")
Back to Journals » OncoTargets and Therapy » Volume 12
Gain-Of-Function E76K-Mutant SHP2 Promotes Cell Proliferation, Metastasis, And Tumor Growth In Glioblastoma Through Activation Of The ERK/CREB Pathway
Authors Yang F, Xu M, Wang S, Song L, Yu D, Li Y, Cao R, Xiong Z, Chen Z, Zhang Q, Zhao B, Wang S
Received 11 July 2019
Accepted for publication 21 September 2019
Published 7 November 2019 Volume 2019:12 Pages 9435—9447
DOI https://doi.org/10.2147/OTT.S222881
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Arseniy Yuzhalin
Fan Yang,1,2 Mo Xu,1 Shiqing Wang,1 Le Song,1 Dandan Yu,1 Yao Li,1 Rui Cao,1 Zhang Xiong,1,2 Zhijun Chen,1 Qian Zhang,1 Bing Zhao,2 Siying Wang1
1School of Basic Medical Sciences, Anhui Medical University, Hefei, People’s Republic of China; 2Department of Neurosurgery, The Second Affiliated Hospital of Anhui Medical University, Hefei, People’s Republic of China
Correspondence: Siying Wang
School of Basic Medical Sciences, Anhui Medical University, 81 Meishan Road, Hefei, Anhui Province 230032, People’s Republic of China
Tel +86 551 6516 1215
Email [email protected]
Bing Zhao
Department of Neurosurgery, The Second Affiliated Hospital of Anhui Medical University, 678 Fu Rong Road, Hefei, Anhui Province 230601, People’s Republic of China
Tel +86 551 6386 9502
Email [email protected]
Purpose: The aim of this study was to investigate the effects of gain-of-function (GOF) E76K-mutant Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2) on the biological behaviors of glioblastoma (GBM) cells, and explore the molecular mechanisms of GBM progression.
Methods: Firstly, a negative control vector and vectors overexpressing SHP2 and E76K-mutant SHP2 were transduced into GBM cells (U87 and A172) using a lentivirus. The effect of GOF-mutant SHP2 on proliferation was measured using the MTT assay, flow cytometry, colony formation assay, and soft agar assay. Moreover, the migration and invasion of GBM cells were determined through the transwell assay. Related proteins of the extracellular signal-regulated kinase/cAMP response element binding protein (ERK/CREB) pathway were detected by Western blotting analysis. A xenograft model was established to confirm the tumor-promoting effect of GOF-mutant SHP2 in vivo. Finally, ERK was inhibited using a mitogen-activated protein kinase/ERK kinase inhibitor (U0126) to further explore the molecular mechanism of GOF-mutant SHP2 affecting GBM cells.
Results: After transduction, the expression of SHP2 in the SHP2-mutant and SHP2-overexpression groups was higher than that observed in the control and normal groups. Our data indicated that GOF-mutant SHP2 enhanced the abilities of GBM cells for proliferation, migration, and invasion in vitro, and promoted tumor growth in vivo. Mechanistically, the ERK/CREB pathway was activated, and the levels of relevant proteins were increased in the SHP2-mutant group. Furthermore, following inhibition of ERK in the GOF-SHP2 mutant group, the activation of CREB was also depressed, and the malignant biological behaviors were weakened accordingly.
Conclusion: The GOF-mutant SHP2 promoted GBM cell proliferation, metastasis, and tumor growth through the ERK/CREB pathway, providing a promising target for the treatment of GBM.
Keywords: glioblastoma, E76K mutation, SHP2, malignant behavior, ERK, CREB, cancer
Introduction
Glioblastoma (GBM) is the most common type of malignant brain tumor. It is associated with rapid growth, low survival, and high mortality rates, particularly in patients for whom tumor removal was incomplete.1 Despite significant efforts over the last several decades to improve combined therapies for GBM, its prognosis remains poor.2 Therefore, the molecular mechanisms underlying the progression of GBM must be investigated to develop new treatment methods.
Src homology-2 (SH2) domain containing protein tyrosine phosphatase (PTP) 2 (SHP2), coded by the PTP non-receptor type 11 (PTPN11) gene, is a widely occurring non-receptor PTP. It is composed of two N-terminal SH2 regions, a PTP region, and a C-terminal region.3 SHP2 is auto-inhibited, as the N-terminal SH2 region binds intramolecularly to the phosphatase region, and becomes catalytically sensitized when interacting with a phosphate tyrosine.4 Accumulating evidence indicated that SHP2 promotes cell survival and proliferation through various signaling pathways, such as the RAS/extracellular signal-regulated kinase (RAS/ERK) and Janus kinase/signal transducer and activator of transcription (JAK/STAT), induced by cytokines, growth factors, and hormones.3 Dominant-negative SHP2 mutants disrupt Xenopus gastrulation and impair fibroblast growth factor-induced ERK activation, mesoderm induction, and elongation in ectodermal explants.5,6 Homozygous SHP2−/− mice (deletion of exon 3 of SHP2) expire in the uterus at mid-gestation due to the lack of ERK activation.7
SHP2 was the first reported oncogenic tyrosine phosphatase.8 Most of its gain-of-function (GOF) mutations are missense mutation and affect the N-SH2 or PTP domain residues involved in basal auto-inhibition.9 Somatic mutations of SHP2 have been identified and associated with higher proliferation rate in Noonan syndrome (~50%), juvenile myelomonocytic leukemia (~35%), and other leukocythaemias.10,11 Although GOF mutations in SHP2 occur at low frequency in certain solid tumors, including GBM, lung cancer, colon cancer, and melanoma, GOF SHP2 appears to play an important pathogenic role in contributing to the occurrence, progression, and metastasis of these diseases.12,13 A recent study found that inhibition of SHP2 phosphatase activity reduced the proliferation of acute myeloid leukemia cells through downregulation of phosphorylated STAT5.14 Sun et al demonstrated that inhibition of SHP2 promoted irradiation-induced radiosensitivity in glioma.15 In gallbladder cancer, SHP2 can be used as a marker of diagnosis and poor prognosis.16 Nevertheless, some studies have demonstrated the tumor-suppressing role of SHP2. Bard-Chapeau et al showed that SHP2 suppresses hepatocellular carcinoma development via downregulation of inflammatory signaling.17 In addition, SHP2 inhibits the growth of esophageal squamous cell cancer cells through dephosphorylation of STAT3.18 Overall, it is generally recognized that SHP2 plays dual roles in specific solid tumors.
Based on data from The Cancer Genome Atlas, ~2% of patients with GBM harbor PTPN11 mutations, and nearly all of these mutations were identified as activating.12 SHP2E76K located in the N-SH2 domain is the most frequently mutated residue in juvenile myelomonocytic leukemia, and was also found in glioma in the Catalog of Somatic Mutations in Cancer databank. On the other hand, SHP2E76K exhibits markedly high phosphatase activity compared with other GOF SHP2 variants, such as SHP2G60V and SHP2D61V.19 However, the effects of GOF E76K-mutant (E76K-MT) SHP2 on the biological behaviors of GBM cells in vitro and in vivo, and the potential molecular mechanisms involved in this process have not been fully elucidated.20 Therefore, it is crucial to understand the role and mechanism of GOF-MT SHP2 in the development of GBM. Since established U87 and A172 cell lines were isolated from patients with GBM, they are suitable for being used as a representative to study biological behaviors and molecular mechanisms of GBM progression.
