")
Back to Journals » Lung Cancer: Targets and Therapy » Volume 5
Gastroesophageal junction adenocarcinoma displays abnormalities in homologous recombination and nucleotide excision repair
Authors Dewalt RI, Kesler K, Hammoud ZT, Baldridge L, Hattab EM, Jalal S
Received 15 November 2013
Accepted for publication 6 December 2013
Published 15 February 2014 Volume 2014:5 Pages 11—20
DOI https://doi.org/10.2147/LCTT.S57594
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Robin I Dewalt,1 Kenneth A Kesler,2 Zane T Hammoud,3 LeeAnn Baldridge,4 Eyas M Hattab,4 Shadia I Jalal1,5
1Division of Hematology/Oncology, Department of Medicine, 2Cardiothoracic Division, Department of Surgery, Indiana University School of Medicine, Indianapolis, IN, USA; 3Henry Ford Hospital, Detroit, MI, USA; 4Department of Pathology and Laboratory Medicine, Indiana University School of Medicine, Indianapolis, IN, USA; 5Indiana University Melvin and Bren Simon Cancer Center, Indianapolis, IN, USA
Objective: Esophageal adenocarcinoma (EAC) continues to be a disease associated with high mortality. Among the factors leading to poor outcomes are innate resistance to currently available therapies, advanced stage at diagnosis, and complex biology. Platinum and ionizing radiation form the backbone of treatment for the majority of patients with EAC. Of the multiple processes involved in response to platinum chemotherapy or ionizing radiation, deoxyribonucleic acid (DNA) repair has been a major player in cancer sensitivity to these agents. DNA repair defects have been described in various malignancies. The purpose of this study was to determine whether alterations in DNA repair are present in EAC compared with normal gastroesophageal tissues.
Methods: We analyzed the expression of genes involved in homologous recombination (HR), nonhomologous end-joining, and nucleotide excision repair (NER) pathways in 12 EAC tumor samples with their matched normal counterparts. These pathways were chosen because they are the main pathways involved in the repair of platinum- or ionizing-radiation-induced damage. In addition, abnormalities in these pathways have not been well characterized in EAC.
Results: We identified increased expression of at least one HR gene in eight of the EAC tumor samples. Alterations in the expression of EME1, a structure-specific endonuclease involved in HR, were the most prevalent, with messenger (m)RNA overexpression in six of the EAC samples. In addition, all EAC samples revealed decreased expression of at least one of numerous NER genes including XPC, XPA, DDB2, XPF, and XPG.
Conclusion: Our study identified DNA repair dysregulation in EAC involving two critical pathways, HR and NER, and is the first demonstration of EME1 upregulation in any cancer. These DNA repair abnormalities have the potential to affect a number of processes such as genomic instability and therapy response, and the consequences of these defects deserve further study in EAC.
Keywords: esophageal adenocarcinoma, DNA repair, MUS81/EME1
Introduction
Esophageal cancer accounts for 4% of cancer deaths in the United States and is the sixth leading cause of cancer deaths among men. Esophageal adenocarcinoma (EAC) incidence has steadily increased during the last 2 decades.1 In 2013, 17,990 new EAC cases will be diagnosed, and the majority of these individuals have or will succumb to the disease.2 The rise in the incidence of esophageal cancer is accompanied by a change in the dominant histology of the disease from squamous cell carcinoma of the esophagus to adenocarcinomas mostly diagnosed in the distal esophagus or gastroesophageal junction. A major risk factor for EAC is chronic gastroesophageal reflux disease, which can lead to Barrett’s metaplasia, a precursor lesion for EAC.3 Progression from metaplasia to adenocarcinoma is thought to be mediated by the development of low-grade followed by high-grade dysplasia (more recently coined intraepithelial neoplasia), yet these histologic abnormalities do not uniformly precede EAC development in all patients.4
It has been suggested that all cancers are expected to display a defect in deoxyribonucleic acid (DNA) repair facilitating the accumulation of multiple mutations, which is a shared characteristic of all cancers.5 DNA repair defects characterizing a number of malignancies are being increasingly identified. For example, homologous recombination (HR) defects were noted in 50% of high-grade serous ovarian tumors.6 Nucleotide excision repair (NER) deficiency has been previously described in sporadic stage 1 breast cancers, testicular cancers, and lung cancers and has been linked to carcinogenesis and genomic instability.7–9 Ataxia telangiectasia mutated deficiency has been found in mantle cell lymphomas and gastric cancers.10,11
Multiple studies have shown that acid induces DNA damage in the esophagus, and multiple culprits leading to the damage have been described, including nitric oxide, bile acids present in duodenal reflux, and low pH.12–14 DNA repair pathways involved in the response to damage induced by these agents have not been fully characterized in EAC. Exploring DNA repair abnormalities in esophageal cancer has largely focused on the analysis of DNA repair polymorphisms and their association with cancer susceptibility or therapy response.15 RAD51, an HR gene, was recently shown to be overexpressed in EAC and was associated with increased genomic instability. Little is known about the expression of other HR genes in addition to NER and nonhomologous end-joining (NHEJ) pathways in EAC. This is the focus of this article.
