")
Back to Journals » OncoTargets and Therapy » Volume 7
Gemcitabine resistance in breast cancer cells regulated by PI3K/AKT-mediated cellular proliferation exerts negative feedback via the MEK/MAPK and mTOR pathways
Authors Yang XL, Lin FJ, Guo YJ, Shao Z, Ou ZL
Received 26 February 2014
Accepted for publication 9 April 2014
Published 13 June 2014 Volume 2014:7 Pages 1033—1042
DOI https://doi.org/10.2147/OTT.S63145
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Xiao Li Yang, Feng Juan Lin, Ya Jie Guo, Zhi Min Shao, Zhou Luo Ou
Key Laboratory of Breast Cancer in Shanghai, Breast Cancer Institute, Cancer Hospital, Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, People's Republic of China
Abstract: Chemoresistance is a major cause of cancer treatment failure and leads to a reduction in the survival rate of cancer patients. Phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) and mitogen-activated protein kinase (MAPK) pathways are aberrantly activated in many malignant tumors, including breast cancer, which may indicate an association with breast cancer chemoresistance. In this study, we generated a chemoresistant human breast cancer cell line, MDA-MB-231/gemcitabine (simplified hereafter as “231/Gem”), from MDA-MB-231 human breast cancer cells. Flow cytometry studies revealed that with the same treatment concentration of gemcitabine, 231/Gem cells displayed more robust resistance to gemcitabine, which was reflected by fewer apoptotic cells and enhanced percentage of S-phase cells. Through the use of inverted microscopy, Cell Counting Kit-8, and Transwell assays, we found that compared with parental 231 cells, 231/Gem cells displayed more morphologic projections, enhanced cell proliferative ability, and improved cell migration and invasion. Mechanistic studies revealed that the PI3K/AKT/mTOR and mitogen-activated protein kinase kinase (MEK)/MAPK signaling pathways were activated through elevated expression of phosphorylated (p)-extracellular signal-regulated kinase (ERK), p-AKT, mTOR, p-mTOR, p-P70S6K, and reduced expression of p-P38 and LC3-II (the marker of autophagy) in 231/Gem in comparison to control cells. However, there was no change in the expression of Cyclin D1 and p-adenosine monophosphate-activated protein kinase (AMPK). In culture, inhibitors of PI3K/AKT and mTOR, but not of MEK/MAPK, could reverse the enhanced proliferative ability of 231/Gem cells. Western blot analysis showed that treatment with a PI3K/AKT inhibitor decreased the expression levels of p-AKT, p-MEK, p-mTOR, and p-P70S6K; however, treatments with either MEK/MAPK or mTOR inhibitor significantly increased p-AKT expression. Thus, our data suggest that gemcitabine resistance in breast cancer cells is mainly mediated by activation of the PI3K/AKT signaling pathway. This occurs through elevated expression of p-AKT protein to promote cell proliferation and is negatively regulated by the MEK/MAPK and mTOR pathways.
Keywords: chemoresistance, gemcitabine, breast cancer
Introduction
Despite advances in the detection and treatment of breast cancer, one in eight women in the United States will develop breast cancer in her lifetime, and this disease is the sixth leading cause of cancer death in females in the People’s Republic of China.1,2 Although there has been development of new targeted drugs, more effective and safe therapeutics for breast cancer are still needed.
Gemcitabine (2′,2′-difluorodeoxycytidine) is a difluorinated analog of deoxycytidine. It is an S-phase–specific drug that requires intracellular transport and activation to exert its cytotoxic effects.3 It is used in combination with other drugs for the treatment of locally advanced or metastatic non-small-cell lung cancer, bladder cancer, and ovarian cancer, and as a single agent for the treatment of adenocarcinoma of the pancreas.4 Clinical studies have also demonstrated its extensive activity against breast carcinomas, including male breast cancer,5 metastatic breast cancer, and triple-negative breast cancer.6,7 In recent years, gemcitabine resistance has emerged and has become a serious concern in clinical practice; however, the exact mechanism of gemcitabine resistance in breast cancer is still unknown.
The phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway is often constitutively activated in drug-resistant breast cancer, and this activation is correlated with increased cell survival and poor prognosis for breast cancer patients. mTOR is a serine/threonine kinase of the PI3K-related kinase family.8 Accumulating data from genetic and cancer biology studies have indicated that the mTOR pathway has a prominent role in both normal physiological development and carcinogenic processes, including cell growth (cell size or mass),9 proliferation (cell number), apoptosis,10 autophagy,11 response to stress such as nutrient starvation (glucose or amino acids), and survival.8,12,13 The downstream effects of mTOR activation include phosphorylation of p70S6 kinase and 4E-binding protein, both of which are critical for protein-synthesis regulation.8,14 Given recent findings that mTOR activation can be controlled by the interplay between AKT kinase and AMPK, there is now extensive evidence validating various components of this pathway as potential molecular targets for cancer treatment. Autophagy can induce both cell survival and cell death under nutrient starvation, tumorigenesis including breast cancer,15 neurodegeneration, and other physiological and pathological processes.16,17 The conversion of LC3-I to LC3-II reflects the occurrence of autophagy.18 Two relatively well-established pathways that regulate autophagy contain the mTOR and IP3 cascades. Inhibition of mTOR activity or a decrease of IP3 level can each induce autophagy.19
The mitogen-activated protein kinase (MAPK) pathway is highly conserved across eukaryotes and transmits signals from the cell surface receptors to nuclear transcription. Three major MAPK pathways are known to function in humans. Activation of Raf–mitogen-activated protein kinase kinase (MEK)–extracellular signal-regulated kinase (ERK) is critical for both normal and cancer cellular development.20 Antiproliferative and proapoptotic effects in breast cancer are mediated through p38 phosphorylation21 and it is also involved in apoptosis of prostate cancer.22 MAPK pathways also take part in many drug resistance patterns in cancer cells.23,24 There is a large level of cross-talk and a number of feedback loops between kinases of these two pathways, and inhibition of one cascade often activates the other. The lack of efficacy of some chemotherapy drugs in cancer may be due to loss of feedback inhibition. In this study, we generated gemcitabine-resistant breast cancer cells to explore the roles of PI3K/AKT/mTOR and MAPK in drug resistance and to search for more-potent molecular targets to overcome breast cancer cell resistance to gemcitabine.
Materials and methods
Cell lines and culture
The human breast cancer cell line MDA-MB-231 (hereafter, “231”) was obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in the recommended medium (Dulbecco’s Modified Eagle’s Medium [DMEM]) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. The cultures were incubated at 37°C in a humidified 5% CO2 atmosphere. Cell culture medium and fetal bovine serum were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Establishment of gemcitabine-resistant breast cancer cell line
Gemcitabine-resistant cells were generated by exposure to gradually increasing concentrations of gemcitabine for 12 months, as described previously.25 Parental 231 cells were exposed to gemcitabine at an initial concentration of 12 nM, and then exposed repetitively to stepwise increased gemcitabine concentration. The final concentrations were 60 μM. Through this process, we successfully established the gemcitabine-resistant breast cancer cell line MDA-MB-231/gemcitabine (hereafter, “231/Gem”).
Drugs and reagents
Gemcitabine was purchased from Lilly France (St-Cloud, France) (Lot No A875303A). Antibodies against β-catenin (51067-2-AP, 1:800), LC3A/B (66139-1-1g, 1:1,000), Cyclin D1 (60186-1-1g, 1:1,000) and GAPDH (60004-1-Ig, 1:1,000) were from Proteintech Group, Inc., (Chicago, IL, USA). Antibodies against MEK1/2 (9126, 1:1,000), Phospho-MEK1/2 (2338, 1:1,000), Phospho-ERK1/2 (4376, 1:1,000), AKT (4691, 1:1,000), Phospho-AKT (4060P, 1:1,000), Phospho-P38 (4511, 1:1,000), Phospho-JNK (4668, 1:1,000), E-Cadherin (3195, 1:1,000), Phospho-p53 (9284, 1:1,000), and mTOR Substrates Sampler kit (CST 9862), which includes Phospho-mTOR, mTOR, Phospho-p70s6389 (Thr389), Phospho-P70389 (Thr371), Phospho-4EBP1 (Thr37/46), and anti-rabbit IgG, were purchased from Cell Signaling Technology, Inc., (Danvers, MA, USA). Antibodies against p38 (sc-7149, 1:1,000), P53 (sc-126, 1:1,000) and JNK (sc-571, 1:1,000) were from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). PD98059 (MEK inhibitor, 9900, 20 UM), rapamycin (mTOR inhibitor, 9904, 10 nM), and LY294002 (PI3K Inhibitor, 9901, 50 μM) were purchased from Cell Signaling Technology.
Growth inhibition assay
The growth inhibition assay was performed using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumanoto, Japan). In brief, cells were incubated for 5 days under several concentrations of gemcitabine in 96-well plates at a density of 2,000 per well (200 μL) and cultured in growth medium. The number of cells was counted according to CCK-8 protocol.
Cell apoptosis analysis
The effect of gemcitabine on cell viability was assessed using flow cytometry by staining with Annexin V/propidium iodide (PI) (V13241; Thermo Fisher Scientific). Briefly, cells were cultured for 3 days after receiving gemcitabine treatment and washed twice in ice-cold phosphate-buffered saline. A total of 1×105 cells were resuspended in 100 μL binding buffer to which 5 μL of 2 mg/mL Annexin V and 5 μL of 50 μg/mL PI was added. Following 15 minutes of incubation in the dark, flow cytometry was performed. All tests were performed three times.
