")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 11
Gastric adenocarcinoma and proximal polyposis of the stomach: diagnosis and clinical perspectives
Authors Rudloff U
Received 13 September 2018
Accepted for publication 6 November 2018
Published 3 December 2018 Volume 2018:11 Pages 447—459
DOI https://doi.org/10.2147/CEG.S163227
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Koulaouzidis
Udo Rudloff
Rare Tumor Initiative, Pediatric Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA
Abstract: Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS) is a recently described, rare gastric polyposis syndrome. It is characterized by extensive involvement of the fundus and body of the stomach with fundic gland polyps sparing the antrum and lesser curvature, an autosomal dominant inheritance pattern with incomplete penetrance, and a significant predisposition for the development of gastric adenocarcinoma. Due to the recent discovery of APC promotor IB mutations (c.-191T>C, c.-192A>G, and c.-195A>C), which reduce binding of the transcription factor Yin Yang 1 (YY1) and transcriptional activity of the promotor, as its underlying genetic perturbation, GAPPS has been added to the growing molecular class of APC-associated disorders. Recent reports on family members afflicted by gastric polyposis due to GAPPS have described the development of metastatic cancer or the presence of invasive gastric adenocarcinoma in total gastrectomy specimens after variable periods of endoscopic surveillance emphasizing the need for an improved understanding of the to-date poorly characterized natural history of the syndrome. There are, however, currently no guidelines on screening, timing of prophylactic gastrectomy, or endoscopic surveillance for GAPPS available. In this review, we summarize the clinical, pathological, and genetic aspects of GAPPS as well as management approaches to this rare cancer predisposition syndrome, highlighting the need for early recognition, a multidisciplinary approach, and the creation of prospective family registries and consensus guidelines in the near future.
Keywords: gastrointestinal polyposis, fundic gland polyp, APC promotor IB variant, APC-associated disorder, gastric adenocarcinoma predisposition
Diagnosis
In 2012, Worthley et al reported in three families the clinicopathological features of a novel gastric polyposis syndrome termed gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS).1 The authors described in a large Australian family and two smaller families from USA and Canada that multiple family members afflicted by fundic gland polyposis (>100 polyps carpeting the fundus and body of the stomach sparing the antrum and lesser curvature in the index proband) multiple cases of intestinal-type adenocarcinoma of the stomach arising in regions of fundal gland polyposis with high-grade dysplasia and adenomatous polyps. Within the same issue, researchers from Japan described two additional families with a similar phenotype.2 Genetic testing for known gastrointestinal (GI) polyposis and gastric cancer predisposition syndromes was negative. Together with an autosomal dominant inheritance pattern, the authors proposed a series of endoscopic and pathological criteria for the diagnosis of GAPPS, which are listed in Table 1.1
 | Table 1 Diagnostic criteria for GAPPS Notes: aGastric polyposis syndromes: MAP, GJPS, PJS, and Cowden syndrome. bIn patients on PPI to be repeated after appropriate off-therapy period. Data from [Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS): a new autosomal dominant syndrome, Worthley DL, Phillips KD, Wayte N, et al, vol 61, pages 774–779, 2012] with permission from BMJ Publishing Group Ltd.1 Abbreviations: FGPs, fundic gland polyps; GAPPS, gastric adenocarcinoma and proximal polyposis of the stomach; GJPS, generalized juvenile polyposis syndrome; MAP, MUTYH-associated polyposis; PJS, Peutz–Jeghers syndrome; PPIs, proton pump inhibitors. |
The initial endoscopic appearance of massive gastric polyposis of family members afflicted by GAPPS can resemble polyposis as a part of other gastric or GI polyposis syndromes. Figure 1 shows the pathognomonic lack of involvement of the antrum by fundic gland polyps (FGPs) on upper endoscopy in GAPPS, which can facilitate differential diagnosis to other GI polyposis syndromes. Generous biopsy sampling for an accurate histopathological diagnosis of the encountered lesions is paramount. In addition, in view of the overlapping phenotype with other GI polyposis syndromes, the criteria proposed by Worthley et al require the exclusion of other heritable gastric polyposis syndromes.1 These should include MUTYH-associated polyposis (due to germline variants in the MUTYH gene; GI phenotype of multiple adenomatous colon polyps with greatly increased lifetime risk of colorectal cancer, followed by duodenal polyps and elevated duodenal cancer risk), juvenile polyposis syndrome (mutations in the BMPR1A or SMAD4 genes; multiple initially benign hamartomatous juvenile polyps across the GI tract with colorectal cancer the most common type of cancer), Peutz–Jeghers syndrome (loss of tumor suppressor STK1 [LKB1], hamartomatous polyps in the GI tract, elevated cancer risk in many other organs), and Cowden syndrome (loss of tumor suppressor PTEN; hamartomatous polyps, colorectal cancer most frequent GI cancer; breast, thyroid, and endometrial cancers and extraintestinal manifestations). Table 2 lists cancer manifestations and initial phenotypes of common hereditary GI polyposis syndromes in comparison to the clinicopathological phenotype of GAPPS. To rule out germline variants associated with other gastric polyposis syndromes, commercially available multi-cancer gene panels for genetic testing are available, which include above polyposis-disposition genes and in addition can capture variants associated with known hereditary gastric cancer predisposition syndromes, which include CDH1 (hereditary diffuse gastric cancer [HDGC]), CTNNA1 (HDGC), TP53 (Li–Fraumeni syndrome), MLH1, PMS2, MSH2, and MSH6 (Lynch syndrome).3 Panel testing should follow current guidelines established by the American College of Medical Genetics and American Society of Clinical Oncology including ethical and policy issues in the genetic testing and screening of children.4–7
