")
Back to Journals » International Journal of Nanomedicine » Volume 12
Gambogic acid-loaded biomimetic nanoparticles in colorectal cancer treatment
Authors Zhang Z, Qian HQ, Yang M, Li RT, Hu J, Li L, Yu LX, Liu BR, Qian XP
Received 10 November 2016
Accepted for publication 12 January 2017
Published 28 February 2017 Volume 2017:12 Pages 1593—1605
DOI https://doi.org/10.2147/IJN.S127256
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Zhen Zhang,1 Hanqing Qian,2 Mi Yang,2 Rutian Li,2 Jing Hu,1 Li Li,1 Lixia Yu,2 Baorui Liu,1,2 Xiaoping Qian1,2
1Comprehensive Cancer Center, Nanjing Drum Tower Hospital Clinical College of Traditional Chinese and Western Medicine, Nanjing University of Traditional Chinese Medicine, 2Comprehensive Cancer Center, Nanjing Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute, Nanjing University, Nanjing, China
Abstract: Gambogic acid (GA) is expected to be a potential new antitumor drug, but its poor aqueous solubility and inevitable side effects limit its clinical application. Despite these inherent defects, various nanocarriers can be used to promote the solubility and tumor targeting of GA, improving antitumor efficiency. In addition, a cell membrane-coated nanoparticle platform that was reported recently, unites the customizability and flexibility of a synthetic copolymer, as well as the functionality and complexity of natural membrane, and is a new synthetic biomimetic nanocarrier with improved stability and biocompatibility. Here, we combined poly(lactic-co-glycolic acid) (PLGA) with red blood-cell membrane (RBCm), and evaluated whether GA-loaded RBCm nanoparticles can retain and improve the antitumor efficacy of GA with relatively lower toxicity in colorectal cancer treatment compared with free GA. We also confirmed the stability, biocompatibility, passive targeting, and few side effects of RBCm-GA/PLGA nanoparticles. We expect to provide a new drug carrier in the treatment of colorectal cancer, which has strong clinical application prospects. In addition, the potential antitumor drug GA and other similar drugs could achieve broader clinical applications via this biomimetic nanocarrier.
Keywords: gambogic acid, nanocarriers, RBCm-GA/PLGA nanoparticles, colorectal cancer
Introduction
Gambogic acid (GA), the major active ingredient in gamboge, is a traditional Chinese medicine that can be potentially developed for various new antitumor drugs. A variety of studies have demonstrated its incredible antitumor effect on multiple types of tumors, including colorectal cancer.1–3 Nevertheless, the appendant poor aqueous solubility (less than 0.5 μg/mL) and accompanying toxic side effects while acquiring satisfactory antitumor effects of GA limit its bioavailability in clinical application.4,5
The appearance of various nanocarriers not only enhances the solubility of hydrophobic antitumor drugs but also controls a slow and stable release of drugs in vivo to reduce side effects and preserve the same therapeutic effect.4,6 Multifarious nanotechnologies have been widely applied in diagnosis, therapeutic, and other biomedical areas.7–11 On one hand, via the enhanced permeability and retention (EPR) effect, nanoparticles (NPs) accumulate at tumor sites, benefiting from the special environment of vascular malformation and lack of lymphatic circumfluence. On the other hand, hydrophobic drug molecules can be encapsulated in NPs with relatively high drug-loading efficiency and excellent stability.12,13
Despite the utilization of biocompatible and biodegradable materials, such as poly(lactic-co-glycolic acid) (PLGA), polylactic acid, and polycaprolactone,14,15 nanocarriers are unavoidably met with quick excretion from the mononuclear phagocyte system. The hydrophilic polymer polyethylene glycol (PEG) can be decorated on the surface of NPs and then the NPs can be shielded by water molecules. PEGylated NPs appear invisible to macrophages, resulting in immunoevasion16–18 in theory. However, an anti-PEG immunological response is observed after the first dose of PEGylated NPs injected in vivo, leading to the accelerated blood clearance phenomenon upon repeated injection of the PEGylated NPs.19
Therefore, more and more attention has been focused on natural lipids20 and cell membranes.21,22 Membrane coating has been tried in an attempt to disguise NPs as autologous cells by a novel nanocoating technology. In consideration of enormous quantity in vivo as well as simple structure and properties, red blood-cell membrane (RBCm) was the earliest and most widely explored for building synthetic biomimetic nanocarriers.23–27
RBCm coating containing membrane proteins (eg, CD47,28 C8bp,29 and HRP30) can retain their original biological properties completely,22 disguise NPs as RBCs,31 evade the immune response from the mononuclear phagocyte system to prolong systemic circulation without the accelerated blood clearance phenomenon,19 and concurrently reduce reticuloendothelial system uptake.32 This provides more opportunity to accumulate at tumor sites. Research has shown that the RBC-NP platform does not induce immune responses on the cellular level or the humoral level,19 highlighting its superior biocompatibility. This conforms to some of the requirements of a desired drug-carrier system, ie, evading the host immune system while delivering its “cargo” safely to its “destination”. Besides, the versatility of the inner polymeric core, which is able to deliver a wide variety of antitumor drugs,25,33 is also equally valuable compared with nanoerythrosomes.34,35
Here, we try to demonstrate that the synthetic RBC-NP platform is able to enhance the efficiency of potential antitumor drug GA in a mouse model of colorectal cancer (Figure 1). Furthermore, the biocompatibility and safety of GA-loaded RBC NPs are included in the exploration scope.
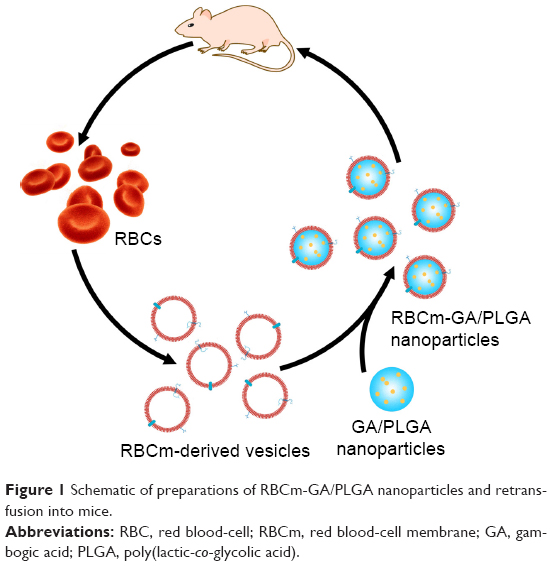 | Figure 1 Schematic of preparations of RBCm-GA/PLGA nanoparticles and retransfusion into mice. |
Materials and methods
Materials
GA (purity ≥99%), PLGA (molecular weight 40,000), polyvinyl alcohol (PVA), and dichloromethane (DCM) were purchased from Aladdin Industrial Corporation (Shanghai, China). 3,3′-Dioctadecyloxacarbocyanine perchlorate (DiO), 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI), phorbol-12-myristate-13-acetate/12-O-tetradecanoylphorbol 13-acetate (PMA/TPA), and a cell-cycle analysis kit were purchased from Beyotime Institute of Biotechnology (Haimen, China). An annexin V–Fluos staining kit was purchased from Hoffman-La Roche Ltd (Basel, Switzerland).
