")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Gastric cancer combination therapy: synthesis of a hyaluronic acid and cisplatin containing lipid prodrug coloaded with sorafenib in a nanoparticulate system to exhibit enhanced anticancer efficacy and reduced toxicity
Authors Yang F, Li A, Liu H, Zhang H
Received 11 June 2018
Accepted for publication 1 September 2018
Published 4 October 2018 Volume 2018:12 Pages 3321—3333
DOI https://doi.org/10.2147/DDDT.S176879
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Feng Yang,1 Aimei Li,2 Han Liu,3 Hairong Zhang1
1Department of Gastroenterology, Affiliated Hospital of Jining Medical University, Jining 272029, Shandong, People’s Republic of China; 2Department of Anesthesiology, Affiliated Hospital of Jining Medical University, Jining 272029, Shandong, People’s Republic of China; 3Department of Gastroenterology, The First Affiliated Hospital of South China, Hengyang 421000, Hunan, People’s Republic of China
Purpose: Gastric cancer is one of the most common human epithelial malignancies, and using nanoparticles (NPs) in the diagnosis and treatment of cancer has been extensively studied. The aim of this study was to develop hyaluronic acid (HA) containing lipid NPs coloaded with cisplatin (CDDP) and sorafenib (SRF) for the treatment of gastric cancer.
Materials and methods: HA and CDDP containing lipid prodrug was synthesized using polyethylene glycol (PEG) as a linker (HA-PEG-CDDP). HA-PEG-CDDP and SRF were entrapped into the lipid NPs by nanoprecipitation method (H-CS-NPs). The physicochemical and biochemical properties such as size, zeta potential, and drug release pattern were studied. In vitro viability was also evaluated with MKN28 and SGC7901 human gastric cancer cells. In vivo testing including biodistribution and accumulation in tumor tissue was applied in gastric tumor-bearing mice to confirm the inhibition of gastric cancer.
Results: H-CS-NP has a particle size of 173.2±5.9 nm, with a zeta potential of -21.5±3.2 mV. At day 21 of in vivo treatment, H-CS-NPs inhibited the tumor volume from 1,532.5±41.3 mm3 to 259.6±16.3 mm3 with no obvious body weight loss. In contrast, mice treated with free drugs had body weight loss from 20 to 15 g at the end of study.
Conclusion: The results indicate that H-CS-NPs enhanced the antitumor effect of drugs and reduced the systemic toxicity effects. It could be used as a promising nanomedicine for gastric cancer combination therapy.
Keywords: nanocarriers, chemotherapy, polyethylene glycol, enhanced permeability and retention effect, reticuloendothelial system
Introduction
Gastric cancer is one of the most common human epithelial malignancies and remains the second leading cause of cancer-associated mortality for clinical cancer.1 To enhance the therapeutic efficiency for gastric cancer, chemotherapy or radiotherapy is often used combined with surgery.2 However, these treatments may inhibit normal cell growth and cause unexpected systemic toxicity. Also, the failure of cancer treatment could be caused by drug resistance, thus leading to low quality of life.3 Additionally, combinations of cytotoxic agents, including cisplatin, docetaxel, irinotecan, capecitabine, and oxaliplatin, have also been reported to prolong survival.4 However, therapeutic efficacy is still limited by two major factors: drug resistance and side effects.5,6 Nowadays, using nanoparticles (NPs) in the diagnosis and treatment of cancer has been extensively studied.7 Anticancer drug-loaded NP drug delivery systems have been extensively utilized and have become an important research area in gastric cancer therapy.8
Novel drug delivery strategies are applied by using nanotechnology in the anticancer therapy field. However, nanomedicines still face several obstacles, including the amount of drugs loaded may be low due to the physicochemical properties of nanocarriers; the release of drugs may not be ideally controlled after administration; and the drugs may be instable in the blood circulation.9 Therefore, prodrug approaches were widely applied to alleviate these disadvantages. Among all the nanosystems, lipid prodrug strategy has received considerable attentions.10
Hyaluronic acid (HA), a polyanionic glycol amino glycan with repetitive disaccharide units of D-glucuronic acid and D-N-acetylglucosamine, has been identified as a potent ligand to target tumor cells that overexpress CD44.11,12 Gastric cancer cells are reported to have CD44 expression and could be used as a receptor for HA targeting.13 Thus, HA-decorated NPs were applied for ligand-receptor-targeted gastric cancer therapy.14 Zhang et al developed an HA containing prodrug and loaded in NPs for synergistic combination therapy of lung cancer.15 Results illustrated that the targeted ability of HA helped the system to enhance the antitumor effects and reduce the systemic toxicity.
Sorafenib (SRF) is an anticancer drug, which is mainly used in the treatment of gastric cancers.16 SRF is a tyrosine kinase inhibitor, which inhibits proliferation, angiogenesis, and invasion of tumor cells. SRF has been administered in NP encapsulation and demonstrated better antitumor effect compared with that of free drug counterpart.17,18 The combination of SRF with chemotherapy is well-tolerated and is associated with encouraging response rates in several malignances.19 SRF plus cisplatin (CDDP) was evaluated in terms of the safety, pharmacokinetics, and preliminary efficacy in patients with advanced gastric cancer by Yamada et al.20 Kudo et al also used SRF combined low-dose CDDP in patients with advanced hepatocellular carcinoma.21 CDDP containing prodrugs was designed in many researches.22–24 So in this study, SRF and CDDP prodrug combination was coloaded in the NPs.
In the present research, HA and CDDP containing lipid prodrug were synthesized using polyethylene glycol (PEG) as a linker (HA-PEG-CDDP). HA-PEG-CDDP and SRF were entrapped into the lipid NPs by nanoprecipitation method. Their physicochemical and biochemical properties such as size, zeta potential, and drug release pattern were studied. Their in vitro viability was also evaluated with MKN28 and SGC7901 human gastric cancer cells. In vivo testing including biodistribution and accumulation in tumor tissue was applied in gastric tumor-bearing mice to confirm the inhibition of gastric cancer.
Materials and methods
Chemicals and reagents
NH2-PEG-NH2 (molecular weight: 3,400) was purchased from Peng Sheng Biological Co., Ltd. (Shanghai, People’s Republic of China). Injectable HA was obtained from Bloomage Freda Biopharm Co., Ltd. (Jinan, People’s Republic of China). CDDP, SRF, and succinic anhydride were purchased from Sigma-Aldrich (St Louis, MO, USA). Egg phosphatidylcholine (PC) was obtained from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). Pluronic F68 was provided by BASF (Ludwigshafen, Germany). 1-Ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl), N-hydroxysuccinimide (NHS), and dimethyl sulfoxide (DMSO) were provided by Aladdin Reagent Database Inc (Shanghai, People’s Republic of China). FBS, DMEM, and MTT were purchased from Invitrogen Corporation (Carlsbad, CA, USA).
Synthesis of HA-PEG-CDDP
HA-PEG was first prepared by amidation of the carboxyl groups of HA with the amine groups of PEG (Figure 1A).25 HA, PEG, and CDDP were dissolved in DMSO separately. EDC·HCl and NHS were added into HA solution and stirred for 1 hour at room temperature (RT) (mixture A). HA-PEG was obtained by adding PEG solution dropwise into mixture A and stirred for 12 hours at RT. H2O2 was then added to CDDP solution to get CDDP-(OH)2. Succinic anhydride was added to CDDP-(OH)2 and stirred at 50°C for 12 hours to obtain CDDP-(COOH)2 (Figure 1A).26 Then, EDC·HCl and NHS were added into CDDP-(COOH)2 solution to get mixture B. Mixture B was added to HA-PEG, stirred for 24 hours at RT, and then dialyzed against water for 12 hours. HA-PEG-CDDP was finally obtained by lyophilization (Figure 1A). The chemical structure of HA-PEG-CDDP was determined by 1H-NMR analysis in the solvent of DMSO-d6.
