolved and no systemic safety issues were observed. In the rAAV.sFLT-1 treated group BCVA improved by a median of 1.0 letters from baseline compared to a median of −5.0 in the control group. Furthermore, the treated group needed fewer ranibizumab intravitreal injections than the control group.
Another Phase I dose-escalating study recruited patients aged 50 years or older with wet AMD and a baseline BCVA score of 20/100 or less. Here the patients were enrolled into 4 dose-ranging cohorts and followed for 52 weeks.53 Although 2 patients in cohort 4 experienced adverse events that were possibly study-drug related (pyrexia and intraocular inflammation) overall the authors concluded injection of AAV2-sFLT01 was safe and well tolerated at all doses.
Further studies include one evaluating aflibercept delivered via gene therapy, ADVM-022 (AAV.7m8-aflibercept)54 and a Phase I/IIa dose-escalation study evaluating the safety and tolerability of gene therapy with RGX-314 (an anti-VEGF) in patients with wet AMD.55
PEDF is a glycoprotein belonging to the serine proteinase inhibitor (SERPIN) superfamily and is a potent antiangiogenic factor.56 In a Phase I trial, 28 patients with advanced wet AMD were given a single intravitreal injection of an adenoviral vector expressing human PEDF (AdPEDF.11).57 In this study, there were no serious adverse events. The results of this study suggest the possibility of antiangiogenic activity that may last for several months after a single intravitreal injection, highlighting the potential of this treatment for wet AMD.
Similarly, endostatin a known inhibitor of tumour angiogenesis has also been used in human studies. A group tested the safety and expression profile of a lentiviral Equine Infectious Anemia Virus (EIAV) vector expressing endostatin and angiostatin (RetinoStat).58 Patients with advanced wet AMD were recruited to this study. Each of the doses was well tolerated. Long-term follow-up demonstrated expression was maintained at last measurement (2.5 years in eight subjects and >4 years in two subjects).
Finally, another study looked at patients with treatment naive wet AMD.59 Patients in this study received an intravitreal anti-VEGF injection at day 0 followed by an intravitreal injection of AAVCAGsCD59 at Day 7 and then patients were monitored monthly for 12 months. They assessed the efficacy and safety of two doses of the adeno-associated viral vector serotype 2 expressing sCD59. The full results are still not available.
Cell Therapy for Macular Degeneration
Stem cells are cells capable of both self-renewal and of differentiating into different cell types. A variety of stem cells are used in cell-based therapies including embryonic stem cells (ESCs), adult stem cells, eg, mesenchymal, and induced pluripotent stem cells (iPSCs).
There are two main mechanisms by which stem cells can be used as therapy in disease. Firstly, either as encapsulated cell therapies whereby a group of cells are formed in order to produce a protein which is lacking from the diseased tissue or secondly via the production of cells to replace diseased and damaged tissues such as the RPE in AMD. When replacing diseased tissue in the neural retina one of the main challenges is to be able to re-establish neural connections in order for cells to function as healthy tissue. Replacement of the RPE in contrast does not suffer this challenge as no synapsis with other cells in the retina is required. However, the timing of RPE transplantation is key. If RPE is replaced too late in the disease course the cells it normally nourishes and protects such as the photoreceptors will have perished making the therapy redundant. However, intervening at an early stage carries the risk of surgery and therapy-related complications when patients still retain relatively good vision. Additionally, it is also important to note that if the retinal disease causing RPE generation is not halted, the transplanted RPE cells will eventually degenerate. Another layer that can be targeted in cell-based therapy is the choroidal endothelial layer. Endothelial progenitor cells can be used to help re-establish vascularisation.60 This may be relevant to AMD as choroidal degeneration is thought to be an aetiological factor in the development of AMD.
Some of the feared complications associated with stem cell therapy include tumour formation, immune rejection and risk of differentiation into unwanted cell types. A recent study of intravitreal injection of mesenchymal stem cells was associated with significant complications including severe intravitreal fibrosis and tractional retinal detachment in RP patients.61 Thus, highlighting the devasting consequences of unscrupulous use of intravitreal stem cells and the need for rigorous evaluation in properly conducted clinical trials. Due to studies such as this the clinicaltrials.gov website recently added a disclaimer to its website stating that “Listing a study does not mean it has been evaluated by the US Federal Government.”