Cells and cell culture
The human colon cancer-cell line SW480, human gastric cancer-cell lines MKN45 and AGS, and the human monocytic cell line THP1 were preserved by our lab and purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Roswell Park Memorial Institute 1640 medium with 10% calf serum at 37°C in a humidified atmosphere with 5% CO2.
Animals
Male BALB/c mice (weight 18-25 g, age 5-8 weeks) were supplied by the Experimental Animal Center of Nanjing Drum Tower Hospital. All animal experiments were approved by the Animal Care Committee, Drum Tower Hospital, Nanjing, China. All animal-handling procedures were performed in compliance with guidelines set by the Animal Care Committee, Drum Tower Hospital.
Preparation of RBCm-GA/PLGA NPs
Preparation of RBCm-derived vesicles
Whole blood was collected from BALB/c mice with heparin solution and centrifuged at 800× g for 5 minutes at 4°C. Then, the serum and buffy coat were removed. Residual RBCs were washed with normal saline (NS) at 4°C three times prior to hypotonic medium treatment for hemolysis. The washed RBCs were suspended in 0.25× phosphate-buffered saline (PBS) in an ice bath for 30 minutes and centrifuged at 17,000× g for 10 minutes at 4°C. After centrifugation, a pink pellet, which was the RBCm, could be observed at the bottom of the tube. RBCm was collected after the hemoglobin was removed, and was then sonicated in a capped glass vial for 6 minutes using a bath sonicator (Elma Schmidbauer GmbH, Singen, Germany) at a frequency of 37 kHz and power of 60 W. The resulting membrane vesicles were subsequently extruded repeatedly through 800, 400, and 200 nm polycarbonate porous membranes using a mini-extruder (Avanti Polar Lipids, Alabaster, AL, USA).
Preparation of GA/PLGA NPs
GA and PLGA were dissolved in 0.5 mL of DCM as the oil phase. The organic solution was then added into 1.5 mL of 450 mM aqueous PVA solution and sonicated to obtain the O/W emulsion. The emulsion was added to 2.5 mL of 115 mM PVA and sonicated again. The final solution was subsequently left under stirring for at least 2 hours to remove DCM. The resulting solution was centrifuged at 20,000× g for 30 minutes and washed three times to remove PVA. After the yellow sediment was resuspended in pure water and filtered with a 0.45 μm microfiltration membrane to remove unloaded GA, the inner GA/PLGA NPs were prepared.
Drug loading (DL) and encapsulation efficiency (EE) of GA in PLGA NPs were detected using high-performance liquid chromatography (HPLC). The GA/PLGA NPs were first dissolved in acetone. After acetone had been volatilized, the resulting yellow sediment was then dissolved in methanol. The GA content was dissolved in methanol, while PLGA was precipitated. The GA content was detected using an HPLC system (Agilent Technologies, Santa Clara, CA, USA) equipped with an HC-C18 column at 30°C and detected at 360 nm. The mobile phase consisted of methanol and 0.1% phosphoric acid (95:5, v/v) and the flow rate was 1.0 mL/min. DL and EE were calculated as follows:
|
|
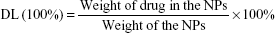
|
|

Fusion of RBCm-derived vesicles with GA/PLGA NPs
The prepared RBCm-derived vesicles and GA/PLGA NPs were mixed and then extruded ten times through a 200 nm polycarbonate porous membrane with the Avanti mini-extruder. Finally, the RBCm-GA/PLGA NPs were obtained as a light-yellow suspension.
Characterization of RBCm-GA/PLGA NPs
Chemical interaction between GA and PLGA through O/W emulsion was analyzed by infrared (IR) spectra. The samples, containing GA, PLGA, and GA/PLGA NPs, were freeze-dried in preparation. Fourier-transform IR spectra were recorded on a Nicolet Nexus 470-ESP (Thermo Fisher Scientific, Waltham, MA, USA). Baseline values were corrected, and the samples were scanned against a blank KBr-pellet background at wave numbers in the range of 400–4,000 cm−1.
The diameter and polydispersity of the NPs in (0.1 M, pH 7.4) were measured by dynamic light scattering (DLS) using a BI-900AT/ζ-potential analyzer (Brookhaven Instruments Corporation, Holtsville, NY, USA). To evaluate the stability of NPs, the RBCm-GA/PLGA NPs were kept at 25°C and the diameter determined by DLS every day for a week.
The structure of the RBCm-GA/PLGA NPs was examined using transmission electron microscopy (JEM-100S; JEOL, Tokyo, Japan). A drop of the NP suspension was deposited onto a cupper grid and air-dried at room temperature. The sample was stained with 1% uranyl acetate before observation. Drug release was studied by dialyzing samples against PBS at different pH values (7.4 and 5.4) using a dialysis bag with a molecular weight cutoff of 10 kDa at 37°C for a week. The GA content was detected using HPLC.
In vitro uptake and cytotoxicity
In vitro uptake
RBCm and PLGA NPs were fluorescence-labeled with red DiI and green DiO dye, respectively. Subsequently, three gastrointestinal cancer-cell lines (SW480, MKN45, and AGS) were seeded into confocal dishes, respectively, at a density of 106. NPs (500 μL) were added to the cell-culture medium. After 30 minutes of incubation, the cells were washed with NS and cellular nuclei stained with DAPI. The cellular uptake of NPs was observed using confocal laser-scanning microscopy (Olympus, Tokyo, Japan).
In vitro cytotoxicity (MTT assay)
MTT assays were carried out to test the in vitro cytotoxicity of RBCm-GA/PLGA NPs against SW480 cells. Cells were seeded in 96-well plates at a density of 5,000 cells per well and incubated at 37°C with 5% CO2 for attachment. After cells had been exposed to various concentrations of RBCm-GA/PLGA NPs and free GA for 24 and 48 hours, 20 μL of 5 mg/mL MTT solution was added to each well, and incubation was carried out for another 4 hours. The supernatant was discarded, and 150 μL of dimethyl sulfoxide was added to dissolve the formazan crystals. Absorption at 490 nm was measured by a Multiskan Spectrum microplate reader (Thermo Fisher Scientific), and cell viability was calculated by the formula:
|
|
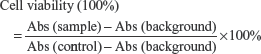
Apoptosis assay
Apoptosis was detected using the annexin V–Fluos staining kit according to the instructions. After being exposed to various concentrations of RBCm-GA/PLGA NPs and free GA for 12, 24, 48, and 72 hours, 106 SW480 cells were collected and washed with PBS. Washed cells were resuspended in 100 μL of annexin V–Fluos labeling solution (100 μL of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer, 2 μL of annexin V–fluorescein and 2 μL of propidium iodide) per sample, and incubated in the dark for 15 minutes at room temperature. The apoptosis of SW480 was analyzed by flow cytometry (EMD Millipore, Billerica, MA, USA).