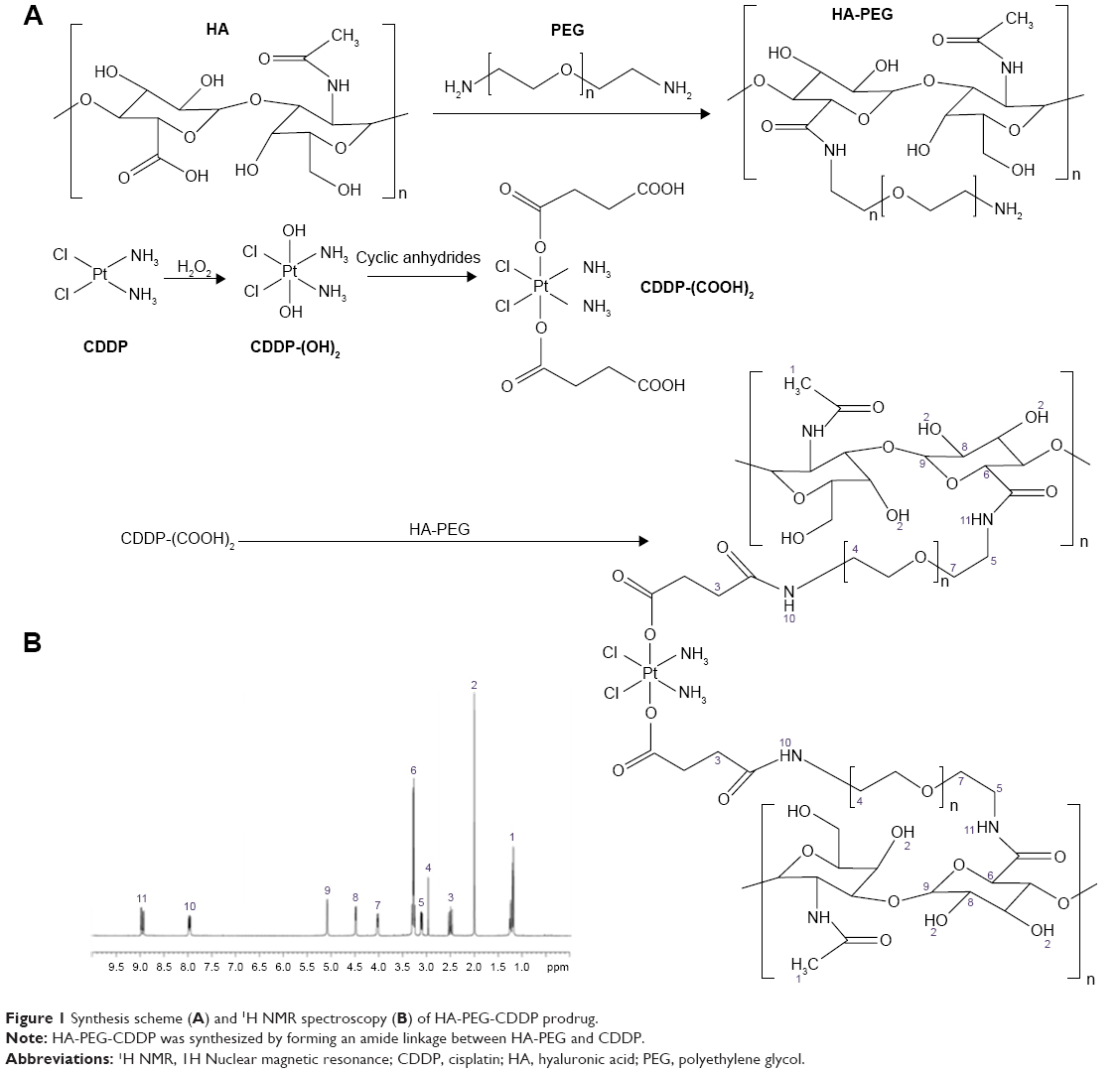 | Figure 1 Synthesis scheme (A) and 1H NMR spectroscopy (B) of HA-PEG-CDDP prodrug. |
NPs preparation
NPs were prepared utilizing a nanoprecipitation technique.27 Briefly, SRF (50 mg) and PC (200 mg) were dissolved together in acetone (10 mL) as the organic phase. The aqueous phase was formed by dissolving HA-PEG-CDDP (200 mg) in F68 solution (1%, w/v). Ten milliliter of the organic phase was added into 100 mL of aqueous phase drop by drop and homogenized (22,000 rpm, 30 seconds). The organic solvent was evaporated by stirring the suspension for 12 hours at RT. Then suspension was centrifuged at 15,000 rpm for 15 minutes to collect the NPs, washed twice with purified water, and lyophilized at −40°C for 24 hours to get HA-PEG-CDDP and SRF coloaded NPs (H-CS-NPs, Figure 2A). The lyophilized H-CS-NPs were stored at 4°C until use. For non-HA-contained CDDP and SRF-coloaded NPs (CS-NPs), PEG-CDDP was added into the aqueous phase instead of HA-PEG-CDDP. For single HA-PEG-CDDP-loaded NPs (H-C-NPs), SRF was not added into the organic phase. For single SRF-loaded NPs (H-S-NPs), HA-PEG was added into the aqueous phase instead of HA-PEG-CDDP. For blank NPs (NPs), SRF was not added into the organic phase, and HA-PEG was added into the aqueous phase instead of HA-PEG-CDDP.
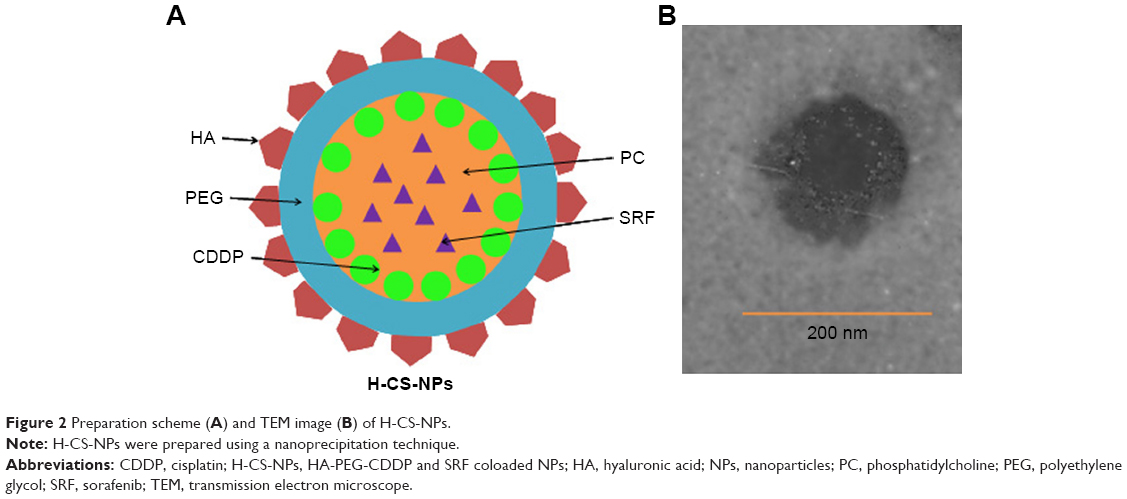 | Figure 2 Preparation scheme (A) and TEM image (B) of H-CS-NPs. |
NPs characterization
Particle size and zeta potential of NPs were determined in purified water using a Zetasizer Nano ZS system (Malvern Instruments, Malvern, UK). Particle size and morphology of H-CS-NPs were observed by transmission electron microscope (TEM) with a JEOL electron microscope (Tokyo, Japan).28
The amount of CDDP in the NPs was tested by an Inductively Coupled Plasma Optical Emission Spectrometer (ICP-OES, PerkinElmer Inc, Waltham, MA, USA).29 The amount of SRF was measured by ultraviolet–visible spectroscopy spectrophotometer at a wavelength of 267 nm.30 The drug encapsulation efficiency (EE) as well as the loading capacity (LC) of NPs were calculated by the formulas:
|
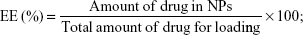
|
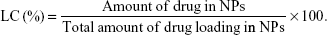
Stability of the NPs
Various kinds of NPs were placed in Cl (NaCl 150 mM) and FBS (10%) cocontaining PBS solution separately.31 The particle size and EE were monitored to evaluate the stability of NPs.