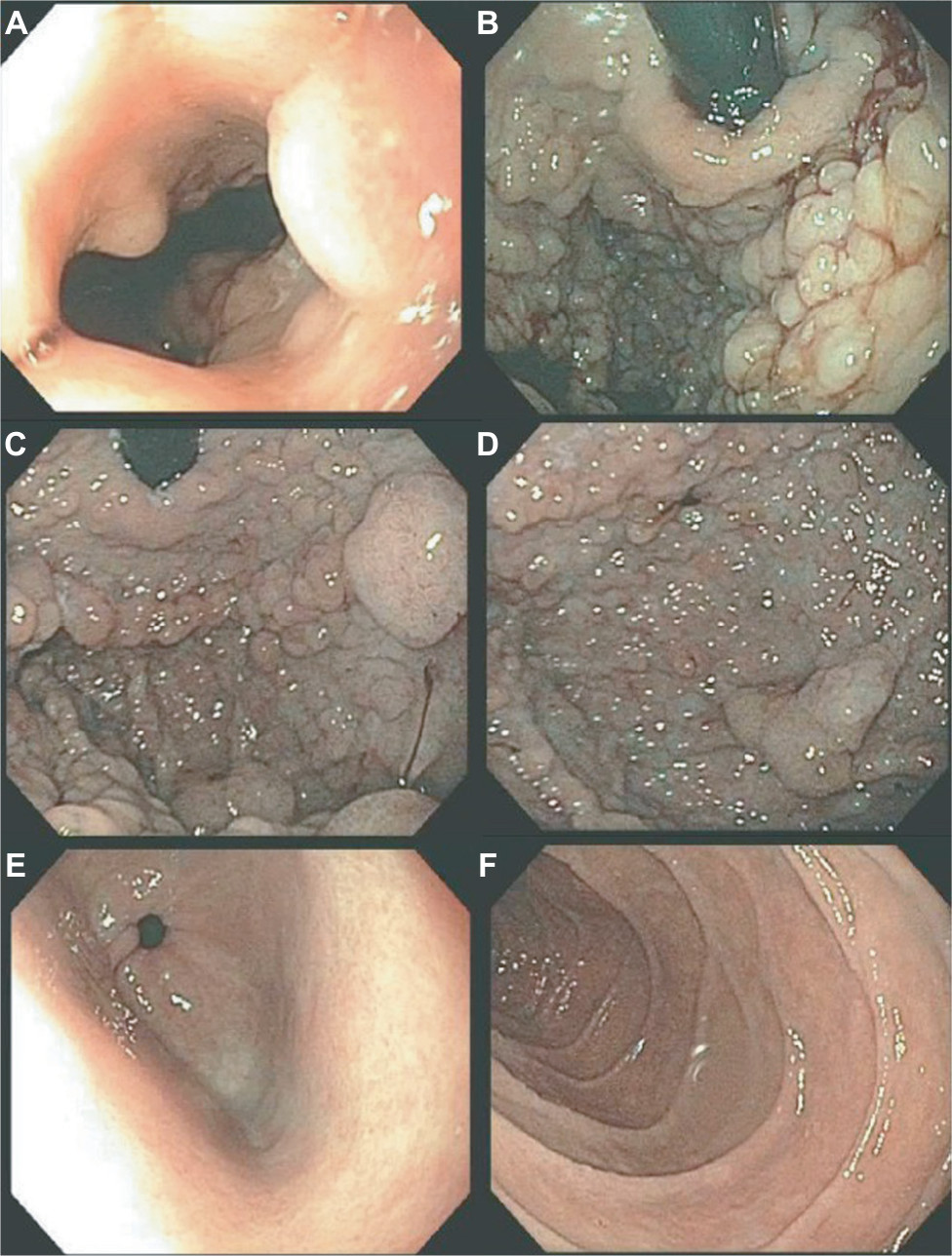 | Figure 1 Endoscopy findings of gastric polyposis due to GAPPS. Notes: (A) Polyps immediately visible after passage through gastroesophageal junction. (B) Gastric cardia (retroflexed scope), (C) fundus, and (D) corpus of the stomach involved by extensive polyposis with heterogenous polyp size. (E) Pathognomonic feature of sparing of the gastric antrum from gastric polyps. (F) No duodenal involvement. Abbreviation: GAPPS, gastric adenocarcinoma and proximal polyposis of the stomach. |
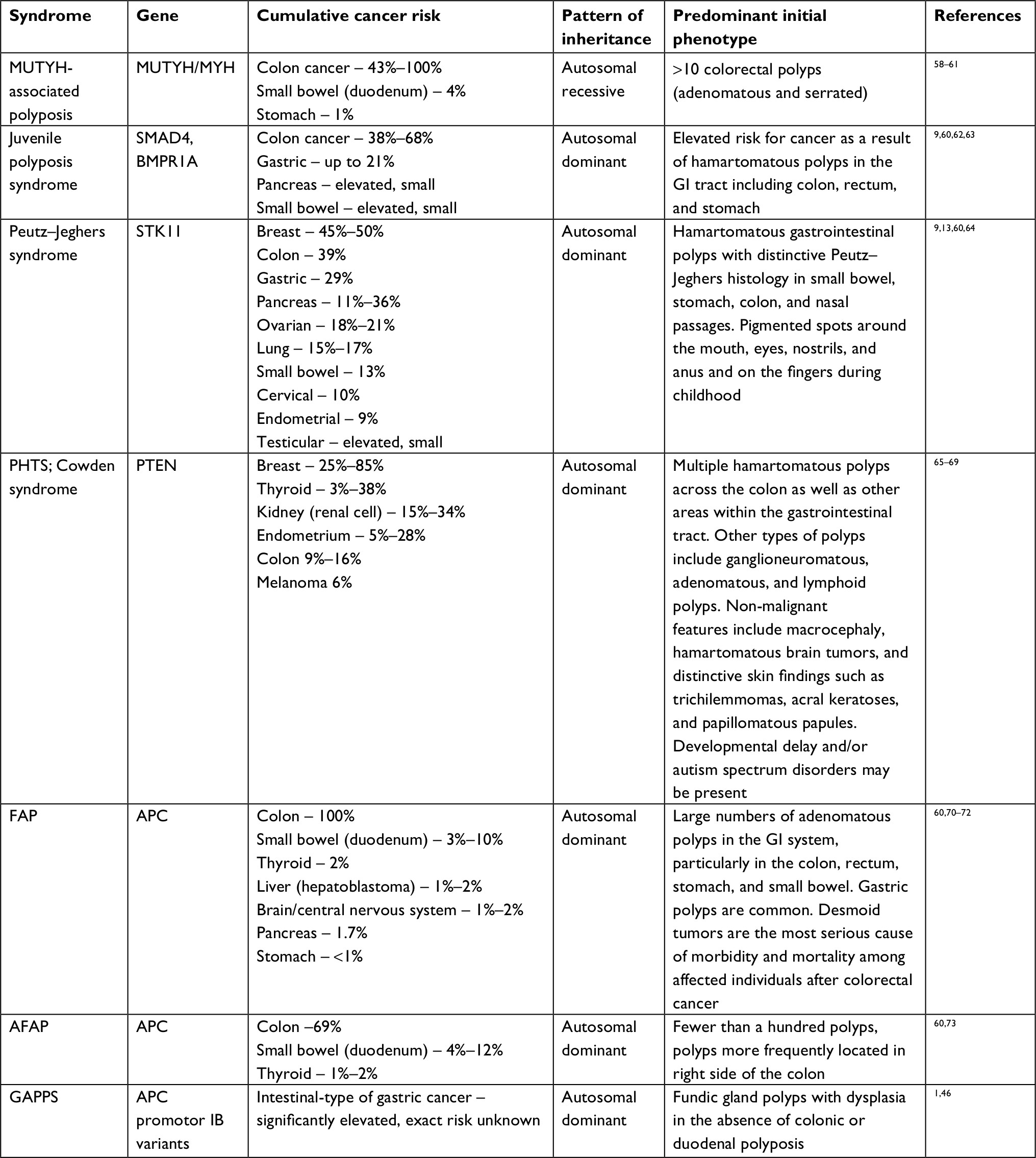 | Table 2 Hereditary GI polyposis syndromes Note: Data from Snygal et al.57 Abbreviations: AFAP, attenuated FAP; FAP, familial adenomatous polyposis; GAPPS, gastric adenocarcinoma and proximal polyposis of the stomach; GI, gastrointestinal; PHTS, PTEN hamartoma tumor syndrome. |
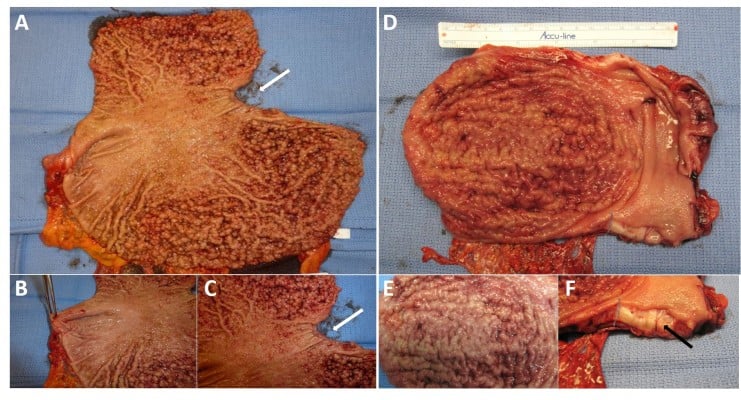
While awaiting results of genetic testing, the initial clinicopathological information from the proband can be used to aid differential diagnosis of other gastric polyposis syndromes: for example, hamartomatous polyps in Peutz–Jeghers, juvenile polyposis, or Cowden syndrome are distinct from FGPs with dysplasia due to their disorganized growth of tissue and initially benign character. However, securing a diagnosis of the underlying GI polyposis syndrome based on morphological characteristics of gastric polypectomy specimens alone is unlikely; for example, histopathological differences of gastric hamartomatous polyps among GI polyposis syndromes are difficult to diagnose, have a low diagnostic accuracy (41% for juvenile polyposis and 54% for Peutz–Jeghers syndrome), and in general, do not allow to distinguish the underlying genetic syndrome without any genetic or clinical corroboration.8,9 Distinctive features reported for Peutz–Jeghers polyps are a cytoarchitectural pattern of pits and glands grouped or packeted together with intervening septations of smooth muscle strands not connected to the muscularis mucosa and an unremarkable lamina propria mucosae, whereas juvenile polyps more frequently have an overall disorganized pit and gland architecture with varying cystic glands and form spherical, club-shaped, or irregular villiform structures due to overgrowth of an edematous lamina propria with a mononuclear inflammatory cell infiltrate.8,9 Of note, hybrid polyps with varying degrees of hamartomatous and FGP features have been described in FGPs arising in familial adenomatous polyposis (FAP) and attenuated FAP (AFAP) patients similarly underscoring the need of clinical and genetic corroboration to secure a diagnosis.10,11 Another entity with histopathological similarities to hamartomatous polyps are hyperplastic polyps where pits are connected to deeper portions in a linear trajectory with overall reduced architectural disorganization and varying degrees of stromal edema and inflammatory cell infiltrate.8,12 In contrast to GAPPS, MUTYH-associated polyposis is an autosomal recessive condition and is frequently associated with colonic polyposis, an exclusion criteria of GAPPS, with rare FGP involvement of the stomach.9,13 Other conditions with possible endoscopic resemblance of GAPPS are Ménétrier’s disease or hypoproteinemic hypertrophic gastropathy forming giant folds in the stomach lining, which are due to an overgrowth of mucous cells in the stomach wall. Cronkhite–Canada syndrome is another GI polyposis syndrome, which is characterized by protein-losing gastroenterocolopathy, which typically affects adults older than 50 years.9,14 Two-thirds of cases are reported from Japan with a male:female ratio of 2:1.14 The histological hallmark of Cronkhite–Canada syndrome polyps resembles juvenile polyposis syndrome or hyperplastic polyps histologically characterized by expanded edematous lamina propria containing a predominantly mononuclear inflammatory cell infiltrate and tortuous, dilated to cystic glands/foveolae or crypts.9,15 Rather than occurring due to a hereditary predisposition, its etiopathogenesis is suspected to be of autoimmune origin.16 Thus, in contrast to hereditary GI polyposis syndromes, the intestinal mucosa not involved by hamartomatous polyps is histologically not normal but shows lamina propria edema, inflammatory cell infiltration, and gland/crypt dilation.9,15 Clinical symptoms are driven by the GI symptoms of protein losing enteropathy or complications from hamartomatous polyps and include unique skin changes in the form of alopecia, hyperpigmentation, onychodystrophy (loss of fingernails), or the loss of taste (ageusia). Patients afflicted by Cronkhite–Canada syndrome carry a poor prognosis predominantly due to the difficult to control enteropathy. Linitis plastica, the submucosal invasion of the diffuse form of gastric cancer, can predominantly involve the upper parts of the stomach and resemble “polypoid” enlargement of the stomach mucosa. Figure 2 shows total gastrectomies specimens of patients with florid gastric polyposis due to GAPPS and with linitis plastic due to HDGC involving the upper four-fifth of the stomach. The endoscopic appearance will depend on the extent of the polyposis, and in case of confluent, “carpet-like” involvement with limited disruption of the mucosa, the endoscopist may need to rely on the unique localization of the disease sparing the antrum and parts of the lower curvature and the histopathological diagnosis of FGPs on obtained biopsies to make the connection to GAPPS. Most reports on families afflicted by GAPPS report the presence of small fundal and body polyps (3–12 mm in size).