Cell-cycle analysis
SW480 cells were incubated with RBCm-GA/PLGA NPs and free GA for 24 and 48 hours. Then, the cells were collected and washed with PBS and fixed in 70% alcohol overnight at 4°C. Fixed cells (106) were washed with PBS again, resuspended in 500 μL of propidium iodide-staining solution per sample, and incubated in the dark at 37°C for 30 minutes. The samples were analyzed by flow cytometry.
In vivo antitumor efficacy
SW480 cells (5×106) were implanted subcutaneously into the right flank of BALB/c (nu/nu) mice. The tumors were allowed to grow for a week. Then, SW480 tumor-bearing nude mice were divided into five groups (n=7 per group) and given injections of RBCm-GA/PLGA NPs (6 mg/kg, RBCm-GA/PLGA group), GA/PLGA NPs (6 mg/kg, GA/PLGA group), GA solution (6 mg/kg, GA group), RBCm-PLGA NPs (RBCm-PLGA group), and NS (NS group) via the tail vein once a week a total of twice. Tumor size and mouse weight were measured on day 7 postimplantation and every other day after injection. Tumor volume was calculated as V = D × d2/2, where D and d are the longest and shortest diameter of the tumor in millimeters, respectively. Survival was predefined as tumor size <2,000 mm3 prior to the initiation of the study. In order to reduce the impact of initial tumor volume and weight differences among different groups, relative tumor volume and relative weight were calculated by the formulae:
|
|

|
|

where V represents absolute tumor volume and V0 average tumor volume of the group on day 7. Similarly, W represents the absolute weight and W0 average weight of the group on day 7.
Biocompatibility and safety studies
PLGA and RBCm-PLGA NPs were both fluorescence-labeled with DiO. THP1 macrophages, which had been stimulated with PMA/TPA previously, were incubated with PLGA and RBCm-PLGA NPs, respectively, for 30 minutes at 37°C. Then, the macrophages were washed and digested by trypsin. Fluorescence quantitation was performed by flow cytometry to reflect the biocompatibility of NPs indirectly. In antitumor-efficacy experiments, mice were killed on day 22, and heart, lungs, liver, spleen, and kidneys of mice in each group were collected for hematoxylin and eosin (H&E) staining to assess systemic toxicity.
Statistical analysis
Statistical analysis was performed using Statistical Package for the Social Sciences (SPSS) version 23.0 software (IBM Corporation, Armonk, NY, USA). One-way analysis of variance or Student’s t-test was used to determine the differences among various treatments. Data are presented as mean ± standard deviation. Statistically significant results were subjected to Tukey’s post hoc test (GraphPad Prism 5.0; GraphPad Software Inc, La Jolla, CA, USA). All quantitative data reported represent the mean of at least three independent experiments. Differences were considered statistically significant if P<0.05.
Results
Preparation and physicochemical characterization of RBCm-GA/PLGA NPs
Blank PLGA or GA/PLGA NPs were prepared using O/W emulsion. The loading content of GA in PLGA core was controlled by varying the ratio between the drug and PLGA (Figure 2A). By increasing the input of GA, the loading of the drug in NPs was also increased. DL was approximately 20 wt% when the ratio of GA:PLGA in weight was higher than 30%. Moreover, when drug-input concentration was 30 wt%, EE reached its maximum value. Therefore, 30 wt% GA was used for subsequent in vitro and in vivo studies.
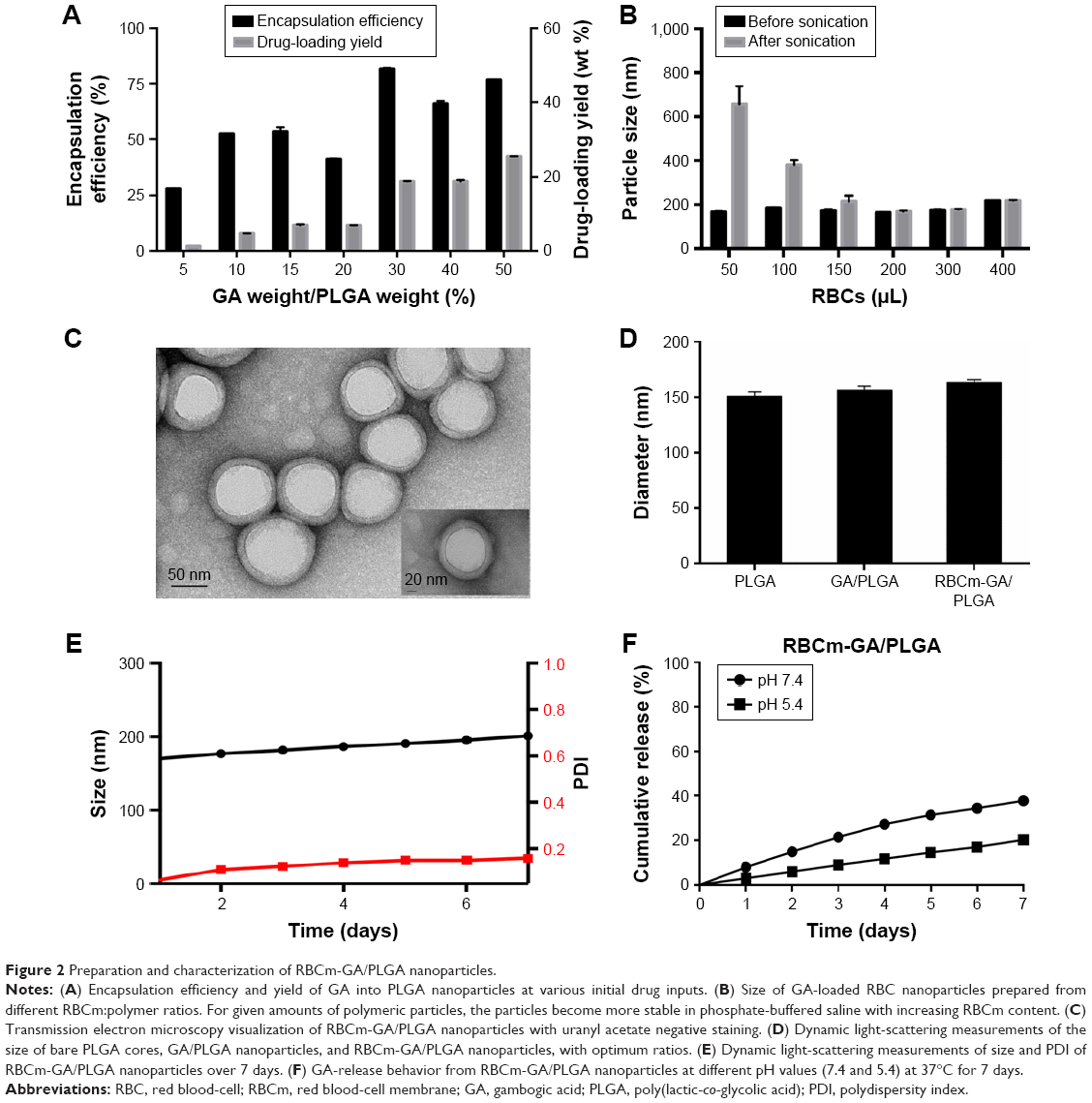 | Figure 2 Preparation and characterization of RBCm-GA/PLGA nanoparticles. |
Fourier-transform IR spectrometry was used to evaluate the chemical interaction between GA and PLGA through O/W emulsion. As shown in Figure S1, the characteristic peak of PLGA at 3,508 cm−1 and peaks of GA at 1,593.3 cm−1 and 1,633.5 cm−1 were changed. In the spectra of GA/PLGA NPs, emerging peaks of 1,594.7 cm−1, 1,634.9 cm−1, and 3,349.7 cm−1 proved the existence of the interaction between GA and the PLGA.