In this study, we analyzed the DNA repair transcript expression in 12 EAC tumor samples and individually matched normal gastroesophageal tissue. We focused on the HR, NHEJ, and NER pathways, as these are the most relevant to the therapies used in the clinic in the treatment of EAC because of their major role in repair of cisplatin- and/or ionizing-radiation-induced DNA damage. Our data highlight a number of DNA repair alterations that can have a critical effect on both EAC biology and DNA repair.
Materials and methods
Patients and samples
Primary EAC tumor samples and paired normal gastroeso-phageal tissue of 12 patients who had not received prior chemotherapy or radiation and who underwent esophagectomy at the Indiana University Hospital, Division of Cardiothoracic Surgery, were procured from the Indiana University Melvin and Bren Simon Cancer Center Tissue procurement facility through an institutional review board-approved protocol. All tumor samples originating from the gastroesophageal area had been snap-frozen immediately on resection in liquid nitrogen and stored until the time of analysis. Consistent with the epidemiology of EAC, all patients except one were men (n=11). The age of the patients ranged from 48 to 83 years, with a median of 74.5 years. Histologic examination was performed by an experienced pathologist on the formalin-fixed paraffin embedded (FFPE) blocks of both normal gastroesophageal and EAC samples to confirm the histology was accurate. Only EAC samples with 70% or more tumors were included in the analysis. Tumor note metastasis (TNM) stage was available for 11 patients, and stage distribution was as follows: one patient had stage 1A carcinoma, four patients had stage 2B disease, five patients were stage 3A, and one patient had stage 3C disease. Patient characteristics are outlined in Table 1.
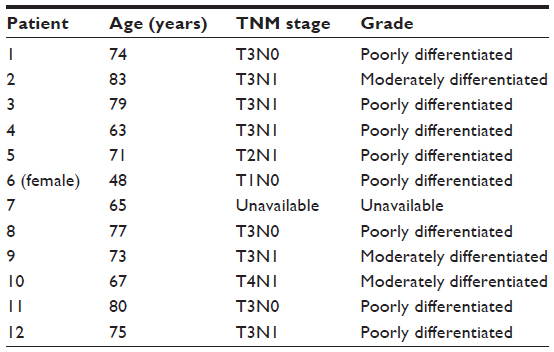 | Table 1 Characteristics of the patients who were included in the analysis |
RNA extraction and complementary DNA synthesis
Total RNA was isolated from 100 mg tissue samples using Trizol (Life Technologies, Carlsbad, CA, USA) and using an RNeasy minikit (Qiagen, Hilden, Germany) per manufacturer’s instructions. A nanodrop spectrophotometer was used to determine the quantity of RNA. Complementary (c)DNA was generated using a High Capacity RNA-to-cDNA Kit (Life Technologies, Carlsbad, CA, USA).
Transcript expression measurement by quantitative reverse transcription polymerase chain reaction
The expression levels of 16 reference genes were measured using the Taqman endogenous control array from ABI (catalog no 4367563; Applied Biosystems, Grand Island, NY, USA) to determine the most stably expressed gene between EAC samples and their matched normal tissue (Figure S1). Seven samples (four tumors and three normal) were included in the endogenous control assay selection. Validated gene primer/probe sets for each gene of interest were purchased from ABI. Taqman reactions were analyzed using ABI’s 7900HT system. For each target gene, fold change in expression levels between normal and tumor sample was evaluated using ABI’s ΔΔCT Relative Quantification methodology. Data were normalized to the housekeeping gene GUSB (glucuronidase, beta) (Hs99999908_m1), and each tumor sample was calibrated to its normal matched control. Results presented are the average of two quantitative reverse transcription polymerase chain reaction (qRT-PCR) analyses with triplicate Taqman assays per RNA sample set. Genes analyzed and assays used are outlined in Table 2.
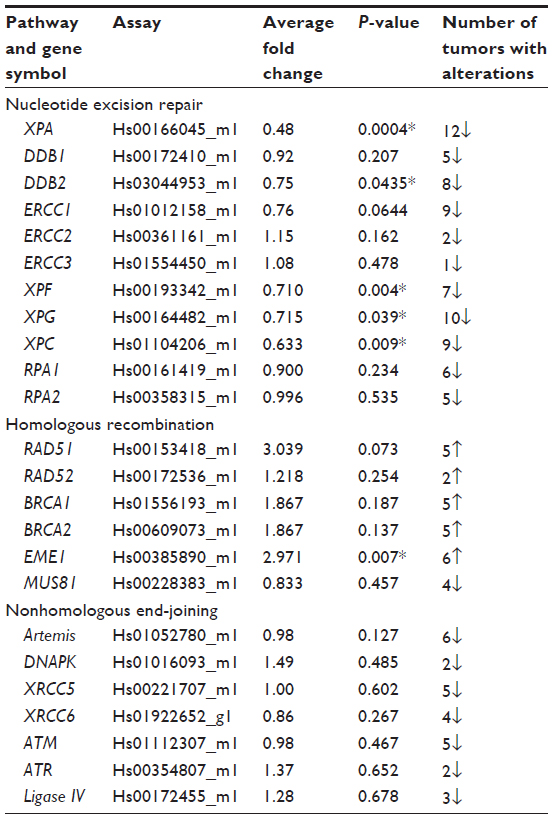 | Table 2 Summary of DNA repair genes analyzed with their average fold changes and P-values |
Statistical analysis
Statistical analysis was performed using a paired Student’s two-tailed t-test (SigmaStat, Systat, Point Richmond, CA, USA). Differences between groups at P≤0.05 were considered statistically significant.