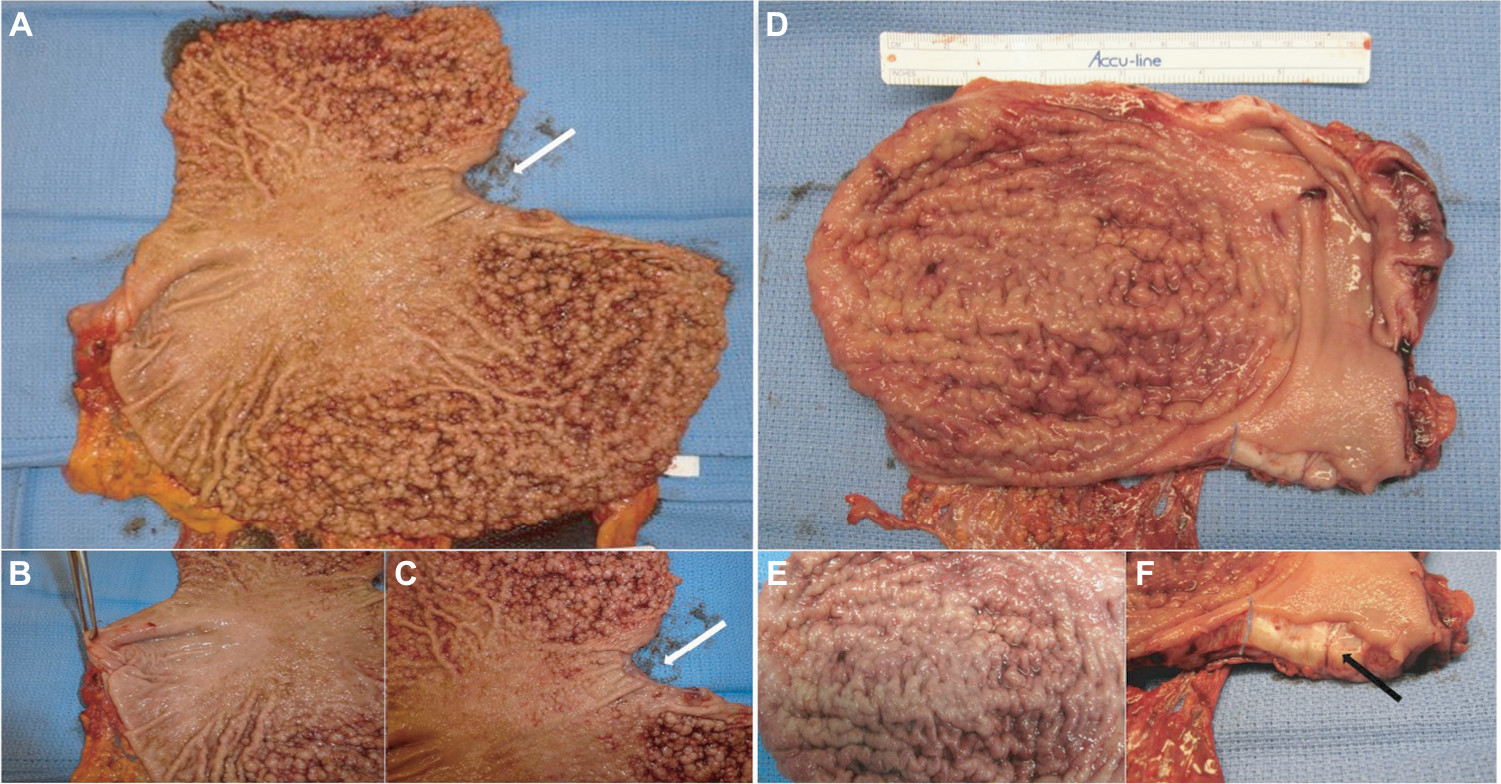 | Figure 2 Gastric polyposis of the fundus and body of the stomach with pathognomonic sparing of the antrum and lesser curvature in a GAPPS proband with confirmed c.-191T>C APC gene promotor IB variant. Notes: (A) Carpet of polyps involving upper four-fifth of the stomach including cardia, fundus, and body; arrow indicates gastroesophageal junction. (B) Uninvolved antrum, forceps lifts pylorus. (C) Gastric polyps at cardia not extending into esophagus (arrow indicates esophageal mucosa). (D) Linitis plastica due to HDGC causing enlarged folds and “polypoid” appearance without involvement of the distal stomach. (E) Irregular enlarged folds with intact mucosa due to HDGC. (F) Sagittal section shows tumor submucosal tumor infiltration (arrow). Abbreviations: GAPPS, gastric adenocarcinoma and proximal polyposis of the stomach; HDGC, hereditary diffuse gastric cancer. |
An important other benign cause for gastric polyposis on endoscopy is the chronic use of proton pump inhibitors (PPIs) inducing oxyntic cell hyperplasia, glandular dilatations, and FGPs.17,18 In this case, endoscopy is recommended to be repeated after discontinuation and an appropriate off-treatment interval.19 There is currently insufficient evidence to consider PPI therapy, a precipitating factor for the development of GAPPS. Serum gastrin levels, which are usually elevated in chronic PPI usage, were found to be normal in a few family members harboring the GAPPS phenotype arguing against a connection of PPI usage and GAPPS.1 In contrast, there are several clinical observations, which imply gastric acid homeostasis and milieu in the natural history of FGPs in family members with a genetic predisposition to GI cancers: while chronic PPI usage has been observed as a risk factor for the development of FGPs, inversely Helicobacter pylori (H. pylori) infections have been reported to be negatively associated with the occurrence of FGPs both in healthy individuals and FAP patients.20–23 In family members afflicted by FAP, regression of FGPs has been described upon acquisition of H. pylori infection.24 A possible protective role of H. pylori infection in the APC-related disorder GAPPS has also been suggested in the initially described large Australian GAPPS family when comparing H. pylori status of affected and unaffected family members (P=0.007).1 Thus, in light of opposing associations of PPI usage and H. pylori infections on FGP incidence and in the absence of more natural history information derived from families afflicted by GAPPS, an approach of caution including early discontinuance of PPIs if clinically feasible and heightened surveillance of family members at risk for GAPPS with a history of chronic PPI usage appears to be the safest course of action. Discontinuation of PPI therapy and regression of FGPs may aid cases of diagnostic uncertainty where a florid polyposis picture with the unique sparing of the gastric antrum and lesser curvature with dysplasia on final pathology of generous biopsy sampling of FGPs is not present. FGPs due to PPI usage are significantly fewer, rarely harbor dysplasia, and do not spare the antrum. With the recent identification of APC promotor IB variants and ability to conduct timely germline genetic testing after genetic counseling stoppage and re-scoping of PPIs will rarely be required to secure the diagnosis and is not a must when other criteria have been met.