Next, RBCm derived from the blood of BALB/c mice was coated onto the polymeric cores using an extrusion approach, as previously described.23–27 According to the parameters, including density of polymeric cores, the mean particle sizes before and after the RBCm coating, mean surface area of mouse RBCs, membrane thickness of RBCs, we estimated that RBCm collected from 200 μL mouse RBCs could fully coat 10 mg of PLGA NPs. A series of RBCm-GA/PLGA formulations were prepared using RBCm collected from 50 μL, 100 μL, 150 μL, 200 μL, 300 μL, and 400 μL of RBCs. The change in particle diameter was determined using DLS before and after sonication for 30 minutes (Figure 2B). The results reflected higher particle stability with increased RBCm content. Besides, NPs showed negligible size increase following sonication, when the content of RBCm was above 200 μL (the theoretical value for full particle coverage).
After the ratio of GA, PLGA, and RBCm had been confirmed, the core–shell structure of the RBCm-GA/PLGA NPs was visualized by transmission electron microscopy with uranyl acetate negative staining (Figure 2C). The thickness of the outer lipid shell was approximately 7–8 nm, consistent with the membrane width of RBCs, indicating a successful RBCm coating of PLGA NPs.
Drug loading and membrane coating increased particle size (Figure 2D). When GA was loaded into PLGA NPs, particle size increased from 151 nm to 156 nm. Coating of GA/PLGA cores with RBCm resulted in a size increase to approximately 163 nm. The resulting RBC-mimicking NPs showed long-term stability in vitro (Figure 2E). Particle size and polydispersity index of RBCm-GA/PLGA NPs suspended in PBS were monitored by DLS, and little change was observed in 7 days.
Whether in a neutral (pH 7.4) or acidic (pH 5.4) environment, the RBCm-GA/PLGA NPs displayed a slow and sustained release in 7 days (Figure 2F). Approximately 40% of GA was released from RBCm-GA/PLGA NPs in neutral PBS (pH 7.4) over 7 days, while about 20% of GA was released from the RBCm-GA/PLGA NPs in acidic PBS (pH 5.4). This observed prolonged release was partly due to the RBCm coating, which acted as a diffusional barrier for GA release. Moreover, when RBCm-GA/PLGA NPs were exposed to the acidic environment (PBS, pH 5.4), yellow sediment was observed in the dialysis bag, indicating an increase in particle size. This was likely to bring slower release.
In vitro uptake and cytotoxicity
Uptake of RBCm-PLGA NPs in vitro was confirmed by laser-scanning confocal microscopy after incubation of tumor cells with RBCm-PLGA NPs. It can be seen from the fluorescence images that green punctate (polymeric cores were visualized with DiO dye) and red punctate (RBCm were visualized with red DiI dye) points were around the nucleus (visualized with blue DAPI dye), overlapping each other (Figure 3A). The images also suggested colocalization of RBCm and polymeric cores after being internalized by gastrointestinal cancer cells.
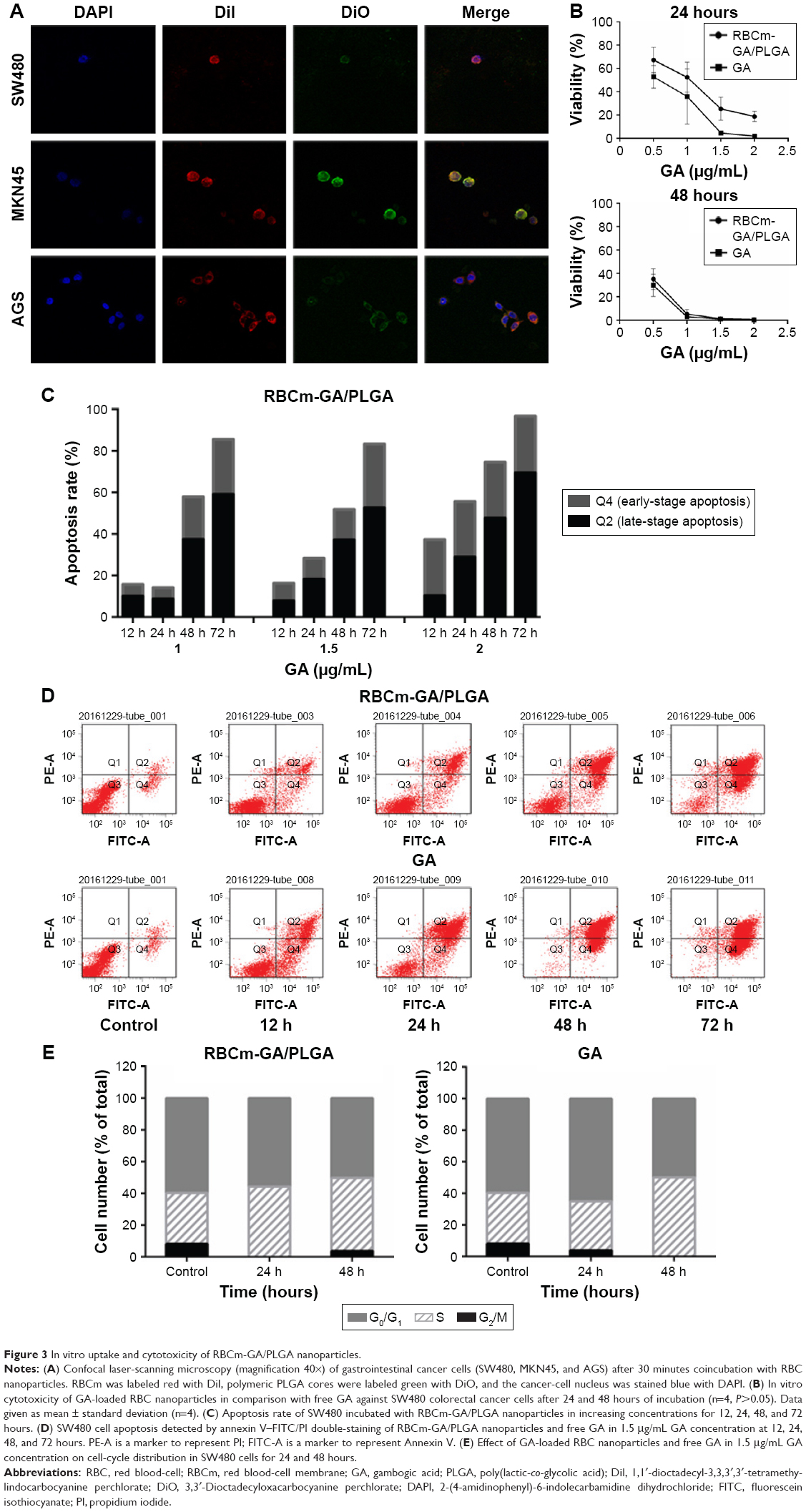 | Figure 3 In vitro uptake and cytotoxicity of RBCm-GA/PLGA nanoparticles. |
Based on the result that the NPs could be taken up by tumor cells in vitro, MTT assays were used to evaluate whether the GA encapsulated within the RBCm-GA/PLGA NPs could retain its antitumor activity against SW480 cells. The results showed a dose-dependent cytotoxicity of both RBCm-GA/PLGA NPs and GA against SW480 cells for 24 and 48 hours incubation (Figure 3B). Moreover, compared with cell viability, free GA demonstrated slightly better efficacy (P>0.05, n=4) against SW480 cells for 24 hours, but almost no difference for 48 hours. This further expounds the therapeutic potential of GA-loaded RBCm-cloaked NPs with its continuous antitumor efficacy in vitro.
