")
Back to Journals » International Medical Case Reports Journal » Volume 10
Fusion of lower limbs with severe urogenital malformation in a newborn, a rare congenital clinical syndrome: case report
Authors Al Hadhoud F , Kamal AH, Al Anjari A, Diejomaoh MF
Received 6 April 2017
Accepted for publication 4 August 2017
Published 21 September 2017 Volume 2017:10 Pages 313—317
DOI https://doi.org/10.2147/IMCRJ.S139067
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Fatemah Al Hadhoud,1 Abeer H Kamal,1 Abdulmohsen Al Anjari,1 Michael FE Diejomaoh1,2
1Department of Obstetrics and Gynecology, Maternity Hospital, 2Department of Obstetrics and Gynecology, Faculty of Medicine, Kuwait University, Kuwait City, Kuwait
Background: Fused lower limbs combined with severe urogenital malformation, also known as sirenomelia, is a rare congenital clinical syndrome. The etiology is unknown, and the outcome for the affected fetus is rather uncertain.
Case report: Mrs RB, a Kuwaiti woman primigravida, married to a non-consanguineous husband, had uneventful antenatal care in a private health service, until she was admitted to the Maternity Hospital, Kuwait, at 31 weeks of gestation with a 3-hour history of ruptured membranes. She had a negative family history for diabetes mellitus, and her past surgical/medical/gynecological history was noncontributory. General physical examination revealed a healthy parturient with normal vital signs, clear lungs and normal heart sounds. Obstetric examination revealed a fundal height compatible with the gestational age; there was a single living fetus in breech presentation; she was not in labor. The mother was managed conservatively with antibiotics and dexamethasone injections. Labor ensued later, progressing rapidly to the second stage. Assisted breech delivery was performed, and a live baby, birth weight 1570 g and Apgar score 3/9, was delivered. The neonate had multiple congenital abnormalities, including fusion of both lower limbs, ambiguous genitalia, cloacal anomaly and tracheoesophageal fistula. The neonate was admitted to the neonatal intensive care unit, fully investigated, managed medically and surgically and eventually discharged home after a hospital stay of 123 days for further management.
Conclusion: A case of peculiar fetal anomaly called sirenomelia, the first case in Kuwait, has been presented. Survival of such babies requires costly management with average results.
Keywords: sirenomelia, premature labor, conservative management, outcome
Introduction
Sirenomelia, a rare congenital malformation, has been reported as being frequently fatal.1 The low incidence of 1 in 100,000 pregnancies2 has been reported in some studies. Cases have been reported from various ethnic groups all over the world.3 The malformation has been described in monozygotic twins, and it has been reported as having a male preference in presentation with a report of sex ratio of 2.7:1.4 The most predominant feature of this malformation is the complete or partial fusion of both lower limbs into what appears as a single lower limb.3 Other synonyms describing the disorder include monopodia, sympodia, synmelia and sympus, with the fusion of the lower limbs giving an appearance that looks like a mermaid of Greek mythology.4 Multiple visceral anomalies have been described in the fetuses presenting with these limb malformations, and the entire syndrome is not compatible with life,3,5 although a few cases of surviving infants with this malformation have been described in the English literature.2,6
The first case of a fetus with fused lower limbs with severe urogenital malformation was recently diagnosed in Kuwait. As far as we are aware, there has been no such clinical case in Kuwait, and this is the first such case report in Kuwait. In this case report, we present our findings and compare these with similar cases previously reported.
Case report
Mrs RB, a 25-year-old Kuwaiti woman primigravida, married for 18 months, presented to the Maternity Hospital, Kuwait, at 31 weeks of gestation with rupture of membranes of 3 hours duration. This pregnancy was spontaneous and resulted from a non-consanguineous marriage and was followed up in the private health care service. The antenatal period had been uneventful until this emergency presentation. There was no evidence of antenatal ultrasonography in the private health services as the patient did not present any information or reports of such an investigation. She was not known to have diabetes mellitus. Her past surgical/medical/gynecological history was not contributory.