In this study, we found that GOF-MT SHP2 significantly promoted the proliferation, migration, and invasion of GBM cells, and accelerated tumor growth in a xenograft model. Furthermore, our results proved that GOF-MT SHP2 promotes the malignant behaviors of GBM cells through the ERK/cAMP response element binding protein (ERK/CREB) signaling pathway.
Materials And Methods
Cell Culture And Lentivirus (LV) Transduction
The human GBM cell lines A172 and U87 were purchased from the Chinese Academy of Sciences (Shanghai, China) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT, USA) in an incubator at 37°C with 5% carbon dioxide. A recombinant LV-negative control (LV-NC) vector, and vectors overexpressing SHP2 (LV-SHP2) and E76K MT SHP2 (LV-E76K MT-SHP2) were obtained from GenePharma (Shanghai, China). Virus solution and polybrene (Sigma–Aldrich, St Louis, MO, USA) were added into complete medium for gene transduction in A172 and U87 cells. Following transduction for 48 h, stable cell lines were subjected to treatment with 2 µg/mL puromycin (MedChem Express, Monmouth Junction, NJ, USA) for 14 days. U0126 (MedChem Express), a mitogen-activated protein kinase/ERK kinase inhibitor, was dissolved in dimethyl sulphoxide (DMSO) (Sigma–Aldrich), and added into the cell culture at a concentration of 10 μmol/l. In the control group, only DMSO was added.
Western Blotting
Total protein from the different cell groups were collected in radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors (Sigma–Aldrich). The protein samples (~15 µg) were electrophoresed in a sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, and subsequently transferred onto polyvinylidene difluoride membranes. Non-fat milk (5%) was used to block the membranes at room temperature, followed by incubation with primary antibodies against SHP2, phosphorylated ERK (p-ERK), ERK, p-CREB, CREB, cyclin D1, matrix metallopeptidase 9 (MMP9: 1:1,000; Cell Signaling Technology, Danvers, MA, USA), proliferating cell nuclear antigen (PCNA: 1:1,000; Elabscience Biotechnology, Wuhan, China), cyclin E1, cyclin A (1:1,000; Abcam, Cambridge, UK), and β-actin (1:5,000; Abcam) at 4°C overnight. Finally, the polyvinylidene difluoride membranes were incubated with a horseradish peroxidase-conjugated IgG secondary antibody (1:5,000; GE Healthcare, Little Chalfont, UK). Signals were detected through electrochemiluminescence (GE Healthcare). Quantification of protein bands was performed using the ImageJ 1.51 software (National Institutes of Health, Bethesda, MD, USA). The levels of β-actin were used to ensure equal loading of proteins. Each experiment was performed in triplicate.
MTT Assay
A172 and U87 cells were seeded in a 96-well plate at a density of 4,000 cells/well to test cell viability. After growth for 24, 48, and 72 h, MTT solution (1 mg/mL) (50 µl) was added into each well for 4 h. Subsequently, DMSO (150 µl) was used to terminate the reaction, and the optical density was determined at 490 nm using a multi-well plate reader (Thermo Fisher Scientific, Waltham, MA, USA). Each experiment was performed in triplicate.
Cell Cycle Analysis
The cellular DNA content was measured using flow cytometry (FCM) to determine the cell cycle distribution. According to the protocol provided by the manufacturer of the reagent (Beyotime, Shanghai, China), glioma cells, grown to 70–80% confluence in 60-mm plates, were collected after two washes with phosphate-buffered saline (PBS), fixed in 75% cold ethanol overnight. After fixation, cells were washed with cold PBS and stained with 50 μg/mL propidium iodide in the presence of 20 μg/mL RNase A at 37 °C for 30 min in darkness. The FACSAria III (BD Biosciences, San Jose, CA, USA) was used to analyze the samples. The cell cycle distribution was analyzed using the ModFit LT software (BD Biosciences). Each experiment was performed in triplicate.
Colony Formation And Soft Agar Assay
In the plate-colony experiment, cells (100 cells/well) were incubated in six-well plates containing complete DMEM supplemented with 10% FBS. After 2 weeks, the colonies were fixed with 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet solution for 30 min. Colonies containing >50 cells were counted under a microscope. For the soft agar assay, ~2,000 cells/well were cultured in six-well plates containing DMEM supplemented with 10% FBS and 0.35% agar in the upper layer and 0.5% agar in the bottom layer. After culturing in agar for 2 weeks, visible cell colonies (size: >1 mm) were imaged and counted at ×100 magnification under an Axio Observer.A1 microscope (Zeiss, Jena, Germany). Each experiment was performed in triplicate.
Cell Migration And Invasion Assay
Cell migration and invasion were measured using transparent polyethylene terephthalate membranes (8-μm pore size; BD Biosciences). For the migration test, 4×104 GBM cells were placed in the upper chambers of a Transwell plate. For the invasion test, 4×104 cells were placed in the upper chambers of a Transwell plate with a 1 mg/mL matrigel-coated membrane. Cells in the upper Transwell chambers were cultured in serum-free medium, while medium with 20% serum was added to the lower chambers as a chemoattractant. After culture for 24 h, the cells remaining in the upper chambers were removed using cotton-tipped swabs, whereas those that had migrated/invaded to the underside of the chamber membranes were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. More than 5 randomly selected microscopic fields were analyzed to count the number of migrating/invading cells. Each experiment was performed in triplicate.
Tumorigenicity Assay In Vivo
A total of 24 BALB/c mice were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China) and randomly assigned to four groups (n=6 per group). Normal, LV-NC, LV-SHP2, and LV-E76K MT-SHP2 U87 cells (1×106) were suspended in 0.1 mL PBS and injected into the right armpit of nude mice. The mice were maintained under specific pathogen-free conditions at the Core Facility Centre for Life Sciences of the University of Science and Technology of China (Hefei, China). All the animal tests were processed in accordance with the guide for the care and use of laboratory animals and approved by the Institutional Research Ethics Committee of the University of Science and Technology of China. The nude mice were sacrificed at 4 weeks following inoculation, according to the volume of the growing tumor [(length × width2)/2] measured using Vernier calipers twice per week. The tumor tissues were preserved in 10% neutral buffered formalin for further experiments.
Hematoxylin And Eosin (H&E) Staining And Immunohistochemistry (IHC) Assay
The paraffin blocks of the xenografts were cut into 5-μm slices. For H&E staining, the slides were dewaxed and dyed with hematoxylin for 10 min, placed in hydrochloric acid alcohol differentiation solution, and subsequently stained with eosin for 1 min. For the IHC assay, endogenous peroxidase activity was blocked using 0.35% hydrogen peroxide in PBS. Subsequently, antigens were retrieved by microwaving, and non-specific binding was blocked with 5% bovine serum albumin. The prepared tissue slices were incubated with anti-SHP2 and anti-p-ERK (1:200; Cell Signaling Technology), anti-p-CREB (1:800; Cell Signaling Technology), and anti-Ki-67 (1:200; ZSBIO, Beijing, China) antibodies overnight at 4°C. Signals were detected using a DAB Detection kit (ZSBIO) and images were captured under an Axio Observer.A1 microscope (Zeiss). For Ki-67 and p-CREB nuclear staining, the “count small-cells” function of the Image-Pro Plus version 6.0 software (Media Cybernetics, Rockville, MD, USA) was used to count the positive cells. For p-ERK staining, the integrated optical density was measured using the “measure stain” function. At least six random fields in each section were measured and three sections were selected from each xenograft.