GA may cause the apoptosis of cancer cells to create cytotoxicity. To illustrate the possible mechanism of cytotoxicity in vitro, further analysis of apoptosis and the cell cycle was also conducted. Apoptotic rate (Q2 + Q4) obviously increased when SW480 cells were exposed to different concentrations of RBCm-GA/PLGA NPs from 12 to 72 hours (Figure 3C). When the concentration was above 1.5 μg/mL, an apparent apoptosis of SW480 cells was observed. Therefore, flow-cytometry analysis conducted at 1.5 μg/mL of RBCm-GA/PLGA NPs and GA-induced apoptosis. Representative results at 12, 24, 48, and 72 hours are shown in Figure 3D. The result reflected the rate of both early (Q4)- and late (Q2)-stage apoptotic SW480 cells increased with time. In the previous 24 hours, free GA induced more apoptosis of SW480 than RBCm-GA/PLGA NPs, while the two groups both showed similar apoptosis of SW480 after 48 hours. This matched with the results of MTT assays. In addition, cell-cycle distribution was also affected after exposure to 1.5 μg/mL GA-loaded NPs and free GA at 24 and 48 hours (Figure 3E). The distribution showed an increased accumulation of cells at the S phase with time in both GA-loaded NPs and the GA group, which meant RBCm-GA/PLGA NPs and GA caused S arrest in SW480 cells.
In vivo antitumor efficacy
After verifying the antitumor activity of RBCm-GA/PLGA NPs in vitro, we sequentially determined whether the activity could act as effective therapy against tumor growth in vivo. BALB/c nude mice bearing subcutaneous inoculated SW480 tumors were used to observe the long-term tumor burden in a colorectal cancer model. Treatments were administered via tail vein with RBCm-GA/PLGA NPs, GA/PLGA NPs, GA, RBCm-PLGA NPs, and NS and repeated once a week a total of twice. After the injection of RBCm-GA/PLGA NPs, a majority of subcutaneous tumors of this group successively began to necrose from the middle of tumors, and then tumor volumes were diminished. As shown in Figure 4A and B, compared with the control group, the tumor volumes and relative tumor volumes of the RBCm-GA/PLGA group were markedly decreased (P≤0.001, n=7), while the GA group showed a negligible tumor-inhibition effect (P>0.05). More detailed information about tumors in different groups was captured (Figure 4C). Moreover, the GA/PLGA group also showed certain efficacy in terms of tumor-growth inhibition, reflecting the advantage of this nanocarrier for tumor therapy.
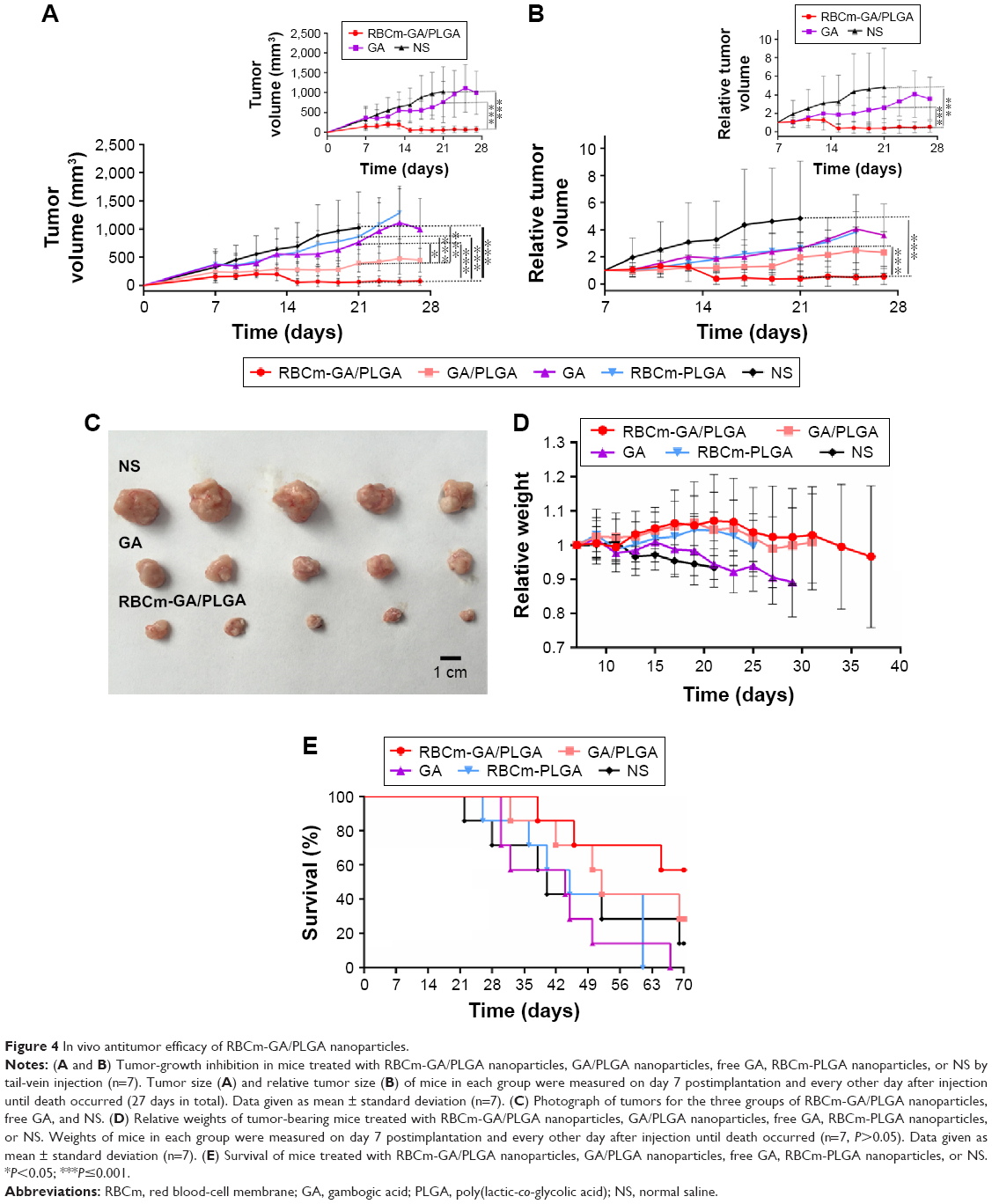 | Figure 4 In vivo antitumor efficacy of RBCm-GA/PLGA nanoparticles. |
The weights of mice in each group were also recorded (Figure 4D). Along with increasing tumor sizes in the control and GA groups, the weight of mice decreased or remained steady, which reflected that mice in these groups suffered from the increasing tumor burden or toxicity of free GA. This was also reflected in survival results (Figure 4E): when compared with NP formulations (both RBCm-GA/PLGA and GA/PLGA groups), other groups showed worse survival. Mice treated with RBCm-GA/PLGA NPs showed extended median survival of at least 30 days more compared with the no-treatment group. However, the free-GA group was extended by only 4 days.