At admission, the patient was calm, afebrile and the vital signs were normal. There was no abnormality in the respiratory and cardiovascular systems. Obstetric examination confirmed a 31 weeks of gestation, the fetus in breech presentation and the presence of normal fetal heart sounds. She was managed conservatively, and she was placed on prophylactic antibiotics. A course of dexamethasone injections was given, and she was transferred to the antenatal ward. After 8 hours, the patient was readmitted to the labor ward complaining of labor pains, and pelvic examination revealed that the cervix was fully dilated with a frank breech presentation at station 1 cm below the ischial spine. An assisted breech delivery of a premature baby, birth weight 1570 g, was conducted. The Apgar scores of 3 and 9 were recorded, and an endotracheal tube was inserted into the baby.
The baby was discovered to have the following abnormalities:
- fusion of both lower limbs (Figure 1);
- ambiguous genitalia (Figure 2);
- cloacal anomaly (Figure 3) and
- tracheoesophageal fistula with atresia
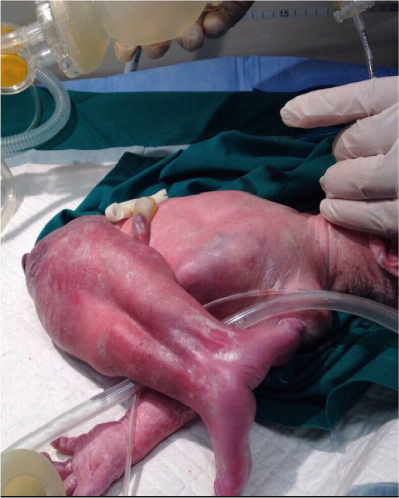 | Figure 1 Fused lower limbs. Note: The fused lower limbs are demonstrated, and they appear in a rather extended abnormal position in relationship to the vertebral/spinal column. |
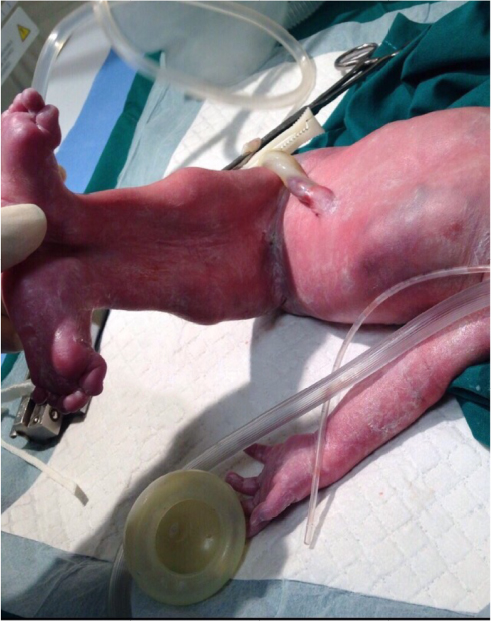 | Figure 2 Ambiguous genitalia. Notes: The fused lower limbs are again demonstrated with the baby in the supine position and are well aligned to the trunk of the baby. The genitalia are ambiguous. |
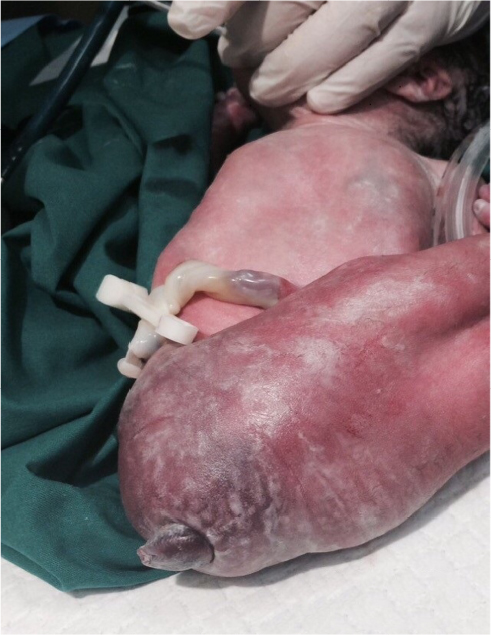 | Figure 3 Cloacal anomaly. Note: The abnormal cloacal orifice is displayed close to the location where anorectal features should have been displayed. |
Active resuscitation of the new born was undertaken, the baby was then transferred to the neonatal intensive care unit. Fluorescent in situ hybridization (FISH) study was conducted on the baby, and it revealed 46 XX chromosome patterns with no numerical or structural abnormalities. Skeletal survey showed that the baby had two femoral bones, two tibias, two fibulas with sacral segmentation defect with 13 bilateral ribs and a normal vertebrae. The baby passed urine through a single partial opening over the sacral area.