Cell cycle analysis
We plated 231 and 231/Gem cells in 60 mm diameter dishes with medium containing 10% fetal bovine serum. The next day, cells were treated with 35 μM gemcitabine for 3 days. Floating and adherent cells were collected and fixed overnight in cold 70% ethanol at 4°C. After washing, the cells were subsequently treated with 100 μg/mL PI and 100 μg/mL RNase A for 30 minutes in the dark and then subjected to flow cytometric analysis to determine the percentage of cells in specific phases of the cell cycle. All the experiments were performed three times.
Proliferation assay
Cell proliferation was detected by using CCK-8. Cells were plated in 96-well plates at a density of 2,500 per well (100 μL) and cultured in growth medium. The number of the cells was counted according to the protocol of the kit from the company.
Western blot
Western blot analysis was performed according to the standard protocols. Briefly, aliquots of total protein (30 μg) were electrophoresed on sodium dodecyl sulfate polyacrylamide Tris-HCl gels. The separated proteins were transferred to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA) and incubated with primary antibodies for 2 hours. Chemiluminescent detection was performed, and images were captured by LAS-3000 system (Fuji Photo Film Co, Ltd, Tokyo, Japan).
Migration assay
Chemotaxis assays were performed using Transwell insert polycarbonate membranes (8 μm pore size, BD, Franklin Lakes, NJ, USA). First, 1×105 cells were seeded in the top compartment and 600 μL DMEM with 10% fetal bovine serum was added to the bottom at 37°C for 13 hours. Nonmigratory cells on the upper membrane surface were removed, and migratory cells attached to the bottom surface of the membrane were fixed with 10% formalin and stained with 0.1% crystal violet for 30 minutes at room temperature. Migrated cells in five random fields were counted using a light microscope at 200× magnification. All assays were performed in triplicate.
Invasion assay
Invasion experiments were carried out with a Matrigel invasion chamber (BD). Each well insert was layered with 75 μL of 1:4 mixture of Matrigel/DMEM. Serum-starved cells (1×105) were added to the upper compartment and 600 μL DMEM with 10% fetal bovine serum was added to the bottom at 37°C for 24 hours. Invasion was assessed by counting the cells that had traveled across the filter and were attached to the bottom side of the filter. Briefly, the filters were fixed in 10% formalin and stained with 1% crystal violet. Cells that had invaded through the Matrigel and reached the lower surface of the filter were counted under a light microscope at 200× magnification. Five fields were counted for each sample.
Statistical analysis
Statistical analysis was performed using Statistical Package for the Social Sciences (SPSS) software Version 16.0 for Windows (SPSS Inc., Chicago, IL, USA). Analysis of variance and Student’s t-test were used to determine the statistical significance of differences between experimental groups in vitro. Values of P<0.05 were considered statistically significant. Graphs were created with GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Establishment of gemcitabine-resistant breast cancer cell line
To investigate the chemosensitivity of 231/Gem cells as compared to parental cells, flow cytometry assay was performed. 231 and 231/Gem were cultured and harvested after 3 days following treatment of 35 μM gemcitabine. As shown in Figure 1A, the number of apoptotic cells induced by gemcitabine was greatly increased in 231 cells compared to 231/Gem cells, as indicated by the quantitative analysis in Figure 1B. Additionally, the half-maximal inhibitory concentration (IC50) of gemcitabine was higher for 231/Gem than for control cells (Figure 1C).
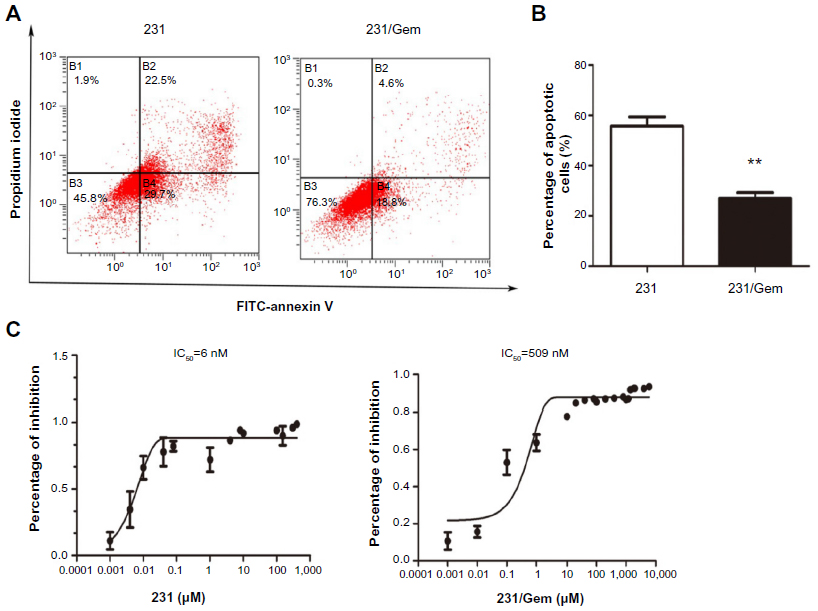 | Figure 1 Establishment of a gemcitabine-resistant breast cancer cell line. |
Gemcitabine resistance increases breast cancer cell proliferation and invasion
To investigate whether 231/Gem cells displayed increased proliferation and invasion, we used the proliferation and invasion assays and compared to control cells. As shown in Figure 2A, the CCK-8 result showed that 231/Gem cells displayed greater proliferation than 231 cells. Further studies indicated that in comparison to parental cells, the percentage of S-phase cells in 231/Gem was increased and G-phase cells were reduced after gemcitabine treatment for 3 days (Figure 2B), as indicated by the quantitative analysis in Figure 2C. Next, we tested the migration and invasiveness of 231 and 231/Gem cells. We first observed morphology alteration by inverted microscopy. As shown in Figure 2D, 231/Gem displayed a greater number of projections than parental cells. Transwell chambers were used to evaluate the invasiveness of 231 and 231/Gem cells, and the results showed that more 231/Gem cells than parental cells intruded into the bottom chamber (Figure 2E and F). We then tested the expression of cell adhesion-associated proteins by Western blot, showing a significant decrease of E-cadherin in 231/Gem cells (Figure 2G). These data suggest that 231/Gem cells gained enhanced proliferative and invasive abilities after developing resistance to gemcitabine.