Biocompatibility and safety studies
RBCm coating may make nanocarriers achieve better biocompatibility theoretically. We identified whether RBCm-coated PLGA NPs had better biocompatibility than bare PLGA NPs by analyzing the uptake of macrophages in vitro. The fluorescent quantitation of two groups showed the quantity of NPs taken up (Figure 5A) indirectly reflected the immunoevasion of the mononuclear phagocyte system by nanocarriers. RBCm coating resulted in fewer NPs being taken up (P≤0.001, n=3) by macrophages, indicating better biocompatibility and immunoevasion of the mononuclear phagocyte system, which may result in long circulation in vivo. In vivo safety was also the object of study for RBCm-GA/PLGA NPs. H&E-stained images of major organs in each group showed that there was no noticeable signal of organ damage (Figure 5B). In vivo safety also suggested good biocompatibility for these RBCm-GA/PLGA NPs.
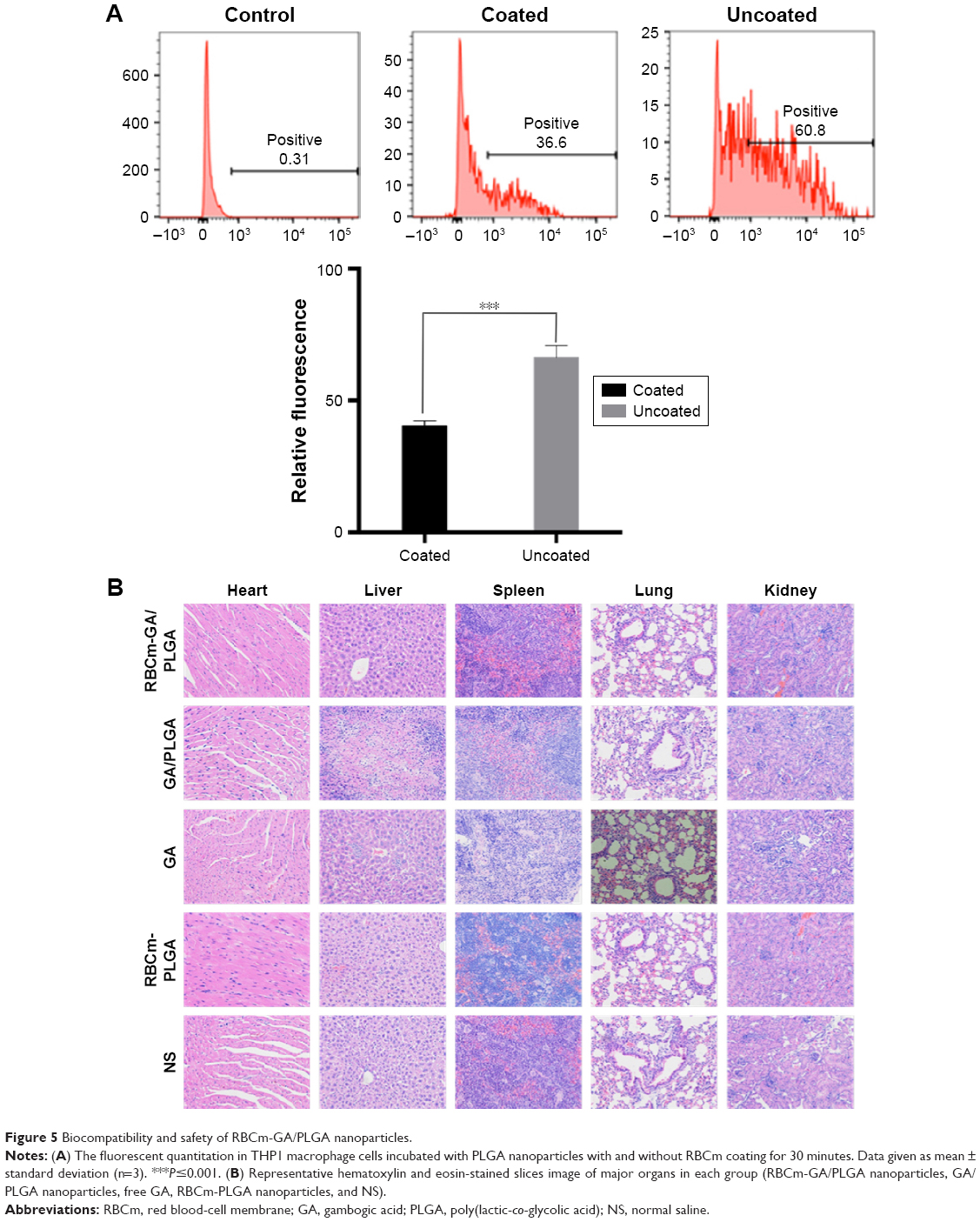 | Figure 5 Biocompatibility and safety of RBCm-GA/PLGA nanoparticles. |
Discussion
The RBC-NP platform we structure here integrates two distinct materials (polymer and cell membrane) and takes advantage of their respective and particular properties, achieving a synthetic biomimetic nanocarrier.21 On the basis of stability and biocompatibility of the RBC-NP platform, multiple types of antineoplastic agents – hydrophilic or hydrophobic – can be carried in by appropriate approaches. Moreover, hydrophobic agents benefit more from this nanocarrier than hydrophilic agents, owing to the satisfactory solution of safe solvent. GA-loaded RBC NPs increased the solubility of GA to some extent. We tried to enhance the solubility of free GA through a sonicated mixture containing ethanol, Tween, and water, but the vascular stimulation led by the cosolvent in GA solution could be observed during vivo test. Just as with the clinical application of some chemotherapy drugs with poor water solubility (eg, paclitaxel), the use of a cosolvent is required, accompanied by unavoidable allergic reaction and vascular stimulation. The introduction of nanocarriers avoids such inconvenience by increasing solubility tactfully. Meanwhile, other potential antitumor drugs similar to GA could also be delivered for broader clinical applications by this biomimetic nanocarrier. In addition, the coating of autologous liposome RBCm enhances biocompatibility through evasion of the immunoreaction of the mononuclear phagocyte system, as shown in the phagocytosis test. This also reflected this synthetic biomimetic nanocarrier still retained biological properties of RBCm to some extent.