The possible association of H. pylori infection rates and the GAPPS phenotype including fundic gland polyposis was evaluated by Worthley et al in greater detail; in 27 family members with known H. pylori status, all evaluated family members with the GAPPS phenotype were negative for H. pylori (0/18) whereas four of the nine unaffected family members including obligate carriers (4/9) tested positive (P=0.007) suggesting a protective role of H. pylori in the development of GAPPS.1 These findings mirror observations in FAP where patients with FGPs had lower rates of H. pylori compared to patients without FGPs (13% vs 67%).22 In contrast, H. pylori infection and degree of atrophic gastritis were positively correlated with the incidence of gastric adenomas in FAP patients.22 It is currently not known whether H. pylori infection rates are associated with gastric adenoma or adenocarcinoma in GAPPS or whether reduced H. pylori infection rates in patients with the GAPPS phenotype are a result of the disturbed intraluminal milieu due to fundic gland polyposis. Without a causative implication of H. pylori in GAPPS progression, clinical management of GAPPS patients is currently not affected by H. pylori status. In addition to the exclusion of other heritable gastric polyposis syndromes listed in Table 2, Worthley et al require as a part of their diagnostic criteria for GAPPS the endoscopic exclusion of colonic and duodenal polyposis as a part of FAP and AFAP. Fundal gland polyps are frequently present in FAP and AFAP, which are GI polyposis syndromes involving the colon and rectum but frequently harbor FGPs.
Histopathology
The polyposis and cancer predisposition syndrome with the greatest phenotypic overlap with GAPPS and most frequently cited as a diagnostic challenge are FAPs. Fundal gland polyps are one of the sentinel features of the GAPPS phenotype; FGPs most commonly occur as sporadic FGPs where they occur in small numbers, are usually small (<5 mm), are associated with chronic PPI therapy, and frequently harbor in the sporadic setting somatic CTNNB1 (β-catenin) mutations (Table 3).25,26 Sporadic FGPs rarely harbor dysplasia and are not connected to an increased risk of cancer.27–29 They are more frequently found in probands in their forties and fifties and more commonly in women.20 Gastric FGPs are also a frequent phenotypic manifestation of the APC-associated disorders FAP and AFAP.11,30–32 While widely varying rates of FGPs and fundal gland polyposis in individuals with FAP and AFAP have been reported, low-grade dysplasia is a common feature (up to 96%) of FAP-associated FGPs.10,30 FAP-associated FGPs preferentially harbor APC mutations and have an, albeit low, potential of malignant transformation toward adenocarcinoma of the stomach.33–35 While gastric carcinoma arising in FGPs of FAP patients has been described, the lifetime risk overall of gastric cancer in FAP patients from Western countries has been reported to be low (0.6%) but has been suggested to be rising.33,36,37 Higher rates of gastric cancer have been described in FAP patients from countries with higher gastric cancer risks including Japan and Korea.38,39 Table 3 summarizes common clinicopathological characterizations and clinical implications of FGPs detected within sporadic vs within polyposis-associated contexts. Figure 3 shows GAPPS-associated FGPs with low-grade and focal high-grade dysplasia.
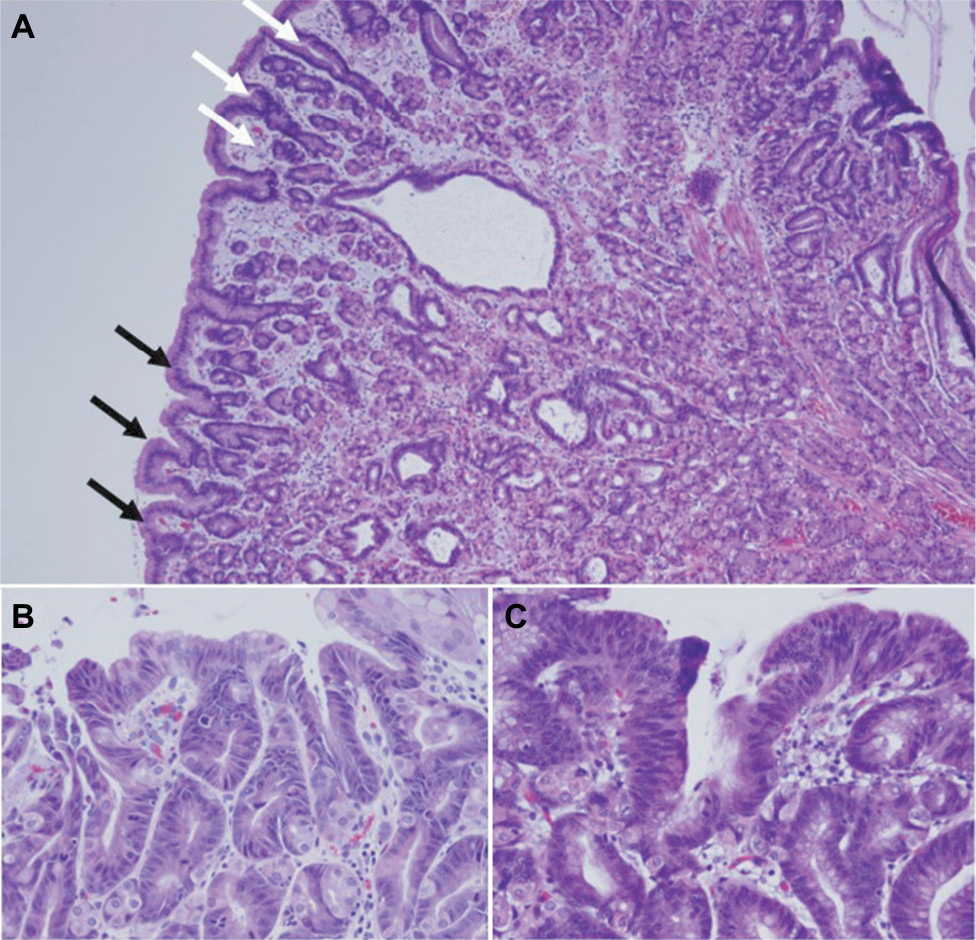 | Figure 3 Histopathology of fundic gland polyps in GAPPS. Notes: (A) Fundic gland polyp with flattened cuboidal epithelium with focal area of low-grade dysplasia (black arrows) and high-grade dysplasia (white arrows) from total gastrectomy specimens of a GAPPS proband with confirmed c.-191T>C APC gene promotor IB variant. (B) Fundic gland polyp with low-grade and (C) high-grade dysplasia in gastrectomy specimens (inset 20×). Abbreviations: GAPPS, gastric adenocarcinoma and proximal polyposis of the stomach; HDGC, hereditary diffuse gastric cancer. |
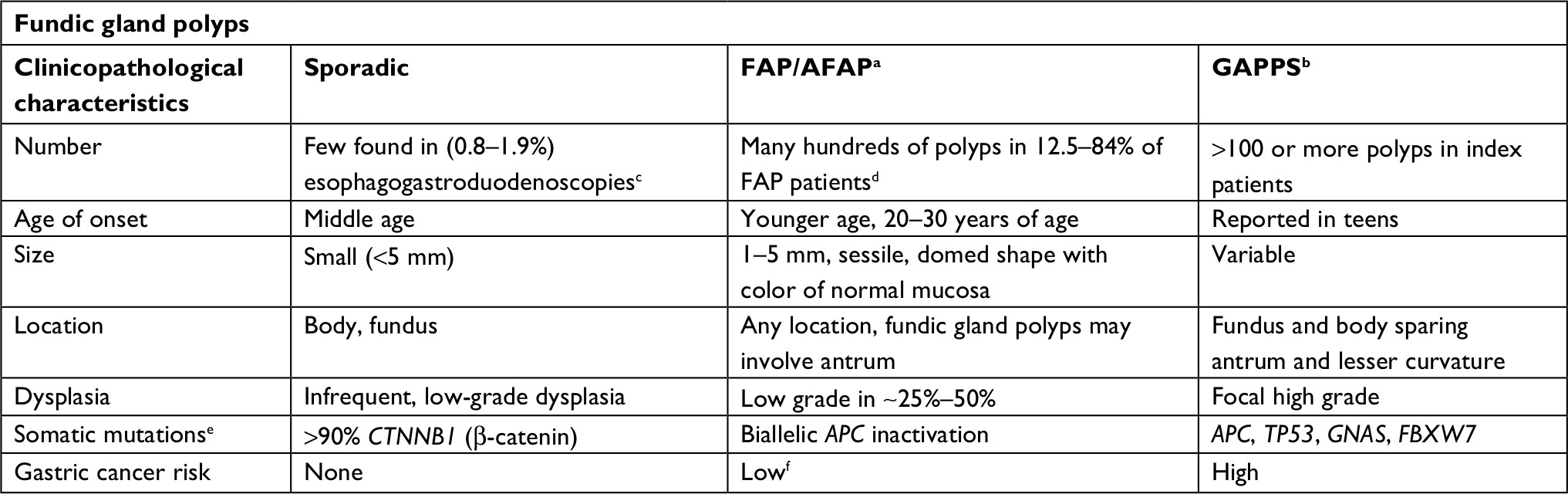 | Table 3 Disease associations determine biological behavior of fundic gland polyps Notes: aFAP/attenuated FAP due to APC germline variants. bGAPPS due to APC promotor IB germline variants. cMore common in middle age women. dMore equal gender distribution. eVariants detected in polyp tissue in addition to the APC germline variants in FAP/AFAP and APC promotor IB variants in GAPPS. fGastric cancer risk reported to be higher in afflicted patients from areas with high gastric cancer rates such as Japan and Korea. Abbreviations: AFAP, attenuated FAP; FAP, familial adenomatous polyposis; GAPPS, gastric adenocarcinoma and proximal polyposis of the stomach. |
A more systematic review on the histopathology of GAPPS has recently been released.40 While limited to the review of multiple biopsies and gastrectomy specimens from the initially described, large Australian family, the detailed pathological and immunohistochemical review made important findings: hyperproliferative aberrant pits (HPAPs) were found to be the most frequent and most early histopathological abnormality in a wider histopathological spectrum of GAPPS.40 HPAPs describe the disorganized hyper-proliferation of oxyntic glands of the stomach mucosa around gastric pits, which give rise to polypoid lesions. The finding of concomitant neoplastic elements in the form of FGPs with multifocal “flat” dysplasia, gastric adenomatous polyps, which were associated with gastric adenocarcinoma together with an immunohistochemical profile, in particular the gastric lineage marker MUC5AC shared by both precancerous lesions and adenocarcinoma, suggests a dysplasia–adenoma–adenocarcinoma sequence in the development of the malignant phenotype of GAPPS. Whether the absence or presence of some these precancerous lesions including their histopathological characteristics (HPAPs, FGPs with or without dysplasia, presence of adenomatous lesions) can be used to triage patients to undergo total gastrectomy vs endoscopic surveillance has to await the collection of larger case series. The concerns of adequate sampling via endoscopic biopsies in the face of hundreds of heterogenous polypoid lesions, which may include FGPs with different degrees of dysplasia, adenomas, or mixed histology polyps, pose significant challenges.