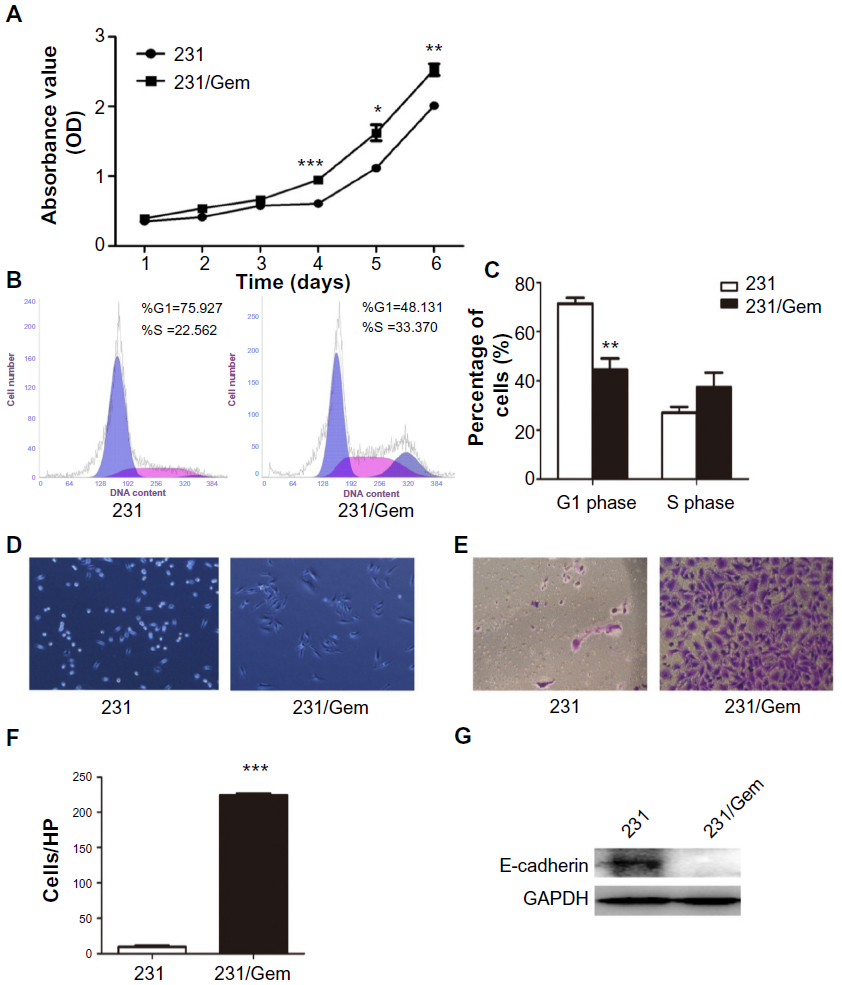 | Figure 2 231/Gem cells display increased proliferation and invasion. |
Involvement of the PI3K/AKT, MAPK, and mTOR pathways in gemcitabine resistance
Because previous studies have shown that the signaling pathways of PI3K/AKT, MAPK, and mTOR may promote cancer chemoresistance and radioresistance, we evaluated the expression of the major proteins of these three pathways by Western blot. As shown in Figure 3, there is no difference in basal level of total AKT expression in both cell lines, whereas phosphorylated (p)-AKT was increased in gemcitabine-resistant 231/Gem cells. These data show that activation of the PI3K/AKT signaling pathway is associated with gemcitabine-resistance in breast cancer cells.