Exploratory laparotomy was performed, which showed distal jejunal atresia with dilated proximal bowel of 10–15 cm. The distal colon was atretic. The esophagus was ligated, 15 cm of dilated proximal loop was resected and jejunostomy along with gastrostomy was performed. Renal ultrasound showed left polycystic kidney, and the right kidney was not visualized (query for right ectopic kidney). No magnetic resonance imaging (MRI) was performed on the baby. The baby was discharged after 123 days to continue medical treatment in the USA.
There was a discussion between two of the authors (FAH and AAA) and the parents about the authors’ intention to make a report of the case using all the information provided and the illustrations/pictures/X-rays of the neonate, and the parents gave their written informed consent for such a publication effort.
Discussion
Complete or partial fusion of the lower limbs in a fetus, also described as sirenomelia, is a rare congenital clinical syndrome in which there may be other severe fetal anomalies. The anomalies that have been described in various previous reports include bilateral renal agenesis, complete or partial agenesis of genitourinary system, imperforate anus, absence of genitalia or ambiguous external genitalia, single umbilical artery, lung hypoplasia and gastrointestinal, vertebral and cardiac anomalies.5–7
In the current case report, the anomalies present in the fetus were genitourinary and gastroenterological anomalies, imperforate anus and ambiguous external genitalia.
Various theories have been proposed to try to explain the etiology of sirenomelia. Some authors have described an embryonic insult to the caudal mesoderm between 28 and 32 days of gestation leading to vascular hypoperfusion, which might explain the fusion process in the limbs.4,5,7–9 Uncontrolled maternal diabetes, especially in early first trimester of pregnancy, which has been linked with sacral agenesis, limb malformations and other anomalies in the fetus, has been described as an important risk factor for caudal malformations in general.3,9,10 However, since only ~0.5%–3.7% of sirenomelia cases have been reported in diabetic mothers,7 the etiological association between maternal diabetes and sirenomelia has been described as weak.7 Our patient was not diabetic. The sacral agenesis referred earlier can be described as being synonymous with caudal regression syndrome, although it could actually be described as part of caudal regression syndrome. Poor diabetic control has been associated with caudal regression syndrome.10 Although the features of sacral agenesis and caudal regression syndrome may be similar to those reported in sirenomelia and the former may be considered in the differential diagnosis of sirenomelia, there is no agreement on the fact of linkage of etiology between the two entities.9,10 Genetic disorders, teratogenic factors including drugs and toxic diseases like infections have all been associated with the etiology of caudal regression syndrome and sirenomelia;9,10 the exact mechanisms are not fully elucidated. Although Lynch and Wright6 described an autosomal form of caudal dysgenesis, no chromosomal abnormalities have been reported in cases of sirenomelia and the fetal anomaly does not recur in families.7 This was a reassuring information for our patient, in whose baby, there was no demonstrable chromosomal anomaly, and this could serve as a counseling guideline for mothers who have fetuses with this unpleasant and unsettling anomaly.
There is still a long-standing debate to establish the complex etiology of sirenomelia. We do not want to dwell at length on all the ramifications of the etiology of sirenomelia in this manuscript, but we highlight some specific areas. In a recent review,9 the authors discussed the multiple etiological factors associated with sirenomelia, such as diabetes mellitus, monozygotic twinning and teratogenicity already referred to earlier, “embryonic insult” and the vascular steal theory previously referred to, as well as “defective blastogenesis”; the authors9 expressed the view that sirenomelia may be due to a primary defect in blastogenesis. The vascular steal theory will be discussed further. Various theories have been postulated to explain the origin of sirenomelia, including a vascular steal theory.7,8,11 This theory, as described by some authors, suggests that there is shunting of blood toward the placenta via an abnormal abdominal artery arising from high up in the aorta. Such shunting, as described, leaves the caudal part of the embryo poorly perfused. This will ultimately lead to hypoplasia of the vasculature distal to the artery, leading to nutritional deficiency of the caudal half of the body.4,7 Such altered blood perfusion could lead to complete or incomplete agenesis of the caudal structures, such as the kidneys, sacrum, and lower portions of the digestive system, except the gonads, which are usually intra-abdominal. There could also be vertebral dysgenesis, lower limb atrophy and variable lower limb fusion.5–7,11 The single umbilical artery in our patient is in favor of this theory. Jaiyesimi et al11 reported a case of sirenomelia in which this vitelline artery steal disorder was not present, therefore suggesting that factors other than vitelline artery steal disorder could be responsible for the malformation, sirenomelia, in human beings. This observation is similar to the findings in the current case report.
