")
Back to Journals » International Journal of Nanomedicine » Volume 17
GA&HA-Modified Liposomes for Co-Delivery of Aprepitant and Curcumin to Inhibit Drug-Resistance and Metastasis of Hepatocellular Carcinoma
Authors Li Y, Wu J , Lu Q, Liu X, Wen J, Qi X, Liu J, Lian B, Zhang B, Sun H, Tian G
Received 19 March 2022
Accepted for publication 26 May 2022
Published 7 June 2022 Volume 2022:17 Pages 2559—2575
DOI https://doi.org/10.2147/IJN.S366180
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lijie Grace Zhang
Yanying Li,1,2 Jingliang Wu,2 Qiao Lu,1 Xuemin Liu,2 Jiaxuan Wen,3 Xiaohui Qi,1 Jianhao Liu,4 Bo Lian,1 Bo Zhang,4 Hengyi Sun,1 Guixiang Tian1
1School of Life Science and Technology, Weifang Medical University, Weifang, 261053, People’s Republic of China; 2School of Nursing, Weifang University of Science and Technology, Weifang, 262700, People’s Republic of China; 3School of Nursing, Weifang Medical University, Weifang, 261053, People’s Republic of China; 4School of Pharmacy, Weifang Medical University, Weifang, 261053, People’s Republic of China
Correspondence: Hengyi Sun; Guixiang Tian, Email [email protected]; [email protected]
Background: Tumor microenvironment (TME) plays a vital role in the development of hepatocellular carcinoma (HCC). Mounting evidence indicates that peripheral nerves could induce a shift from quiescent hepatic stellate cells (HSCs) to cancer-associated fibroblasts (CAFs) by secreting substance P (SP). The anti-tumor strategy by targeting “SP-HSCs-HCC” axis might be an effective therapy to inhibit tumor growth and metastasis.
Objective: In this study, we prepared novel liposomes (CUR-APR/HA&GA-LPs) modified with hyaluronic acid (HA) and glycyrrhetinic acid (GA) for co-delivery aprepitant (APR) and curcumin (CUR), in which APR was chosen to inhibit the activation of HSCs by blocking SP/neurokinin-1 receptor (NK-1R), and CUR was used to induce apoptosis of tumor cells.
Results: To mimic the TME, we established “SP+HSCs+HCC” co-cultured cell model in vitro. The results showed that CUR-APR/HA&GA-LPs could be taken up by CAFs and HCC simultaneously, and inhibit tumor cell migration. Meanwhile, the “SP+m-HSCs+HCC” co-implanted mice model was established to evaluate the anti-tumor effect in vivo. The results showed that CUR-APR/HA&GA-LPs could inhibit tumor proliferation and metastasis, and reduce extracellular matrix (ECM) deposition and tumor angiogenesis, indicating a superior anti-HCC effect.
Conclusion: Overall, the combination therapy based on HA&GA-LPs could be a potential nano-sized formulation for anti-HCC therapy.
Keywords: hepatocellular carcinoma, hepatic stellate cells, substance P, liposomes, co-delivery
Corrigendum for this paper has been published
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignancies in the world, accounting for approximately 75% of all primary liver cancer.1 Surgical resection, liver transplantation, or ablation are the main approaches of HCC treatment.2 While the 5-years overall survival rate of the patients with HCC is only 10% due to its poor prognosis, high recurrence rate, and distant tumor metastasis.3,4 Therefore, it is crucial to design a novel strategy for HCC treatment. Mounting evidence indicates that tumor cells are embedded in complex tumor microenvironment (TME), in which the stromal cells and various cytokines play a crucial role in tumor development.5,6 Hence, much effort has been focused on the combination of TME-remodeling and pro-apoptosis activities to inhibit tumor growth and metastasis.
More recent researches have shown that peripheral nerves have emerged as the important component of TME, and maintained a relationship with the stromal cells and tumor cells by secreting neuropeptides, such as SP and CGRP.7,8 In HCC, SP is an important messenger molecule to regulate TME by SP/NK1-R signal pathway.9,10 Our previous researches have shown that SP could activate the hepatic stellate cells (HSCs) from quiescent phenotype to carcinoma-associated fibroblasts (CAFs), leading to drug resistance and metastasis of HCC.11 Hence, blocking “SP-HSCs-HCC” axis might be a potential strategy to inhibit HCC development.12,13
In this study, we chose aprepitant (APR) and curcumin (CUR) for combination therapy. APR, a non-peptide NK-1R antagonist, is widely used to prevent postoperative nausea and vomiting induced by chemotherapy.14,15 Furthermore, APR can also inhibit tumor cell proliferation, migration and angiogenesis by blocking SP/NK-1R signal pathway, suggesting that APR is a potential anti-tumor drug.16–18 CUR, a highly polyphenolic molecule extracted from the herbal turmeric roots, has a wide range of pharmacological activities, such as anti-inflammatory, antioxidant, and anticancer effects.19,20 Recent researches showed that CUR could inhibit the proliferation of HCC cells by inducing the damage of mitochondrial and nuclear genome DNA.21–24 The combination therapy of APR and CUR was expected to achieve the synergistic anti-HCC effects by TME-remodeling and pro-apoptosis activities.
However, traditional drug formulations were limited in clinic due to their low selectivity, rapid clearance and severe systemic side-effects. Recently, the drug delivery systems based on nano-sized carriers (liposomes, nanoparticles, and so on) have attracted more and more attention.25,26 Among of them, liposomes composed of an aqueous core and lipid bi-layer were long-circulating, biocompatible and biodegradable, and widely used for delivery of antitumor drugs.27,28 Furthermore, in the past few decades, ligand-modified liposomes were prepared for targeted delivery of chemotherapy drugs to tumor cells.29,30 Thinking that glycyrrhetinic acid (GA) receptors were over-expressed in HCC cells, GA-modified liposomes have been prepared for anti-HCC therapy in our previous studies.31 While the therapy targeting to tumor cells alone could not effectively inhibit drug resistance and metastasis of tumor cells due to the cross-talk between CAFs and HCC cells.32 Recent researches have reported that the CD44 receptors were over-expressed in activated hepatic stellate cells (aHSCs), and that hyaluronic acid (HA)-modified nano-sized vehicles could specifically bind to CD44 receptors.33,34 Hence, the novel co-targeting liposomes modified by GA and HA molecules were expected to be taken up by aHSCs and HCC cells simultaneously.
In this paper, the HA&GA-modified liposomes (CUR-APR/HA&GA-LPs) were prepared for co-delivery of CUR and APR to inhibit HCC development (Figure 1). To mimic actual HCC, the novel TME-like models composed of SP, HSCs and HCC were established to evaluate the anti-tumor efficacies in vitro and in vivo. Moreover, the “SP+m-HSCs+H22” subcutaneous tumor-bearing mice and lung metastasis model were used to investigate the anti-proliferation and anti-metastatic effects of CUR-APR/HA&GA-LPs.
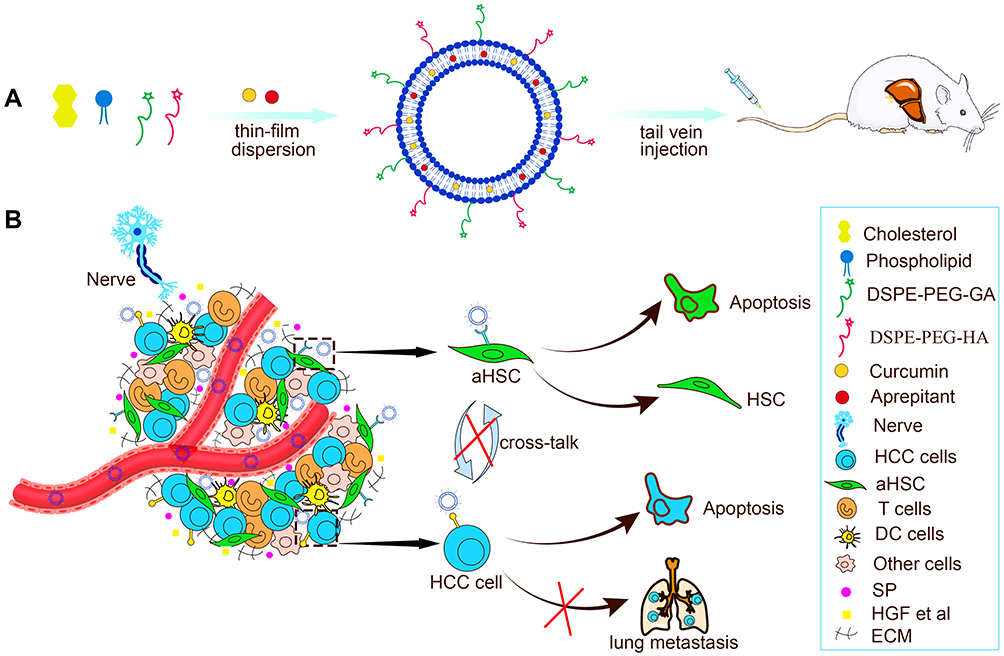 |
Figure 1 (A) The preparation and (B) anti-HCC mechanism of CUR-APR/HA&GA-LPs. |
Materials and Methods
Materials
Curcumin (CUR) was purchased from damas-beta (Shanghai, China). Soybean Phospholipid was obtained from A. V. T. (Shanghai) Pharmaceutical Co., Ltd. Aprepitant (APR), cell membrane near infrared fluorescent dye (DiR) and cell membrane red fluorescent dye (DiD) were acquired from Dalian Meilunbio (China). Fetal bovine serum and MTT were obtained from Solarbio (Beijing, China). DSPE-PEG-FITC and DSPE-PEG2000-NHS were purchased from Xi’an Ruixi Biological Technology Co., Ltd. (China). All other reagents used in this article were analytical grade.