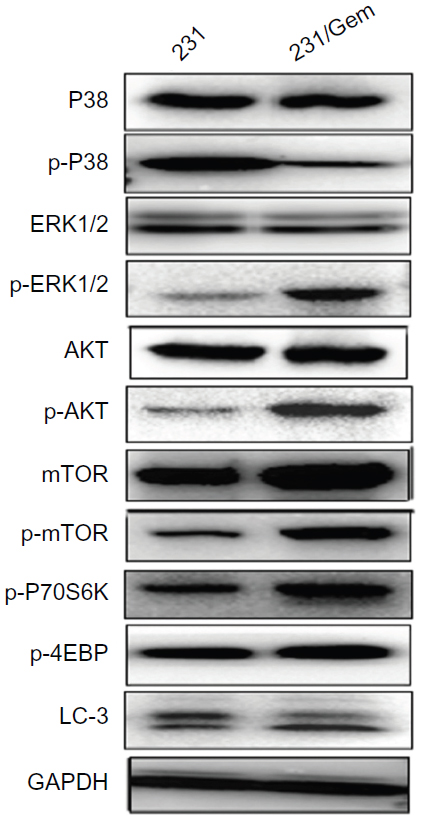 | Figure 3 Changes in activation of proteins in the PI3K/AKT/mTOR and MAPK pathways. |
We then tested the major protein expression of MAPK and mTOR pathways. Though the total ERK level was similar in both cell lines, the level of p-ERK was markedly increased in 231/Gem cells. However, the level of p-P38 was significantly decreased in 231/Gem cells, and no change of total p38 protein expression was found. As for the mTOR pathway, we found that either mTOR or p-mTOR were significantly increased in 231/Gem cells compared with 231 cells, although the downstream proteins of mTOR pathway, p-P70S6K and p-4EBP, were upregulated in 231/Gem cells. Many studies have shown that inhibition of mTOR activity could trigger autophagy. In our study, 231/Gem cells exhibited decreased expression of LC3 protein, a marker of autophagy, suggesting that autophagy may contribute to gemcitabine-resistance of breast cancer cells. Additionally, AMPK as a negative regulator of mTOR and Cyclin D1 were tested by Western blot, which revealed that their expressions were no different.
The PI3K/AKT pathway and mTOR, but not MAPK, regulates gemcitabine resistance-augmented cell proliferation
To further elucidate the mechanisms underlying the growth-promoting effects of acquired gemcitabine resistance, we used the CCK-8 assay to detect whether inhibitors of the above three pathways would reverse increased proliferative ability of 231/Gem. As shown in Figure 4, treatment with PD98059 (a MAPK inhibitor) did not affect cell proliferation. Cell proliferation was significantly inhibited in 231/Gem cells treated with LY294002 (a PI3K inhibitor), while rapamycin (an mTOR inhibitor) had a slight inhibitory effect on cell proliferation in 231/Gem.
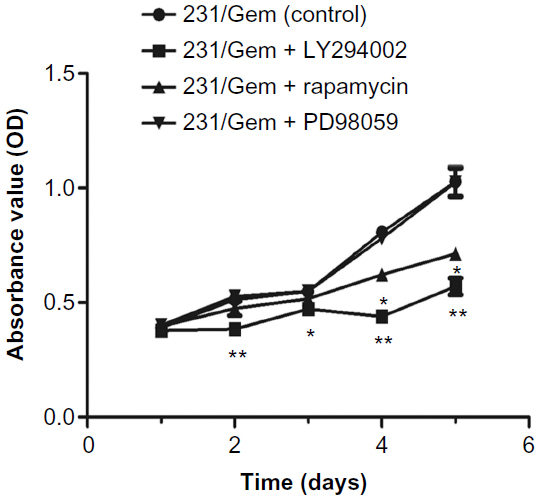 | Figure 4 PI3K/AKT/mTOR pathway, but not MAPK, regulates gemcitabine resistance-augmented cell proliferation. |
Phosphorylation of AKT controls acquired proliferation of gemcitabine- resistant breast cancer cells
The above results demonstrated that suppression of MAPK and mTOR pathways did not significantly influence 231/Gem cell proliferation, which is not consistent with previous studies.20 To examine the exact mechanism, we performed Western blot analysis. Our data show that with treatment of LY294002, PD98059, and rapamycin, expression levels of p-AKT, p-MEK, p-p70S6K, and p-mTOR were decreased, respectively, indicating that the three inhibitors were effective (Figure 5).