Statistical Analysis
The GraphPad Prism 6 software (GraphPad, La Jolla, CA, USA) was employed to analyze the experimental data. All data were obtained from at least three independent experiments and presented as the mean ± standard error of mean (SEM). One-way analysis of variance and Student’s t-test were used to compare differences between the groups. A P<0.05 denoted statistically significant differences.
Results
Overexpression Of SHP2 By The Recombinant LVs In GBM Cells
A172 and U87 cells were transduced with recombinant LVs (LV-NC, LV-SHP2, and LV-E76K MT-SHP2) to determine the effects of GOF-MT SHP2 on GBM cells. The expression levels of SHP2 in the different groups were measured. As expected, we found that the levels of SHP2 were elevated in cells infected with LV-SHP2 and LV-MT-SHP2, compared with those reported in the normal or LV-NC groups (Figure 1A and B).
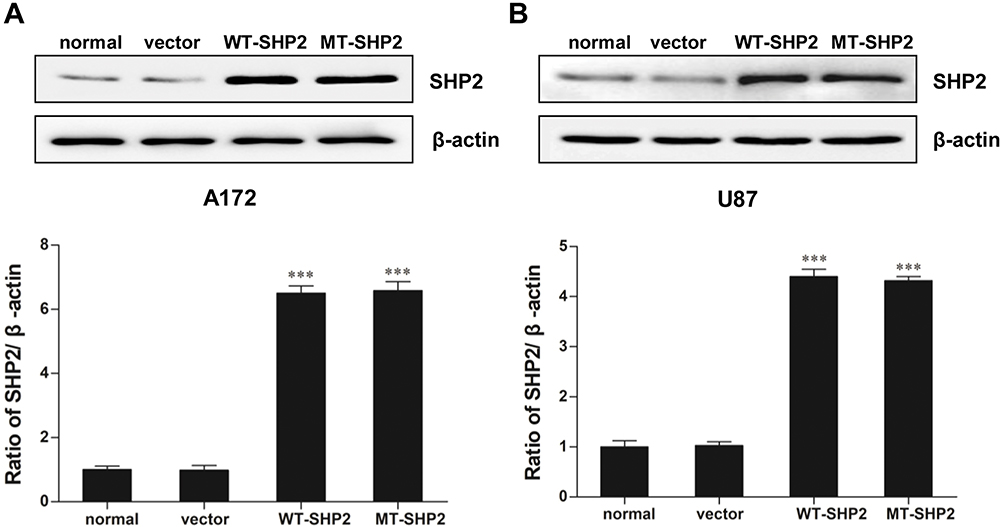 |
Figure 1 SHP2 expression in normal, vector, WT-SHP2, and MT-SHP2 A172 and U87 cells. (A and B) Western blotting analysis was used to measure the expression of SHP2 following transduction with LV-NC, LV-SHP2, and LV-E76K MT-SHP2 in A172 and U87 cells. β-actin was used to ensure equal loading. The data are presented as the means ± SEM (***P<0.001). Abbreviations: SHP2, Src homology-2 domain-containing protein-tyrosine phosphatase-2; WT, wild type; MT, mutant; LV, lentivirus; NC, negative control. |
GOF-MT SHP2 Promotes The Proliferation Of GBM Cells
The effect of transduction on the proliferation of A172 and U87 cells was evaluated. Growth curves produced following the MTT assays revealed that GOF-MT SHP2 significantly increased the viability of A172 and U87 cells compared with SHP2-overexpression and control (Figure 2A and B). Subsequently, FCM was used to assess the cell cycle distribution. The analysis showed that LV-MT-SHP2 resulted in a notable increase in the S phase population and a decrease in the G0/G1 population in both A172 and U87 cells (Figure 2C and D). Additionally, the colony formation and soft agar assays suggested that the density-independent (Figure 2E and F) and anchorage-independent (Figure 2G and H) growth capacity was markedly enhanced in A172 and U87 cells following transduction with LV-MT-SHP2.
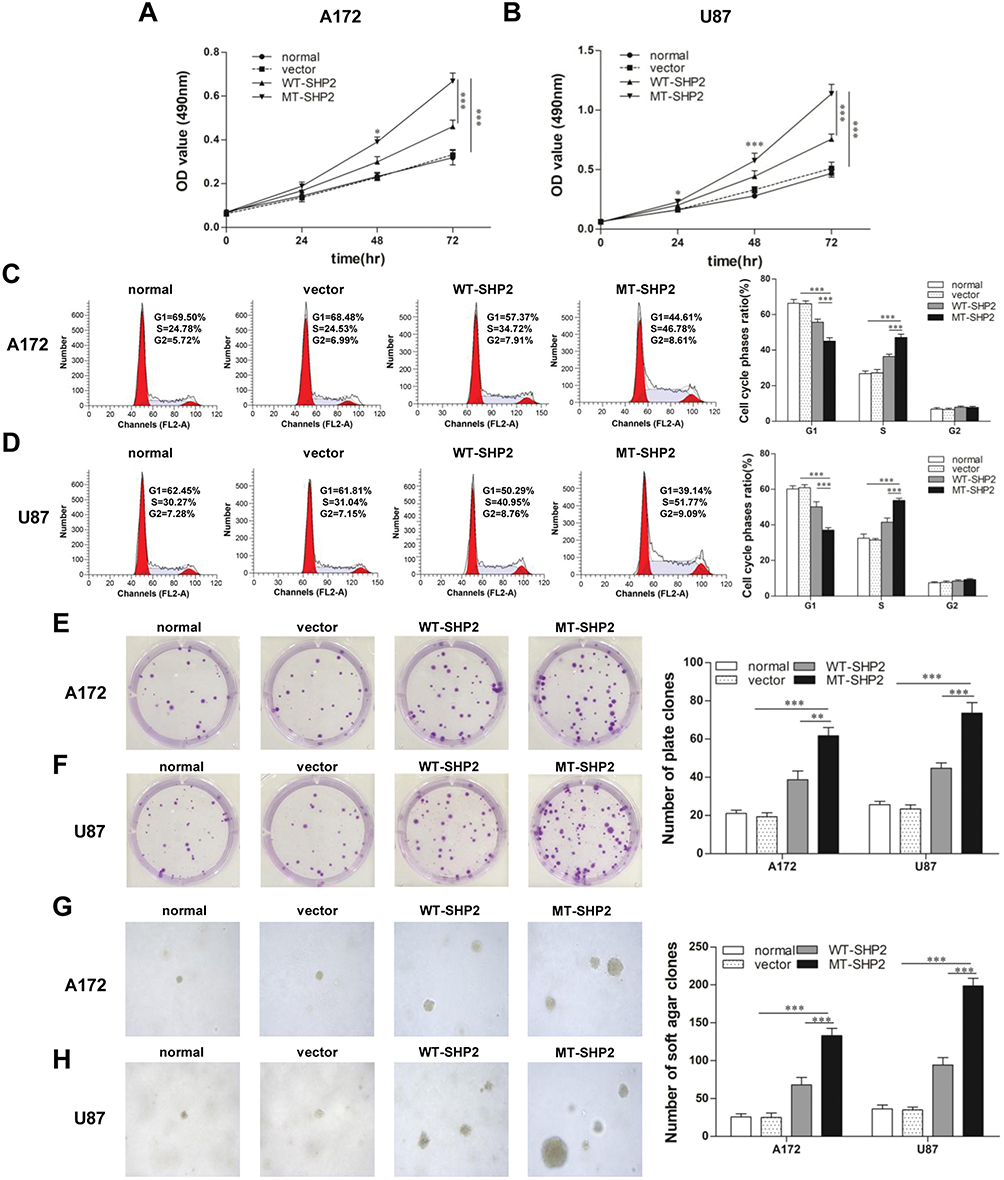 |
Figure 2 GOF-MT SHP2 promotes the proliferation of glioblastoma cells. (A and B) The viability of normal, vector, WT-SHP2 and MT-SHP2 A172 and U87 cells was determined using the MTT assay. (C and D) The cell cycle distribution was detected using flow cytometry. (E and F) A colony formation assay was performed to test the density-independent proliferation of cells in the different groups. Colonies containing >50 cells were counted under a microscope. (G and H) Cell proliferation in agar was used to test the ability of anchorage-independent development. Visible cell colonies (size: >1 mm) were imaged and counted at ×100 magnification. Typical images are shown. The data are presented as the means ± SEM (*P<0.05, **P<0.01, and ***P<0.001). Abbreviations: GOF, gain-of-function; SHP2, Src homology-2 domain-containing protein-tyrosine phosphatase-2; WT, wild type; MT, mutant; MTT, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide. |
GOF-MT SHP2 Promotes The Abilities Of GBM Cells For Migration And Invasion
Subsequently, we examined the metastatic ability of A172 and U87 GBM cells in vitro. Cell migration was tested using a Transwell chamber system. We found that GOF-MT SHP2 promoted the migration of GBM cells compared with SHP2-overexpression and control (Figure 3A and B). In the invasion assay, the cells transduced with LV-MT-SHP2 exhibited a stronger invasive ability compared with that observed in cells transduced with LV-SHP2 or control (Figure 3C and D).