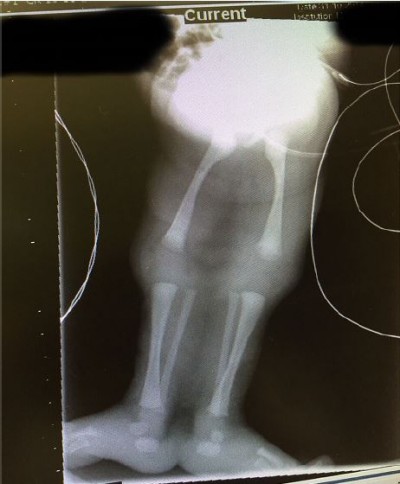
The sirenomelia sequence has been classified into seven types7,8 as shown in Table 1. The radiographs of our patient, (Figure 4), indicate that we should classify our patient as type 1.
 | Table 1 Classification of the sirenomelic sequence Note: Copyright ©1987. S Karger AG, Basel. Reproduced from Stocker JT, Heifetz SA. Sirenomelia. A morphological study of 33 cases and review of the literature. Perspect Pediatr Pathol. 1987;10:7–50.8 |
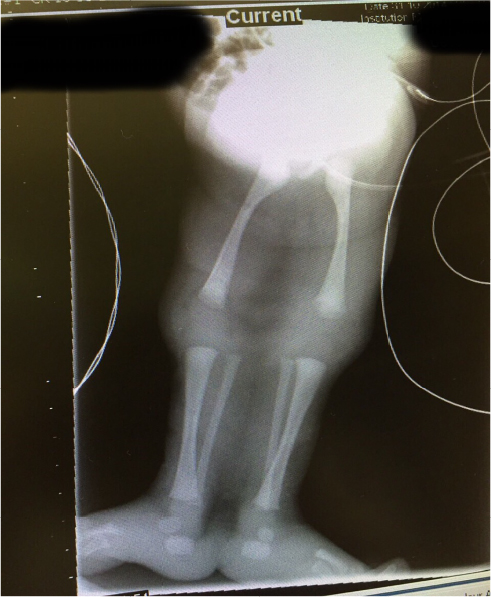 | Figure 4 Radiographs of the limbs of the baby. Notes: All the thigh and leg bones are clearly displayed in the radiograph. The view of X-ray available did not clearly display the chest and vertebral and sacral structures. |
The disorder of sirenomelia is associated with a rather poor prognosis.9 The chances of survival of these fetuses as described by some authors are largely dependent on the extent and may be severity of associated visceral anomalies, especially disorders such as obstructive renal failure due to renal agenesis/dysgenesis.4,5,10 In the cases where antenatal diagnosis may be offered to the patient and a firm diagnosis is made, a voluntary termination of pregnancy is advisable to avoid the mental and psychological disorders for the patients and their family. The decision for termination of pregnancy however depends on the gestational age of the pregnancy at which the diagnosis is made, the severity of the malformations and of course the decision of the patients7 and their religious beliefs. In our case, the offer of voluntary termination was not available since the termination of pregnancy for these types of malformations, which may be compatible with life, albeit, not the best quality of life, is not allowed in Kuwait, on religious grounds, beyond 17 weeks of gestation. The gestational age of our patient at the first presentation in the private clinic could not be ascertained, and there was no evidence of antenatal ultrasonography in our patient who had utilized private services before her presentation as an emergency to our services at Maternity Hospital, Kuwait. Diagnosis of sacrococcygeal dysgenesis, an important component of sirenomelia, could be made by antenatal/prenatal ultrasonography, as early as 9 weeks in the first trimester by transvaginal ultrasonography, although more commonly in the second and third trimesters of pregnancy.9,10 If such an investigative tool had been available to our patient, it could have contributed to the management of our patient depending on the gestational age at diagnosis, because she could have been informed about the presence of malformations and thus enabled to make a proper decision on the subsequent management after appropriate counseling.