Cell Lines and Animals
SMMC-7721 (Human-derived HCC cells), LX-2 (Human hepatic stellate cells), H22 (Mouse-derived HCC cells) and m-HSCs (Mouse hepatic stellate cells) were obtained from Beijing Bnbio research institute (China). SMMC-7721 were cultured in RPMI 1640 medium and LX-2 were cultured in DMEM medium. Female BALB/c mice (6~8 weeks) were purchased from Pengyue Lab animal center (Jinan, China). All animal handling procedures complied with the Animal Management Rules in China (D.N. 55, 2001), and the in vivo anti-tumor studies were approved by the Animal Ethics Committee of Weifang Medical University (2019–045).
Preparation of HA&GA-Modified Liposomes
DSPE-PEG2000-HA was synthesized by cross-linking reaction of DSPE-PEG2000-NH2 with HA in the presence of 1-Ethyl-3(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS), and DSPE-PEG2000-GA was obtained according to our previous researches.31
CUR-APR/HA&GA-LPs were prepared by the thin-film dispersion method.35 Briefly, soybean phospholipid, cholesterol, DSPE-PEG2000-GA and DSPE-PEG2000-HA in mass ratio (30:10:1:1) were dissolved in ethanol (5 mL), and then APR (6 mg) and CUR (6 mg) were added to the solution. The mixed solution was evaporated using a rotating evaporator to form a uniform thin film. Then, 5 mL of water was added to the film, and the mixture was hydrated at 60°C for 1 h. The suspension was sonicated for 8 min in an ultrasonic cell crusher (VCX750, USA), and extruded through poly-carbonate membranes (220 nm) for three times to remove unloaded drugs. Similarly, the FITC-labelled liposomes were prepared according to the above method.
Characterization of CUR-APR/HA&GA-LPs
The particle size and zeta potentials of different liposomal formulations were measured using a Malvin Zetasizer (Nano-ZS90, UK). To evaluate the stability of liposomes, the drug-loaded liposomes were dissolved in PBS (pH 7.4) and RPMI 1640 medium, respectively. The mean particle sizes were measured for seven days. The morphologies of liposomes were observed using a transmission electron microscope (TEM) (JEM 100CX, JEOL Ltd., Japan). The drug loading capacity (DL) and drug encapsulation efficiency (EE) of CUR and APR were measured using UV-Vis spectrophotometer (Shanghai instruments Co., LTD., China) and high performance liquid chromatography (HPLC) (Agilent Technologies Co., LTD., USA), respectively.
In vitro Drug Release
The release of CUR and APR from liposomes was investigated by dialysis method.36 1 mL of CUR-APR/HA&GA-LPs was added into a dialysis bag (MWCO = 3500), and the bag was dialyzed against 50 mL PBS solution containing 1% tween-80 and 20% aqueous ethanol at 100 rpm under 37°C. At the predetermined point of times (0.5, 1, 2, 4, 8, 12, 24, 36, and 48 h), 4 mL of release medium was replaced with equivalent fresh release medium. The cumulative releases of CUR and APR were measured by UV spectrophotometry at the wavelength of 425 nm and HPLC at the wavelength of 215 nm, respectively.
In vitro Cellular Uptake
The in vitro cellular uptake of liposomes was performed to evaluate the liver-targeting property. To track the distribution of liposomes in SMMC-7721 cells, FITC, a fluorescent dye, was used to label the liposomes. In brief, SMMC-7721 cells were incubated with FITC-LPs and FITC/HA&GA-LPs for 1 h, respectively. After washing twice with PBS, the cells were fixed with 4% paraformaldehyde, and stained with DAPI for 10 min. The cells were photographed using the CLSM.
In order to mimic the TME, a “SP+LX-2+SMMC-7721” co-cultured cell model was established by mixing SMMC-7721 and LX-2 cells at the ratio of 5:1 in the presence of SP (10 ng/mL). In this assay, CUR was replaced by coumarin-6 (C6, a fluorescent dye) to investigate the drug retention against tumor cells.37 The co-cultured cell systems were treated with C6, C6+APR, C6-APR/LPs and C6-APR/HA&GA-LPs for 4 h, 12 h, 24 h, respectively. After washing thrice with PBS, the cells were photographed by CLSM, and the intracellular fluorescent intensity was quantified by flow cytometry (FCM).
In vitro Cytotoxicity
Recent researches have shown that CUR has a strong pro-apoptotic effect on HCC cells by blocking the activation of nuclear factor kappa B (NF-kB).38,39 MTT assay was performed to detect the cytotoxicity of different drug formulations. We established 2-level cell models: (1) SMMC-7721 cells, (2) “SP+LX-2+SMMC-7721” co-cultured cells. The cells were seeded into 96-well plates and incubated in the incubator for 24 hrs. Afterward, the cells were treated with free CUR, free APR, CUR+APR, CUR-APR/LPs and CUR-APR/HA&GA-LPs at different concentrations of CUR (0.01 ug/mL~10 ug/mL) for 48 hrs. After incubation with MTT for 4 h, 150 uL of DMSO was added to each well. The absorbance values were measured using a microplate reader (ELX800, BioTek Instruments, Inc. USA) at a wavelength of 490 nm.
Live-Dead Staining
The cell viability was also qualitatively evaluated using a live/death staining kit (Calcein-AM/PI).40 After 48 hrs of drug treatment, the co-cultured cells were stained with Calcein-AM (2 μM) and PI (8 μM) for 20 min, and then imagined by a fluorescence microscope (C-SHG1, Nikon Crop. Japan).
Cell Migration
A wound healing assay was performed to evaluate the anti-migration effects of CUR-APR/HA&GA-LPs. Briefly, SMMC-7721 cells were seeded in 6-well plates for incubation overnight. Then the cells were streaked with a 200 ul pipette tip to form three wound-like lines in each well. After washing with PBS, the cells were treated with CUR, APR, CUR+APR, CUR-APR/LPs and CUR-APR/HA&GA-LPs. The wound width in each well was photographed using a Nikon fluorescence microscope at 0 h and 24 h. Furthermore, we established a “SP+LX-2+SMMC-7721” wound healing model, in which LX-2 cells were labelled with CFSE (a green fluorescent dye) to distinguish them. The cell migration assay is the same as above. In addition, the transwell assay was used to further assess the anti-migration capacity of various drug formulations against “SP+LX-2+SMMC-7721” co-culture cell model.
In vivo Biodistribution and Tumor Penetration
In order to detect the in vivo biodistribution of CUR-APR/HA&GA-LPs, a “SP+m-HSCs+H22” tumor-bearing model was established. Briefly, H22 (2×106), m-HSCs (4×105) and SP (10 ng/mL) were subcutaneously injected into the right flank of female BALB/c mice. When the tumor volume grew to ~150 mm3, the DiR, DiR-LPs, DiR/HA&GA-LPs were injected into mice via tail vein, respectively. The in vivo biodistribution was detected using a fluorescence imaging system at different time points (2, 6, 12, 24, 48 hrs). After 48 hrs, the main organs and tumors of the mice were harvested for further in vitro fluorescence imaging analysis.