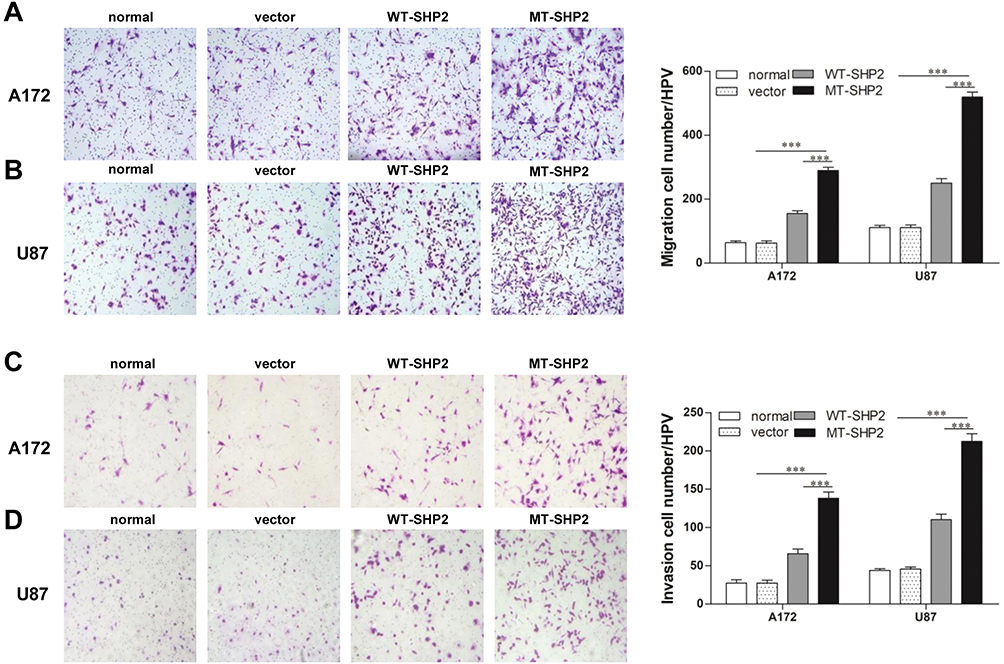 |
Figure 3 The SHP2 mutation enhances the migration and invasion of A172 and U87 cells. (A and B) A migration test was performed for normal, vector, WT-SHP2, and MT-SHP2 A172 and U87 cells using a Transwell chamber with polycarbonate membrane in 24-well plates. (C and D) The invasion ability of cells in the different groups was tested utilizing the Matrigel-coated Boyden chamber. Representative images are shown. The data are presented as the means ± SEM (***P<0.001). Abbreviations: SHP2, Src homology-2 domain-containing protein-tyrosine phosphatase-2; WT, wild type; MT, mutant. |
Regulatory Mechanism Of The Positive Effects Of SHP2 GOF-Mutation
ERK plays an important role in intracellular signaling pathways. Our results demonstrated that, following transduction with LV-MT-SHP2, the phosphorylation of ERK was markedly increased compared with that recorded after transduction with LV-SHP2 or control. Interestingly, we also found that, although the total expression of CREB was not altered, the levels of p-CREB were distinctly increased in the LV-MT-SHP2 group. Furthermore, we examined the expression of related proteins (eg PCNA, cyclin D1, cyclin E1, MMP9 and cyclin A), which are associated with CREB regulation. We found that the expression of these factors was significantly increased in the LV-MT-SHP2 group (Figure 4A and B, Figure S1). All these molecular results confirmed that transduction with LV-MT-SHP2 accelerated the development of GBM compared with LV-SHP2 or control.