RBCm-GA/PLGA NPs and free GA at the same dose exhibited similar cytotoxicity through the occurrence of apoptosis and variation in cell cycle after 48 hours in vitro. However, a significant difference was observed in the inhibition of tumor growth in vivo. Free GA at a dose of 6 mg/kg exerted marginal control of tumor growth and no obvious signal of organ damage in H&E-stained images compared with the control group. This may suggest that a dose of 6 mg/kg did not reach the effective dose for free GA. On the contrary, GA-loaded RBC NPs at the same dose achieved significant inhibition of tumor growth. This can be attributed to the EPR effect, which promoted RBCm-GA/PLGA NPs to accumulate at the tumor site, despite no active target existing, and the local GA concentration at the tumor site was increased. Furthermore, immunoevasion led by membrane coating made the RBCm-GA/PLGA group slightly better than the GA/PLGA group. In future work, achieving a specific active target by utilizing specific membranes derived from selected cells,36 such as cancer cells,37–40 or via targeting ligands inserted into membranes41,42 can be designed to build a nanocarrier with better antitumor effect.
For in vivo antitumor efficacy, we also observed that the decrease in tumor volume size was via necrosis of subcutaneous tumors. This phenomenon is behind our supposition that accumulation of GA at the tumor site led to necrosis by its effect on local blood vessels. More work will be needed to explain this in future.
Conclusion
RBCm-GA/PLGA NPs served as a biomimetic nanocarrier and united the customizability and flexibility of synthetic copolymer PLGA, in addition to the functionality and complexity of natural liposome RBCm. The loading of the potential antitumor drug GA bestowed on the nanocarrier new antitumor efficacy. The combination of RBCm and PLGA has been shown to enhance the stability and biocompatibility of a composite carrier compared with bare nanoerythrosomes43 and a polymeric carrier,44 and we confirmed this in vitro. Moreover, GA-loaded RBC NPs lifted restrictions on the application of GA, which were due to its poor water solubility.
With regard to antitumor effects, it was confirmed that RBCm-GA/PLGA NPs were taken up by gastrointestinal cancer cells and inhibited the proliferation of SW480 cells in vitro via the influence of apoptosis and the cell cycle. A series of the antitumor efficacy of RBCm-GA/PLGA NPs equals bare drug GA over time.
Following a certain antitumor efficacy in vitro, inhibition of tumor growth in vivo is also obvious. RBCm-GA/PLGA NPs notably inhibited the growth of subcutaneous tumors, caused necrosis, and shrank the volume sequentially. However, bare GA at the same dose gained marginal control of tumor growth, far inferior to its activity in vitro. This difference reflects the enormous advantage of this composite biomimetic nanocarrier, and it takes full advantage of the antitumor efficacy of GA. Furthermore, RBCm-GA/PLGA NPs guarantee safety in vivo.
Acknowledgments
This work was supported by grant BK20161107 from the Provincial Natural Science Foundation of Jiangsu and grant 201501013 from the Nanjing Science and Technology Development Project.
Disclosure
The authors report no conflicts of interest in this work.
References
Shi X, Chen X, Li X, et al. Gambogic acid induces apoptosis in imatinib-resistant chronic myeloid leukemia cells via inducing proteasome inhibition and caspase-dependent Bcr-Abl downregulation. Clin Cancer Res. 2014;20(1):151–163. | ||
Wen C, Huang L, Chen J, et al. Gambogic acid inhibits growth, induces apoptosis, and overcomes drug resistance in human colorectal cancer cells. Int J Oncol. 2015;47(5):1663–1671. | ||
Huang GM, Sun Y, Ge X, Wan X, Li CB. Gambogic acid induces apoptosis and inhibits colorectal tumor growth via mitochondrial pathways. World J Gastroenterol. 2015;21(20):6194–6205. | ||
Cai L, Qiu N, Xiang M, et al. Improving aqueous solubility and antitumor effects by nanosized gambogic acid-mPEG2000 micelles. Int J Nanomedicine. 2014;9:243–255. | ||
Yin DK, Yang Y, Cai HX, Wang F, Peng DY, He LQ. Gambogic acid-loaded electrosprayed particles for site-specific treatment of hepatocellular carcinoma. Mol Pharm. 2014;11(11):4107–4117. | ||
Huang W, Wang X, Shi C, et al. Fine-tuning vitamin E-containing telodendrimers for efficient delivery of gambogic acid in colon cancer treatment. Mol Pharm. 2015;12(4):1216–1229. | ||
Minelli C, Lowe SB, Stevens MM. Engineering nanocomposite materials for cancer therapy. Small. 2010;6(21):2336–2357. | ||
Voon SH, Kiew LV, Lee HB, et al. In vivo studies of nanostructure-based photosensitizers for photodynamic cancer therapy. Small. 2014;10(24):4993–5013. | ||
Cheng Z, Al ZA, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338(6109):903–910. | ||
Shi D, Bedford NM, Cho HS. Engineered multifunctional nanocarriers for cancer diagnosis and therapeutics. Small. 2011;7(18):2549–2567. | ||
Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release. 2015;200:138–157. | ||
Pistone A, Iannazzo D, Ansari S, et al. Tunable doxorubicin release from polymer-gated multiwalled carbon nanotubes. Int J Pharm. 2016;515(1–2):30–36. | ||
Natarajan JV, Nugraha C, Ng XW, Venkatraman S. Sustained-release from nanocarriers: a review. J Control Release. 2014;193:122–138. | ||
