")
Back to Archived Journals » Vaccine: Development and Therapy » Volume 4
Vaccines for metabolic diseases: current perspectives
Authors Morais T, Andrade S, Pereira S, Monteiro M
Received 13 May 2014
Accepted for publication 23 June 2014
Published 2 September 2014 Volume 2014:4 Pages 55—72
DOI https://doi.org/10.2147/VDT.S47623
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Tiago Morais, Sara Andrade, Sofia S Pereira, Mariana P Monteiro
Department of Anatomy, Unit for Multidisciplinary Biomedical Research, Institute for Biomedical Sciences Abel Salazar, University of Porto, Porto, Portugal
Abstract: Several metabolic disorders, such as diabetes, hypertension, dyslipidemia, and obesity, represent significant risk factors for cardiovascular disease, which is the leading cause of morbidity and mortality among adult populations in western societies. Understandably, these chronic disorders have now replaced infectious diseases as the most important public health problem and economic burden to society in most countries. Treatment of metabolic risk factors in order to prevent cardiovascular disease requires an enduring approach with multiple drugs, which can be associated with considerable costs, side effects, and a low rate of therapeutic compliance due to lack of symptoms until later stages of the disease. Since vaccines have proven to be a powerful and effective approach to preventing infectious diseases, attempts to expand the therapeutic use of vaccines into the context of highly prevalent diseases has been attracting increased research interest. Vaccination strategies for chronic diseases in particular are an exciting area of research, with new treatment targets and strategies on the horizon. This review discusses the development of innovative therapeutic agents, focusing on the use of molecular vaccines for the treatment of common and highly prevalent chronic metabolic disorders, ie, diabetes, hypertension, dyslipidemia, and obesity.
Keywords: vaccines, diabetes, hypertension, dyslipidemia, obesity
Introduction
The rising prevalence of chronic noninfectious diseases, in particular cardiovascular disease, is well known and a major public health problem in many western countries, being associated with a significant increase in morbidity and mortality.1,2 In addition, chronic metabolic diseases have replaced infectious diseases as the major economic burden to society in most countries throughout the world.3 This phenomenon has been related not only to changes in lifestyle and rates of population ageing, but also to the increasing prevalence of predisposing risk factors, which include diabetes mellitus, hypertension, dyslipidemia, and obesity. Moreover, these risk factors frequently occur as a cluster of clinical findings known as the metabolic syndrome, a complex clinical condition attributed to insulin resistance that includes visceral or abdominal obesity, glucose intolerance, hypertension, and dyslipidemia and is characterized by elevated triglycerides and decreased high-density lipoprotein (HDL) cholesterol,4,5 an entity that has been associated with an increased risk of type 2 diabetes, cardiovascular disease, and certain types of cancer.4,6–8
Treatment of metabolic risk factors in order to prevent cardiovascular disease requires a chronic long-term approach, which often involves multiple pharmacological agents that can be associated with considerable costs, side effects, and lack of adherence, which can limit their efficacy.9
Over the years, vaccines have proven to be a powerful and effective approach to preventing infectious diseases, and attempts to expand the use of vaccines into a therapeutic context have been of increasing interest, particularly for highly prevalent diseases and with an high impact on lifestyle.3 Therefore, the possibility of using vaccination strategies for prevention and treatment of chronic metabolic diseases is an exciting scientific perspective and burgeoning research field, with new treatment targets on the horizon. This review discusses the development of innovative therapeutic agents and focuses on the use of molecular vaccines for the treatment of common and highly prevalent chronic metabolic disorders, ie, diabetes, hypertension, dyslipidemia, and obesity, with the aim of preventing the associated risk of cardiovascular disease.
Vaccines for diabetes mellitus
Diabetes is defined as a chronic condition characterized by hyperglycemia and includes several types of disease,10 among which the most common are type 1 (T1DM), type 2 (T2DM), and gestational diabetes. The prevalence of the disease, T2DM in particular, is expanding, and is expected to affect 592 million people worldwide by 2035.11 The pathogenesis of T1DM and T2DM differs, since T1DM is characterized by insulin deficiency due to autoimmune damage to pancreatic beta cells, whereas the predominant defects in gestational and T2DM are insulin resistance and secondary beta cell failure, which are usually associated with overweight, a sedentary lifestyle, and poor eating habits.10,12,13 Due to the high prevalence of the disease, the development of a therapeutic vaccine represents an attractive and challenging treatment approach, in particular for immune-mediated subtypes of diabetes, eg, T1DM and latent autoimmune diabetes of adults, which is immunologically similar to T1DM but with a protracted clinical evolution.