In addition, to further evaluate the tumor tissue penetration abilities of HA&GA-LPs, free DiD, DiD-LPs and DiD/HA&GA-LPs were injected into “SP+m-HSCs+H22” tumor-bearing mice via tail vein. After 12 hrs, the mice were euthanized and tumor tissues were excised. The tumor tissues were sectioned at a thickness of 10 μm using a cryosectioning machine (Leica CM1950). Then the slices were fixed in 4% paraformaldehyde, and stained with DAPI, followed by photograph under CLSM.
In vivo Antitumor Efficacy
In order to evaluate the antitumor efficacies of different drug formulations and mimic actual TME, we established “SP+m-HSCs+H22” tumor-bearing mice, in which the mixed solution of H22 and m-HSCs cells (the ratio was 5:1) was injected into the right flank of mice in the presence of SP (10 ng/mL). When tumor size reached about 150 mm3, the mice were randomly divided into seven groups (n = 5): H22 (saline, control), SP+m-HSC+H22 (saline, control), free CUR, free APR, CUR+APR, CUR-APR/LPs, CUR-APR/HA&GA-LPs. The mice were injected at the dosage of 5 mg/kg (CUR) and 5 mg/kg (APR) for 7 times. Both tumor volume and body weight were monitored every 2 days. On the 14th day, mice were euthanized, and the major organs (kidney, lung, liver, spleen, heart) and tumors were harvested, followed by fixation by 4% paraformaldehyde for photography and histological study.
Lung Metastasis Inhibition
To investigate the anti-metastatic effects of CUR-APR/HA&GA-LPs, a lung metastasis model was established. 0.2 mL of H22 cells (1×107 cells/mL) were injected into female BALB/c mice via the tail vein. All mice were randomly assigned into seven groups (n = 3): Health, Saline (control), free CUR, free APR, CUR+APR, CUR-APR/LPs, CUR-APR/HA&GA-LPs. The intravenous injection was performed 7 times. After 2 weeks, the mice were sacrificed, and lung tissues were removed for H&E staining. The number of metastatic lung nodules was calculated.
Statistical Analysis
All the data in this study were given by mean ± SD. Student’s t-test was used to compare the differences between the different drug-treated groups. P < 0.05 was considered statistical significance.
Results and Discussion
Preparation and Characterization of CUR-APR/HA&GA-LPs
CUR-APR/HA&GA-LPs were prepared by the thin-film dispersion method. The particle sizes, zeta potentials, polydispersed index (PDI), drug loading capacity (DL) and drug encapsulation efficiency (EE) of different liposomal formulations are described in Table 1. The average particle sizes of drug-loaded liposomes were around 120 nm, and zeta potentials were negative. These properties were beneficial to prolong the circulating time of liposomes in blood, and promote drug accumulation in tumor region by passive-targeting manner.
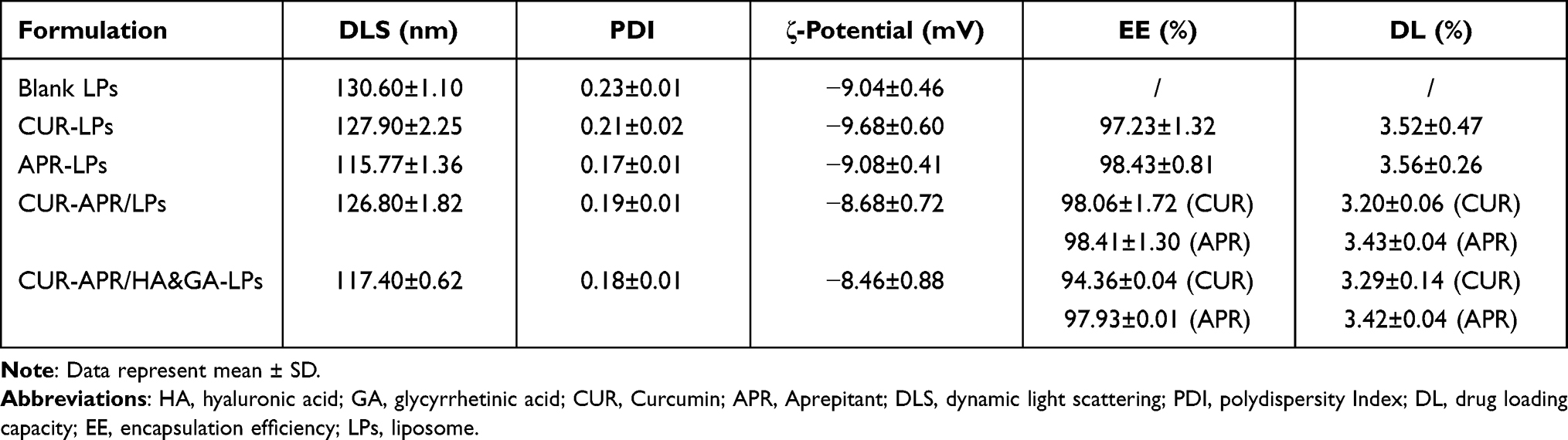 |
Table 1 Characterization of the Different Liposomal Formulations |
As shown in Figure 2A and B, the TEM images revealed that Blank liposomes and CUR-APR/HA&GA-LPs were spherical in shape with the narrow size distribution. The stability assay showed that the mean particle size of CUR-APR/HA&GA-LPs did not change significantly in PBS (pH 7.4) and RPMI 1640 medium for 7 days (Figure 2C), indicating that HA&GA-LPs was a stable nano-sized carrier.
 |
Figure 2 In vitro characteristics of CUR-APR/HA&GA-LPs. TME images (scale bar: 200 nm) and particle size distribution of CUR-APR/HA&GA-LPs (A) and Blank liposomes (B). (C) The stability of CUR-APR/HA&GA-LPs in PBS (PH 7.4) and RPMI 1640 medium, respectively. (D) The cumulative release of CUR and APR from CUR-APR/HA&GA-LPs. |
The in vitro drug release of APR and CUR from liposomes is showed in Figure 2D. It showed an initial drug release of 11.43% (CUR) and 39.53% (APR) at first 4 hrs, and an accumulative release of 47.27% (CUR) and 76.43% (APR) at 48 hrs. Compared with CUR, APR showed faster drug release from liposomes.
In vitro Cellular Uptake
To monitor the cellular uptake of HA&GA-LPs in SMMC-7721 cells (Figure 3A), FITC (green fluorescent dye) was used as a probe to label liposomes, and DAPI (blue dye) was chosen to dye the nuclei. As shown in Figure 3C, compared with FITC-LPs, FITC/HA&GA-LPs showed stronger green fluorescent signals in the cells, suggesting that the dual-ligand modified liposomes could promote cellular uptake by GA receptor-mediated endocytosis. In Figure 3D–E, FCM assay showed similar results, in which the fluorescent intensity of FITC/HA&GA-LPs group was higher than that of FITC/LPs group.
 |
Figure 3 Cellular uptake of (A) and drug retention assay (B) of HA&GA-LPs. (C) CLSM images of SMMC-7721 cells uptake (green: FITC, blue: DAPI). (D and E) FCM analysis of cell uptake in SMMC-7721 cells. The drug retention assay using CLSM (F) and FCM (G) against co-cultured cell model (green: C6, blue: DAPI). (H) Quantitative analysis of drug retention by FCM. Scale bar: 50 μm, *P < 0.05, **P < 0.01, ***P < 0.001. |
Drug efflux plays an important role in the development of drug resistance of tumor cells.41 In the TME, the stromal cells, such as HSCs, secret various cytokines to up-regulate MDR-related proteins, leading to low drug detention.42 In this study, we established a “SP+LX-2+SMMC-7721” co-cultured cell model to mimic TME (Figure 3B), and C6 (a fluorescent dye) was chosen to replace CUR for drug detention assays via CLSM and FCM. As shown in Figure 3F, compared with other groups, more fluorescent signals were detected in the C6-APR/HA&GA-LPs, indicating that the modification of GA and HA molecules could improve liposomes internalization. Furthermore, the retained drug in cells were quantitatively analyzed by FCM (Figure 3G). The fluorescent intensity of liposomal formulations at 24 hrs was higher than that of free C6 and C6+APR, indicating that liposomes improved drug detention. As expected, C6-APR&/HA&GA-LPs group exhibited stronger fluorescent signals than C6-APR/LPs (Figure 3H). The result indicated that HA&GA-LPs could increase drug retention in tumor cells.