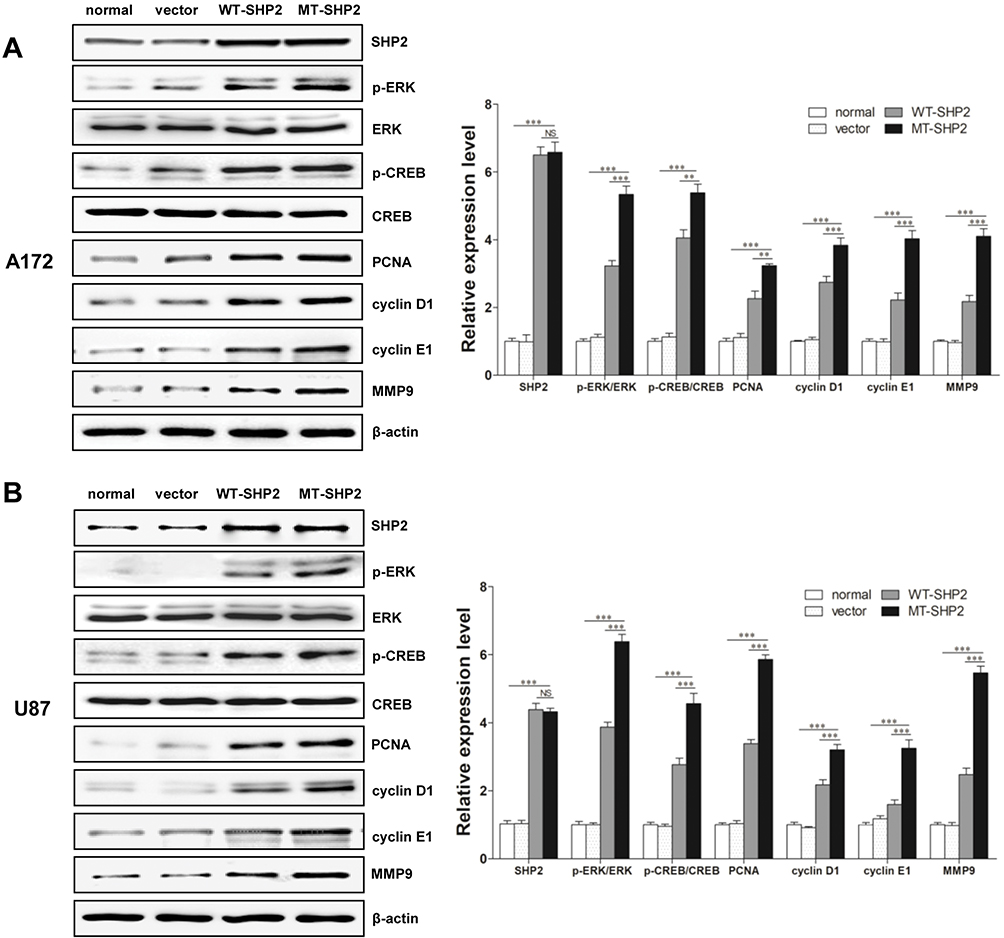 |
Figure 4 Regulatory mechanisms of GOF-MT SHP2 on cell malignancy. (A and B) Western blotting demonstrated the expression of SHP2, total ERK, p-ERK, total CREB, p-CREB, and several related downstream proteins (ie PCNA, cyclin D1, cyclin E1, MMP9) in normal, vector, WT-SHP2 and MT-SHP2 A172 and U87 cells. NS: no significant. The data are presented as the means ± SEM (**P<0.01 and ***P<0.001). Abbreviations: GOF, gain-of-function; SHP2, Src homology-2 domain-containing protein-tyrosine phosphatase-2; WT, wild-type; MT, mutant; ERK, extracellular signal-regulated kinase; CREB, cAMP-response element-binding protein; MMP, matrix metallopeptidase; PCNA, proliferating cell nuclear antigen. |
GOF-MT SHP2 Promotes Tumor Growth In Vivo
We used a tumor xenograft model to investigate the function of GOF-MT SHP2 in vivo. Tumor size was measured twice a week. The mice were sacrificed 4 weeks post inoculation. The tumors in the LV-MT-SHP2 group were noticeably larger compared with those observed in the LV-SHP2 or control groups (Figure 5A and B). Analysis of continuous measurement data for tumor size in the different groups revealed that LV-MT-SHP2 markedly accelerated tumor growth (Figure 5C). Consistently, the tumor weight was also higher in the SHP2 MT group (Figure 5D).
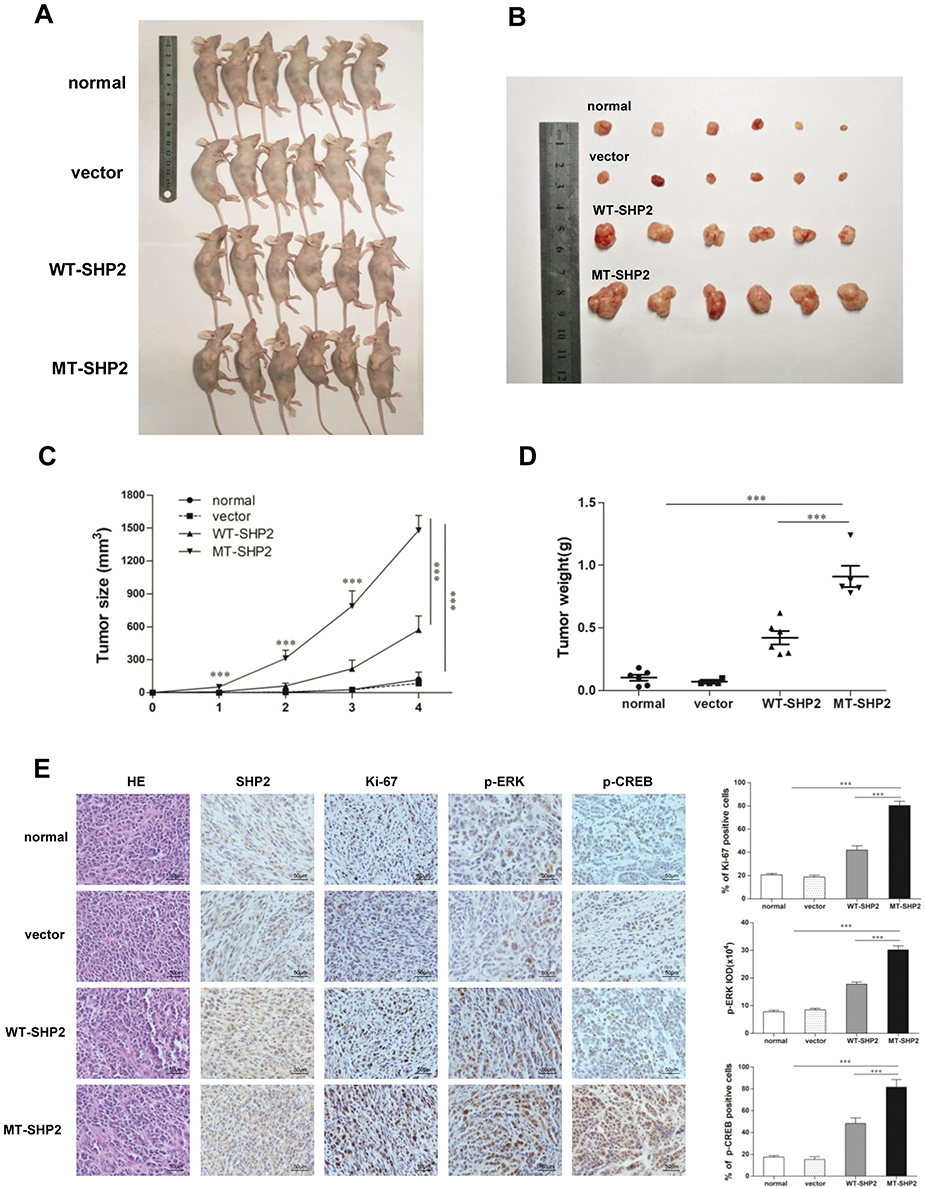 |
Figure 5 GOF-MT SHP2 promotes glioblastoma tumor growth; immunohistochemical staining of tumor specimens. (A and B) Normal, control, WT-SHP2, and MT-SHP2 U87 cells were hypodermically injected into nude mice (n=6 per group). At 4 weeks post injection, the mice were sacrificed, and solid tumors were dissected and imaged. (C and D) After the injection, the tumor size and weight were measured twice per week. (E) H&E staining and IHC assay for SHP2, Ki-67, p-ERK, and p-CREB in tumor tissues obtained from normal, control, WT-SHP2, and MT-SHP2 mice. Images were captured from random fields of view at a magnification of ×400, Scale bar: 50 µm; representative images are shown. The data are presented as the means ± SEM (***P<0.001). Abbreviations: GOF, gain-of-function; SHP2, Src homology-2 domain-containing protein-tyrosine phosphatase-2; WT, wild type; MT, mutant, IHC, immunohistochemistry; H&E, hematoxylin and eosin; CREB, cAMP-response element-binding protein; ERK, extracellular signal-regulated kinase. |
IHC Analysis Of The Expression Of SHP2 And Related Proteins In Xenograft Tissues
IHC was used to detect the levels of related proteins in the tissues of xenograft tumors to investigate the potential molecular mechanism underlying the effects of GOF-MT SHP2 on GBM cells in vivo. The results indicated that the expression of p-ERK and p-CREB was elevated in the LV-MT-SHP2 group compared with that reported in the LV-SHP2 or control groups. Furthermore, there was a markedly higher number of Ki-67-positive cells in the LV-MT-SHP2 group, indicating enhanced proliferative activity (Figure 5E). These data demonstrated that GOF-MT SHP2 conferred an increased tumor growth ability, which was, at least partly, mediated by the increased levels of p-ERK and p-CREB.