In vitro drug release
CDDP and/or SRF-loaded NPs were placed in separate dialysis bags with a molecular mass cutoff of 1 kDa.32 The dialysis bags were incubated in 20 mL of PBS (pH =7.4) at 37°C with gentle shaking (100 rpm), and the incubation buffer was collected and replaced by fresh incubation buffer at every designated time point. The collected incubation buffer containing the released drugs was analyzed by the methods mentioned in Section “NPs characterization.”
Cells and animals
Human gastric cancer cell lines: MKN28 and SGC7901 cells were purchased from ScienCell Research Laboratories (San Diego, CA, USA) and were cultured in Roswell Park Memorial Institute medium with 10% FBS, and 10% penicillin at 37°C, 5% CO2.
BALB/cA nude mice (6–8 weeks old) were purchased from Shandong University Laboratory Animal Center (Jinan, People’s Republic of China). The study protocol was approved by the Committee for the Care and Use of Laboratory Animals of Jining Medical University (No JNMU20180116-001) and followed the National Institutes of Health guide for the care and use of laboratory animals.
In vitro cytotoxicity
MKN28 and SGC7901 cells were cultured overnight (3,000 cells/well) in a 96-well plate, and cytotoxicity was assessed by the MTT assay.33 The cells were added to each well, incubated at 37°C for 6 hours, and followed by replacement of the solution with complete growth medium. After cell attachment and growth resumption, growth media were replaced with various concentrations of CDDP and/or SRF-loaded NPs, free CDDP plus SRF (free CS), free CDDP (free C), and free SRF (free S). Control cells were added with equivalent volume of fresh media. After 48 hours, medium was replaced with 20 mL of MTT solution (5 mg/mL), followed by incubation for another 4 hours at 37°C. Then the medium was replaced by 100 mL of DMSO. Absorbance of the solution was measured at 560 nm.
In vivo blood analysis and tissue distribution
MKN28 cells (107) were injected subcutaneously in the dorsal skin of mice to obtain the gastric cancer model.34 When tumor size reached 100 mm3, CDDP and/or SRF-loaded NPs, free CDDP, and/or SRF were administered via the tail vein of mice at a dose of 5 mg drugs per kg body weight. After 3 weeks, mice were killed and blood samples were collected into heparinized tubes. Blood was centrifuged at 15,000 rpm for 20 minutes at 4°C to isolate plasma, which was immediately flash frozen in liquid nitrogen until processing. Clinical chemical parameters, including alanine transaminase (ALT), creatine phosphokinase (CPK), lactate dehydrogenase (LDH), and blood urea nitrogen (BUN) were assayed.35 The plasma was decanted and serum levels of ALT, CPK, LDH, and BUN with an autoanalyzer (COBAS C111, Roche Diagnostics, Basel, Switzerland) were measured to obtain various biochemical data. Then, tissue including tumor, liver, spleen, kidney, stomach, colon, heart, and lung were collected and weighed.
In vivo antitumor activity and toxicity
MKN28 cell-induced gastric cancer model was used for the evaluation of antitumor activity and toxicity. When tumor size reached 100 mm3, the mice were treated five times at 3-day intervals with CDDP and/or SRF-loaded NPs, free CDDP, and/or SRF via the tail vein of mice at a dose of 5 mg drugs per kg body weight.36 The antitumor activity was evaluated in terms of the tumor size, which was estimated by the following formula below: V = (major and minor axes)×(minor axes of the tumor)2/2.
The physical conditions and body weight change of mice were monitored for 3 weeks to evaluate the in vivo toxicity during the treatment.
Statistical analysis
All the results were expressed as mean ± SD from at least three independent runs. Comparisons between groups were made using Student’s t test and with more than two groups, ANOVA was used to compare results. Statistical significance was set at P<0.05. All statistical analysis was performed using SPSS, version 21.0.
Results
Characterization of HA-PEG-CDDP
The 1H NMR spectrum of HA-PEG-CDDP was analyzed and marked one by one in Figure 1B. The peaks at 1.26 (1), 1.93 (2), 3.26 (6), 4.47 (8), and 5.05 (9) ppm belong to the −CH3, −OH, and sugar rings of HA, respectively. The peaks at 2.91 (4) and 3.98 (7) ppm are the protons of PEG. The peaks of 7.98 (10) and 8.93 (11) ppm belong to the −NH in the amide linkage. The peaks of 2.48 (3), 2.91(4), 3.07 (5), and 3.26 (6) ppm are the characteristic peaks of the −CH2 and −CH next to the amide linkage. These peaks could prove the existence of the amide linkage.
Characterization of NPs
The H-CS-NP has a particle size of 173.2±5.9 nm, with a zeta potential of –21.5±3.2 mV (Table 1). The size of CS-NPs (118.3±5.1 nm) is smaller than H-CS-NPs. The zeta potential of CS-NPs changed to –8.4±0.9 mV, which may be explained by the anionic HA on the surface of the NPs, thus brought about more negative charges to the system. Blank NPs exhibit similar size and zeta potential as drug-loaded NPs, means the loading of drugs did not enlarge the size and influence the surface charge. The CDDP and SRF EE were measured to be over 80%. TEM images revealed that H-CS-NPs had a spherical shape with obvious coat on the outer layer (Figure 2B). This may be produced by the HA coating on the NPs. The size of H-CS-NPs is <200 nm according to the bar.
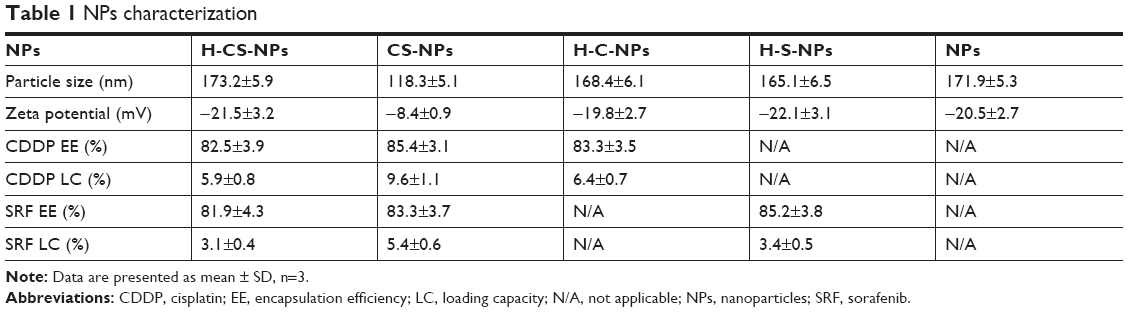 | Table 1 NPs characterization |
Serum stability of NPs
To investigate the serum stability of NPs, the size and EE change of the particles were measured along with the time. It was found that the average sizes of the NPs remained unchanged during the test (Figure 3A), this phenomenon means the particles that did not aggregate in the serum and the serum proteins were also not adsorbed on the NP surfaces. The constant EE during the tested time could be the evidence of the stable drug encapsulation capacity of the NPs constructed (Figure 3B and C).