Immunohistochemistry
FFPE tissue sections corresponding to the EAC tumor samples were available for nine of the 12 samples. Immunohistochemistry (IHC) was performed with the assistance of the IHC pathology laboratory at Indiana University. FFPE blocks were cut into 4 mm thick sections and collected on charged slides, and immunostaining was performed using the Autostainer Plus platform from Dako (Carpinteria, CA, USA). Slides were heated at 65°C for 30 minutes, deparaffinized with xylene, and rehydrated with graded alcohol. Endogenous peroxidase activity was blocked using 3% peroxidase blocking reagent (Dako K8002) for 5 minutes. Antigen retrieval was performed by immersing sections in Dako’s PT module filled with FLEX Target Retrieval Solution, low pH (K800521), for 20 minutes. Sections were incubated for 60 minutes with a 1:10 dilution of EME1 primary antibody (MTA31 7h2/1) from Santa Cruz Biotechnology (Dallas, TX, USA; sc-53275). Detection was performed using an Envision FLEX+ mouse kit from Dako. Normal uterine tissue was used as a positive control to optimize IHC conditions. All sections were examined by a surgical pathologist who confirmed the histology of normal/nonneoplastic esophageal tissue and EAC samples by reviewing hematoxylin and eosin sections. IHC staining for EME1 was scored on the basis of the intensity of nuclear staining (0, absent; 1, faint; 2, moderate; and 3, strong staining) and the extent of staining (0, 0%; 1, 1%–10%; 2, 10%–50%; and 3, >50% of tumor cells staining).
Western blot
Western blots were used to ensure specificity of the primary EME1 antibody, as this is the first report of the assessment of EME1 expression by IHC. HeLa cell, WI-38 (normal lung fibroblasts), and SK-GT4 (esophageal adenocarcinoma) cell line extracts were prepared as previously described.16 Protein concentration was determined using the Bio-Rad protein assay system (Bio-Rad Laboratories, Hercules, CA, USA) with bovine serum albumin standards. Forty micrograms of protein were loaded onto sodium dodecyl sulfate polyacrylamide gel electrophoresis, and after electrophoresis, proteins were transferred to polyvinylidene difluoride membranes. EME1 protein was detected using EME1 antibody (MTA31 7h2/1) at a 1:1000 ratio, with secondary immunoglobulin (Ig)G horseradish peroxidase anti-mouse antibody (sc-2005, Santa Cruz) at a 1:4000 dilution.
Oncomine database search
The Oncomine Cancer Profiling Database version 4.4.3, September 2012 data release (http://www.oncomine.org; Life Technologies, Carlsbad, CA, USA), was used to profile the gene EME1. Expression data and statistics were obtained directly through the Oncomine 4.4.3 software.
Results
EAC shows messenger RNA overexpression of HR genes
Defects in a number of HR genes have been described in solid organ malignancies and have been shown to mediate sensitivity to a variety of chemotherapy agents including platinum, crosslinking agents, and poly(ADP-ribose) polymerase inhibitors.17–19 We initiated our work by screening 16 endogenous housekeeping genes to determine the optimal endogenous control and identified GUSB as an excellent housekeeping gene with small differences in expression between tumor and normal tissues (Figure S1). All genes of interest were therefore normalized to GUSB transcript levels, and each tumor sample was compared with its matched normal sample. Eight of the 12 EACs showed messenger (m)RNA overexpression of at least one of the HR genes, including RAD51, RAD52, BRCA1, BRCA2, and EME1 (twofold cutoff or greater; Figure 1). EME1 was overexpressed in six of the 12 EACs, and the difference in expression was statistically significant compared with normal tissue (P<0.001). Three of these tumor samples showed greater than fourfold EME1 mRNA overexpression (Figure 2).