In vitro Cytotoxicity Assay
The in vitro cytotoxicity of different drug formulations against SMMC-7721 cells was evaluated by MTT assay (Figure 4A). As shown in Figure 4C, all drug formulations exhibited concentration-dependent cytotoxicity. Compared with CUR+APR and CUR-APR/LPs, the cell viability of CUR-APR/HA&GA-LPs was lower. The possible explanation was that HA&GA-LPs improved drug internalization, leading to enhanced pro-apoptotic effects.
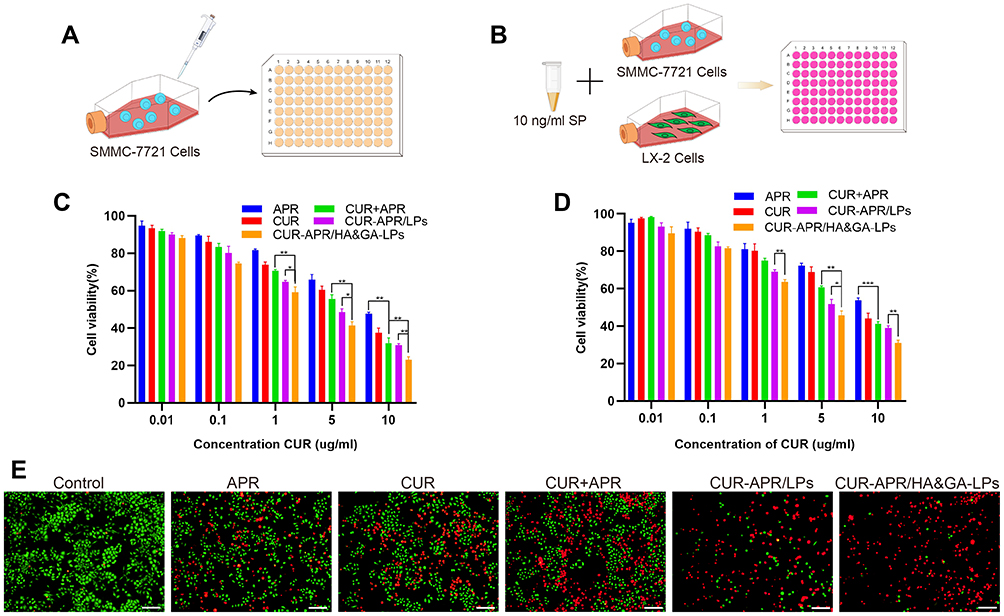 |
Figure 4 In vitro cytotoxicity assay. MTT assay against SMMC-7721 cells (A) and co-cultured cell model (B). Cytotoxicity of SMMC-7721 cells (C) and co-cultured cells (D) incubated with different drug formulations for 48 h. (E) The live/dead staining assay of co-cultured cell model (PI: red, Calcein-AM: green). Scale bar: 100 µm, *P < 0.05, **P < 0.01, ***P < 0.001. |
However, tumor cells were located in complex microenvironment.43 To mimic TME, “SP+LX-2+SMMC-7721” co-culture cell model was established to evaluate the cytotoxicity of different drug formulations (Figure 4B). The IC50 of free CUR in the co-cultured cells was obviously higher than that of SMMC-7721 cells alone. Interestingly, compared to free CUR or APR, CUR+APR showed higher toxicity, indicating that the combination therapy of CUR and APR had the potential synergistic effect (Figure 4D). As expected, CUR-APR/HA&GA-LPs displayed stronger cytotoxicity than CUR+APR and CUR-APR/LPs. Our previous researches have proven that GA receptor and CD44 receptor were over-expressed on HCC cells and aHSCs, respectively.11,44 The result might be due to the fact that HA&GA-LPs could be taken up by LX-2 and SMMC-7721 cells simultaneously, resulting in greater anti-HCC effects.
To qualitatively detect the cytotoxicity of CUR-APR/HA&GA-LPs against the co-cultured cell model, the live/dead staining was performed, in which live cells were stained by Calcein-AM (green color), and dead cells were labelled by propidium iodide (PI, red color). As shown in Figure 4E, compared with the control, only few red spots (dead cells) were observed in the single drug groups. While there were more dead cells occurred in the combined drug groups. CUR-APR/HA&GA-LPs showed less green spots than other groups, suggesting that HA&GA-LPs promoted pro-apoptotic effects of drugs. The results showed that HA&GA/LPs were an effective carrier for anti-HCC drugs.
Cell Migration Assay
To assess the anti-migration effects of different drug formulations, a wound healing assay was performed against SMMC-7721 cells (Figure 5A). As shown in Figure 5B, the cell migrations in drug-treated groups were inhibited. Compared to free CUR and APR, the combined drug groups showed greater wound width (Figure 5C). Obviously, the migration rate of CUR-APR/HA&GA-LPs was 3.47%, which was 0.33 fold and 0.35 fold of CUR+APR and CUR-APR/LPs, respectively. The result suggested that HA&GA-modified liposomes could improve anti-migration effects of drugs.
 |
Figure 5 In vitro anti-migration effect. A wound healing assay against SMMC-7721 cells (A) and co-cultured cells model (D). Cell migration images (C) and the migration ratio (B) of SMMC-7721 cells. Cell migration images (F) and the migration ratio (E) of co-cultured cells model. (G) Images of transwell assay in co-cultured cell model after treatment of various drug formulations. Scale bar: 100 µm. (H) Quantification of migrated cells in transwell assay. *P < 0.05, **P < 0.01. |
However, recent studies have shown that the cross-talk between stromal cells and tumor cells plays an important role in tumor migration and metastasis.45 The in vitro cell model composed of tumor cells alone could not affect the actual TME. In this study, we established a novel “SP+LX-2+SMMC-7721” co-cultured migration model to mimic TME, in which CFSE was used to label LX-2 cells (Figure 5D). Interestingly, the migration rate in the co-cultured model was higher than SMMC-7721 cells alone (Figure 5E), suggesting that the addition of SP and LX-2 cells promoted tumor cell migration. After drug treatment, the wound healing speed was slightly inhibited in single drug groups (CUR or APR), and obviously restrained in the three combined drug groups (Figure 5F). The possible explanation was that CUR+APR exhibited the synergistic anti-metastasis effect by pro-apoptotic and anti-SP activities, respectively. As expected, CUR-APR/HA&GA-LPs showed lower cell migration rate than CUR-APR/LPs. This was due to the fact that HA&GA-modified liposomes promoted active uptake of drugs by LX-2 and SMMC-7721 cells, leading to the greater anti-tumor effect.
To further detect the anti-metastatic capacity of CUR-APR/HA&GA-LPs, the transwell migration assay was performed against “SP+LX-2+SMMC-7721” co-culture cell model. As shown in Figure 5G and H, compared with other drug formulation groups, the number of metastatic cells was significantly reduced in CUR-APR/HA&GA-LPs, which was consistent with the result of wound healing assay.
In vivo Bio-Distribution Imaging and Tumor Penetration
To investigate the liver-targeting property of HA&GA-LPs, a “SP+m-HSCs+H22” tumor-bearing mice model was established, in which m-HSCs and H22 cells were mixed at the ratio of 1:5. DiR, a fluorescent dye, was used as a drug to track bio-distribution of HA&GA/LPs using a real-time imaging system (IVIS). As shown in Figure 6A, fluorescent signals were easily observed in the three groups at 6 hrs. While the fluorescence in tumor region declined quickly in free DiR group, and the two liposomal groups, especially HA&GA-LPs, kept strong fluorescent signals until 48 hrs, suggesting that liposomes could prolong the blood circulation time of drug.