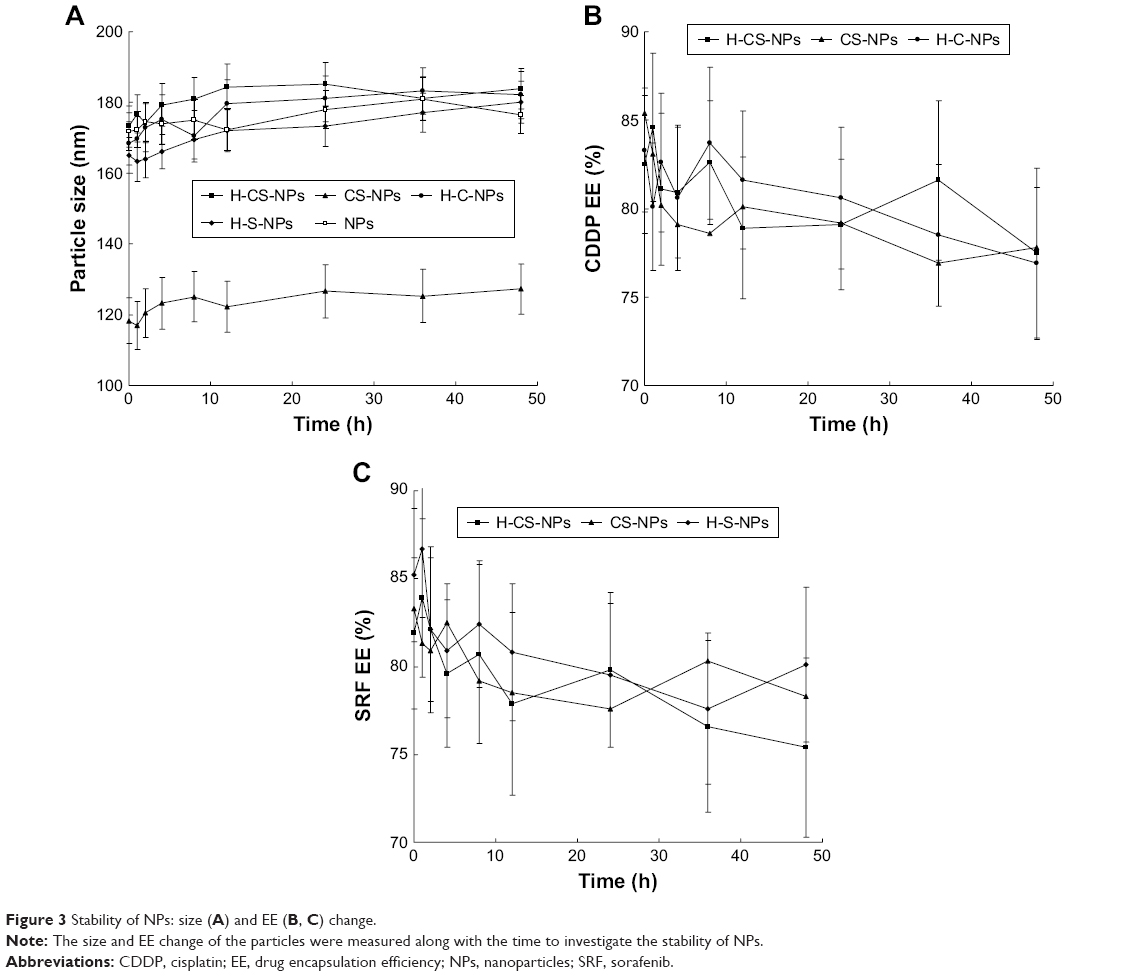 | Figure 3 Stability of NPs: size (A) and EE (B, C) change. |
In vitro drug release
The in vitro CDDP and SRF release profile of NPs showed that the release of drugs from various kinds of NPs all followed a sustained manner (Figure 4). However, the release rates of CDDP and SRF are different. CDDP had faster release than SRF, nearly complete drug releases were achieved by CDDP and SRF at 36 and 48 hours, respectively. These results may be caused by the different position of drugs loaded in the NPs, which will be discussed later.
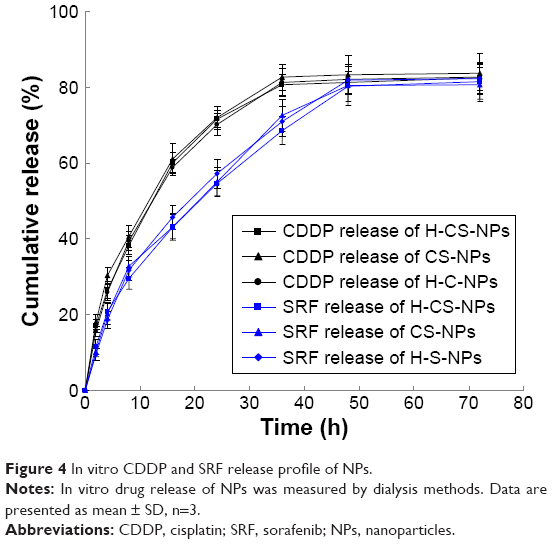 | Figure 4 In vitro CDDP and SRF release profile of NPs. |
In vitro cytotoxicity
Cytotoxicity of drug-loaded NPs and free drugs were evaluated on two kinds of gastric cancer cell lines. Blank NPs did not show obvious toxicity against both MKN28 and SGC7901 cells (Figure 5). All the drugs containing groups exerted concentration-dependent cytotoxicity on gastric cancer cells. On MKN28 cells, H-CS-NPs showed obvious enhanced cytotoxic activity compared with CS-NPs, H-C-NPs, and H-S-NPs (P<0.05). Free CS exhibited lower cytotoxicity than H-CS-NPs and CS-NPs (P<0.05). Similar results were found in SGC7901 cells.
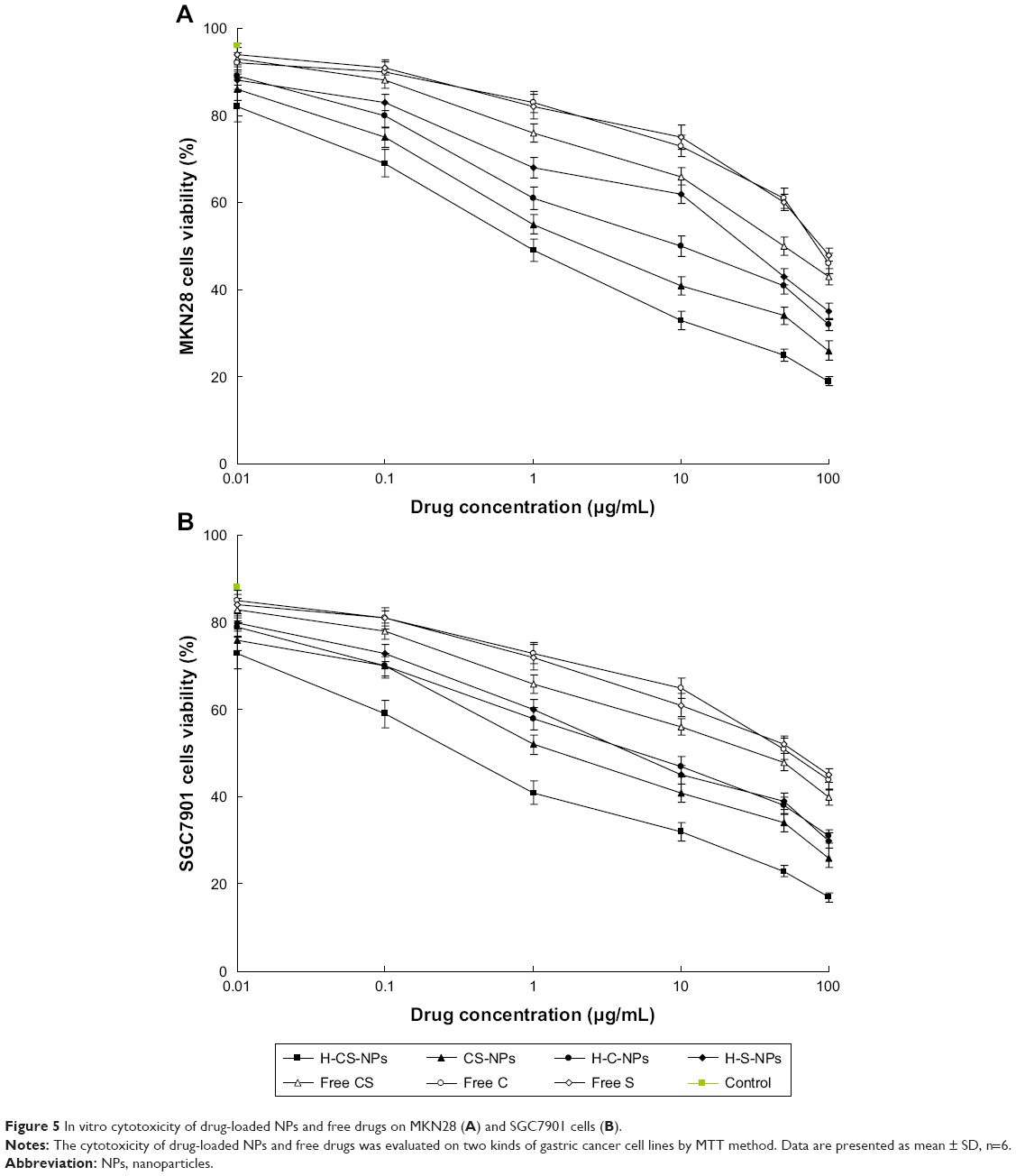 | Figure 5 In vitro cytotoxicity of drug-loaded NPs and free drugs on MKN28 (A) and SGC7901 cells (B). |
In vivo blood analysis
Blood analysis was performed to analyze the clinical chemical parameters (Figure 6). The differences in ALT, CPK, LDH, and BUN between the NPs groups were not statistically significant (P>0.05). While significantly lower blood enzyme levels than free drug groups suggested the lower toxicity of drug-loaded NPs treatment in vivo (P<0.05).