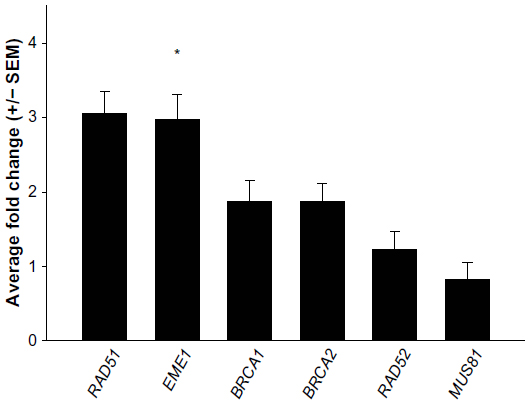 | Figure 1 Quantitative reverse transcription polymerase chain reaction analyses of messenger (m)RNA expression changes of homologous recombination genes. Data shown are the average of two separate quantitative reverse transcription polymerase chain reaction assays. Genes of interest were normalized to GUSB transcript levels and are presented as fold changes relative to normal matched samples. EME1 transcripts are increased in EAC samples. |
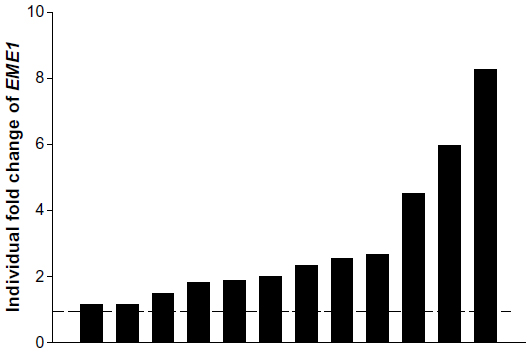 | Figure 2 EME1 messenger (m)RNA expression in individual esophageal adenocarcinoma samples, showing three tumor samples display more than fourfold overexpression of EME1. |
The MUS81/EME1 protein complex is an endonuclease in the HR pathway with cleaving DNA intermediates as its main function. It plays an important role in maintaining genomic integrity, yet its role in human disease and cancer is still undefined. As EME1 is present in complex with MUS81, we analyzed mRNA expression of MUS81 in the EAC samples and found that four tumor samples showed underexpression of MUS81 when compared with a matched normal sample. RAD51 overexpression approached statistical significance with a threefold change in EAC samples compared with normal counterparts. NHEJ genes evaluated showed a slight reduction in transcript expression in a few EAC samples when compared with normal counterparts, but none of them were statistically significant (Table 2). Three of the patients had two samples from separate areas of the tumor available, and with the recent data revealing tumor heterogeneity in drug target expression and genomic differences within the same renal cell carcinoma sample, we were interested in whether we would be able to identify the heterogeneity of EME1 expression in different areas of the tumor.20 Transcript expression of the HR genes was reproducible between these biological duplicates (Table S1), suggesting EAC may not be as heterogeneous as renal cell carcinoma. We observed that all HR genes followed similar patterns in each tumor sample, where the majority of them either showed increased transcript expression or no change. Table 2 outlines genes evaluated in this analysis, along with fold change results.
EME1 overexpression in EAC by IHC
As this is the first published report of EME1 upregulation in cancer, we were interested in assessing whether changes in the mRNA expression of MUS81/EME1 are accompanied by changes in its protein expression. Therefore, EME1 IHC was performed on FFPE sections corresponding to the fresh frozen samples analyzed by qRT-PCR for mRNA expression. Western blot assessment confirmed that the commercially available EME1 antibody used in these analyses did not show cross-reactivity and was highly specific for EME1, showing one dominant band at the expected size of 65 kDa noted in the HeLa and SK-GT4 cell lines, both of which represent positive controls (Figure S2). The WI-38 cell line, in contrast, does not display increased expression of EME1 with a weaker band observed.
Normal uterine tissue was used as a control to optimize IHC conditions and showed an appropriate nuclear staining pattern of EME1 (Figure 3, insert). IHC analysis revealed that EME1 was overexpressed at varying intensities in nine of nine tumor samples available for testing compared with normal tissues (Figure 3). Of note, EME1 staining intensity was noticeably less evident in the less-differentiated areas of the tumor, but the significance of this observation is unclear. Of note, gene and protein expression of EME1 did not necessarily correlate, as adenocarcinoma samples numbered 1, 2, and 11 showed less than twofold increase in EME1 mRNA expression but moderately increased EME1 IHC staining, and tumor sample 10 had intense IHC staining that was not associated with any change in EME1 mRNA expression (Table S2).
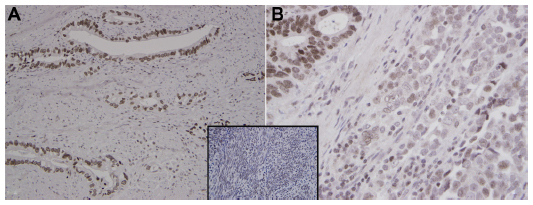 | Figure 3 Eme1 immunohistochemistry in gastroesophageal junction adenocarcinoma. (A) Well-differentiated gastroesophageal junction adenocarcinoma shows uniform strong nuclear staining pattern with EME1 (250×). (B) EME1 staining intensity was noticeably less in the less differentiated areas (500×). Note the nuclear immunoreactivity for EME1 in a control of benign endometrial stroma (inset). |
EACs display defects in NER
The NER pathway plays a significant role in repairing a variety of DNA lesions, including those resulting from ultraviolet radiation, chemotherapy agents, and mutagens.21 Our analysis of the NER pathway showed that each of the 12 tumor samples displayed an abnormality in transcript expression of at least one of the eleven NER genes analyzed (Table 2). XPC, XPA, DDB2, XPF, and XPG genes were statistically significantly underexpressed in EACs compared with normal samples (Table 2). XPA was underexpressed in all 12 EAC samples, with fold changes in the range of 0.2 to 0.7, and this was statistically significant across all samples. This finding is consistent with the increased incidence of cancer in patients with xeroderma pigmentosum.22 The observation of NER deficiency in all EAC samples analyzed is extremely intriguing, especially with the role of this pathway in repair of DNA damage resulting from various sources.