Dry AMD Cell-Based Therapies
The safety and tolerability of subretinal transplantation of human ESC-derived RPE was assessed in 9 patients with Stargardt dystrophy and 9 with dry AMD.62 Three dose cohorts of 50,000, 100,000, and 150,000 cells were injected. There was no evidence of serious ocular or systemic safety issues related to the transplanted tissue. This study was a landmark study as it provided the first evidence of medium to long-term safety and graft survival in patients with any disease. In patients with advanced dry AMD vision-related quality of life measures improved after treatment.
In another study, patients with GA were injected with subretinal human umbilical tissue-derived cells (palucorcel [CNTO-2476]).63 Although the treatment was well tolerated, the method of delivery of the cells was associated with a high rate of retinal perforations and detachments. It was noted that when palucorcel was sequestered in the subretinal space it was associated with some gain in VA.
Another type of cell therapy is in the form of encapsulated cell technology (ECT). Ciliary neurotrophic factor (CNTF) was delivered by ECT implant in patients with retinitis pigmentosa and GA.64 The implant is a small capsule that contains human RPE cells. These cells have been engineered to make CNTF and subsequently release it through the capsule membrane into the surroundings. The implants produced CNTF consistently over a 2-year period and the implant was well tolerated.
Most recently, bone marrow derived stem cells (BMSCs) have been used in trials for dry AMD with positive outcomes. The Stem Cell Ophthalmology Treatment Study (SCOTS) was conducted to assess the effect of bone marrow-derived stem cells (BMSCs) in dry AMD.65 The treatment of dry AMD with BMSC using the protocols developed in the SCOTS clinical trial showed a statistically significant improvement in VA. The mean improvement in LogMAR vision was 0.963 post intervention with a standard deviation (SD) of 0.42. This has now opened up a new avenue of possible treatment for dry AMD. In an earlier study, the therapeutic potential and safety of intravitreal injections of bone marrow mononuclear fraction (BMMF) containing CD34+ stem cells in patients with dry AMD was evaluated.66 A volume of 0.1 mL suspension of BMMF CD34+ cells was injected into the vitreous cavity of patients with GA. The intravitreal BMMF injections were safe and associated with significant improvement in BCVA and macular sensitivity threshold. Mean BCVA improved from 1.18 logMAR prior to injection to 1.0 logMAR at 12 months.
Wet AMD Cell-Based Therapy
In a Phase I clinical study, an RPE patch comprising a fully differentiated, human ESC-derived-RPE monolayer was grown on a coated, synthetic basement membrane and implanted subretinally in 2 patients with severe wet AMD.67 The researchers concluded that successful delivery was possible and survival of the RPE patch was demonstrated by biomicroscopy and on OCT. There was a visual acuity gain of 29 and 21 letters in the two patients, respectively, over a 12-month period. This study, however, lacked a control group.
In a world first a Japanese woman in her 70s was the first person to receive tissue derived from induced pluripotent stem cells.68 The iPSC-RPE were derived from her own skin cells. This was a significant milestone in cell-based therapy treatment for AMD. However, the trial was halted due to unexpected genetic changes in the iPSCs. The cost of the entire process means it is not feasible to carry this process out on a large number of patients. Current thinking is that HLA-matched RPE cell banks could be used to bring costs down. Furthermore, it took a significant period of time to generate the RPE cells before implantation making it difficult to implement this as an established treatment for AMD. In 2017, the procedure was performed in a Japanese man in his 60s with wet AMD using IPSC cells donated from another person.69 The surgery included the removal of the neovascular membrane and transplantation of the autologous iPSC derived RPE cell sheet under the retina. At 1 year after surgery, the transplanted sheet remained intact, BCVA unchanged, and cystoid macular oedema was present.
In the future, multipotent stem cells within the human retina called RPE stem cells can be explored as a therapeutic target.70 If these cells could be stimulated to proliferate, differentiate and restore function in patients with AMD or other retinal degenerative conditions prior to the loss of photoreceptors they could potentially be sight saving.
Conclusion
In this review, we have discussed 3 different treatment options that can be potentially used in the treatment of AMD – cell, gene and antibody-based therapies. Currently, the most successful and clinically used treatment is antibody therapy with anti-VEGFs such as ranibizumab, bevacizumab and aflibercept in the treatment of wet AMD. Unfortunately, there are currently no established treatments for dry AMD. However, ongoing research on antibody, gene and cell therapies may lead to treatment for dry AMD and also improved therapies for patients with wet AMD. Current research suggests these approaches are promising and should be explored further. The results of current clinical trials are eagerly awaited.