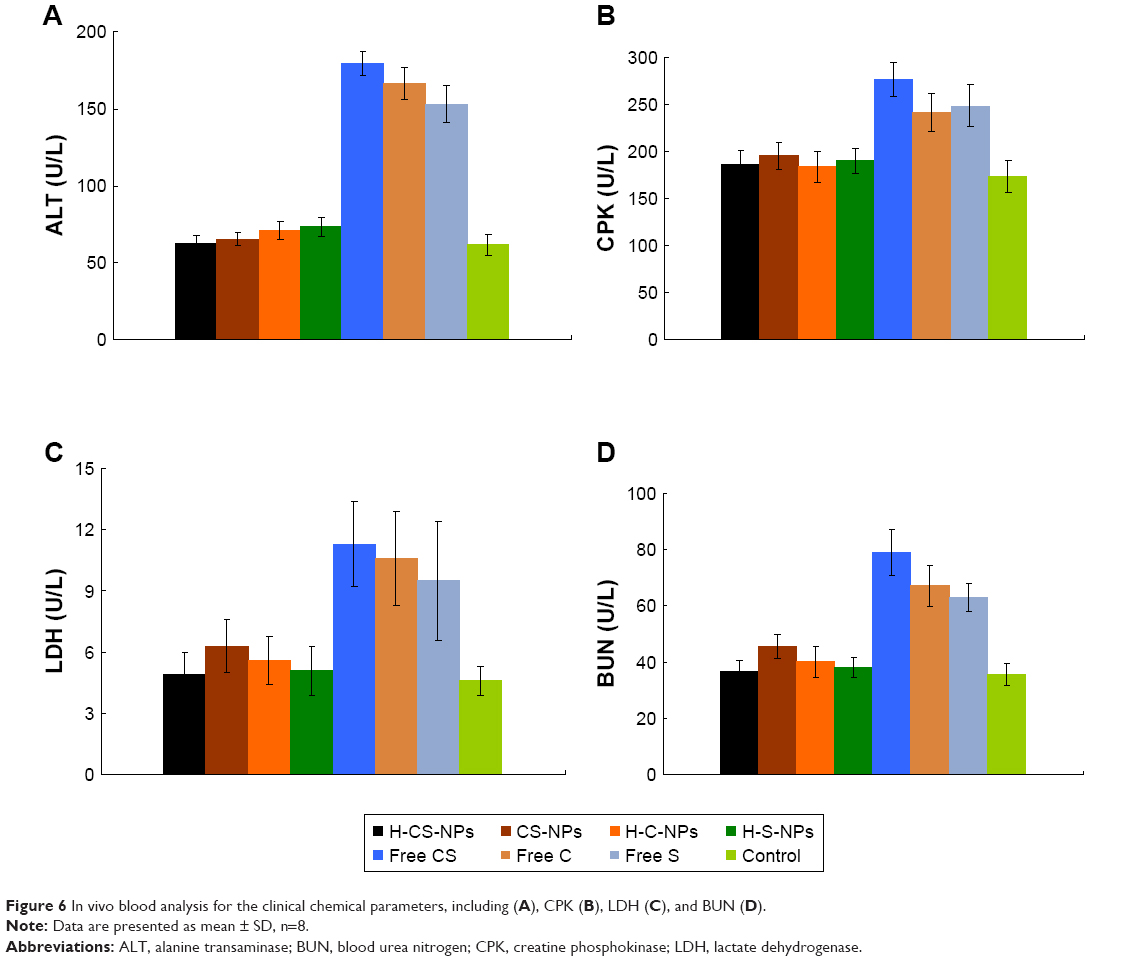 | Figure 6 In vivo blood analysis for the clinical chemical parameters, including (A), CPK (B), LDH (C), and BUN (D). |
In vivo tissue distribution
Figure 7 exhibited the in vivo tissue distribution results. Compared with free CS, higher accumulation of CDDP and SRF was observed in the tumor, liver, and spleen for H-CS-NPs and CS-NPs (P<0.05). The heart and kidney drug distribution of free CS was higher than H-CS-NPs and CS-NPs (P<0.05). However, no significant increase of CDDP and SRF accumulation was observed in most normal tissues for NPs groups except liver and spleen. More important, H-CS-NPs showed obvious higher distribution compared with CS-NPs in the tumor (P<0.05).