Gastric hyperplastic polyps (GHPs) account for the most frequent type of polyp in the stomach, and based on the above unique features of FGPs due to GAPPS, an expeditious distinction to GHPs based on clinicopathological criteria should be possible. GHPs more frequently occur as solitary lesions (65%–75%).12,41 Common sites include the antrum, and in more recent reports, the body of the stomach; however, GHPs can occur anywhere.12,41 GHPs are intimately linked to gastric inflammation and gastric mucosal injury with chronic gastritis (with or without H. pylori) being the most common (up to 85%).12 Other conditions associated with GHPs include autoimmune gastritis, bile reflux, and post-gastrectomy status. GHPs are understood as a hyper-regenerative, hyperproliferative repair response to an underlying mucosal injury. GHPs carry a risk of intraepithelial dysplasia and cancer, which is most strongly related to polyp size and age.42,43 Rates of dysplasia in removed GHPs range from 0.4% to 10%, and gastric adenocarcinoma is found less frequent (<1%).12,44 GHPs greater than 5 mm should be removed, and oncologic post-polypectomy surveillance is guided by findings on histopathological examination of the polyp and the stage of the surrounding chronic gastritis stage. GHPs result from excessive proliferation of foveolar cells and are histopathologically characterized by surface pits connected to deeper portions of glands in a linear trajectory, which can take on a serrated or star-like appearance on cross-sections.12,45 In addition, GHPs not infrequently have infiltration of the stroma by a variable mononucleolar cell infiltrate. The antral involvement, presentation as solitary or multifocal lesions, and the unique histopathological characteristics of foveolar cell hyperplasia with cystic dilatations, smooth muscle strands extending from muscularis mucosa towards surface, and a variable acute and chronic inflammatory stromal infiltrate are key differences to FGPs occurring in GAPPS.
Genetics
The underlying genetic aberration of GAPPS was recently discovered by Investigators from the Queensland Institute of Medical Research, Adelaide, Australia.46 This advancement already had significant implications both for an improved understanding of the molecular perturbations present in GAPPS and for clinical management with regard to the screening of family members at risk for GAPPS. Initial whole exome and whole genome analyses of affected family members in the first two families described in the original study in 2012 failed to produce a variant, which co-segregated with afflicted family members by GAPPS.46 A loss of heterozygosity analysis in FGPs on a 46 Mb region of chromosome 5 previously identified by linkage analysis narrowed the region to 12.7 Mb, which was consequently subject to Sanger sequencing. Sequencing identified variants c.-195A>C and c.-125delA in the IB promotor of the APC gene, which completely co-segregated with the multiple affected family members in the large Australian family.46 APC promotor IB variant c.-191T>C co-segregated in affected family members of families 2, 4, 5, and 6, and promotor variant c.-192A>C co-segregated in family 3 with the GAPPS phenotype. These hotspot APC promotor IB variants were consequently identified in other GAPPS families reported on more recently.47–49 Molecularly, the identified point mutations in the APC IB promotor significantly reduce the binding of the transcription factor Yin Yang 1 (YY1) in gastric and colon cancer cells providing functional validation of the identified germline mutation.46 Importantly, methylation studies on both uninvolved stomach mucosa and FGPs in GAPPS specimens showed methylation of the promotor IA isoform of the APC gene, which the authors discussed as a possible cause for the selective gastric phenotype of the syndrome compared to the other APC-associated disorders FAP and AFAP.46 It is estimated that promotor IB-driven transcription of the APC gene is ~15-fold higher than transcripts originating from the IA promotor, which is nearly universally methylated in gastric cancer and noninvolved gastric mucosa.50,51 Thus, reduced promotor IB activity due to the loss of the enhancer function of the transcriptional enhancer YY1 leads to a selective tumor suppressive phenotype in the stomach, which does not occur in the colon where intact promotor IA-driven APC gene transcription is preserved and able to compensate for the lost promotor IB function explaining why APC promotor IB point mutations are not associated with a colonic phenotype typically found in FAP or AFAP. Structural variants differently affecting APC promotor IA and IB function hence may form the basis for important unique phenotypic traits in APC-associated disorders adding to the many described genotype–phenotype associations described for FAP. Figure 4 illustrates the position of promotor IA and IB on the APC gene locus, which are ~29 kb apart, in relation to several large genomic deletions described in FAP families. FAP families with underlying noncoding deletions in the APC gene promotor regions more commonly affect the IB isoform and have been reported to have a greater variability of upper GI manifestations, possibly including a higher prevalence of FGPs, compared to the FAP families with the common exomic APC mutations.52 It is thought that interference with predominantly promotor IB regulated enhancer elements and thus IB activity in APC-mutation carriers with large genomic deletions manifests with different upper GI phenotypes including a greater prevalence of FGPs.52,53 These promotor IB structural variants, either in the form of select point mutations in the YY1 binding motif in GAPPS or large deletions affecting regulatory elements of promotor IB, have a unique gastric phenotype due to methylation of the IA promotor in the upper GI tract but preserved promotor IA function in the colon.51 APC gene perturbations due to variants in the codon regions or rare structural variants affecting the IA promotor in contrast present with the classic FAP and AFAP phenotypes with the classic colonic polyposis and extracolonic manifestations.54 That APC promotor IB variants are part of a greater spectrum of APC-associated disorders and are related to FAP and AFAP with its classical colonic polyposis phenotype is supported by the presence of a mild colonic phenotype in the form of a greater frequency of colonic polyps or a prior report of a point mutation in the APC IB promotor in a FAP family with colonic phenotype.55,56 It is tempting to speculate that the colonic phenotype in this family was due to promotor IA function not completely compensating for the loss of the impaired IB promotor.