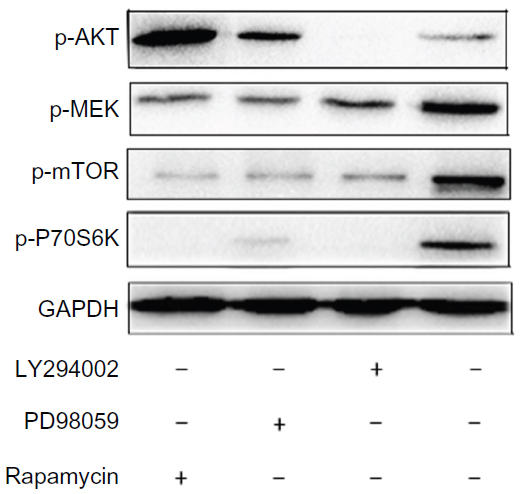 | Figure 5 Phosphorylation of AKT controls the proliferation of gemcitabine-resistant breast cancer cells. |
Treatment with LY294002 downregulated expression of p-MEK, p-mTOR and p-p70S6K. We found that after treatment with either PD98059 or rapamycin, p-AKT protein expression markedly increased while p-mTOR and p-p70S6K expression levels were reduced. Our results demonstrate that p-AKT is the upstream regulatory protein in the pathway, which could activate downstream pathway components including MAPK and mTOR. In addition, the downstream pathway demonstrated negative feedback for upstream regulation of PI3K/AKT.
Discussion
Chemotherapy resistance is commonly associated with multiple genetic and epigenetic abnormalities; however, changes in one or a few main genes remain crucial for maintaining cell survival and malignant phenotype. In recent years, increasing evidence has demonstrated that the PI3K/AKT/mTOR and ERK/MAPK signaling pathways are involved in many drug-resistance patterns among different types of cancer.26–29
In this study, we generated a gemcitabine-resistant human breast cancer cell model (231/Gem) to investigate whether PI3K/AKT/mTOR and ERK/MAPK pathways were involved in the chemoresistance. The long-term culture of MDA-MB-231 to establish MDA-MB-231/Gem cell line did not activate AKT, mTOR and ERK pathways (Figure S1). Flow cytometry, apoptosis assay, and CCK-8 demonstrated that gemcitabine-resistant breast cancer cells (231/Gem) were successfully established. The results showed that 231/Gem displays increased proliferation, a larger number cells in S phase of the cell cycle, and more cellular projections and invasion than parental cells. Western blot E-cadherin protein was reduced, which can lead to enhanced cellular invasion as compared to parental cells.
These data suggested that 231/Gem cells gained increased proliferative and invasive abilities after obtaining resistance to gemcitabine. Further mechanistic studies confirmed that PI3K/AKT/mTOR and MEK/MAPK signaling pathways were activated through elevated expression of p-ERK, p-AKT, mTOR, p-mTOR, and p-P70S6K, and reduced expression of p-P38 and LC3-II (the marker of autophagy) in 231/Gem as compared to control cells. However, no significant change of cyclin D1 and p-AMPK can be found between MDA-MB-231 and MDA-MB-231/Gem cell lines (Figure S2). In culture, inhibitors of PI3K/AKT and mTOR but not MEK/MAPK could reverse the enhanced proliferative ability of 231/Gem. Further Western blot analysis showed that with treatment of PI3K/AKT inhibitor, the expression of p-AKT, p-MEK, p-mTOR, and p-P70S6K was decreased; however, treatment with either a MEK/MAPK or an mTOR inhibitor significantly increased p-AKT expression. Thus, our data suggest that gemcitabine resistance in breast cancer cells is mediated by activation of the PI3K/AKT signaling pathway through elevation of expression of p-AKT protein to promote cell proliferation and is negatively regulated by MEK/MAPK and mTOR pathways.
In recent years, many mechanisms have been reported with respect to gemcitabine resistance in various cancers. Zheng et al30 reported that ERK1/2 activity contributed to gemcitabine resistance in pancreatic cancer cells. Mei et al31 indicated that the gene therapy of antiangiogenesis by intramuscular delivery of plasmid DNA encoding chemokine CXCL10 combined with gemcitabine has synergistic effects on tumor by inhibiting the proliferation of endothelial cells, inducing the apoptosis of tumor cells, and recruiting lymphocytes to tumor in murine models. CXCL12–CXCR4 signaling axis can play a role in conferring gemcitabine resistance to pancreatic cancer cells and suggest that it could serve as a novel therapeutic target for pancreatic cancer therapy, alone and in combination with the cytotoxic drug.32
In breast cancer studies, Liang et al33 showed that 3-phosphoinositide-dependent protein kinase-1 (PDK1) may be a superior alternative to AKT1 as a target for sensitizing breast cancer cells to chemotherapeutic agents, particularly gemcitabine. Moreover, Hernandez-Vargas et al34 performed gene expression analysis to shed light on nuclear factor-kappa B pathway activation as a potential mechanism of resistance. Although the AKT/mTOR pathway has been reported to be involved in gemcitabine resistance induced by Annexin II in pancreatic cancer cells,35 the role of PI3K/AKT/mTOR and MAPK signaling pathways and their cross-talk and feedback loops in gemcitabine resistance in breast cancer cells is still unknown. To receive highly effective clinical benefit, dual inhibition of the target plus the subsequent feedback loop is necessary. In this study, we have not detected the inhibition of the combination of AKT, mTOR, or MEK and this hypothesis also needs to be tested in future clinical trials. Our study suggests that targeting PI3K/AKT/mTOR and MAPK signaling pathways may help to develop more effective therapies in patients with gemcitabine-resistant breast cancer.