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63(3):170–183. | ||
Gunatillake PA, Adhikari R. Biodegradable synthetic polymers for tissue engineering. Eur Cell Mater. 2003;5:1–16. | ||
Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed Engl. 2010;49(36):6288–6308. | ||
Liu Z, Robinson JT, Sun X, Dai H. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs. J Am Chem Soc. 2008;130(33):10876–10877. | ||
Aryal S, Hu CM, Zhang L. Polymeric nanoparticles with precise ratiometric control over drug loading for combination therapy. Mol Pharm. 2011;8(4):1401–1407. | ||
Rao L, Bu LL, Xu JH, et al. Red blood cell membrane as a biomimetic nanocoating for prolonged circulation time and reduced accelerated blood clearance. Small. 2015;11(46):6225–6236. | ||
Gao WW, Hu CM, Fang RH, Zhang LF. Liposome-like nanostructures for drug delivery. J Mater Chem B Mater Biol Med. 2013;1(48):6569. | ||
Luk BT, Zhang L. Cell membrane-camouflaged nanoparticles for drug delivery. J Control Release. 2015;220(Pt B):600–607. | ||
Fang RH, Luk BT, Hu CM, Zhang L. Engineered nanoparticles mimicking cell membranes for toxin neutralization. Adv Drug Deliv Rev. 2015;90:69–80. | ||
Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci U S A. 2011;108(27):10980–10985. | ||
Luk BT, Hu CM, Fang RH, et al. Interfacial interactions between natural RBC membranes and synthetic polymeric nanoparticles. Nanoscale. 2014;6(5):2730–2737. | ||
Aryal S, Hu CM, Fang RH, et al. Erythrocyte membrane-cloaked polymeric nanoparticles for controlled drug loading and release. Nanomedicine (Lond). 2013;8(8):1271–1280. | ||
Piao JG, Wang L, Gao F, You YZ, Xiong Y, Yang L. Erythrocyte membrane is an alternative coating to polyethylene glycol for prolonging the circulation lifetime of gold nanocages for photothermal therapy. ACS Nano. 2014;8(10):10414–10425. | ||
Guo Y, Wang D, Song Q, et al. Erythrocyte membrane-enveloped polymeric nanoparticles as nanovaccine for induction of antitumor immunity against melanoma. ACS Nano. 2015;9(7):6918–6933. | ||
Tsai RK, Rodriguez PL, Discher DE. Self inhibition of phagocytosis: the affinity of ‘marker of self’ CD47 for SIRPα dictates potency of inhibition but only at low expression levels. Blood Cells Mol Dis. 2010;45(1):67–74. | ||
Schönermark S, Rauterberg EW, Shin ML, Löke S, Roelcke D, Hänsch GM. Homologous species restriction in lysis of human erythrocytes: a membrane-derived protein with C8-binding capacity functions as an inhibitor. J Immunol. 1986;136(5):1772–1776. | ||
Zalman LS, Wood LM, Müller-Eberhard HJ. Isolation of a human erythrocyte membrane protein capable of inhibiting expression of homologous complement transmembrane channels. PNAS. 1986;83(18):6975–6979. | ||
Fang RH, Hu CM, Zhang L. Nanoparticles disguised as red blood cells to evade the immune system. Expert Opin Biol Ther. 2012;12(4):385–389. | ||
Rao L, Xu JH, Cai B, et al. Synthetic nanoparticles camouflaged with biomimetic erythrocyte membranes for reduced reticuloendothelial system uptake. Nanotechnology. 2016;27(8):085106. | ||
Luk BT, Fang RH, Hu CM, et al. Safe and immunocompatible nanocarriers cloaked in RBC membranes for drug delivery to treat solid tumors. Theranostics. 2016;6(7):1004–1011. | ||
Lejeune A, Moorjani M, Gicquaud C, Lacroix J, Poyet P, Gaudreault R. Nanoerythrosome, a new derivative of erythrocyte ghost: preparation and antineoplastic potential as drug carrier for daunorubicin. Anticancer Res. 1994;14(3A):915–919. | ||
Gupta N, Patel B, Ahsan F. Nano-engineered erythrocyte ghosts as inhalational carriers for delivery of fasudil: preparation and characterization. Pharm Res. 2014;31(6):1553–1565. | ||
Hu CM, Fang RH, Wang KC, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526(7571):118–121. | ||
Fang RH, Hu CM, Luk BT, et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14(4):2181–2188. | ||
Rao L, Bu LL, Cai B, et al. Cancer cell membrane-coated upconversion nanoprobes for highly specific tumor imaging. Adv Mater. 2016;28(18):3460–3466. | ||
Zhu JY, Zheng DW, Zhang MK, et al. Preferential cancer cell self-recognition and tumor self-targeting by coating nanoparticles with homotypic cancer cell membranes. Nano Lett. 2016;16(9):5895–5901. | ||
Sun HP, Su JH, Meng QS, et al. Cancer-cell-biomimetic nanoparticles for targeted therapy of homotypic tumors. Adv Mater. 2016;28(43):9581–9588. | ||
Fang RH, Hu CM, Chen KN, et al. Lipid-insertion enables targeting functionalization of erythrocyte membrane-cloaked nanoparticles. Nanoscale. 2013;5(19):8884–8888. | ||
Fu QA, Lv PP, Chen ZK, et al. Programmed co-delivery of paclitaxel and doxorubicin boosted by camouflaging with erythrocyte membrane. Nanoscale. 2015;7(9):4020–4030 | ||
Bhateria M, Rachumallu R, Singh R, Bhatta RS. Erythrocytes-based synthetic delivery systems: transition from conventional to novel engineering strategies. Expert Opin Drug Deliv. 2014;11(8):1219–1236. | ||
Mahmoodi NO, Ghavidast A, Amirmahani N. A comparative study on the nanoparticles for improved drug delivery systems. J Photochem Photobiol B. 2016;162:681–693. |
Supplementary material
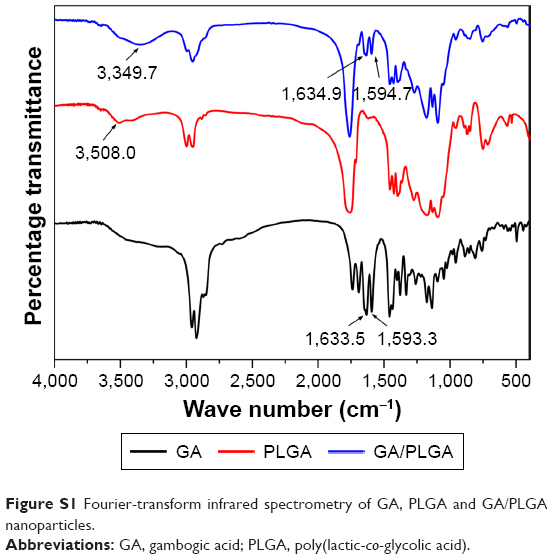 | Figure S1 Fourier-transform infrared spectrometry of GA, PLGA and GA/PLGA nanoparticles. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.