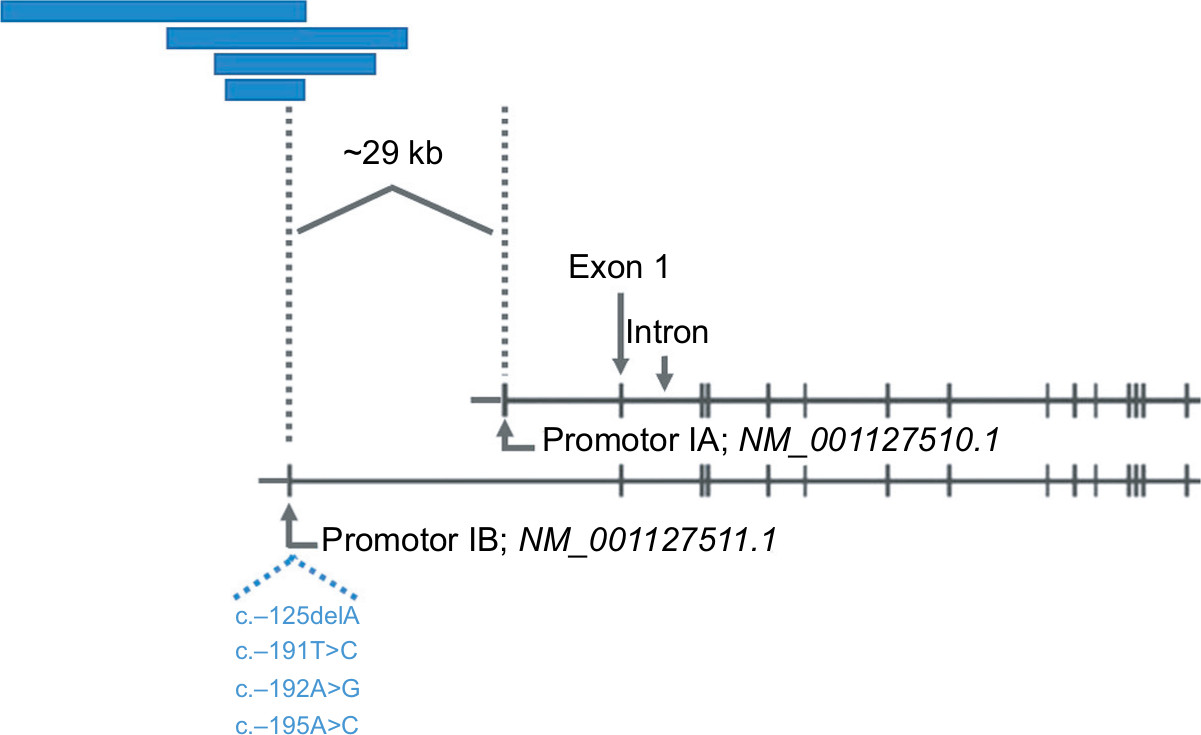 | Figure 4 Selective loss of APC gene promotor IB function by large genomic deletions or point mutations. Notes: Schematic representation showing inactivation of promoter 1B of APC by genomic deletions (blue bars) or hotspot mutations in the YY1-binding motif (listed by nucleotide position relative to promotor, in blue). As examples, length and position of previously described large deletions in relation to the promotor IB reported (blue bars, from top to bottom) by Rohlin et al,52 Snow et al,53 Kadiyska et al,74 and Lin et al75 are shown. Reprinted from The American Journal of Human Genetics, vol 98, Li J, Woods SL, Healey S, et al., Point mutations in exon 1B of APC reveal gastric adenocarcinoma and proximal polyposis of the stomach as a familial adenomatous polyposis variant, pages 830–842, copyright (2016), with permission from Elsevier.46 |
There are several phenotypic variabilities in GAPPS, most notably age of onset, penetrance, or degree of dysplasia to name the clinically most relevant ones. The recent genetic and molecular pathological findings may now provide some explanation for the observed heterogeneity in the clinical phenotype and manifestations of GAPPS. For example, the synchronous promotor IB variants c.-125delA and c.-195A>C found in the original large Australian family had a greater impact on APC IB promotor activity in gastric and colonic cancer cells than the c.-191T>C variant.46 In addition, a number of second hits have been identified in GAPPS specimens, which might differently augment the impact of APC gene germline haploinsufficiency. Reported second hit events in GAPPS in the form of somatic mutations in FGPs or gastric adenocarcinoma of individuals affected by GAPPS include truncating APC mutations, or mutations in TP53, GNAS, or FBXW7.46,49,55 It is tempting to speculate that the presence and type of these second hit events might be responsible for different rates of malignant transformation possibly manifesting in the accelerated development of high-grade FGP dysplasia, the development of adenomatous changes, and the clinical expression of the syndrome in general.
Clinical course
There currently is a large void in the understanding of the natural history of GAPPS, and at least two disconcerting reports have emerged, which in general, do not support prolonged endoscopic surveillance in family members with gastric polyposis due to GAPPS. Most recently, Repak et al described as a part of their European family a 54-year-old index proband with GAPPS who was diagnosed after a 19-month of endoscopic surveillance with liver metastasis and who died shortly thereafter.47 Of note, there was no change in the endoscopic appearance or the histopathology of the proband’s gastric polyposis. Similarly, one of the daughters of the proband after being surveilled for 5 years showed an increase in the size of FGPs with low-grade dysplasia and focal high-grade dysplasia on repeat endoscopy.47 While awaiting total gastrectomy, the patient was diagnosed with widespread metastatic disease and succumbed thereafter. Both her sisters when undergoing gastrectomy after a variable period of surveillance were found to harbor invasive stage I gastric adenocarcinoma showing again FGPs with low-grade and focal high-grade dysplasia on their last endoscopy. Worthley et al described a 33- and 48-year-old relative of the initial proband in their large Australian GAPPS family who died from metastatic intestinal-type adenocarcinoma of the stomach after being diagnosed with GAPPS previously and undergone a period of prior endoscopic observation.1 In the case of the 33-year-old index patient, both adenomatous polyps and polyps with mixed FGP and adenomatous histopathology were found at total gastrectomy 2 years after the initial diagnosis. While histopathological findings as known to-date indicate a dysplasia–adenoma–carcinoma sequence in the syndrome, these reports suggest that reliance on unchanged endoscopic appearance and histopathology of sampled polyps carries the risk to miss occult sites of malignant transformation and focal progression. The inability to adequately sample the upper four-fifth of the stomach covered by hundreds of polyps by endoscopy, which are frequently large, and harbor a heterogeneous pattern of dysplasia and adenomatous changes should trigger an expedited referral to a clinical geneticist and surgical oncologist. Total gastrectomy should be considered in all GAPPS patients with fundal gland polyposis and the presence of dysplasia on gastric biopsy or polypectomy specimens, who are able to undergo major surgery. Heterogeneity in the impact of the individual primary promotor variants (c.-125 delA c.-195A>C vs c.-191T>C) differently affecting the recruitment of YY1, differences in the frequency and type of second hit events, and any underlying perturbations of the APC promotor IA are likely drivers of the malignant phenotype including rate of progression toward invasion and metastasis. As their impact on malignant transformation and cancer progression is currently unknown, the interindividual heterogeneity within GAPPS will require an individual approach to family members at risk and affected by the phenotype.