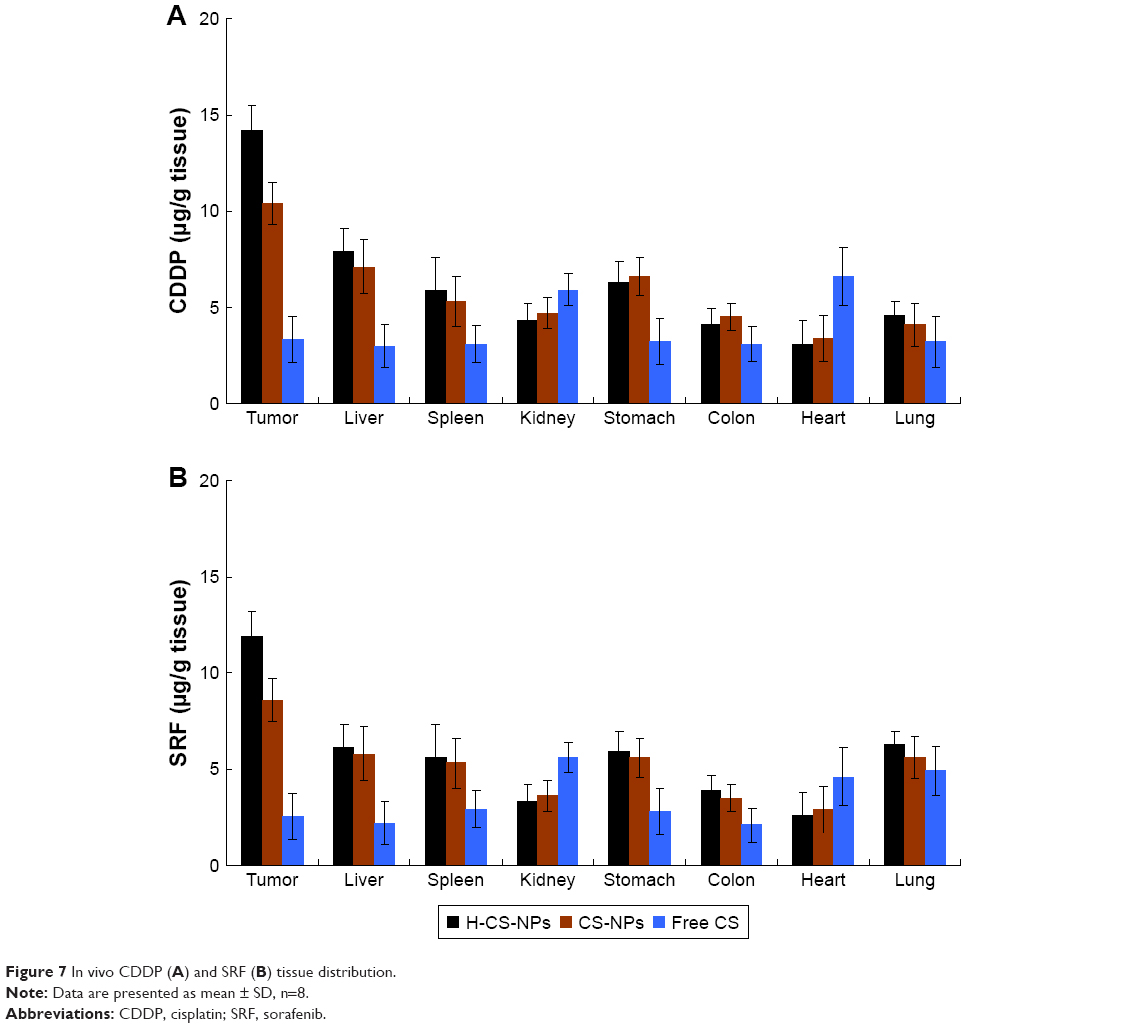 | Figure 7 In vivo CDDP (A) and SRF (B) tissue distribution. |
In vivo antitumor activity and toxicity
Although both CDDP and/or SRF-loaded NPs, free CDDP, and/or SRF showed in vivo antitumor activity in gastric tumor bearing (Figure 8A), the most significant in vivo antitumor activity was presently observed when H-CS-NPs was used (P<0.05). At day 21, H-CS-NPs inhibited tumor volume from 1,532.5±41.3 mm3 to 259.6±16.3 mm3. This inhibition efficiency was remarkably better than CS-NPs (412.3±26.5 mm3) and free CS (846.4±33.6 mm3) (P<0.05). As shown in Figure 8B, the mice treated with free drugs had body weight loss from 20 to 15 g at day 21. In contrast, there was no obvious body weight loss in NPs groups. No animal death was observed in all the tested groups.
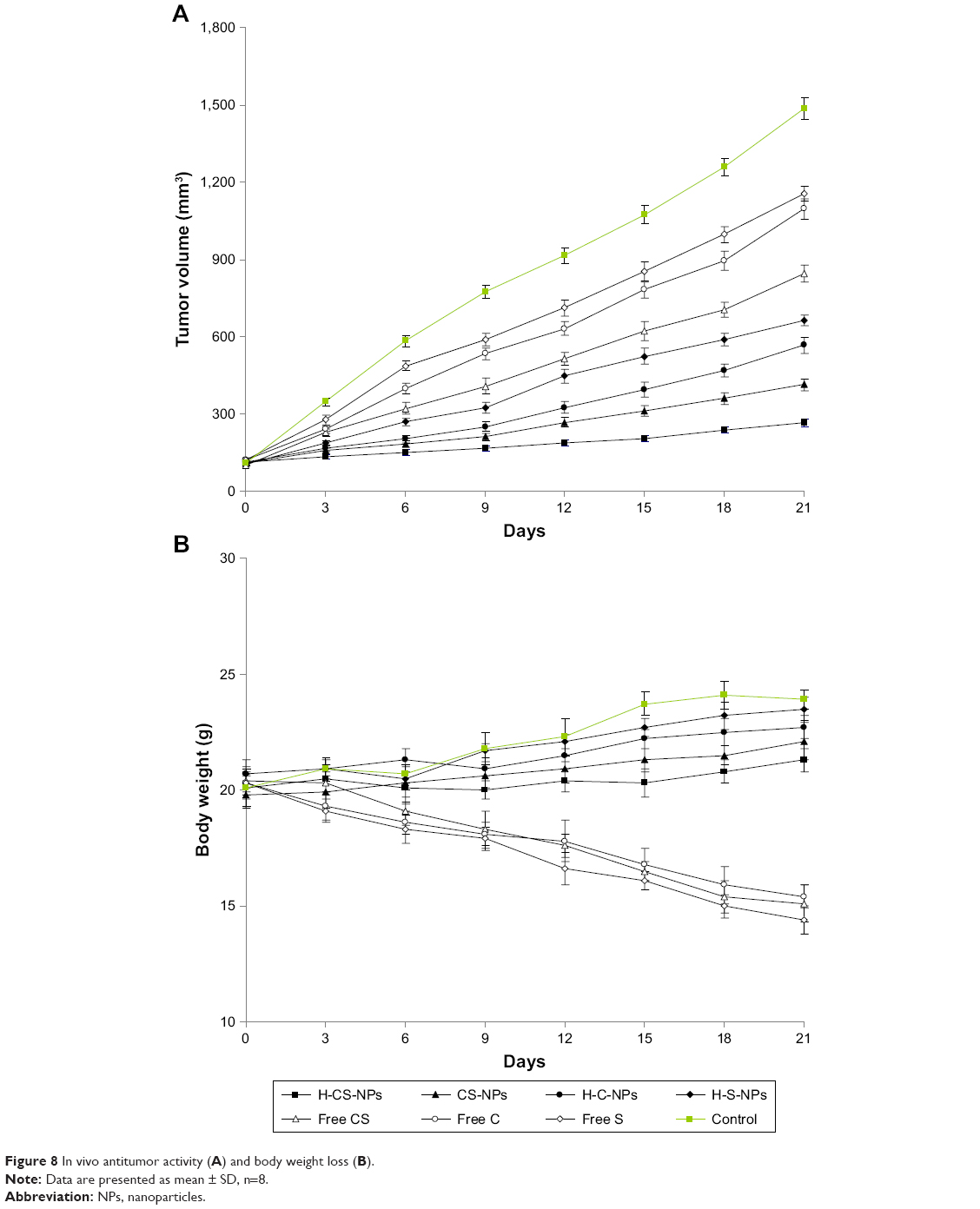 | Figure 8 In vivo antitumor activity (A) and body weight loss (B). |
Discussion
With the rapid development of nanoscience and technology, nanoparticulate drug delivery systems have been widely used in health care applications, especially for highly efficient drug delivery of chemotherapeutic agents.37 Macromolecular prodrugs are among the most studied nanomedicines, several of which are currently in clinical trials.38 However, macromolecular prodrugs usually display inferior tumor accumulation and low stability, thus hindering their applications. So nanocarriers need to cooperate with prodrugs to get a better therapeutic effect.
This research begins with the synthesis of HA and CDDP containing lipid prodrug using PEG as a linker (HA-PEG-CDDP). HA is a natural polysaccharide, which plays an important role in the tumorigenesis process.39 Strong affinity to CD44 receptors overexpressed on different cancer cells as well as cancer stem cells made HA an ideal tumor-targeted ligand. Among polymers commonly used for drug conjugation, PEG is currently considered as the most successful and promising modifier.40 Through PEG modification, the physicochemical and biopharmaceutical properties of drugs might be greatly improved, thus resulting in enhancement of therapeutic efficacy and reduction in side effects. In this study, the peaks of 2.48, 2.91, 3.07, and 3.26 ppm are the characteristic peaks which could prove the existence of the amide linkage.