Underexpression of NER components has been tied to poor prognosis in cancer, with clinical data suggesting it is more likely observed in higher stages of cancer as compared with earlier stages.23 As only one patient had stage 1 disease and still displayed decreased expression of NER components, it is unclear whether alterations in NER expression are more frequently encountered in more advanced EAC disease stages. It is potentially possible, however, that underexpression of NER in EAC contributes to its unfavorable prognosis.
Eme1 is overexpressed in a number of cancers
EME1 overexpression noted in EAC in this study has not been previously described. In fact, EME1 overexpression has not been published in any cancer. This led us to conduct a search of the Oncomine gene expression database to evaluate whether other tumors overexpress EME1, and we chose a twofold change as threshold. This search showed that EME1 is overexpressed in head and neck, breast, and central nervous system tumors when compared with normal tissues. In addition, in one data set, papillary renal cell carcinoma and lymphoma each displayed mRNA overexpression of EME1 (Table 3). There are a limited number of data sets for EAC in the Oncomine database, but our search did show EME1 was overexpressed in a portion of EACs compared with normal esophageal tissue in the Kim Esophagus data set. EME1 seemed to be more highly expressed in our EAC samples when compared with data sets of other tumors reported in Oncomine, where the fold change was between 2 and 3. This observation might suggest that EME1 upregulation in EAC is unique, possibly as a response to acid damage.
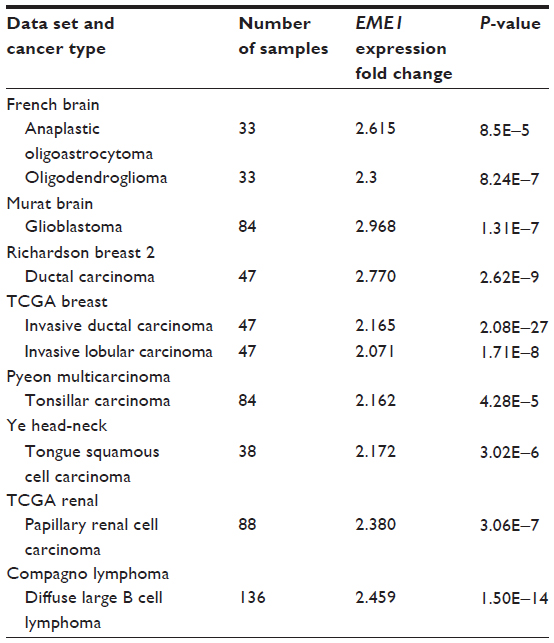 | Table 3 Summary of Oncomine database search results of tumors with increased EME1 expression |
Discussion
Multiple studies have highlighted genetic alterations in EAC, revealing mutations in the frequently mutated genes such as TP53 and PIK3CA, as well as less common mutations in SPG20, TLR4, ELMO1, and DOCK2.24,25 Limited data exist describing the DNA repair alterations present in EAC and with the fundamental role of chemotherapy and radiation in this disease, understanding those alterations has the potential of further refining therapies and improving patient outcomes. In this study, we performed gene expression analysis of a focused group of DNA repair genes in EAC samples arising in the gastroesophageal junction area. The genes were carefully selected because of their involvement in the repair of damage induced by platinum or ionizing radiation in addition to existing data showing alterations in these genes in other malignancies. Our findings highlight a number of important observations including NER deficiency and HR overexpression in EAC.
Our analysis of 12 EAC samples shows that alterations in transcript expression of at least one NER gene are detected in all tumor samples, with XPC, XPA, DDB2, XPF, and XPG showing significant underexpression in tumors compared with their normal counterparts. Defective NER, through reduced expression of some of its components, especially XPA and ERCC1, has been postulated to be one of the main mechanisms of hypersensitivity of testicular cancer, including in the metastatic stage to cisplatin therapy.26 There is an abundance of data demonstrating the connection between platinum resistance and NER function. XPA is an essential NER component and a rate-limiting factor in excision repair, and its decreased expression in all EAC samples evaluated would suggest potential EAC sensitivity to cisplatin. However, clinical data are not consistent with that, as cisplatin has modest activity in EAC and, as a single agent, has response rates of only 20%.27 The patients included in this analysis were not candidates for chemotherapy, and we are therefore unable to assess the effect of NER alterations via underexpression of XPA on their response to platinum-based therapy. However, on the basis of outcomes of patients with T3N1 EAC (the same stage as nine of 12 patients included in this analysis), estimated cure rates with the combination of platinum-based chemotherapy, radiation, and surgery are only 50%.28 These observations highlight the complexity of predicting response to platinum, which is orchestrated by multiple steps and proteins in the NER pathway in addition to other pathways such as HR playing a role, making it doubtful that the expression of a single DNA repair factor will dictate response to platinum.