Some recent reports have indicated that ~50% of these infants with sirenomelia are born alive after 8–9 months of gestation.7 However, many of these neonates die within 5 days of life.7 Our patient delivered before the gestational age of 8–9 months, and the baby survived for >123 days with some specialized medical and surgical management. We could not confirm MRI examination in our neonate, an important diagnostic tool in such neonates.10 The definitive management of sirenomelia is difficult, time-consuming, complex and expensive, and a satisfactory outcome is not always guaranteed.2,7 The main therapeutic modality involves surgical and medical management aimed mainly at carrying out ameliorative surgery and maintaining satisfactory systemic function. Initial surgery, mainly in the gastrointestinal tract, had been performed in the neonate in our case port. We anticipated that further surgery may have been performed on the baby on arrival to the USA, although, such information was not available in our records. Surgery and medical facilities are available in the developed world, albeit at high costs, which could lead to survival of some sirenomelic babies. The results from such interventions are usually limited as only mediocre results have been achieved. Some reports9,12 have documented surviving sirenomelic fetuses where costly conservative management measures have been implemented on visceral anomalies discovered in the fetuses.
Conclusion
Sirenomelia is a rare congenital malformation syndrome with severe urogenital abnormality and many other visceral anomalies, which are incompatible with life; there may be a low chance of survival with optimal medical and surgical management. The etiology remains unknown.
Prenatal diagnosis should be offered in such cases, and termination is recommended.
With the availability of modern technology in developed countries, some optimal management could be offered, which could lead to some enhanced, though uncertain, quality of life for a few surviving fetuses. In our report, the neonate survived for 123 days and was offered further treatment in the USA.
Disclosure
The authors report no conflicts of interest in this work.
References
Taori KB, Mitra K, Ghonga NP, et al. Sirenomelia sequence (mermaid): report of three cases. Indian J Radiol Imaging. 2002;12(3):399–401. | ||
Martinez-Frias ML, Garcia A, Bermejo E. Cyclopia and sirenomelia in a live born infant. J Med Genet. 1998;35(3):263–264. | ||
Garrido-Allepuz C, Haro E, González-Lamuño D, Martínez-Frías ML, Bertocchini F, Ros MA. A clinical and experimental overview of sirenomelia: insight into the mechanisms of congenital limb malformations. Dis Model Mech. 2011;4(3):289–299. | ||
Valenzano M, Paoletti R, Rossi A, Farinini D, Garlaschi G, Fulcheri E. Sirenomelia. Pathological features, antenatal ultrasonographic clues, and a review of current embryogenic theories. Hum Reprod Update. 1999;5(1):82–86. | ||
Contu R, Zoppi MA, Axiana C, Ibba RM, Monni G. First trimester diagnosis of sirenomelia by 2D and 3D ultrasound. Fetal Diagn Ther. 2009;26(1):41–44. | ||
Lynch SA, Wright C. Sirenomelia, limb reduction defects, cardiovascular malformation, renal agenesis in an infant born to a diabetic mother. Clin Dysmorphol. 1997;6(1):75–80. | ||
Fadhlaoui A, Khrouf M, Gaigi S, Zhioua F, Chaker A. The sirenomelia sequence a case history. Clin Med Insights Case Rep. 2010;3:41–49. | ||
Stocker JT, Heifetz SA. Sirenomelia. A morphological study of 33 cases and review of the literature. Perspect Pediatr Pathol. 1987;10:7–50. | ||
Boer LL, Morava E, Klein WM, Schepens-Franke AN, Oostra RJ. Sirenomelia: a multi-systemic polytopic field defect with ongoing controversies. Birth Defects Res. 2017;109(10):791–804. | ||
Kumar Y, Gupta N, Hooda K, et al. Caudal regression syndrome: a case series of a rare congenital anomaly. Pol J Radiol. 2017;82:188–192. | ||
Jaiyesimi F, Gomathinayagam T, Dixit A, Amer M. Sirenomelia without vitelline artery steal. Ann Saudi Med. 1998;18(6):542–544. | ||
Messineo A, Innocenti M, Gelli R, Pancani s, Lo Piccolo R, Martin A. Multidisciplinary surgical approach to a surviving infant with sirenomelia. Pediatrics. 2006;118(1);e220–e223. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.