Acknowledgments
The authors wish to acknowledge funding from the National Natural Science Foundation of China (Grant No 81172506), and the Shanghai Committee of Science and Technology, China (Grant No 12DZ2260100).
Disclosure
The authors report no conflicts of interest in this work.
References
DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64(1):52–62. | |
Zhang ML, Huang ZZ, Zheng Y. [Estimates and prediction on incidence, mortality and prevalence of breast cancer in China, 2008]. Zhonghua Liu Xing Bing Xue Za Zhi. 2012;33(10):1049–1051. Chinese. | |
Ali S, El-Rayes BF, Aranha O, Sarkar FH, Philip PA. Sequence dependent potentiation of gemcitabine by flavopiridol in human breast cancer cells. Breast Cancer Res Treat. 2005;90(1):25–31. | |
Heinemann V. Gemcitabine in the treatment of advanced pancreatic cancer: a comparative analysis of randomized trials. Semin Oncol. 2002;29(6 Suppl 20):9–16. | |
Kohail H, Shehata S, Mansour O, et al. A phase 2 study of the combination of gemcitabine and cisplatin in patients with locally advanced or metastatic breast cancer previously treated with anthracyclines with/without taxanes. Hematol Oncol Stem Cell Ther. 2012;5(1):42–48. | |
Erten C, Demir L, Somali I, et al. Cisplatin plus gemcitabine for treatment of breast cancer patients with brain metastases; a preferential option for triple negative patients? Asian Pac J Cancer Prev. 2013;14(6):3711–3717. | |
Franchina T, Adamo B, Ricciardi GR, et al. Activity of pegylated liposomal doxorubicin in combination with gemcitabine in triple negative breast cancer with skin involvement: two case reports. Cancer Biol Ther. 2012;13(7):472–476. | |
Seto B. Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer. Clin Transl Med. 2012;1(1):29. | |
Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16(12):1472–1487. | |
Wang L, Liu Q, Li F, et al. Apoptosis induced by PGC-1β in breast cancer cells is mediated by the mTOR pathway. Oncol Rep. 2013;30(4):1631–1638. | |
Kumar D, Shankar S, Srivastava RK. Rottlerin induces autophagy and apoptosis in prostate cancer stem cells via PI3K/Akt/mTOR signaling pathway. Cancer Lett. 2014;343(2):179–189. | |
Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23(18):3151–3171. | |
Pola C, Formenti SC, Schneider RJ. Vitronectin-αvβ3 integrin engagement directs hypoxia-resistant mTOR activity and sustained protein synthesis linked to invasion by breast cancer cells. Cancer Res. 2013;73(14):4571–4578. | |
Dragowska WH, Verreault M, Yapp DT, et al. Decreased levels of hypoxic cells in gefitinib treated ER+ HER-2 overexpressing MCF-7 breast cancer tumors are associated with hyperactivation of the mTOR pathway: therapeutic implications for combination therapy with rapamycin. Breast Cancer Res Treat. 2007;106(3):319–331. | |
Jung CH, Kim H, Ahn J, et al. Anthricin Isolated from Anthriscus sylvestris (L.) Hoffm. Inhibits the Growth of Breast Cancer Cells by Inhibiting Akt/mTOR Signaling, and Its Apoptotic Effects Are Enhanced by Autophagy Inhibition. Evid Based Complement Alternat Med. 2013;2013:385219. | |
Cho YH, Han KM, Kim D, et al. Autophagy regulates homeostasis of pluripotency-associated proteins in hESCs. Stem Cells. 2014;32(2):424–435. | |
Lorin S. [Leucine sensing in the regulation of autophagy: glutamate dehydrogenase is on the way]. Med Sci (Paris). 2013;29(10):839–842. French. | |
Wei PF, Zhang L, Nethi SK, et al. Accelerating the clearance of mutant huntingtin protein aggregates through autophagy induction by europium hydroxide nanorods. Biomaterials. 2014;35(3):899–907. | |
Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6(2):239–247. | |