Disclosure
Professor Andrew Lotery reports an equity interest in Gyroscope Therapeutics and personal fees from Novartis and Allergan outside the submitted work. The authors report no other conflicts of interest in this work. f,
References
1. Friedman DS, O’Colmain BJ, Munoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122(4):564–572.
2. Owen CG, Jarrar Z, Wormald R, Cook DG, Fletcher AE, Rudnicka AR. The estimated prevalence and incidence of late stage age related macular degeneration in the UK. Br J Ophthalmol. 2012;96(5):752–756. doi:10.1136/bjophthalmol-2011-301109
3. Wong WL, Su X, Li X, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106–e116. doi:10.1016/S2214-109X(13)70145-1
4. Ferris FL
5. Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246(4935):1306–1309. doi:10.1126/science.2479986
6. Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1432–1444. doi:10.1056/NEJMoa062655
7. Chakravarthy U, Harding SP, Rogers CA, et al. Alternative treatments to inhibit VEGF in age-related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet. 2013;382(9900):1258–1267. doi:10.1016/S0140-6736(13)61501-9
8. Martin DF, Maguire MG, Fine SL, et al.; Comparison of Age-related Macular Degeneration Treatments Trials Research G. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology. 2012;119(7):1388–1398. doi:10.1016/j.ophtha.2012.03.053.
9. Heier JS, Brown DM, Chong V, et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology. 2012;119(12):2537–2548. doi:10.1016/j.ophtha.2012.09.006
10. Regillo CD, Brown DM, Abraham P, et al. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER Study year 1. Am J Ophthalmol. 2008;145(2):239–248. doi:10.1016/j.ajo.2007.10.004
11. Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1419–1431. doi:10.1056/NEJMoa054481
12. Tufail A, Patel PJ, Egan C, et al. Bevacizumab for neovascular age related macular degeneration (ABC Trial): multicentre randomised double masked study. BMJ. 2010;340:c2459. doi:10.1136/bmj.c2459
13. Schmidt-Erfurth U, Chong V, Loewenstein A, et al. Guidelines for the management of neovascular age-related macular degeneration by the European Society of Retina Specialists (EURETINA). Br J Ophthalmol. 2014;98(9):1144–1167. doi:10.1136/bjophthalmol-2014-305702
14. Sarks SH. Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol. 1976;60(5):324–341. doi:10.1136/bjo.60.5.324
15. Hogan MJ. Role of the retinal pigment epithelium in macular disease. Trans Am Acad Ophthalmol Otolaryngol. 1972;76(1):64–80.
16. Owsley C, McGwin G
17. Young RW. The Bowman Lecture, 1982. Biological renewal. Applications to the eye. Trans Ophthalmol Soc U K. 1982;102(Pt 1):42–75.
18. Sivaprasad S, Bird A, Nitiahpapand R, et al. Perspectives on reticular pseudodrusen in age-related macular degeneration. Surv Ophthalmol. 2016;61(5):521–537. doi:10.1016/j.survophthal.2016.02.005
19. Khanifar AA, Koreishi AF, Izatt JA, Toth CA. Drusen ultrastructure imaging with spectral domain optical coherence tomography in age-related macular degeneration. Ophthalmology. 2008;115(11):1883–1890. doi:10.1016/j.ophtha.2008.04.041
20. Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73(6):887–896. doi:10.1006/exer.2001.1094
21. Thornton J, Edwards R, Mitchell P, Harrison RA, Buchan I, Kelly SP. Smoking and age-related macular degeneration: a review of association. Eye (Lond). 2005;19(9):935–944. doi:10.1038/sj.eye.6701978
22. Esparza-Gordillo J, Soria JM, Buil A, et al. Genetic and environmental factors influencing the human factor H plasma levels. Immunogenetics. 2004;56(2):77–82. doi:10.1007/s00251-004-0660-7
23. Scholl HP, Charbel Issa P, Walier M, et al. Systemic complement activation in age-related macular degeneration. PLoS One. 2008;3(7):e2593. doi:10.1371/journal.pone.0002593
24. Edwards AO, Ritter R
25. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308(5720):419–421. doi:10.1126/science.1110359
26. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–389. doi:10.1126/science.1109557
27. Cipriani V, Lores-Motta L, He F, et al. Increased circulating levels of factor H-related protein 4 are strongly associated with age-related macular degeneration. Nat Commun. 2020;11(1):778. doi:10.1038/s41467-020-14499-3
28. Fritsche LG, Loenhardt T, Janssen A, et al. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet. 2008;40(7):892–896. doi:10.1038/ng.170
29. Fritsche LG, Igl W, Bailey JN, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016;48(2):134–143. doi:10.1038/ng.3448
30. Holz FG, Sadda SR, Busbee B, et al. Efficacy and safety of lampalizumab for geographic atrophy due to age-related macular degeneration: chroma and spectri Phase 3 randomized clinical trials. JAMA Ophthalmol. 2018;136(6):666–677. doi:10.1001/jamaophthalmol.2018.1544
31. Yehoshua Z, de Amorim Garcia Filho CA, Nunes RP, et al. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology. 2014;121(3):693–701. doi:10.1016/j.ophtha.2013.09.044
32. Pharmaceuticals N. Intravitreal LFG316 in patients with age-related macular degeneration (AMD). ClinicalTrialsgov. NCT01527500. 2019
33. Corporation O. Zimura in subjects with geographic atrophy secondary to dry age-related macular degeneration. ClinicalTrialsgov. NCT02686658; 2020
34. Rosenfeld PJ, Berger B, Reichel E, et al. A randomized Phase 2 study of an anti-amyloid β monoclonal antibody in geographic atrophy secondary to age-related macular degeneration. Ophthalmol Retina. 2018;2(10):1028–1040. doi:10.1016/j.oret.2018.03.001
35. Pharmaceuticals A. Study of of APL-2 therapy in patients geographic atrophy (FILLY). ClinicalTrialsgov. NCT02503332; 2019.