Another finding in our analysis is that two thirds of EACs display increased expression of at least one of the HR genes analyzed, specifically BRCA1, BRCA2, RAD51, RAD52, and EME1. The HR pathway is mainly involved in the repair of DNA double strand breaks, yet consistently, HR defects have been shown to increase sensitivity to a wide variety of chemotherapy agents, including cisplatin.29 This more general effect of HR on therapy response could be related to the many proteins and subpathways involved in HR. The HR pathway is critical for the maintenance of genomic integrity, and further studies are needed to assess the consequences of upregulation of HR in EAC described here on the genomic instability noted in EAC. We have demonstrated no change in expression of the NHEJ genes evaluated; however, mRNA expression alone does not reflect the function of the NHEJ pathway in EAC. Both NHEJ and HR activity are regulated by protein interactions and posttranslational modifications, rather than transcription alone. The efficiency of NHEJ renders it less affected by transcription alone, and therefore, normal NHEJ gene expression does not necessarily mean normal function. Potential limitations of our study include the relatively small sample size.
Of the genes analyzed in this study, EME1, a component of the structure-specific endonuclease complex MUS81/EME1, demonstrated the most frequent and statistically significant overexpression in EAC. EME1 shows increased transcript expression in 50% of EACs, with three tumor samples showing more than fourfold overexpression. In addition, EME1 protein expression is increased at varying intensities when analyzed by IHC in all EAC samples. Our study illustrates that although EME1 shows increased expression in EAC, its partner MUS81 showed either decreased or unchanged mRNA expression. We are unaware of other reports evaluating the mRNA expression of both components of the MUS81/EME1 complex, but in another study, correlation patterns of EME1 and MUS81 protein levels in vitro varied according to the cell lines analyzed.30 In addition, our data demonstrate that EME1 mRNA levels do not correlate with the protein expression absolutely (Table S1). These observations lead to a number of possibilities, including increased EME1 protein stability or posttranscriptional regulation of EME1 in some tumors.
A variety of biochemical and structural aspects of the MUS81/EME1 complex have been elucidated during the last decade, but its role in carcinogenesis remains to be defined. MUS81/EME1’s contribution to the recovery of stalled replication forks, a crucial step in preserving genome stability, suggests MUS81/EME1 defects would possibly mediate genomic instability, a bona fide hallmark of cancer.31,32 This is supported by a number of observations including the associated increase of spontaneous gross chromosomal rearrangements in the setting of defective MUS81 or EME1.33 Our search of the Oncomine Database showed that increased EME1 mRNA overexpression is not limited to EAC, as a number of malignancies display two- to threefold increased expression of EME1 (Table 3).