Saini KS, Loi S, de Azambuja E, et al. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev. 2013;39(8):935–946. | |
Woo CC, Hsu A, Kumar AP, Sethi G, Tan KH. Thymoquinone inhibits tumor growth and induces apoptosis in a breast cancer xenograft mouse model: the role of p38 MAPK and ROS. PLoS One. 2013;8(10):e75356. | |
Gil-Araujo B, Toledo Lobo MV, Gutiérrez-Salmerón M, et al. Dual specificity phosphatase 1 expression inversely correlates with NF-κB activity and expression in prostate cancer and promotes apoptosis through a p38 MAPK dependent mechanism. Mol Oncol. 2014;8(1):27–38. | |
Ko JC, Chiu HC, Wo TY, et al. Inhibition of p38 MAPK-dependent MutS homologue-2 (MSH2) expression by metformin enhances gefitinib-induced cytotoxicity in human squamous lung cancer cells. Lung Cancer. 2013;82(3):397–406. | |
Zhang B, Wang X, Cai F, Chen W, Loesch U, Zhong XY. Antitumor properties of salinomycin on cisplatin-resistant human ovarian cancer cells in vitro and in vivo: involvement of p38 MAPK activation. Oncol Rep. 2013;29(4):1371–1378. | |
Davidson JD, Ma L, Flagella M, Geeganage S, Gelbert LM, Slapak CA. An increase in the expression of ribonucleotide reductase large subunit 1 is associated with gemcitabine resistance in non-small cell lung cancer cell lines. Cancer Res. 2004;64(11):3761–3766. | |
Gadgeel SM, Wozniak A. Preclinical rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for epidermal growth factor receptor inhibitor-resistant non-small-cell lung cancer. Clin Lung Cancer. 2013;14(4):322–332. | |
Hoang B, Benavides A, Shi Y, et al. The PP242 mammalian target of rapamycin (mTOR) inhibitor activates extracellular signal-regulated kinase (ERK) in multiple myeloma cells via a target of rapamycin complex 1 (TORC1)/eukaryotic translation initiation factor 4E (eIF-4E)/RAF pathway and activation is a mechanism of resistance. J Biol Chem. 2012;287(26):21796–21805. | |
McCubrey JA, Steelman LS, Chappell WH, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012;3(10):1068–1111. | |
Xu ZH, Hang JB, Hu JA, Gao BL. RAF1-MEK1-ERK/AKT axis may confer NSCLC cell lines resistance to erlotinib. Int J Clin Exp Pathol. 2013;6(8):1493–1504. | |
Zheng C, Jiao X, Jiang Y, Sun S. ERK1/2 activity contributes to gemcitabine resistance in pancreatic cancer cells. J Int Med Res. 2013;41(2):300–306. | |
Mei K, Wang L, Tian L, Yu J, Zhang Z, Wei Y. Antitumor efficacy of combination of interferon-gamma-inducible protein 10 gene with gemcitabine, a study in murine model. J Exp Clin Cancer Res. 2008;27:63. | |
Singh S, Srivastava SK, Bhardwaj A, Owen LB, Singh AP. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer. 2010;103(11):1671–1679. | |
Liang K, Lu Y, Li X, et al. Differential roles of phosphoinositide-dependent protein kinase-1 and akt1 expression and phosphorylation in breast cancer cell resistance to Paclitaxel, Doxorubicin, and gemcitabine. Mol Pharmacol. 2006;70(3):1045–1052. | |
Hernández-Vargas H, Rodríguez-Pinilla SM, Julián-Tendero M, et al. Gene expression profiling of breast cancer cells in response to gemcitabine: NF-kappaB pathway activation as a potential mechanism of resistance. Breast Cancer Res Treat. 2007;102(2):157–172. | |
Kagawa S, Takano S, Yoshitomi H, et al. Akt/mTOR signaling pathway is crucial for gemcitabine resistance induced by Annexin II in pancreatic cancer cells. J Surg Res. 2012;178(2):758–767. |
Supplementary materials
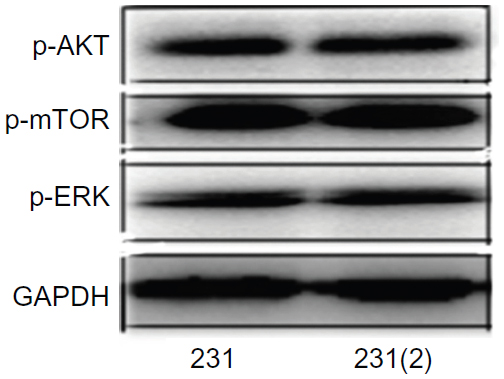 | Figure S1 Change of signaling pathway after long time culture. |
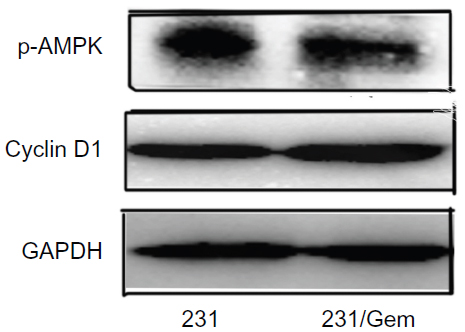 | Figure S2 Change of cyclin D1 and p-AMPK after treatment by gemcitabine. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.