36. Research A. CLG561 proof-of-concept study as a monotherapy and in combination with LFG316 in subjects with geographic atrophy (GA). ClinicalTrialsgov. NCT02515942; 2019.
37. Lalwani GA, Rosenfeld PJ, Fung AE, et al. A variable-dosing regimen with intravitreal ranibizumab for neovascular age-related macular degeneration: year 2 of the PrONTO Study. Am J Ophthalmol. 2009;148(1):43–58. doi:10.1016/j.ajo.2009.01.024
38. Singer MA, Awh CC, Sadda S, et al. HORIZON: an open-label extension trial of ranibizumab for choroidal neovascularization secondary to age-related macular degeneration. Ophthalmology. 2012;119(6):1175–1183. doi:10.1016/j.ophtha.2011.12.016
39. Hykin P, Chakravarthy U, Lotery A, McKibbin M, Napier J, Sivaprasad S. A retrospective study of the real-life utilization and effectiveness of ranibizumab therapy for neovascular age-related macular degeneration in the UK. Clin Ophthalmol. 2016;10:87–96. doi:10.2147/OPTH.S92627
40. Campochiaro PA, Marcus DM, Awh CC, et al. The port delivery system with ranibizumab for neovascular age-related macular degeneration: results from the randomized Phase 2 ladder clinical trial. Ophthalmology. 2019;126(8):1141–1154. doi:10.1016/j.ophtha.2019.03.036
41. Ltd. IBSC. A dose escalation study of IBI302 in patients with wet age-related macular degeneration. ClinicalTrialsgov. NCT03814291; 2020.
42. Dugel PU, Koh A, Ogura Y, et al. HAWK and HARRIER: Phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology. 2020;127(1):72–84. doi:10.1016/j.ophtha.2019.04.017
43. NOVARTIS. Safety of Beovu® (brolucizumab); 2020, June 11. Available from: https://www.brolucizumab.info/.
44. Kunimoto D, Yoon YH, Wykoff CC, et al. Efficacy and safety of abicipar in neovascular age-related macular degeneration: 52-week results of phase 3 randomized controlled study. Ophthalmology. 2020. doi:10.1016/j.ophtha.2020.03.035
45. Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849–860. doi:10.1016/S0140-6736(17)31868-8
46. Heier J Treatment of advanced dry age related macular degeneration with AAVCAGsCD59. ClinicalTrialsgov. NCT03144999; 2019.
47. Biosciences H. Intravitreal AAVCAGsCD59 for advanced dry age-related macular degeneration (AMD) with geographic atrophy (GA). ClinicalTrialsgov. NCT04358471; 2020.
48. Therapeutics G. First in human study to evaluate the safety and efficacy of GT005 administered in subjects with dry AMD. ClinicalTrialsgov. NCT03846193; 2019.