In conclusion, our study highlights a number of alterations in the DNA repair expression profile of EACs, including NER deficiency, HR upregulation, and the novel finding of increased expression of EME1. These defects possibly contribute to the genomic instability of EAC and potentially mediate resistance to chemotherapy and radiation that remains a challenge in caring for these patients. Discovering the implications of DNA repair abnormalities in EAC carcinogenesis and therapy response has the potential to improve outcomes of EAC. We are currently pursuing further studies to dissect the role of EME1 overexpression in EAC pathogenesis.
Acknowledgment
This work was funded by American Cancer Society Institutional Research Grant 84-002-2.
Disclosure
The authors report no conflicts of interest in this work.
References
Devesa SS, Blot WJ, Fraumeni JF Jr. Changing patterns in the incidence of esophageal and gastric carcinoma in the United States. Cancer. 1998;83(10):2049–2053. | |
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | |
Shaheen N, Ransohoff DF. Gastroesophageal reflux, Barrett esophagus, and esophageal cancer: clinical applications. JAMA. 2002;287(15):1982–1986. | |
Rugge M, Zaninotto G, Parente P, et al. Barrett’s esophagus and adenocarcinoma risk: the experience of the North-Eastern Italian Registry (EBRA). Ann Surg. 2012;256(5):788–794. | |
Alberts B. Redefining cancer research. Science. 2009;325(5946):1319. | |
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. | |
Latimer JJ, Johnson JM, Kelly CM, et al. Nucleotide excision repair deficiency is intrinsic in sporadic stage I breast cancer. Proc Natl Acad Sci U S A. 2010;107(50):21725–21730. | |
Ide F, Iida N, Nakatsuru Y, Oda H, Tanaka K, Ishikawa T. Mice deficient in the nucleotide excision repair gene XPA have elevated sensitivity to benzo[a]pyrene induction of lung tumors. Carcinogenesis. 2000;21(6):1263–1265. | |
Saviozzi S, Ceppi P, Novello S, et al. Non-small cell lung cancer exhibits transcript overexpression of genes associated with homologous recombination and DNA replication pathways. Cancer Res. 2009;69(8):3390–3396. | |
Williamson CT, Muzik H, Turhan AG, et al. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol Cancer Ther. 2010;9(2):347–357. | |
Kang B, Guo RF, Tan XH, Zhao M, Tang ZB, Lu YY. Expression status of ataxia-telangiectasia-mutated gene correlated with prognosis in advanced gastric cancer. Mutat Res. 2008;638(1–2):17–25. | |
Jolly AJ, Wild CP, Hardie LJ. Acid and bile salts induce DNA damage in human oesophageal cell lines. Mutagenesis. 2004;19(4):319–324. | |
Burnat G, Majka J, Konturek PC. Bile acids are multifunctional modulators of the Barrett’s carcinogenesis. J Physiol Pharmacol. 2010;61(2):185–192. | |
Clemons NJ, McColl KE, Fitzgerald RC. Nitric oxide and acid induce double-strand DNA breaks in Barrett’s esophagus carcinogenesis via distinct mechanisms. Gastroenterology. 2007;133(4):1198–1209. | |
Pan J, Lin J, Izzo JG, et al. Genetic susceptibility to esophageal cancer: the role of the nucleotide excision repair pathway. Carcinogenesis. 2009;30(5):785–792. | |
Wood RD, Robins P, Lindahl T. Complementation of the xeroderma pigmentosum DNA repair defect in cell-free extracts. Cell. 1988;53(1):97–106. | |
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. | |
Alli E, Sharma VB, Hartman AR, Lin PS, McPherson L, Ford JM. Enhanced sensitivity to cisplatin and gemcitabine in Brca1-deficient murine mammary epithelial cells. BMC Pharmacol. 2011;11:7. | |