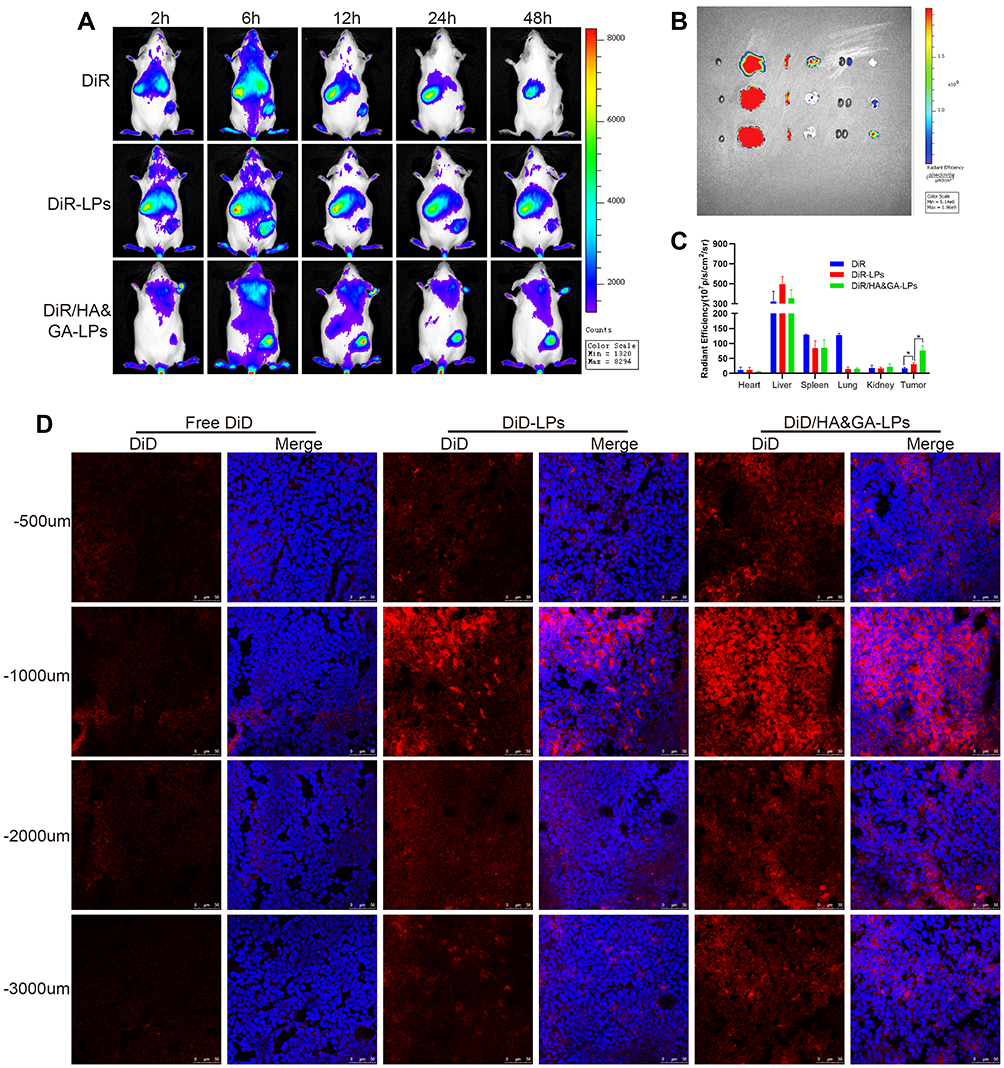 |
Figure 6 The in vivo biodistribution and drug penetration of HA&GA-LPs. (A) The in vivo biodistribution imaging of different formulations at different time points (2, 6, 12, 24, and 48 h). The ex vitro fluorescence imaging (B) and fluorescence quantitative analysis (C) of major organs and tumors. (fluorescence: DiR). (D) CLSM images of tumor penetration (red: DiD, blue: DAPI). Scale bar: 50 µm, *P < 0.05. |
After 48 hrs, mice were sacrificed and the main organs and tumors were harvested for ex vivo fluorescence imaging analysis. In Figure 6B, compared with free DiR group and DiR-LPs group, the tumor in DiR/HA&GA-LPs group exhibited stronger fluorescence signals, which was consistent with the in vivo fluorescence images. The quantitative analysis is shown in Figure 6C, and the fluorescence intensity of DiR/HA&GA-LPs in the tumor region was 2.68 fold and 4.47 fold higher than that of in the DiR-LPs and free DiR, respectively. This might be due to the fact that DiR//HA&GA-LPs could accumulate in tumor by EPR-mediated passive targeting effects and HA&GA receptor-mediated active drug delivery.
To further evaluate the penetration ability of different drug formulations in the tumor tissue, the “SP+m-HSCs+H22” tumor-bearing mice were intravenously injected with free DiD, DiD-LPs and DiD/HA&GA-LPs. The fluorescence images of tumor tissue slices were captured by CLSM. In Figure 6D, fluorescent signals were mainly distributed at the edge of the tumor tissue, suggesting that free drug could not reach the core of tumor, resulting in weak anti-tumor effects. By contrast, the liposomal groups, especially HA&GA-LPs, exhibited obvious red fluorescent signals from the edge to the core, indicating that DiD-LPs and DiD/HA&GA-LPs could effectively penetrate the inner region of tumor tissue. As expected, compared with DiD-LPs group, the fluorescence intensity in the DiD/HA&GA-LPs was higher at the tumor depth of 500–3000 um, suggesting that the modification of HA and GA molecules on liposomes was beneficial for drug penetration in tumors.
In vivo Anti-Tumor Activity of “Sp+m-HSCs+H22” Subcutaneous Tumor-Bearing Mice Model
It has been reported that SP secreted by nerve fibers in TME might prompt the shift of HSCs from quiescent phenotype to aHSCs.46,47 To evaluate the anti-tumor effect of CUR-APR/HA&GA-LPs in actual TME, a subcutaneous “SP+m-HSCs+H22” tumor-bearing mice model was established, in which the ratio of H22 to m-HSCs was 5:1 and the concentration of SP was 10 ng/mL (Figure 7A).
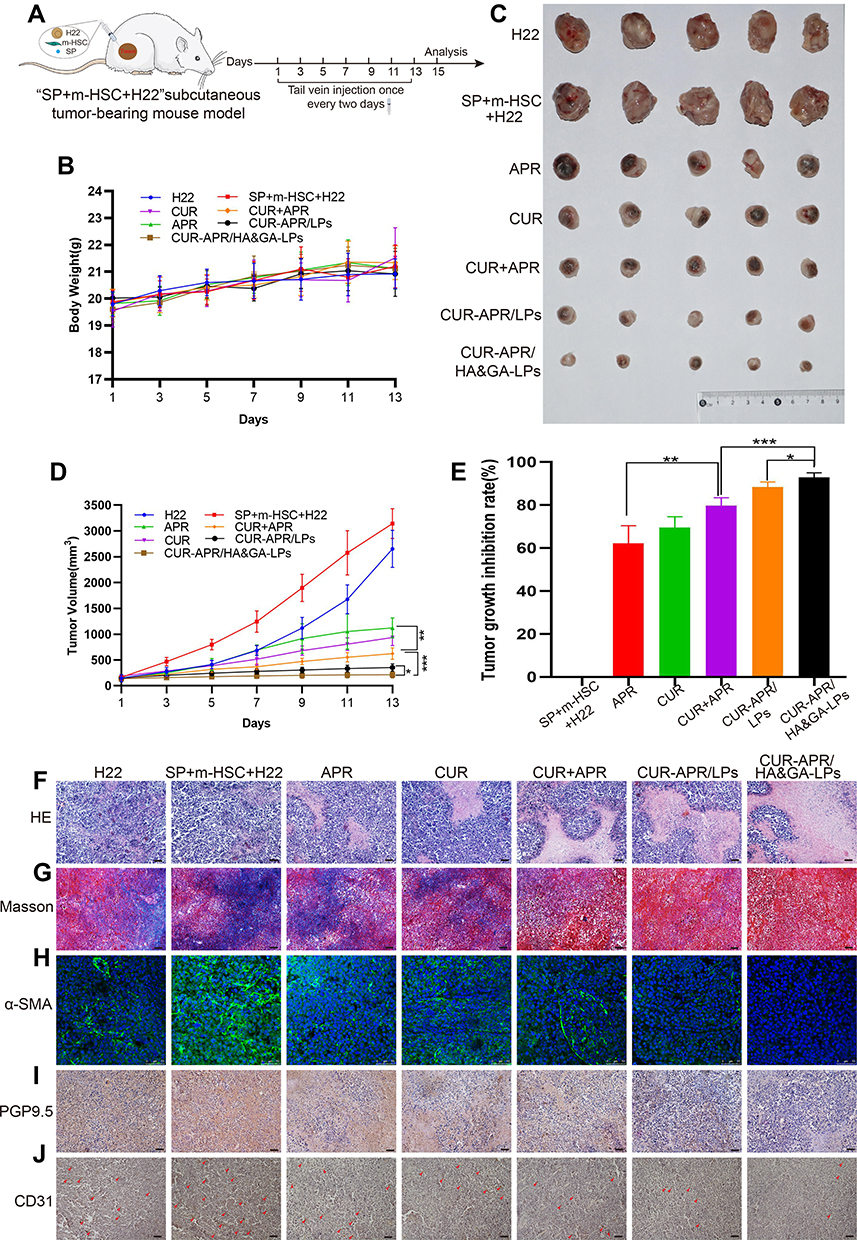 |
Figure 7 In vivo antitumor activity in “SP+m-HSCs+H22” tumor-bearing mice. (A) Schematic illustration of treatment scheme on mice. (B) Tumor images of mice treated with different drug formulations. (C) The body weight in mice. (D) Tumor growth inhibition curves. (E) Tumor growth inhibition rate. H&E (F), and Masson trichromatic staining (G) analysis of tumor tissues slices. (H) α-SMA immunofluorescence staining (green fluorescence: α-SMA protein). Immunohistochemical assay of PGP9.5 proteins (I) and CD31 proteins (J). (red arrow: CD31 proteins). Scale bar: 50 μm, *P < 0.05, **P < 0.01, ***P < 0.001. |
As shown in Figure 7B, there was no significant change in the body weight between saline group and liposomal groups, indicating that HA&GA-LPs was a safe nano-sized carrier for drug delivery. After 7 times of drug administration, the mice were sacrificed, and the tumors were collected and photographed (Figure 7C). Interestingly, in Figure 7D and E, the tumor volume of “SP+m-HSCs+H22” tumor-bearing mice was larger than that of H22 tumor-bearing mice, suggesting that the addition of SP and m-HSCs could promote tumor proliferation. Given that HSCs and SP were present in the TME, the co-implanted mice model was a more appropriate model than traditional H22 tumor-bearing mice model. After drug treatment, the tumor growth was obviously inhibited. Compared with single drug groups (APR, CUR), the combined groups, especially co-loaded liposomal groups, exhibited lower tumor volume, suggesting that the combination therapy of APR and CUR could achieve a synergistic anti-tumor effect. As expected, the tumor growth inhibition rate (IR) of CUR-APR/HA&GA-LPs group was 92.98%, which was higher than that of CUR+APR (79.75%) and CUR-APR/LPs (88.43%). This might be due to the fact that the HA&GA-modified liposomes promoted drug accumulation in tumor region by EPR effect and GA and/or CD44 receptor-medicated endocytosis. Similar results were observed in H&E staining assay of tumor tissue sections. In Figure 7F, obvious karyolysis and cytoplasmic vacuolation were observed in the drug-treated groups. As expected, CUR-APR/HA&GA-LPs exhibited superior pro-apoptotic effects.