The other area of uncertainty is the incomplete understanding of other organs at risk for cancer. Table 4 lists previously described GAPPS families with the age of youngest affected family member by gastric polyposis and gastric adenocarcinoma. With regard to extra gastric manifestations of GAPPS, colonic involvement in GAPPS has undergone additional investigation. McDuffie et al reported an increased incidence of colonic polyps in afflicted family members, and there is agreement on the need of colonoscopic screening with regular follow-up colonoscopic examinations guided by the initial findings in these patients.1,55 It is thought that the mild colonic phenotype generally with small, <20 in number hyperplastic polyps or small adenomas in GAPPS family members is due to an incomplete protection of APC promotor IA activity.46 In contrast, it is interesting to note that colon cancer has been described in almost half of the GAPPS families reported to-date where family history information was available (including families with reported but not confirmed colon cancer incidences). While consistent with the phenotype of other APC-associated disorders, further follow-up of current GAPPS families and the identification of new families have to be awaited prior to including adenocarcinoma of the colon into the cancer phenotype of GAPPS. Pathological examination of colonic polyps from patients with GAPPS did not reveal any unique features, and there has been no detailed pathological review of colon cancers arising within the context of GAPPS.55
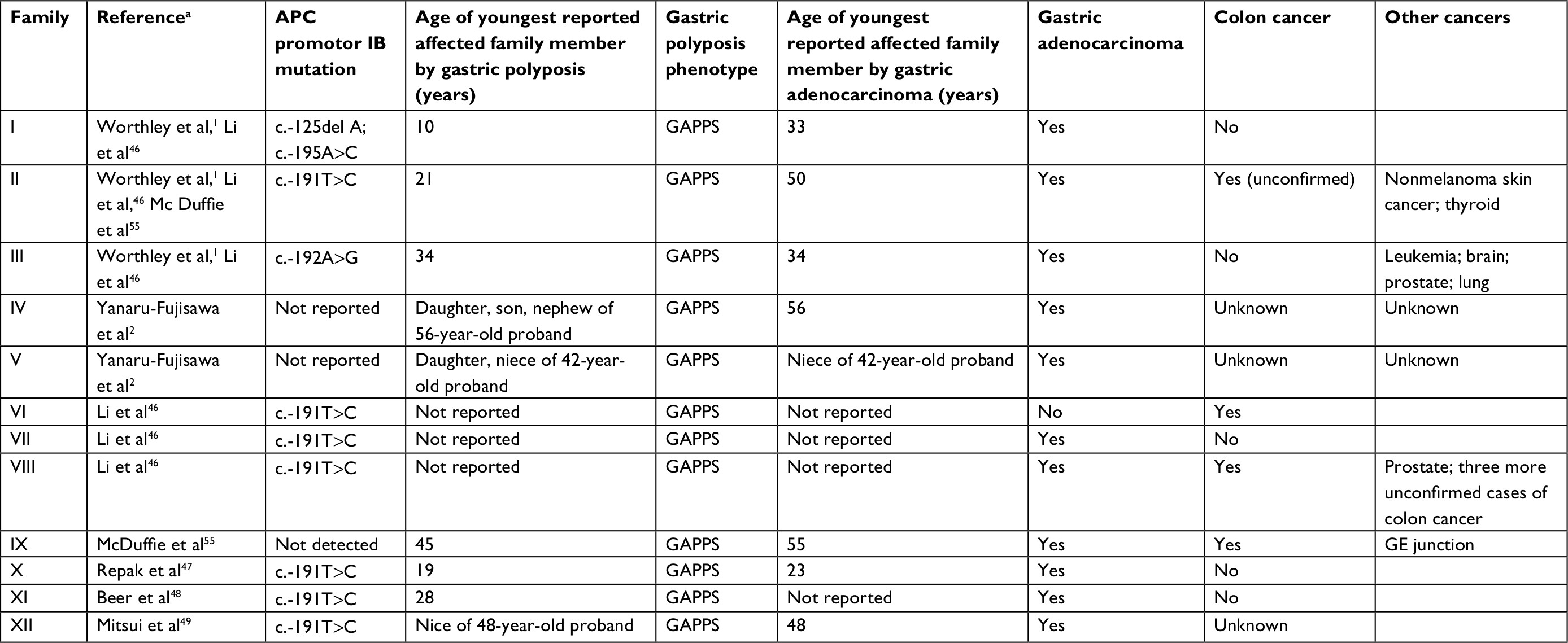 | Table 4 Clinicopathological characteristics of previously reported families with GAPPS Note: aIf family reported more than once, most recent and most comprehensive report was used. Abbreviations: GAPPS, gastric adenocarcinoma and proximal polyposis of the stomach; GE, gastroesophageal. |
Conclusion
GAPPS is a novel, autosomal dominant gastric polyposis syndrome with a significant predisposition for adenocarcinoma of the stomach, metastasis, and death. With the recent identification of APC gene promotor IB variants as its underlying genetic aberration and a phenotypic overlap with FAP and AFAP caused by the loss of the tumor suppressor APC, GAPPS has been included as the most recent addition into APC-associated disorders. Diagnosis is made by the presence of FGPs sparing the antrum and lesser curvature of the stomach, the absence of colonic and duodenal polyposis, an autosomal dominant inheritance pattern, and the exclusion of other gastric polyposis syndromes and chronic use of PPIs. Family members at risk should be screened by germline mutation testing for the presence of APC promotor IB variants to initiate timely endoscopic evaluation and surveillance. GAPPS has a to-date poorly defined natural history and the rate of malignant transformation, and endoscopic surveillance in family members with gastric polyposis with high-grade dysplasia on biopsy and polypectomy specimens can miss invasive cancer. Colonoscopic surveillance is prudent while risk estimates of colon and other cancer phenotypes possibly associated with GAPPS are awaiting larger sample numbers. The creation of family registries and the formation of updated guidelines by expert consensus panels similar to the International Gastric Cancer Linkage Consortium for the screening and management of HDGC, together with enhanced education, are likely to reduce mortality in the future.
Disclosure
The author reports no conflicts of interest in this work.
References
Worthley DL, Phillips KD, Wayte N, et al. Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS): a new autosomal dominant syndrome. Gut. 2012;61(5):774–779. | ||
Yanaru-Fujisawa R, Nakamura S, Moriyama T, et al. Familial fundic gland polyposis with gastric cancer. Gut. 2012;61(7):1103–1104. | ||
Slavin T, Neuhausen SL, Rybak C, et al. Genetic gastric cancer susceptibility in the international clinical cancer genomics community research network. Cancer Genet. 2017;216–217:111–119. | ||
Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17(1):70–87. | ||
Lu KH, Wood ME, Daniels M, et al. American Society of Clinical Oncology Expert Statement: collection and use of a cancer family history for oncology providers. J Clin Oncol. 2014;32(8):833–840. | ||
Robson ME, Storm CD, Weitzel J, Wollins DS, Offit K, American Society of Clinical Oncology. American society of clinical oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2010;28(5):893–901. | ||
Stoffel EM, Mangu PB, Gruber SB, et al. Hereditary colorectal cancer syndromes: american society of clinical oncology clinical practice guideline endorsement of the familial risk-colorectal cancer: european society for medical oncology clinical practice guidelines. J Clin Oncol. 2015;33(2):209–217. | ||
Lam-Himlin D, Park JY, Cornish TC, Shi C, Montgomery E. Morphologic characterization of syndromic gastric polyps. Am J Surg Pathol. 2010;34(11):1–1662. | ||
Vyas M, Yang X, Zhang X. Gastric hamartomatous polyps-review and update. Clin Med Insights Gastroenterol. 2016;9:3–10. | ||
Arnason T, Liang WY, Alfaro E, et al. Morphology and natural history of familial adenomatous polyposis-associated dysplastic fundic gland polyps. Histopathology. 2014;65(3):353–362. | ||
Wood LD, Salaria SN, Cruise MW, Giardiello FM, Montgomery EA. Upper GI tract lesions in familial adenomatous polyposis (FAP): enrichment of pyloric gland adenomas and other gastric and duodenal neoplasms. Am J Surg Pathol. 2014;38(3):389–393. | ||
Markowski AR, Markowska A, Guzinska-Ustymowicz K. Pathophysiological and clinical aspects of gastric hyperplastic polyps. World J Gastroenterol. 2016;22(40):8883–8891. | ||
Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59(7):975–986. | ||
Watanabe C, Komoto S, Tomita K, et al. Endoscopic and clinical evaluation of treatment and prognosis of Cronkhite-Canada syndrome: a Japanese nationwide survey. J Gastroenterol. 2016;51(4):327–336. | ||
Slavik T, Montgomery EA. Cronkhite–Canada syndrome six decades on: the many faces of an enigmatic disease. J Clin Pathol. 2014;67(10):891–897. | ||
Sweetser S, Ahlquist DA, Osborn NK, et al. Clinicopathologic features and treatment outcomes in Cronkhite-Canada syndrome: support for autoimmunity. Dig Dis Sci. 2012;57(2):496–502. | ||
Burt RW. Gastric fundic gland polyps. Gastroenterology. 2003;125(5):1462–1469. | ||
Freeman HJ. Proton pump inhibitors and an emerging epidemic of gastric fundic gland polyposis. World J Gastroenterol. 2008;14(9):1318–1320. | ||
Malfertheiner P, Kandulski A, Venerito M. Proton-pump inhibitors: understanding the complications and risks. Nat Rev Gastroenterol Hepatol. 2017;14(12):697–710. | ||
Huang CZ, Lai RX, Mai L, Zhou HL, Chen HJ, Guo HX. Relative risk factors associated with the development of fundic gland polyps. Eur J Gastroenterol Hepatol. 2014;26(11):1217–1221. | ||
Tran-Duy A, Spaetgens B, Hoes AW, de Wit NJ, Stehouwer CD. Use of Proton Pump Inhibitors and Risks of Fundic Gland Polyps and Gastric Cancer: Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol. 2016;14(12):e1705:1706–1719. | ||
Nakamura S, Matsumoto T, Kobori Y, Iida M. Impact of Helicobacter pylori infection and mucosal atrophy on gastric lesions in patients with familial adenomatous polyposis. Gut. 2002;51(4):485–489. | ||
Straub SF, Drage MG, Gonzalez RS. Comparison of dysplastic fundic gland polyps in patients with and without familial adenomatous polyposis. Histopathology. 2018;72(7):1172–1179. | ||
Watanabe N, Seno H, Nakajima T, et al. Regression of fundic gland polyps following acquisition of Helicobacter pylori. Gut. 2002;51(5):742–745. | ||
Abraham SC, Nobukawa B, Giardiello FM, Hamilton SR, Wu TT, Tt W. Sporadic fundic gland polyps: common gastric polyps arising through activating mutations in the beta-catenin gene. Am J Pathol. 2001;158(3):1005–1010. | ||
Torbenson M, Lee JH, Cruz-Correa M, et al. Sporadic fundic gland polyposis: a clinical, histological, and molecular analysis. Mod Pathol. 2002;15(7):718–723. | ||
Genta RM, Schuler CM, Robiou CI, Lash RH. No association between gastric fundic gland polyps and gastrointestinal neoplasia in a study of over 100,000 patients. Clin Gastroenterol Hepatol. 2009;7(8):849–854. | ||