U0126 Reduces The Expression Of P-ERK And P-CREB, And Significantly Reduces The Proliferation And Migration Of GOF-MT SHP2 GBM Cells
We subsequently verified whether GOF-MT SHP2 increases the malignant behaviors of GBM cells by activating the ERK/CREB pathway. U0126 was added to the cell culture medium and led to a significant reduction in the expression of p-ERK and p-CREB in GBM cells belonging to the GOF-MT SHP2 group (Figure 6A and B). In addition, the MTT and plate-colony assays revealed that the proliferation capacity was significantly decreased in the GOF group (Figure 6C–F). Furthermore, U0126 suppressed the migration ability in the GOF group (Figure 6G and H). These data suggested that GOF-MT SHP2 promoted the growth and migration of U87 and A172 cells in a partially ERK/CREB-dependent manner.
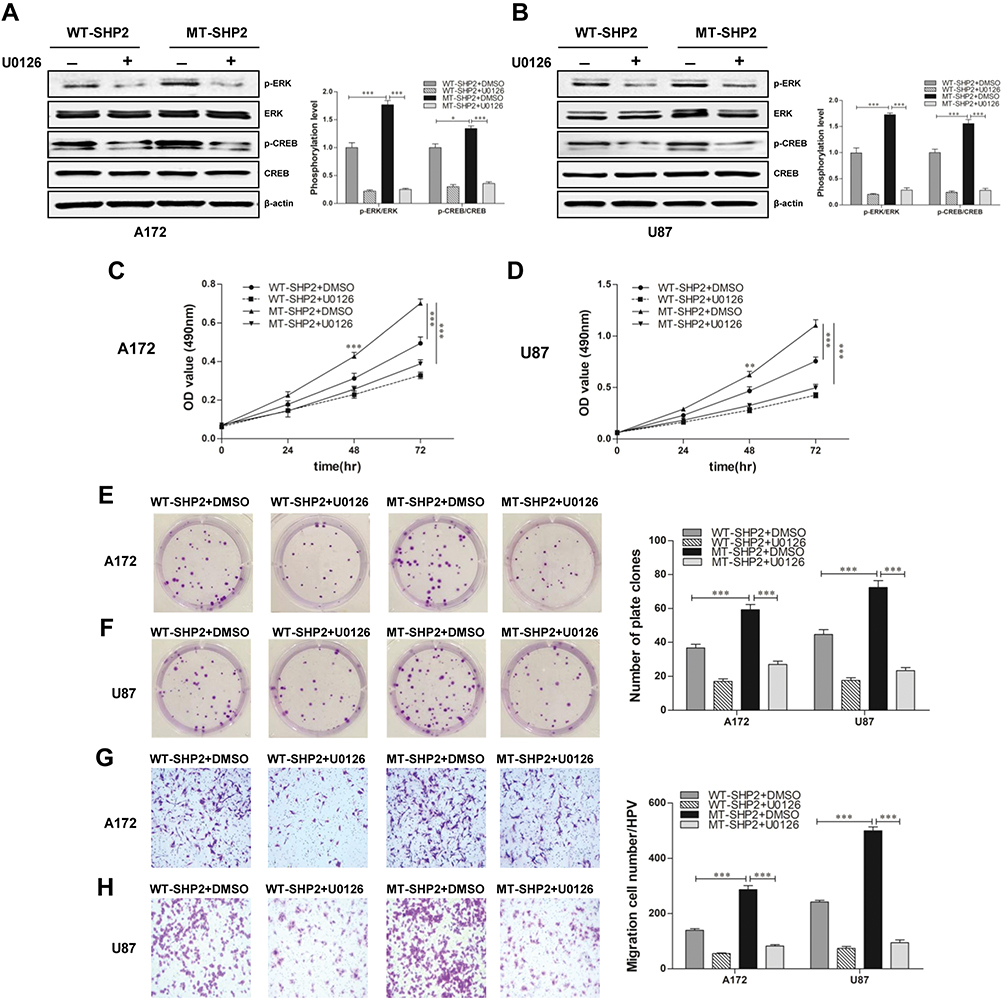 |
Figure 6 U0126 partially diminishes the effects of GOF-SHP2 on glioblastoma cells. (A and B) WT-SHP2 and MT-SHP2 U87 and A172 cells were treated with U0126 (10 μmol/l) or DMSO for 24 h. Western blotting analysis was used to measure the levels of p-ERK, ERK, p-CREB, and CREB. β-actin was used as a loading control. (C and D) The MTT assay was employed to test the viability of glioblastoma cells in the WT-SHP2 and MT-SHP2 groups in the presence of U0126 or DMSO. (E and F) The cloning experiment was used to test the proliferation ability of cells. The WT-SHP2 and MT-SHP2 groups were treated with U0126 or DMSO twice per week for 2 weeks. (G and H) The migration ability of cells was measured using the Transwell chamber assay in the presence of U0126 or DMSO. The data are presented as the means ± SEM (*P<0.05 and ***P<0.001). Abbreviations: GOF, gain-of-function; CREB, cAMP-response element-binding protein; ERK, extracellular signal-regulated kinase; SHP2, Src homology-2 domain-containing protein-tyrosine phosphatase-2; WT, wild type; MT, mutant; DMSO, dimethyl sulphoxide; MTT, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide. |
Discussion
The PTP SHP2 is widely expressed in various types of cells, and plays a key role in multiple signaling pathways induced by cytokines and extracellular matrix proteins.21,22 Approximately half of patients with the Noonan syndrome harbor mutations in the PTPN11 gene, and these patients are linked to an increased risk of developing leukemia.10,23 Previous studies have demonstrated that GOF-MT SHP2 promotes the development and progression of leukemia through its phosphatase catalytic activity and effects on the bone marrow microenvironment.24–26 GOF-MTs have also been identified in certain solid tumors, including GBM, lung cancer, colon cancer, and melanoma. GOF-MT SHP2 promotes lung tumorigenesis in transgenic mice, and Src family kinases may be involved in a positive feed-forward loop with SHP2E76K and Gab1.27 However, the role and mechanism of activating-MT SHP2 in solid tumors warrant further investigation.