An NP system is expected to be stable during circulation in the bloodstream for the good drug delivery capacity.41 It was found that the average sizes of the NPs remained unchanged during the test, this phenomenon means the particles did not aggregate in the serum and the serum proteins were also not adsorbed on the NPs surfaces. The constant EE during the tested time could be the proof of the stable drug encapsulation capacity of the NPs constructed. It may also be concluded that the PEG on the surface of the NPs acted as the stabilizer.42
The sustained release of CDDP and SRF is due to the lipid NPs core and the PEG shell. To be noticed, CDDP had faster release than SRF, nearly complete drug releases were achieved by CDDP and SRF at 36 and 48 hours, respectively. These results may be caused by the different position of drugs loaded in the NPs. This may due to the hydrophilic HA and PEG that make the HA-PEG-CDDP prodrugs present nearer to the surface than SRF. The HA-PEG shell and lipid core through which the drugs are released may provide a sustained release of drug molecules at the tumor site. This result will prevent the total drug release in the bloodstream and promote the drug release in endosomes or lysosomes in cancer cells.43
The anticancer ability of drug-loaded NPs was demonstrated by evaluating cell viability and tumor xenograft growth in cultured gastric cancer cell lines in vitro and gastric tumor xenograft mice model in vivo. In vitro cytotoxicity of drug-loaded NPs and free drugs was evaluated on two kinds of gastric cancer cell lines: MKN28 and SGC7901 cells. Similar results were found in both cell lines. H-CS-NPs showed the highest tumor cell inhibition ability by inhibiting the viability of the cancer cell lines at low concentrations, which is significantly lower than other samples tested.44 Higher toxicity of H-CS-NPs in comparison with CS-NPs may be attributed to the attendance of HA that provided the targeted ability to the system. In contrast, free drug formulations illustrated poorer in vitro antitumor efficiency than the drug(s) encapsulated NPs. This may be explained by the protection effect of NPs that could reduce the degradation of drugs in blood circulation and increase the amount of drugs accumulated within the tumor cells and showed better cytotoxicity. In vivo antitumor study, H-CS-NPs inhibited tumor volume to 259.6±16.3 mm3, which was remarkably better than CS-NPs (412.3±26.5 mm3) and free CS (846.4±33.6 mm3). In vivo results suggest a remarkably increased therapeutic efficacy of CDDP and/or SRF-loaded NPs in gastric tumor-bearing mice compared with free CDDP and/or SRF. H-CS-NPs showed obvious better tumor inhibition efficiency than H-C-NPs and H-S-NPs, this may due to the synergistic effect of the two drugs. The in vivo antitumor results are in accordance with the in vitro results, proved the promising efficacy of H-CS-NPs.
Compared with free CS, higher CDDP and SRF accumulation of H-CS-NPs and CS-NPs were observed in the reticuloendothelial system (RES) active tissue: liver and spleen. This result indicated that some of the NPs samples were trapped by the RES after systemic administration.45 Significantly higher accumulations of H-CS-NPs and CS-NPs than free CS tumor tissue were noticed, which may be explained by the enhanced permeability and retention effect on the tumor site that let the NPs accumulated easily in the tumor. Blood analysis was performed to analyze the clinical chemical parameters. Blood enzyme levels were measured at day 21 (3 weeks after first treatment).46 The differences in blood enzyme activities among the NPs or the free drug treatment groups were not statistically significant, while the trends in blood enzyme levels obviously suggested lower toxicity of NPs than free drug treatment in tumor models. The mice treated with NP samples showed no significant body weight loss, whereas free drug administration groups exhibited over 25% body weight loss at the end of study (day 21). There is no obvious difference on behaviors of mice treated with NPs compared with the control group. However, considerably less active and weaker movements were found on the mice treated with free drugs. The above results indicate that the NPs enhanced the antitumor effect of drugs and reduced the systemic toxicity effects in vivo.
Conclusions
HA and CDDP containing lipid prodrug were synthesized and coloaded with SRF into lipid NPs. H-CS-NPs have a small size and high drug-loading efficacy. H-CS-NPs showed obvious better tumor inhibition efficiency than H-C-NPs and H-S-NPs in vivo, this may due to the synergistic effect of the two drugs. Higher toxicity of H-CS-NPs in comparison with CS-NPs may be the evidence of the targeted ability of HA that enhanced the efficiency of the system. Blood enzyme levels obviously suggested lower toxicity of NPs than free drug treatment in tumor models. The mice treated with NP samples showed no significant body weight loss, indicating that the NPs could enhance the antitumor effect of drugs and reduce the systemic toxicity effects in vivo.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhang K, du X, Yu K, Zhang K, Zhou Y. Application of novel targeting nanoparticles contrast agent combined with contrast-enhanced computed tomography during screening for early-phase gastric carcinoma. Exp Ther Med. 2018;15(1):47–54. | ||
Lee JH, Kim JG, Jung HK, et al. Clinical practice guidelines for gastric cancer in Korea: an evidence-based approach. J Gastric Cancer. 2014;14(2):87–104. | ||
Xin J, Wang S, Wang B, et al. AlPcS4-PDT for gastric cancer therapy using gold nanorod, cationic liposome, and Pluronic® F127 nanomicellar drug carriers. Int J Nanomedicine. 2018;13:2017–2036. | ||
Liu D, Li X, Chen C, et al. Target-specific delivery of oxaliplatin to HER2-positive gastric cancer cells in vivo using oxaliplatin-au-fe3o4-herceptin nanoparticles. Oncol Lett. 2018;15(5):8079–8087. | ||
Dank M, Zaluski J, Barone C, et al. Randomized phase III study comparing irinotecan combined with 5-fluorouracil and folinic acid to cisplatin combined with 5-fluorouracil in chemotherapy naive patients with advanced adenocarcinoma of the stomach or esophagogastric junction. Ann Oncol. 2008;19(8):1450–1457. | ||
Cunningham D, Starling N, Rao S, et al. Upper Gastrointestinal Clinical Studies Group of the National Cancer Research Institute of the United Kingdom. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008;358(1):36–46. | ||
Nasr R, Hasanzadeh H, Khaleghian A, Moshtaghian A, Emadi A, Moshfegh S. Induction of apoptosis and inhibition of invasion in gastric cancer cells by titanium dioxide nanoparticles. Oman Med J. 2018;33(2):111–117. | ||
Xin L, Zhang HT, Yang WF, Li YF, Liu C. Evaluation of METase-pemetrexed-loaded PEG-PLGA nanoparticles modified with anti-CD133-scFV for treatment of gastric carcinoma. Biosci Rep. 2018;38(1):BSR20171001. | ||
Mura S, Bui DT, Couvreur P, Nicolas J. Lipid prodrug nanocarriers in cancer therapy. J Control Release. 2015;208:25–41. | ||
Liu B, Han L, Liu J, Han S, Chen Z, Jiang L. Co-delivery of paclitaxel and TOS-cisplatin via TAT-targeted solid lipid nanoparticles with synergistic antitumor activity against cervical cancer. Int J Nanomedicine. 2017;12:955–968. | ||
Qu CY, Zhou M, Chen YW, Chen MM, Shen F, Xu LM. Engineering of lipid prodrug-based, hyaluronic acid-decorated nanostructured lipid carriers platform for 5-fluorouracil and cisplatin combination gastric cancer therapy. Int J Nanomedicine. 2015;10:3911–3920. | ||