Recent researches showed that the compact ECM in TME played a key role in the generation of tumor drug resistance.48 Masson staining assay was performed to examine collagen fiber deposition in tumor tissue. As shown in Figure 7G, obvious collagen deposition was observed in the control group of “SP+m-HSCs+H22” model. Compared with the free drug groups, the liposomal groups, especially CUR-APR/HA&GA-LPs, showed less ECM deposition, indicating that the combination therapy based on HA&GA-LPs could effectively inhibit the production of collagen. Previous studies have reported that aHSCs can promote ECM formation by synthesizing collagen and fibronectin.49,50 The result showed that the combination therapy based on HA&GA-LPs could inhibit aHSCs-induced ECM deposition. Furthermore, alpha smooth muscle actin (α-SMA) was a marker protein of aHSCs,51 and the immunofluorescence staining of α-SMA was performed to examine the activation of HSCs. As shown in Figure 7H, there were obvious green fluorescent signals observed in the novel co-implanted mice model (saline group), indicating that the HSCs were activated to become CAF-like phenotype. After drug treatment, nearly no α-SMA+ cells were observed in the CUR-APR/HA&GA-LPs group, suggesting that CUR-APR/HA&GA-LPs effectively inhibited the activation of HSCs.
Our previous studies have shown that peripheral nerves can promote tumor development by “SP-HSCs-HCC” axis.11 In the study, the immunohistochemistry assay of PGP9.5 proteins (the marker protein of nerve fiber) was performed. As shown in Figure 7I, the PGP9.5 proteins were over-expressed in the tumor region, indicating that nerve fibers were actually distributed in the TME. Compared with other groups, the expressions of PGP9.5 proteins were significantly inhibited in CUR-APR/HA&GA-LPs group. Considering that nerve fibers could secrete SP and promote the activation of HSC through SP/NK-1R signal pathway,46 the result suggested that the combination therapy of CUR and APR could effectively inhibit “SP-HSC-HCC” axis-mediated tumor growth. Moreover, tumor angiogenesis plays an important role in tumor proliferation and metastasis. In this study, the expression of CD31 proteins in the vascular endothelial cells was examined by immunohistochemical staining assay. In Figure 7J, the vascular density was higher in “SP+m-HSCs+H22” tumor-bearing mice than that in H22 tumor-bearing mice. CUR-APR/HA&GA-LPs showed lower microvascular density than other groups, suggesting that CUR-APR/HA&GA-LPs significantly inhibited tumor angiogenesis.
Additionally, the pathologic changes of the main organs (liver, heart, spleen, lung, kidney) were investigated using H&E staining. In Figure 8, there was no obvious damage in the CUR-APR/HA&GA-LPs in comparison with the saline group, indicating that HA&GA-LPs were biocompatible, and could be used as a vehicle for delivery of anti-tumor drugs.
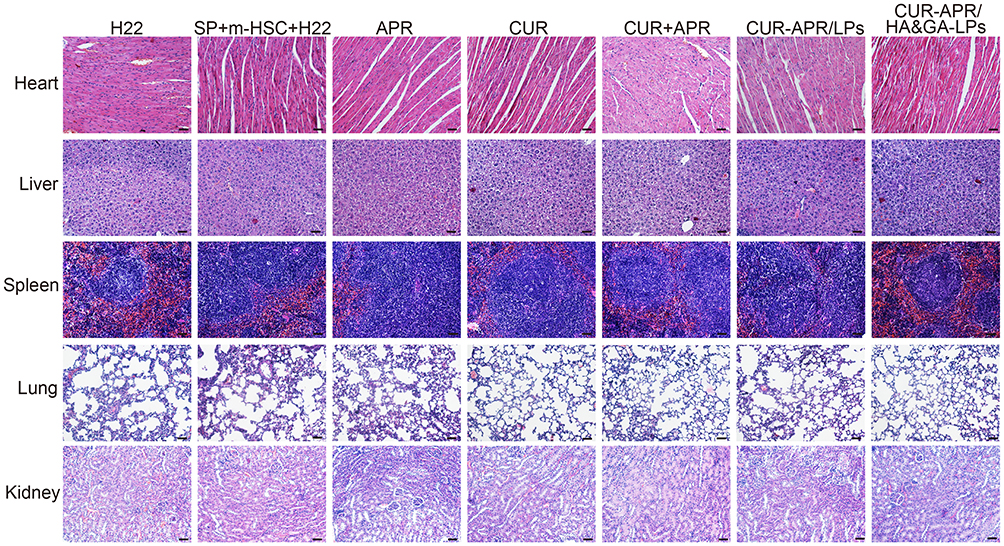 |
Figure 8 H&E staining images of heart, liver, spleen, lung and kidney of mice treated with different drug formulations. Scale bar: 50 μm. |
Inhibition of Lung Metastasis in vivo
Distant metastasis is the main cause of poor prognosis and high recurrence rate of hepatocellular carcinoma.52 To evaluate the inhibitory effect of CUR-APR/HA&GA-LPs on lung metastasis in vivo, a lung metastasis model was established by injecting H22 cells into the tail vein (Figure 9A). During the treatment period, there was no significant change in the bodyweight between the control and liposomal groups, indicating that HA&GA-LPs was a safe carrier for delivery of anti-tumor drugs (Figure 9B). As shown in Figure 9C and D, amounts of metastatic nodules were observed on the lung in the saline group, indicating that the lung metastasis model was successfully established. Compared with free APR or CUR, the three combined drug groups exhibited less pulmonary metastatic nodules, suggesting that the combination therapy of APR and CUR improved anti-metastasis effects. Obviously, CUR-APR/HA&GA-LPs showed greater inhibitory of tumor metastasis than CUR-APR/LPs. H&E staining images showed that the tumor region significantly decreased in drug-treated groups in comparison with the saline group (control group) in Figure 9E. Similarly, CUR-APR/HA&GA-LPs showed stronger anti-metastasis effects than other groups. The possible explanation was that HA&GA-LPs could be effectively taken up by metastatic tumor cells in lung tissue, leading to higher bioavailability of anti-tumor drugs.