49. Ferrara N, Gerber HP. The role of vascular endothelial growth factor in angiogenesis. Acta Haematol. 2001;106(4):148–156. doi:10.1159/000046610
50. Constable IJ, Pierce CM, Lai CM, et al. Phase 2a randomized clinical trial: safety and post hoc analysis of subretinal rAAV.sFLT-1 for wet age-related macular degeneration. EBioMedicine. 2016;14:168–175. doi:10.1016/j.ebiom.2016.11.016
51. Rakoczy EP, Lai CM, Magno AL, et al. Gene therapy with recombinant adeno-associated vectors for neovascular age-related macular degeneration: 1 year follow-up of a phase 1 randomised clinical trial. Lancet. 2015;386(10011):2395–2403. doi:10.1016/S0140-6736(15)00345-1
52. Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci U S A. 1993;90(22):10705–10709. doi:10.1073/pnas.90.22.10705
53. Heier JS, Kherani S, Desai S, et al. Intravitreous injection of AAV2-sFLT01 in patients with advanced neovascular age-related macular degeneration: a phase 1, open-label trial. Lancet. 2017;390(10089):50–61. doi:10.1016/S0140-6736(17)30979-0
54. Adverum Biotechnologies I. ADVM-022 intravitreal gene therapy for wet AMD (OPTIC). ClinicalTrialsgov. NCT03748784; 2018.
55. Inc. R. A Phase I/IIa, open-label, multiple-cohort, dose-escalation study to evaluate the safety and tolerability of gene therapy with RGX-314 in subjects with neovascular AMD (nAMD). ClinicalTrialsgov. NCT03066258; 2017
56. Stellmach V, Crawford SE, Zhou W, Bouck N. Prevention of ischemia-induced retinopathy by the natural ocular antiangiogenic agent pigment epithelium-derived factor. Proc Natl Acad Sci U S A. 2001;98(5):2593–2597. doi:10.1073/pnas.031252398
57. Fischer PG. Study of AdGVPEDF.11D in neovascular age-related macular degeneration (AMD). ClinicalTrialsgov. NCT00109499; 2005.
58. Campochiaro PA, Lauer AK, Sohn EH, et al. Lentiviral vector gene transfer of endostatin/angiostatin for macular degeneration (GEM) study. Hum Gene Ther. 2017;28(1):99–111. doi:10.1089/hum.2016.117
59. Biosciences H. AAVCAGsCD59 for the Treatment of Wet AMD. ClinicalTrialsgov. NCT03585556; 2020.
60. Lois N, McCarter RV, O’Neill C, Medina RJ, Stitt AW. Endothelial progenitor cells in diabetic retinopathy. Front Endocrinol (Lausanne). 2014;5:44. doi:10.3389/fendo.2014.00044
61. Satarian L, Nourinia R, Safi S, et al. Intravitreal injection of bone marrow mesenchymal stem cells in patients with advanced retinitis pigmentosa; a safety study. J Ophthalmic Vis Res. 2017;12(1):58–64. doi:10.4103/2008-322X.200164
62. Schwartz SD, Regillo CD, Lam BL, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet. 2015;385(9967):509–516. doi:10.1016/S0140-6736(14)61376-3
63. Ho AC, Chang TS, Samuel M, Williamson P, Willenbucher RF, Malone T. Experience with a subretinal cell-based therapy in patients with geographic atrophy secondary to age-related macular degeneration. Am J Ophthalmol. 2017;179:67–80. doi:10.1016/j.ajo.2017.04.006
64. Kauper K, McGovern C, Sherman S, et al. Two-year intraocular delivery of ciliary neurotrophic factor by encapsulated cell technology implants in patients with chronic retinal degenerative diseases. Invest Ophthalmol Vis Sci. 2012;53(12):7484–7491. doi:10.1167/iovs.12-9970
65. Weiss JN, Levy S. Stem cell ophthalmology treatment study (SCOTS): bone marrow-derived stem cells in the treatment of age-related macular degeneration. Medicines (Basel). 2020;7(4).
66. Cotrim CC, Toscano L, Messias A, Jorge R, Siqueira RC. Intravitreal use of bone marrow mononuclear fraction containing CD34(+) stem cells in patients with atrophic age-related macular degeneration. Clin Ophthalmol. 2017;11:931–938. doi:10.2147/OPTH.S133502
67. da Cruz L, Fynes K, Georgiadis O, et al. Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nat Biotechnol. 2018;36(4):328–337. doi:10.1038/nbt.4114
68. Cyranoski D. Japanese woman is first recipient of next-generation stem cells. Nature. 2014. doi:10.1038/nature.2014.15915
69. Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038–1046. doi:10.1056/NEJMoa1608368
70. Salero E, Blenkinsop TA, Corneo B, et al. Adult human RPE can be activated into a multipotent stem cell that produces mesenchymal derivatives. Cell Stem Cell. 2012;10(1):88–95. doi:10.1016/j.stem.2011.11.018
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.