Cho HJ, Yoon HY, Koo H, et al. Self-assembled nanoparticles based on hyaluronic acid-ceramide (HA-CE) and Pluronic® for tumor-targeted delivery of docetaxel. Biomaterials. 2011;32(29):7181–7190. | ||
Ghaffarzadehgan K, Jafarzadeh M, Raziee HR, et al. Expression of cell adhesion molecule CD44 in gastric adenocarcinoma and its prognostic importance. World J Gastroenterol. 2008;14(41):6376–6381. | ||
Gao Z, Li Z, Yan J, Wang P, Irinotecan WP. Irinotecan and 5-fluorouracil-co-loaded, hyaluronic acid-modified layer-by-layer nanoparticles for targeted gastric carcinoma therapy. Drug Des Devel Ther. 2017;11:2595–2604. | ||
Zhang R, Ru Y, Gao Y, Li J, Mao S. Layer-by-layer nanoparticles co-loading gemcitabine and platinum (IV) prodrugs for synergistic combination therapy of lung cancer. Drug Des Devel Ther. 2017;11:2631–2642. | ||
Li T, Zhang Y, Meng YP, Bo LS, Ke WB. miR-542-3p appended sorafenib/all-trans retinoic acid (ATRA)-loaded lipid nanoparticles to enhance the anticancer efficacy in gastric cancers. Pharm Res. 2017;34(12):2710–2719. | ||
Yang YC, Cai J, Yin J, Zhang J, Wang KL, Zhang ZT. Heparin-functionalized pluronic nanoparticles to enhance the antitumor efficacy of sorafenib in gastric cancers. Carbohydr Polym. 2016;136:782–790. | ||
Gao DY, Lin T, Sung YC, et al. CXCR4-targeted lipid-coated PLGA nanoparticles deliver sorafenib and overcome acquired drug resistance in liver cancer. Biomaterials. 2015;67:194–203. | ||
Sun W, Powell M, O’Dwyer PJ, Catalano P, Ansari RH, Benson AB. Phase II study of sorafenib in combination with docetaxel and cisplatin in the treatment of metastatic or advanced gastric and gastroesophageal junction adenocarcinoma: ECOG 5203. J Clin Oncol. 2010;28(18):2947–2951. | ||
Yamada Y, Kiyota N, Fuse N, et al. A phase I study of sorafenib in combination with S-1 plus cisplatin in patients with advanced gastric cancer. Gastric Cancer. 2014;17(1):161–172. | ||
Kudo M, Ueshima K, Yokosuka O, et al. Sorafenib plus low-dose cisplatin and fluorouracil hepatic arterial infusion chemotherapy versus sorafenib alone in patients with advanced hepatocellular carcinoma (SILIUS): a randomised, open label, phase 3 trial. Lancet Gastroenterol Hepatol. 2018;3(6):424–432. | ||
Mi Y, Zhao J, Feng SS. Vitamin E TPGS prodrug micelles for hydrophilic drug delivery with neuroprotective effects. Int J Pharm. 2012;438(1–2):98–106. | ||
Yoong SL, Wong BS, Zhou QL, et al. Enhanced cytotoxicity to cancer cells by mitochondria-targeting MWCNTs containing platinum(IV) prodrug of cisplatin. Biomaterials. 2014;35(2):748–759. | ||
Xiao H, Noble GT, Stefanick JF, et al. Photosensitive Pt(IV)-azide prodrug-loaded nanoparticles exhibit controlled drug release and enhanced efficacy in vivo. J Control Release. 2014;173:11–17. | ||
Tan S, Wang G. Lung cancer targeted therapy: folate and transferrin dual targeted, glutathione responsive nanocarriers for the delivery of cisplatin. Biomed Pharmacother. 2018;102:55–63. | ||
Song H, Wang R, Xiao H, et al. A cross-linked polymeric micellar delivery system for cisplatin (IV) complex. Eur J Pharm Biopharm. 2013;83(1):63–75. | ||
Yu K, Zhao J, Yu C, et al. Role of four different kinds of polyethylenimines (PEIs) in preparation of polymeric lipid nanoparticles and their anticancer activity study. J Cancer. 2016;7(7):872–882. | ||
Cui X, Liang T, Liu C, Yuan Y, Qian J. Correlation of particle properties with cytotoxicity and cellular uptake of hydroxyapatite nanoparticles in human gastric cancer cells. Mater Sci Eng C Mater Biol Appl. 2016;67:453–460. | ||
Cai L, Xu G, Shi C, Guo D, Wang X, Luo J. Telodendrimer nanocarrier for co-delivery of paclitaxel and cisplatin: a synergistic combination nanotherapy for ovarian cancer treatment. Biomaterials. 2015;37:456–468. | ||
Xiong Q, Cui M, Yu G, Wang J, Song T. Facile fabrication of reduction-responsive supramolecular nanoassemblies for co-delivery of doxorubicin and sorafenib toward hepatoma cells. Front Pharmacol. 2018;9:61. | ||
Ding Y, Zhai K, Pei P, et al. Encapsulation of cisplatin in a pegylated calcium phosphate nanoparticle (CPNP) for enhanced cytotoxicity to cancerous cells. J Colloid Interface Sci. 2017;493:181–189. | ||
Mi Y, Zhao J, Feng SS. Vitamin E TPGS prodrug micelles for hydrophilic drug delivery with neuroprotective effects. Int J Pharm. 2012;438(1–2):98–106. | ||
Nascimento AV, Singh A, Bousbaa H, Ferreira D, Sarmento B, Amiji MM. Overcoming cisplatin resistance in non-small cell lung cancer with Mad2 silencing siRNA delivered systemically using EGFR-targeted chitosan nanoparticles. Acta Biomater. 2017;47:71–80. | ||
Fan X, Zhao X, Qu X, Fang J. pH sensitive polymeric complex of cisplatin with hyaluronic acid exhibits tumor-targeted delivery and improved in vivo antitumor effect. Int J Pharm. 2015;496(2):644–653. | ||
Hu TM, Lee RP, Lee CJ, Subeq YM, Lin NT, Hsu BG. Heavy ethanol intoxication increases proinflammatory cytokines and aggravates hemorrhagic shock-induced organ damage in rats. Mediators Inflamm. 2013;2013:121786. | ||
Nishiyama N, Okazaki S, Cabral H, et al. Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res. 2003;63(24):8977–8983. | ||
Zhang S, Guan J, Sun M, et al. Self-delivering prodrug-nanoassemblies fabricated by disulfide bond bridged oleate prodrug of docetaxel for breast cancer therapy. Drug Deliv. 2017;24(1):1460–1469. | ||
Zhong Y, Goltsche K, Cheng L, et al. Hyaluronic acid-shelled acid-activatable paclitaxel prodrug micelles effectively target and treat CD44-overexpressing human breast tumor xenografts in vivo. Biomaterials. 2016;84:250–261. | ||
Yang C, Wang X, Yao X, Zhang Y, Wu W, Jiang X. Hyaluronic acid nanogels with enzyme-sensitive cross-linking group for drug delivery. J Control Release. 2015;205:206–217. | ||
Sun G, Lin X, Hong Y, Feng Y, Ruan K, Xu D. PEGylation for drug delivery to ischemic myocardium: pharmacokinetics and cardiac distribution of poly(ethylene glycol)s in mice with normal and ischemic myocardium. Eur J Pharm Sci. 2012;46(5):545–552. | ||
Hu Y, Hoerle R, Ehrich M, Zhang C. Engineering the lipid layer of lipid-PLGA hybrid nanoparticles for enhanced in vitro cellular uptake and improved stability. Acta Biomater. 2015;28:149–159. | ||
Bhimji S, Meneghini RM. Micromotion of cementless tibial baseplates: keels with adjuvant pegs offer more stability than pegs alone. J Arthroplasty. 2014;29(7):1503–1506. | ||
Shirakura T, Kelson TJ, Ray A, Malyarenko AE, Kopelman R. Hydrogel nanoparticles with thermally controlled drug release. ACS Macro Lett. 2014;3(7):602–606. | ||
Song Z, Shi Y, Han Q, Dai G. Endothelial growth factor receptor-targeted and reactive oxygen species-responsive lung cancer therapy by docetaxel and resveratrol encapsulated lipid-polymer hybrid nanoparticles. Biomed Pharmacother. 2018;105:18–26. | ||
Yu T, Hubbard D, Ray A, Ghandehari H. In vivo biodistribution and pharmacokinetics of silica nanoparticles as a function of geometry, porosity and surface characteristics. J Control Release. 2012;163(1):46–54. | ||
Shuhendler AJ, Prasad P, Zhang RX, et al. Synergistic nanoparticulate drug combination overcomes multidrug resistance, increases efficacy, and reduces cardiotoxicity in a nonimmunocompromised breast tumor model. Mol Pharm. 2014;11(8):2659–2674. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.