Levy MD, Bhattacharya B. Sporadic fundic gland polyps with low-grade dysplasia: a large case series evaluating pathologic and immunohistochemical findings and clinical behavior. Am J Clin Pathol. 2015;144(4):592–600. | ||
Kishikawa H, Kaida S, Takarabe S, et al. Fundic gland polyps accurately predict a low risk of future gastric carcinogenesis. Clin Res Hepatol Gastroenterol. 2014;38(4):505–512. | ||
Stolte M, Vieth M, Ebert MP. High-grade dysplasia in sporadic fundic gland polyps: clinically relevant or not? Eur J Gastroenterol Hepatol. 2003;15(11):1153–1156. | ||
Groves C, Lamlum H, Crabtree M, et al. Mutation cluster region, association between germline and somatic mutations and genotype-phenotype correlation in upper gastrointestinal familial adenomatous polyposis. Am J Pathol. 2002;160(6):2055–2061. | ||
Sample DC, Samadder NJ, Pappas LM, et al. Variables affecting penetrance of gastric and duodenal phenotype in familial adenomatous polyposis patients. BMC Gastroenterol. 2018;18(1):115. | ||
Garrean S, Hering J, Saied A, Jani J, Espat NJ. Gastric adenocarcinoma arising from fundic gland polyps in a patient with familial adenomatous polyposis syndrome. Am Surg. 2008;74(1):79–83. | ||
Ngamruengphong S, Boardman LA, Heigh RI, Krishna M, Roberts ME, Riegert-Johnson DL. Gastric adenomas in familial adenomatous polyposis are common, but subtle, and have a benign course. Hered Cancer Clin Pract. 2014;12(1):4. | ||
Abraham SC, Park SJ, Mugartegui L, Hamilton SR, Wu TT, Tt W. Sporadic fundic gland polyps with epithelial dysplasia: evidence for preferential targeting for mutations in the adenomatous polyposis coli gene. Am J Pathol. 2002;161(5):1735–1742. | ||
Mankaney G, Leone P, Cruise M, et al. Gastric cancer in FAP: a concerning rise in incidence. Fam Cancer. 2017;16(3):371–376. | ||
Walton SJ, Frayling IM, Clark SK, Latchford A. Gastric tumours in FAP. Fam Cancer. 2017;16(3):363–369. | ||
Park SY, Ryu JK, Park JH, et al. Prevalence of gastric and duodenal polyps and risk factors for duodenal neoplasm in korean patients with familial adenomatous polyposis. Gut Liver. 2011;5(1):46–51. | ||
Yamaguchi T, Ishida H, Ueno H, et al. Upper gastrointestinal tumours in Japanese familial adenomatous polyposis patients. Jpn J Clin Oncol. 2016;46(4):310–315. | ||
De Boer WB, Ee H, Kumarasinghe MP. Neoplastic lesions of gastric adenocarcinoma and proximal polyposis syndrome (GAPPS) are gastric phenotype. Am J Surg Pathol. 2018;42(1):1–8. | ||
Carmack SW, Genta RM, Schuler CM, Saboorian MH. The current spectrum of gastric polyps: a 1-year national study of over 120,000 patients. Am J Gastroenterol. 2009;104(6):1524–1532. | ||
Imura J, Hayashi S, Ichikawa K, et al. Malignant transformation of hyperplastic gastric polyps: An immunohistochemical and pathological study of the changes of neoplastic phenotype. Oncol Lett. 2014;7(5):1459–1463. | ||
ASGE Standards of Practice Committee, Evans JA, Chandrasekhara V, et al. The role of endoscopy in the management of premalignant and malignant conditions of the stomach. Gastrointest Endosc. 2015;82(1):1–8. | ||
Jain R, Chetty R. Gastric hyperplastic polyps: a review. Dig Dis Sci. 2009;54(9):1839–1846. | ||
Han AR, Sung CO, Kim KM, et al. The clinicopathological features of gastric hyperplastic polyps with neoplastic transformations: a suggestion of indication for endoscopic polypectomy. Gut Liver. 2009;3(4):271–275. | ||
Li J, Woods SL, Healey S, et al. Point mutations in exon 1B of APC reveal gastric adenocarcinoma and proximal polyposis of the stomach as a familial adenomatous polyposis variant. Am J Hum Genet. 2016;98(5):830–842. | ||
Repak R, Kohoutova D, Podhola M, et al. The first European family with gastric adenocarcinoma and proximal polyposis of the stomach: case report and review of the literature. Gastrointest Endosc. 2016;84(4):718–725. | ||
Beer A, Streubel B, Asari R, Dejaco C, Oberhuber G. Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS) - a rare recently described gastric polyposis syndrome - report of a case. Z Gastroenterol. 2017;55(11):1131–1134. | ||
Mitsui Y, Yokoyama R, Fujimoto S, et al. First report of an Asian family with gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS) revealed with the germline mutation of the APC exon 1B promoter region. Gastric Cancer. 2018;21(6):1058–1063. | ||
Tsuchiya T, Tamura G, Sato K, et al. Distinct methylation patterns of two APC gene promoters in normal and cancerous gastric epithelia. Oncogene. 2000;19(32):3642–3646. | ||
Hosoya K, Yamashita S, Ando T, Nakajima T, Itoh F, Ushijima T. Adenomatous polyposis coli 1A is likely to be methylated as a passenger in human gastric carcinogenesis. Cancer Lett. 2009;285(2):182–189. | ||
Rohlin A, Engwall Y, Fritzell K, et al. Inactivation of promoter 1B of APC causes partial gene silencing: evidence for a significant role of the promoter in regulation and causative of familial adenomatous polyposis. Oncogene. 2011;30(50):4977–4989. | ||
Snow AK, Tuohy TM, Sargent NR, Smith LJ, Burt RW, Neklason DW. APC promoter 1B deletion in seven American families with familial adenomatous polyposis. Clin Genet. 2015;88(4):360–365. | ||
Jasperson KW, Patel SG, Ahnen DJ. APC-associated polyposis conditions. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews R. Seattle, WA: University of Washington; 1993. | ||
Mcduffie LA, Sabesan A, Allgäeuer M, et al. β-Catenin activation in fundic gland polyps, gastric cancer and colonic polyps in families afflicted by ‘gastric adenocarcinoma and proximal polyposis of the stomach’ (GAPPS). J Clin Pathol. 2016;69(9):826–833. | ||
Lagarde A, Rouleau E, Ferrari A, et al. Germline APC mutation spectrum derived from 863 genomic variations identified through a 15-year medical genetics service to French patients with FAP. J Med Genet. 2010;47(10):721–722. | ||
Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223–262; quiz 263. | ||
Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362(9377):39–41. | ||
Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology. 2009;1371985(6):e1971–1985. | ||
Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010;138(6):2044–2058. | ||
Lubbe SJ, di Bernardo MC, Chandler IP, Houlston RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol. 2009;27(24):3975–3980. | ||
Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol. 1998;5(8):751–756. | ||
Brosens LA, van Hattem A, Hylind LM, et al. Risk of colorectal cancer in juvenile polyposis. Gut. 2007;56(7):965–967. | ||
van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105(6):1258–1264. | ||
Schreibman IR, Baker M, Amos C, Mcgarrity TJ. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol. 2005;100(2):476–490. | ||
Heald B, Mester J, Rybicki L, Orloff MS, Burke CA, Eng C. Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of PTEN mutation carriers. Gastroenterology. 2010;139(6):1927–1933. | ||
Stanich PP, Owens VL, Sweetser S, et al. Colonic polyposis and neoplasia in cowden syndrome. Mayo Clin Proc. 2011;86(6):489–492. | ||
Riegert-Johnson DL, Gleeson FC, Roberts M, et al. Cancer and lhermitte-duclos disease are common in cowden syndrome patients. Hered Cancer Clin Pract. 2010;8(1):6. | ||
Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18(2):400–407. | ||
Bertario L, Russo A, Sala P, et al. Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. J Clin Oncol. 2003;21(9):1698–1707. | ||
Bulow Set al. Duodenal adenomatosis in familial adenomatous polyposis. Gut. 2004;53(3):381–386. | ||
Gibbons DC, Sinha A, Phillips RK, Clark SK. Colorectal cancer: no longer the issue in familial adenomatous polyposis? Fam Cancer. 2011;10(1):11–20. | ||
Burt RW, Leppert MF, Slattery ML, et al. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology. 2004;127(2):444–451. | ||
Kadiyska TK, Todorov TP, Bichev SN, et al. APC promoter 1B deletion in familial polyposis-implications for mutation-negative families. Clin Genet. 2014;85(5):452–457. | ||
Lin Y, Lin S, Baxter MD, et al. Novel APC promoter and exon 1B deletion and allelic silencing in three mutation-negative classic familial adenomatous polyposis families. Genome Med. 2015;7(1):42. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.