Yun J, Zhong Q, Kwak JY, Lee WH. Hypersensitivity of Brca1-deficient MEF to the DNA interstrand crosslinking agent mitomycin C is associated with defect in homologous recombination repair and aberrant S-phase arrest. Oncogene. 2005;24(25):4009–4016. | |
Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. | |
Jalal S, Earley JN, Turchi JJ. DNA repair: from genome maintenance to biomarker and therapeutic target. Clin Cancer Res. 2011;17(22):6973–6984. | |
de Boer J, Hoeijmakers JH. Nucleotide excision repair and human syndromes. Carcinogenesis. 2000;21(3):453–460. | |
Wu YH, Cheng YW, Chang JT, Wu TC, Chen CY, Lee H. Reduced XPC messenger RNA level may predict a poor outcome of patients with nonsmall cell lung cancer. Cancer. 2007;110(1):215–223. | |
Agrawal N, Jiao Y, Bettegowda C, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012;2(10):899–905. | |
Dulak AM, Stojanov P, Peng S, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45(5):478–486. | |
Köberle B, Masters JR, Hartley JA, Wood RD. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr Biol. 1999;9(5):273–276. | |
Grünberger B, Raderer M, Schmidinger M, Hejna M. Palliative chemotherapy for recurrent and metastatic esophageal cancer. Anticancer Res. 2007;27(4C):2705–2714. | |
van Hagen P, Hulshof MC, van Lanschot JJ, et al; CROSS Group. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med. 2012;366(22):2074–2084. | |
Cavallo F, Graziani G, Antinozzi C, et al. Reduced proficiency in homologous recombination underlies the high sensitivity of embryonal carcinoma testicular germ cell tumors to Cisplatin and poly (adp-ribose) polymerase inhibition. PLoS One. 2012;7(12):e51563. | |
Tomoda Y, Katsura M, Okajima M, Hosoya N, Kohno N, Miyagawa K. Functional evidence for Eme1 as a marker of cisplatin resistance. Int J Cancer. 2009;124(12):2997–3001. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Abraham J, Lemmers B, Hande MP, et al. Eme1 is involved in DNA damage processing and maintenance of genomic stability in mammalian cells. EMBO J. 2003;22(22):6137–6147. | |
McPherson JP, Lemmers B, Chahwan R, et al. Involvement of mammalian Mus81 in genome integrity and tumor suppression. Science. 2004;304(5678):1822–1826. | |
Life Technologies (webpage on the Internet). Oncomine™ Research Edition. 2013. Available from: https://www.oncomine.org/resource/login.html. Accessed February 2, 2014. |
Supplementary materials
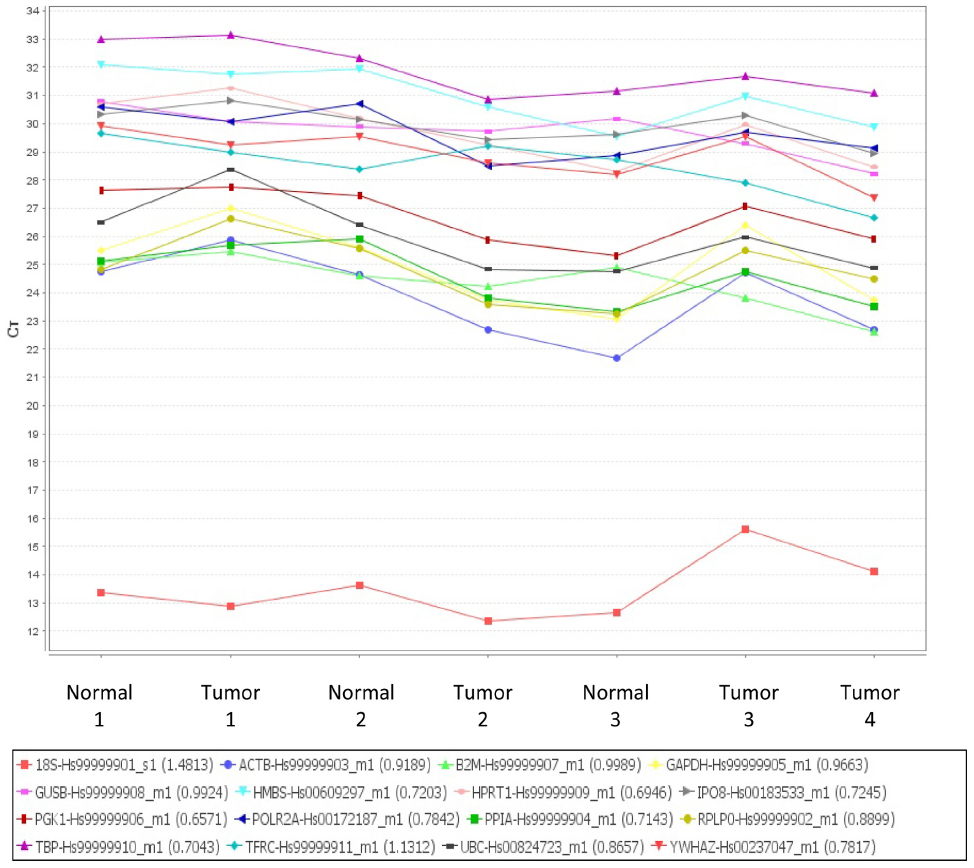 | Figure S1 Taqman endogenous control assay used to determine the optimal housekeeping gene. X axis shows samples used, Y axis showed Ct values of each gene. |
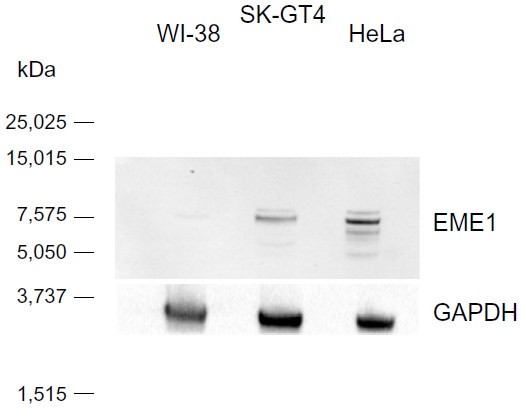 | Figure S2 Western blot analysis evaluating EME1 primary antibody in HeLa, WI-38, and SK-GT4 cell extracts. One major band can be seen at expected size of 65 kDa in both HeLa and SK-GT4 cell extracts. The band is present but much weaker in the WI-38 cell line. |
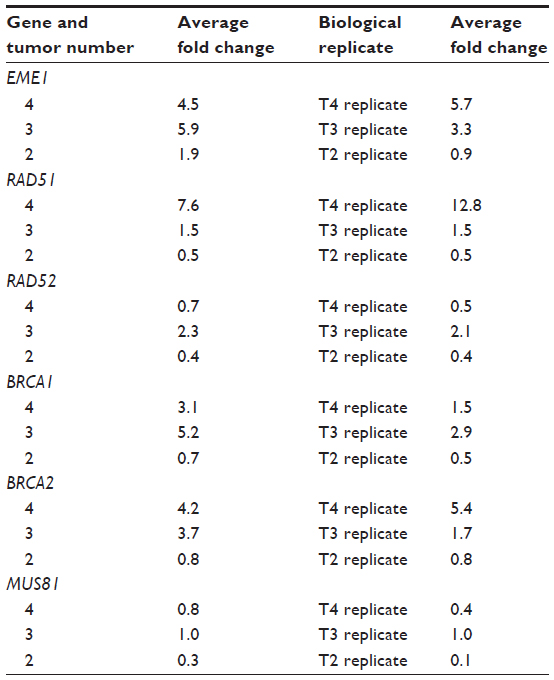 | Table S1 Summary of messenger RNA expression of homologous recombination genes in three tumor samples and their biological duplicates |
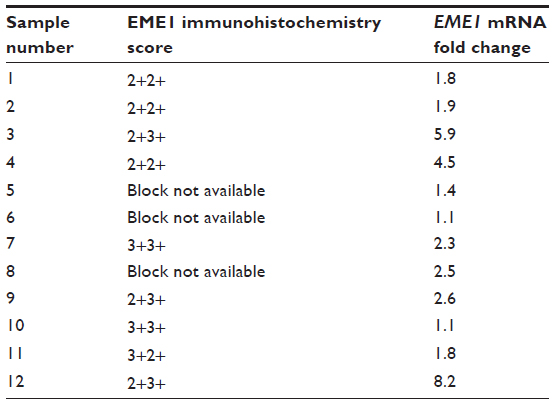 | Table S2 Summary of results of EME1 messenger (m)RNA and EME1 protein expression in each of the individual esophageal adenocarcinoma samples |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.