Our experiments demonstrated that LV-MT-SHP2 promoted the proliferation of GBM cells compared with LV-SHP2 and control. The results of the FCM analysis indicated that the PTPN11 mutation was implicated in disrupting the G1-to-S phase transition, which is crucial for the control of cell proliferation.28 Dysregulation of the cell cycle and abnormal expression of pro-survival and -proliferation proteins are essential factors in the regulation of SHP2-activating mutations.29 Additional functional studies demonstrated that the abilities for migration and invasion were significantly elevated in the LV-MT-SHP2 group, suggesting that the SHP2 mutation is associated with the invasion and poor prognosis of GBM. Zhang et al showed that dominant-active SHP2 promoting epithelial-to-mesenchymal transition may be involved in prostate cancer metastasis.30 As the first reported oncogenic tyrosine phosphatase, SHP2 may play a role in signal transmission in phosphatase-dependent and -independent manners.31–33 The critical role of SHP2 is to promote the activation of the RAS/ERK signaling pathway, a classic signaling cascade involved in various cellular processes, including proliferation, survival, differentiation, migration, or metabolism.9 Apart from the RAS/ERK pathway, SHP2 also promotes the phosphatidylinositol 3 kinase/protein kinase B (PI3K/AKT), JAK/STAT, c-Jun N-terminal kinase, and nuclear factor-κB pathways.34 Goodwin et al have demonstrated that the PI3K-AKT and RAS-ERK pathways work together to promote granulocyte-macrophage colony-stimulating factor hypersensitivity of MT SHP2-expressing cells.35 Recent research found that GBM tumors exhibit significantly higher levels of phosphorylated SHP2 compared with low-grade gliomas and their normal counterparts.36 The dysregulation of SHP2 in GBM has potently stronger proliferation and transformation ability.37 Sturla et al described the ability of SHP2 to suppress cellular senescence in the GBM cell lines.12 In addition, SHP2 also plays an important role in glioma radiosensitivity.15 However, the underlying molecular mechanism through which the GOF-MT SHP2 is involved in the regulation of GBM progression remains unclear and must be elucidated.
Of note, a genomic analysis using data from The Cancer Genome Atlas has revealed that PTPN11 is connected with different mutated genes in GBM, as a member of the six major concatenated genes.38 Therefore, treatments targeting SHP2 may be more effective against GBM. It has been reported that abnormal expression of SHP2 leads to deregulation of ERK hyper-activation.39,40 We measured the levels of total ERK and p-ERK in normal, LV-NC, LV-SHP2, and LV-MT-SHP2 cells, and found that the activation of ERK was significantly higher in the GOF-MT SHP2 group. Furthermore, CREB (a general transcriptional activator) is associated with accelerated progression of numerous cancers, including GBM, leukemia, non-small-cell lung cancer, and pancreatic cancer.41–44 The cAMP-responsive elements of the promoters of several genes are found in the CREB-binding site, including genes that are involved in proliferation or associated with metastasis.45–47 We hypothesized that the activating role of GOF-MT SHP2 in intracellular molecular signaling pathways is related to the function of CREB in GBM. Our results revealed that the levels of p-CREB were indeed increased following the stimulation of GBM cells with GOF-MT SHP2. These findings suggested that the upregulation of p-ERK and p-CREB may be responsible for the malignant biological behaviors of LV-MT-SHP2 cells. Further studies demonstrated that the expression of PCNA, cyclin D1, cyclin E1, and MMP9, which is directly or indirectly modulated by CREB, was elevated.48,49
Furthermore, we found that U0126, a selective inhibitor of ERK1/2 phosphorylation, markedly reduced the expression of p-ERK and p-CREB in GBM cells belonging to the GOF-MT group. Our data showed that the proliferation and migration abilities of cells were also diminished. Collectively, these findings indicated that GOF-MT SHP2 may promote the malignant behaviors of GBM cells by activating the ERK/CREB signaling pathway. However, the detailed mechanisms underlying the regulation of GBM growth and metastasis by GOF-MT SHP2 require further investigation. Consistent with the aforementioned findings, the growth rate and weight of xenograft tumors in the LV-MT-SHP2 group were significantly higher compared with those reported in the LV-SHP2 or control groups in vivo. IHC staining of solid tumors obtained from nude mice revealed high numbers of Ki-67-positive cells in the GOF-MT SHP2 group. Moreover, the protein levels of p-ERK and p-CREB in the GOF-MT SHP2 group were also upregulated compared with those measured in the LV-SHP2 or control groups.
Our study demonstrated that E76K-MT SHP2 promoted the proliferation and metastasis of GBM cells, and the in-vivo results revealed that MT SHP2 markedly promoted xenograft tumor growth. Moreover, the activating pathway of ERK/CREB was associated with the malignant behaviors of GBM cells, and related downstream proteins were notably upregulated. Therefore, these findings provide novel insights into the mechanism of action of GOF-MT SHP2.
Conclusion
Collectively, the results of this study revealed that GOF-MT SHP2 promotes the proliferation, metastasis, and growth of GBM cells. According to our data, these effects were associated with the ERK/CREB pathway, indicating that SHP2 may be a promising therapeutic target for the treatment of GBM.
Acknowledgments
This project received grants from the National Natural Science Foundation of China (Beijing, China; Nos. 30973424 and 81272258).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60(3):166–193. doi:10.3322/caac.20069
2. Zanganeh S, Georgala P, Corbo C, et al. Immunoengineering in glioblastoma imaging and therapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2019;e1575.
3. Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28(6):284–293. doi:10.1016/S0968-0004(03)00091-4
4. Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92(4):441–450. doi:10.1016/s0092-8674(00)80938-1
5. O’Reilly AM, Neel BG. Structural determinants of SHP-2 function and specificity in Xenopus mesoderm induction. Mol Cell Biol. 1998;18(1):161–177. doi:10.1128/mcb.18.1.161
6. Tang TL, Freeman RM
7. Saxton TM, Henkemeyer M, Gasca S, et al. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. Embo J. 1997;16(9):2352–2364. doi:10.1093/emboj/16.9.2352
8. Loh ML, Vattikuti S, Schubbert S, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103(6):2325–2331. doi:10.1182/blood-2003-09-3287
9. Tajan M, de Rocca Serra A, Valet P, Edouard T, Yart A. SHP2 sails from physiology to pathology. Eur J Med Genet. 2015;58(10):509–525. doi:10.1016/j.ejmg.2015.08.005
10. Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29(4):465–468. doi:10.1038/ng772
11. Tartaglia M, Niemeyer CM, Fragale A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34(2):148–150. doi:10.1038/ng1156
12. Sturla LM, Zinn PO, Ng K, et al. Src homology domain-containing phosphatase 2 suppresses cellular senescence in glioblastoma. Br J Cancer. 2011;105(8):1235–1243. doi:10.1038/bjc.2011.345
13. Bentires-Alj M, Paez JG, David FS, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64(24):8816–8820. doi:10.1158/0008-5472.CAN-04-1923
14. Richine BM, Virts EL, Bowling JD, et al. Syk kinase and Shp2 phosphatase inhibition cooperate to reduce FLT3-ITD-induced STAT5 activation and proliferation of acute myeloid leukemia. Leukemia. 2016;30(10):2094–2097. doi:10.1038/leu.2016.131
15. Sun Q, Mu L, Qiao W, et al. Inhibition of SHP-2 promotes radiosensitivity in glioma. Mol Med Rep. 2015;12(3):3563–3568. doi:10.3892/mmr.2015.3829
16. Wang Q, Yang ZL, Zou Q, et al. SHP2 and UGP2 are biomarkers for progression and poor prognosis of gallbladder cancer. Cancer Invest. 2016;34(6):255–264. doi:10.1080/07357907.2016.1193745
17. Bard-Chapeau EA, Li S, Ding J, et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer cell. 2011;19(5):629–639. doi:10.1016/j.ccr.2011.03.023
18. Qi C, Han T, Tang H, et al. Shp2 inhibits proliferation of esophageal squamous cell cancer via dephosphorylation of Stat3. Int J Mol Sci. 2017;18(1):134. doi:10.3390/ijms18010134
19. LaRochelle JR, Fodor M, Vemulapalli V, et al. Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat Commun. 2018;9(1):4508. doi:10.1038/s41467-018-06823-9
20. Martinelli S, Carta C, Flex E, et al. Activating PTPN11 mutations play a minor role in pediatric and adult solid tumors. Cancer Genet Cytogenet. 2006;166(2):124–129. doi:10.1016/j.cancergencyto.2005.10.003
21. Liu X, Qu CK. Protein tyrosine phosphatase SHP-2 (PTPN11) in hematopoiesis and leukemogenesis. J Signal Transduct. 2011;2011:195239. doi:10.1155/2011/195239
22. Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;27(2):179–192. doi:10.1007/s10555-008-9126-y
23. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381(9863):333–342. doi:10.1016/S0140-6736(12)61023-X
24. Chan G, Kalaitzidis D, Usenko T, et al. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood. 2009;113(18):4414–4424. doi:10.1182/blood-2008-10-182626
25. Mohi MG, Williams IR, Dearolf CR, et al. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 2005;7(2):179–191. doi:10.1016/j.ccr.2005.01.010
26. Dong L, Yu WM, Zheng H, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539(7628):304–308. doi:10.1038/nature20131
27. Schneeberger VE, Luetteke N, Ren Y, et al. SHP2E76K mutant promotes lung tumorigenesis in transgenic mice. Carcinogenesis. 2014;35(8):1717–1725. doi:10.1093/carcin/bgu025
28. Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14(8):518–528. doi:10.1038/nrm3629
29. Yang Z, Li Y, Yin F, Chan RJ. Activating PTPN11 mutants promote hematopoietic progenitor cell-cycle progression and survival. Exp Hematol. 2008;36(10):1285–1296. doi:10.1016/j.exphem.2008.04.016
30. Zhang K, Zhao H, Ji Z, et al. Shp2 promotes metastasis of prostate cancer by attenuating the PAR3/PAR6/aPKC polarity protein complex and enhancing epithelial-to-mesenchymal transition. Oncogene. 2016;35(10):1271–1282. doi:10.1038/onc.2015.184
31. Huang WQ, Lin Q, Zhuang X, et al. Structure, function, and pathogenesis of SHP2 in developmental disorders and tumorigenesis. Curr Cancer Drug Targets. 2014;14(6):567–588.
32. Bentires-Alj M, Gil SG, Chan R, et al. A role for the scaffolding adapter GAB2 in breast cancer. Nat Med. 2006;12(1):114–121. doi:10.1038/nm1341
33. Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc Natl Acad Sci U S A. 1994;91(15):7335–7339. doi:10.1073/pnas.91.15.7335
34. Frankson R, Yu ZH, Bai Y, Li Q, Zhang RY, Zhang ZY. Therapeutic targeting of oncogenic tyrosine phosphatases. Cancer Res. 2017;77(21):5701–5705. doi:10.1158/0008-5472.CAN-17-1510
35. Goodwin CB, Li XJ, Mali RS, et al. PI3K p110d uniquely promotes gain-of-function Shp2-induced GM-CSF hypersensitivity in a model of JMML. Blood. 2014;123(18):2838–2842. doi:10.1182/blood-2013-10-535104
36. Bunda S, Burrell K, Heir P, et al. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat Commun. 2015;6:8859. doi:10.1038/ncomms9859
37. Roccograndi L, Binder ZA, Zhang L, et al. SHP2 regulates proliferation and tumorigenicity of glioma stem cells. J Neurooncol. 2017;135(3):487–496. doi:10.1007/s11060-017-2610-x
38. Cerami E, Demir E, Schultz N, Taylor BS, Sander C. Automated network analysis identifies core pathways in glioblastoma. PLoS One. 2010;5(2):e8918. doi:10.1371/journal.pone.0008918
39. Li K, Leung AW, Guo Q, Yang W, Li JY. Shp2-dependent ERK signaling is essential for induction of Bergmann glia and foliation of the cerebellum. J Neurosci. 2014;34(3):922–931. doi:10.1523/JNEUROSCI.3476-13.2014
40. Taniguchi T, Tanaka S, Ishii A, et al. A brain-specific Grb2-associated regulator of extracellular signal-regulated kinase (Erk)/mitogen-activated protein kinase (MAPK) (GAREM) subtype, GAREM2, contributes to neurite outgrowth of neuroblastoma cells by regulating Erk signaling. J Biol Chem. 2013;288(41):29934–29942. doi:10.1074/jbc.M113.492520
41. Daniel PM, Filiz G, Tymms MJ, et al. Intratumor MAPK and PI3K signaling pathway heterogeneity in glioblastoma tissue correlates with CREB signaling and distinct target gene signatures. Exp Mol Pathol. 2018;105(1):23–31. doi:10.1016/j.yexmp.2018.05.009
42. Chae HD, Cox N, Capolicchio S, et al. SAR optimization studies on modified salicylamides as a potential treatment for acute myeloid leukemia through inhibition of the CREB pathway. Bioorg Med Chem Lett. 2019;29(16):2307–2315. doi:10.1016/j.bmcl.2019.06.023
43. Xia Y, Zhan C, Feng M, et al. Targeting CREB pathway suppresses small cell lung cancer. Mol Cancer Res. 2018;16(5):825–832. doi:10.1158/1541-7786.MCR-17-0576
44. Srinivasan S, Totiger T, Shi C, et al. Tobacco carcinogen-induced production of GM-CSF activates CREB to promote pancreatic cancer. Cancer Res. 2018;78(21):6146–6158.
45. Zhang X, Odom DT, Koo SH, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102(12):4459–4464. doi:10.1073/pnas.0501076102
46. Daniel P, Filiz G, Brown DV, et al. Selective CREB-dependent cyclin expression mediated by the PI3K and MAPK pathways supports glioma cell proliferation. Oncogenesis. 2014;3:e108. doi:10.1038/oncsis.2014.21
47. Yang SF, Lee WJ, Tan P, et al. Upregulation of miR-328 and inhibition of CREB-DNA-binding activity are critical for resveratrol-mediated suppression of matrix metalloproteinase-2 and subsequent metastatic ability in human osteosarcomas. Oncotarget. 2015;6(5):2736–2753. doi:10.18632/oncotarget.3088
48. Impey S, McCorkle SR, Cha-Molstad H, et al. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119(7):1041–1054. doi:10.1016/j.cell.2004.10.032
49. Tsai LN, Ku TK, Salib NK, Crowe DL. Extracellular signals regulate rapid coactivator recruitment at AP-1 sites by altered phosphorylation of both CREB binding protein and c-jun. Mol Cell Biol. 2008;28(13):4240–4250. doi:10.1128/MCB.01489-07
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.