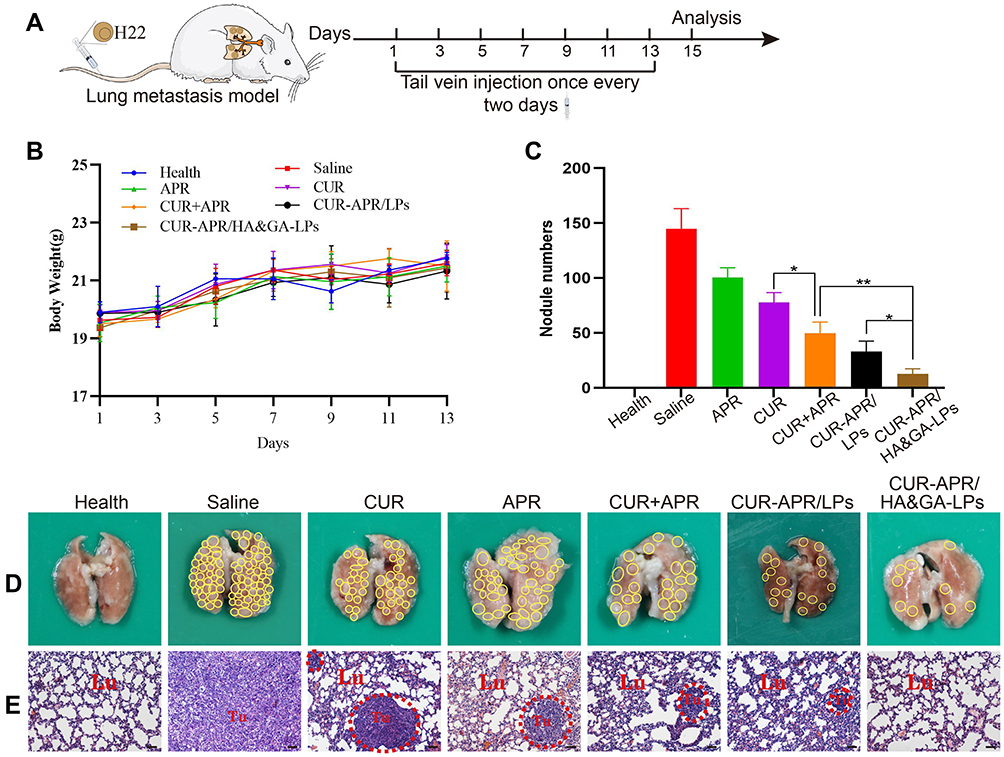 |
Figure 9 In vivo anti-lung metastasis analysis in mice. (A) Establishment of mice model of lung metastasis and schematic illustration of treatment scheme. (B) Variations of body weights in mice. (D) Lung images. (The yellow circles indicated lung nodules). (C) Quantitative analysis of lung nodule numbers. (E) H&E staining of lung tissue slices (scale bar: 50 μm). (The red circles showed tumor region). *P < 0.05, **P < 0.01. |
Conclusion
In summary, SP secreted by peripheral nerves in the TME could induce a shift from quiescent HSCs to CAFs, leading to drug-resistance and metastasis of HCC cells. In this study, we prepared HA&GA-modified liposomes (CUR-APR/HA&GA-LPs) for co-delivery APR and CUR to inhibit the “SP-HSCs-HCC” activated axle. The in vitro studies showed that HA&GA-LPs could be taken up simultaneously by HCC and HSCs. Moreover, the anti-HCC results against “SP+m-HSCs+H22” tumor-bearing mice model showed that CUR-APR/HA&GA-LPs could effectively remodel TME, and exhibit superior anti-HCC efficacy. Hence, CUR-APR/HA&GA-LPs could inhibit tumor development by blocking “SP-HSCs-HCC” axis, and be a potential nano-sized formulation for anti-HCC therapy.
Acknowledgments
This work was supported by the National Science Foundation of China (81803464), Natural Science Foundation of Shandong Province (ZR2020MC078), the Science and Technology Development Program in Weifang (2021GX053), and Scientific Research Project of Weifang University of Science and Technology (KJRC2021004, 2021XKJS12, and 2021XKJS19).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Frager SZ, Schwartz JM. Hepatocellular carcinoma: epidemiology, screening, and assessment of hepatic reserve. Curr Oncol. 2020;27(13):138–143. doi:10.3747/co.27.7181
2. Anwanwan D, Singh SK, Singh S, Saikam V, Singh R. Challenges in liver cancer and possible treatment approaches. Biochim Biophys Acta Rev Cancer. 2020;1873(1):188314. doi:10.1016/j.bbcan.2019.188314
3. Song MJ. Hepatic artery infusion chemotherapy for advanced hepatocellular carcinoma. World J Gastroenterol. 2015;21(13):3843. doi:10.3748/wjg.v21.i13.3843
4. Marengo A, Rosso C, Bugianesi E. Liver cancer: connections with obesity, fatty liver, and cirrhosis. Annu Rev Med. 2016;67(1):103–117. doi:10.1146/annurev-med-090514-013832
5. Williamson T, Sultanpuram N, Sendi H. The role of liver microenvironment in hepatic metastasis. Clin Transl Med. 2019;8(1). doi:10.1186/s40169-019-0237-6
6. Brodt P. Role of the microenvironment in liver metastasis: from pre- to prometastatic niches. Clin Cancer Res. 2016;22(24):5971–5982. doi:10.1158/1078-0432.Ccr-16-0460
7. Gysler SM, Drapkin R. Tumor innervation: peripheral nerves take control of the tumor microenvironment. J Clin Invest. 2021;131(11). doi:10.1172/jci147276
8. Zahalka AH, Frenette PS. Nerves in cancer. Nat Rev Cancer. 2020;20(3):143–157. doi:10.1038/s41568-019-0237-2
9. Rosso M, Muñoz M, Berger M. The role of neurokinin-1 receptor in the microenvironment of inflammation and cancer. Sci World J. 2012;2012:1–21. doi:10.1100/2012/381434
10. Nizam E, Köksoy S, Erin N. NK1R antagonist decreases inflammation and metastasis of breast carcinoma cells metastasized to liver but not to brain; phenotype-dependent therapeutic and toxic consequences. Cancer Immunol Immunother. 2020;69(8):1639–1650. doi:10.1007/s00262-020-02574-z
11. Li Z, Wang F, Li Y, et al. Combined anti-hepatocellular carcinoma therapy inhibit drug-resistance and metastasis via targeting “substance P-hepatic stellate cells-hepatocellular carcinoma” axis. Biomaterials. 2021;276:121003. doi:10.1016/j.biomaterials.2021.121003
12. Berger M, Neth O, Ilmer M, et al. Hepatoblastoma cells express truncated neurokinin-1 receptor and can be growth inhibited by aprepitant in vitro and in vivo. J Hepatol. 2014;60(5):985–994. doi:10.1016/j.jhep.2013.12.024
13. Ge C, Huang H, Huang F, et al. Neurokinin-1 receptor is an effective target for treating leukemia by inducing oxidative stress through mitochondrial calcium overload. Proc Nat Acad Sci. 2019;116(39):19635–19645. doi:10.1073/pnas.1908998116
14. Singh PM, Borle A, Rewari V, et al. Aprepitant for postoperative nausea and vomiting: a systematic review and meta-analysis. Postgrad Med J. 2016;92(1084):87–98. doi:10.1136/postgradmedj-2015-133515
15. He A, Alhariri JM, Sweren RJ, Kwatra MM, Kwatra SG. Aprepitant for the treatment of chronic refractory pruritus. Biomed Res Int. 2017;2017:1–6. doi:10.1155/2017/4790810
16. Muñoz M, Coveñas R, Esteban F, Redondo M. The substance P/NK-1 receptor system: NK-1 receptor antagonists as anti-cancer drugs. J Biosci. 2015;40(2):441–463. doi:10.1007/s12038-015-9530-8
17. Muñoz M, Coveñas R. The neurokinin-1 receptor antagonist aprepitant, a new drug for the treatment of hematological malignancies: focus on acute myeloid leukemia. J Clin Med. 2020;9(6):1659. doi:10.3390/jcm9061659
18. Muñoz M, Rosso M. The NK-1 receptor antagonist aprepitant as a broad spectrum antitumor drug. Invest New Drugs. 2009;28(2):187–193. doi:10.1007/s10637-009-9218-8
19. Priyadarsini KI. The chemistry of curcumin: from extraction to therapeutic agent. Molecules. 2014;19(12):20091–20112. doi:10.3390/molecules191220091
20. Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4(6):807–818. doi:10.1021/mp700113r
21. Zhang X, Dai F, Chen J, et al. Antitumor effect of curcumin liposome after transcatheter arterial embolization in VX2 rabbits. Cancer Biol Ther. 2019;20(5):642–652. doi:10.1080/15384047.2018.1550567
22. Giordano A, Tommonaro G. Curcumin and cancer. Nutrients. 2019;11(10):2376. doi:10.3390/nu11102376
23. Hanafy NAN, Leporatti S, El-Kemary M. Mucoadhesive curcumin crosslinked carboxy methyl cellulose might increase inhibitory efficiency for liver cancer treatment. Mater Sci Eng C Mater Biol Appl. 2020;116:111119. doi:10.1016/j.msec.2020.111119
24. Hanafy NAN. Optimally designed theranostic system based folic acids and chitosan as a promising mucoadhesive delivery system for encapsulating curcumin LbL nano-template against invasiveness of breast cancer. Int J Biol Macromol. 2021;182:1981–1993. doi:10.1016/j.ijbiomac.2021.05.149
25. Pérez-Herrero E, Fernández-Medarde A. Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015;93:52–79. doi:10.1016/j.ejpb.2015.03.018
26. Hanafy N, Leporatti S, El-Kemary M. Mucoadhesive hydrogel nanoparticles as smart biomedical drug delivery system. Appl Sci. 2019;9(5):825. doi:10.3390/app9050825
27. Fan Y, Marioli M, Zhang K. Analytical characterization of liposomes and other lipid nanoparticles for drug delivery. J Pharm Biomed Anal. 2021;192:113642. doi:10.1016/j.jpba.2020.113642
28. Kiaie SH, Mojarad-Jabali S, Khaleseh F, et al. Axial pharmaceutical properties of liposome in cancer therapy: recent advances and perspectives. Int J Pharm. 2020;581:119269. doi:10.1016/j.ijpharm.2020.119269
29. Liu C, Liu X-N, Wang G-L, et al. A dual-mediated liposomal drug delivery system targeting the brain: rational construction, integrity evaluation across the blood–brain barrier, and the transporting mechanism to glioma cells. Int J Nanomedicine. 2017;12:2407–2425. doi:10.2147/ijn.S131367
30. Lakkadwala S, Dos Santos Rodrigues B, Sun C, Singh J. Dual functionalized liposomes for efficient co-delivery of anti-cancer chemotherapeutics for the treatment of glioblastoma. J Control Release. 2019;307:247–260. doi:10.1016/j.jconrel.2019.06.033
31. Jiang H, Li ZP, Tian GX, et al. Liver-targeted liposomes for codelivery of curcumin and combretastatin A4 phosphate: preparation, characterization, and antitumor effects. Int J Nanomedicine. 2019;14:1789–1804. doi:10.2147/ijn.S188971
32. Myojin Y, Hikita H, Sugiyama M, et al. Hepatic stellate cells in hepatocellular carcinoma promote tumor growth via growth differentiation factor 15 production. Gastroenterology. 2021;160(5):1741–1754.e1716. doi:10.1053/j.gastro.2020.12.015
33. Castilho-Fernandes A, de Almeida DC, Fontes AM, et al. Human hepatic stellate cell line (LX-2) exhibits characteristics of bone marrow-derived mesenchymal stem cells. Exp Mol Pathol. 2011;91(3):664–672. doi:10.1016/j.yexmp.2011.09.002
34. Luo J, Zhang P, Zhao T, et al. Golgi apparatus-targeted chondroitin-modified nanomicelles suppress hepatic stellate cell activation for the management of liver fibrosis. ACS nano. 2019;13(4):3910–3923. doi:10.1021/acsnano.8b06924
35. Zhang H. Thin-film hydration followed by extrusion method for liposome preparation. Methods Mol Biol. 2017;1522:17–22. doi:10.1007/978-1-4939-6591-5_2
36. Baghbani F, Moztarzadeh F. Bypassing multidrug resistant ovarian cancer using ultrasound responsive doxorubicin/curcumin co-deliver alginate nanodroplets. Colloids Surf B Biointerfaces. 2017;153:132–140. doi:10.1016/j.colsurfb.2017.01.051
37. Sun Y, Lu J, Yan D, Shen L, Hu H, Chen D. Cellular uptake mechanism and clearance kinetics of fluorescence-labeled glycyrrhetinic acid and glycyrrhetinic acid-modified liposome in hepatocellular carcinoma cells. Environ Toxicol Pharmacol. 2017;53:46–56. doi:10.1016/j.etap.2017.05.003
38. Sun Y, Li X, Zhang L, et al. Cell permeable NBD peptide-modified liposomes by hyaluronic acid coating for the synergistic targeted therapy of metastatic inflammatory breast cancer. Mol Pharm. 2019;16(3):1140–1155. doi:10.1021/acs.molpharmaceut.8b01123
39. Sauraj VK, Kumar B, Priyadarshi R, et al. Redox responsive xylan-SS-curcumin prodrug nanoparticles for dual drug delivery in cancer therapy. Mater Sci Eng C Mater Biol Appl. 2020;107:110356. doi:10.1016/j.msec.2019.110356
40. Yang Y, Liu X, Ma W, et al. Light-activatable liposomes for repetitive on-demand drug release and immunopotentiation in hypoxic tumor therapy. Biomaterials. 2021;265:120456. doi:10.1016/j.biomaterials.2020.120456
41. Wang H, Ellipilli S, Lee WJ, et al. Multivalent rubber-like RNA nanoparticles for targeted co-delivery of paclitaxel and MiRNA to silence the drug efflux transporter and liver cancer drug resistance. J Control Release. 2021;330:173–184. doi:10.1016/j.jconrel.2020.12.007
42. Erin N, Grahovac J, Brozovic A, Efferth T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist Updat. 2020;53:100715. doi:10.1016/j.drup.2020.100715
43. Aponte-López A, Muñoz-Cruz S. Mast cells in the tumor microenvironment. Adv Exp Med Biol. 2020;1273:159–173. doi:10.1007/978-3-030-49270-0_9
44. Qiu N, Liu Y, Liu Q, et al. Celastrol nanoemulsion induces immunogenicity and downregulates PD-L1 to boost abscopal effect in melanoma therapy. Biomaterials. 2021;269:120604. doi:10.1016/j.biomaterials.2020.120604
45. Kubo N, Araki K, Kuwano H, Shirabe K. Cancer-associated fibroblasts in hepatocellular carcinoma. World J Gastroenterol. 2016;22(30):6841. doi:10.3748/wjg.v22.i30.6841
46. Wan Y, Meng F, Wu N, et al. Substance P increases liver fibrosis by differential changes in senescence of cholangiocytes and hepatic stellate cells. Hepatology. 2017;66(2):528–541. doi:10.1002/hep.29138
47. Grässel S, Muschter D. Peripheral nerve fibers and their neurotransmitters in osteoarthritis pathology. Int J Mol Sci. 2017;18(5):931. doi:10.3390/ijms18050931
48. Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20(4):840. doi:10.3390/ijms20040840
49. Sakai M, Yoshimura R. Mechanotransduction-targeting drugs attenuate stiffness-induced hepatic stellate cell activation in vitro. Biol Pharm Bull. 2021;44(3):416–421. doi:10.1248/bpb.b20-00815
50. Musetti S, Huang L. Nanoparticle-mediated remodeling of the tumor microenvironment to enhance immunotherapy. ACS nano. 2018;12(12):11740–11755. doi:10.1021/acsnano.8b05893
51. Liu M, Song W, Huang L. Drug delivery systems targeting tumor-associated fibroblasts for cancer immunotherapy. Cancer Lett. 2019;448:31–39. doi:10.1016/j.canlet.2019.01.032
52. Sun W, Fu S, Wu S, Tu R. Growing evidence of exosomal MicroRNA-related metastasis of hepatocellular carcinoma. Biomed Res Int. 2020;2020:4501454. doi:10.1155/2020/4501454
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.