Vaccines for autoimmune-mediated diabetes
The pathogenesis of autoimmune diabetes is characterized by pancreatic beta cell loss due to direct destruction by CD8+ cytotoxic T-cells and macrophages, and is mediated through several cytokines, including tumor necrosis factor alpha, interleukin (IL)-1β, and interferon gamma.14 This phenomenon is preceded by the presentation of specific beta cell antigens by dendritic cells or macrophages to CD4+ cells that activate the CD8+ cluster.15 The selective beta cell loss leads to a predominance of glucagon-secreting alpha cells, with the end result of absolute insulin deficiency and secondary hyperglucagonemia.16 Furthermore, the role of regulatory T-cells (Tregs) is thought to be impaired and to contribute to deregulation of the immune system. Given that individuals born with a mutation of the gene that encodes an essential Treg transcription factor, Foxp3, develop T1DM,17 and a gene polymorphism in the IL-2/IL-2RA pathway that is fundamental for maintaining and expanding a functional Treg population has also been found in T1DM individuals,18 a genetic individual propensity for development of T1DM is probable.19 The individual genotype does not by itself explain the rise in prevalence of the disease, suggesting that environmental factors must also be involved.20 Among the several proposed triggers are included cow’s milk proteins, viral infections or toxins,21 and the most commonly referred possibilities as initiating antigen comprise the insulinoma-associated protein tyrosine phosphatase antibody (IA2-A), glutamic acid decarboxylase 65 (GAD 65)22 and insulin/pro-insulin.23 Therefore, amongst the several vaccination approaches for T1DM, some of these specific antigens have been used as a strategy to provoke antigen-dependent modulation of the immune system and to reduce the immune response against a specific target, a strategy similar to that widely used in the clinical field of allergy. Another strategy involves use of DNA to downregulate production of the endogenous antigen. Antigen-dependent immune modulation can be accomplished by either repeated injection of the antigen over a long period of time in order to achieve “allergen desensitization”24 or a more recent approach using a tailored epitope of the target antigen that has the advantage of avoiding anaphylactic reactions.25 These vaccination approaches rely on production of Tregs and increase the release of potent anti-inflammatory cytokines, such as IL-10 or transforming growth factor beta, in response to repeated administration of an antigen; unfortunately, when the regulatory mechanism of Tregs is defective, such as in T1DM, these approaches can be ineffective.26 Nevertheless, nonclassical fully functional Tregs have recently been found to be present in the pancreas, and can be activated and help to modulate the immunologic response.27 Herein, we review the antigen targets most commonly used for development of prophylactic and therapeutic vaccines for diabetes that have been trialed with varying degrees of success.
Vaccines targeting proinsulin/insulin
Insulin has been used as an antigen target in several trials for the development of prophylactic and therapeutic vaccines for autoimmune diabetes. A promising proof-of-concept vaccination approach for diabetes has included use of combined nasal proinsulin therapy with anti-CD3. In murine diabetic models, this approach has been demonstrated to induce strong expansion of the pool of Tregs, as well as an increase in IL-10, transforming growth factor beta, and IL-4, which are surrogate markers of a successful CD8+ suppressing response. The efficacy of this combined therapy may be explained by the decrease in number of aggressive T-cells brought about by anti-CD3+, allowing expansion of Tregs induced by proinsulin.28
Based on this principle, a large clinical trial of a prophylactic vaccine for diabetes has been conducted, and consisted of daily intranasal administration of insulin to 264 children with an HLA genotype associated with a greater risk of developing T1DM. After a median follow-up of 1.8 years, this strategy did not prove to be able to decrease the incidence of T1DM, since 56 of the “vaccinated” children developed the T1DM phenotype versus 63 children in the placebo group.29 The spacing of the insulin dose with daily administration has been hypothesized to be the main reason for the failure of that trial, due to an excess of antigen exposure that might not favor activation of Tregs or even lead to their death, as previously described.30
In contrast, most of the proinsulin-based and insulin-based vaccination strategies were intended to develop therapeutic vaccines for T1DM. In a Phase I clinical trial that consisted of administering three monthly intradermal doses of proinsulin epitopes (C19-A3) to patients with longstanding disease, the vaccine was well tolerated. However, IL-10 levels that increased 3 months after vaccination decreased in the ensuing months, reaching basal levels after 6 months.31 A specific insulin B (insulin beta chain) antigen, NBI-6024, injected subcutaneously in T1DM patients at monthly doses for 24 months, was not able to modify levels of C-peptide, which is cosecreted with insulin and used as a surrogate marker for beta cell function.32 In contrast, a Phase I trial that used insulin B as an antigen administered with incomplete Freund’s adjuvant as a single intramuscular dose demonstrated this treatment to be very well tolerated, and although there were no significant variations in C-peptide levels, insulin B chain-specific CD4+ T cells in vaccinated subjects acquired phenotypic and functional characteristics of Tregs for up to 24 months.33
Newer strategies for antigen delivery using modified bacteria to produce the antigen are being tested in mice. Lactococcus lactis has been engineered to produce whole proinsulin along with IL-10, which was complemented with a low systemic dose of anti-CD3+. This approach was able to increase Tregs in pancreatic tissue and revert the T1DM phenotype in nonobese diabetic (NOD) mice while retaining the capacity to respond to other antigens, showing specificity and demonstrating that does not induce excessive immunosuppression.34 Attenuated live Salmonella producing preproinsulin fused with the secretory effector protein of pathogenicity island-2 achieved the goal of having the antigen only expressed inside the host immune cells and then translocated to the cell cytosol, while the same approach was also used to deliver the gene for transforming growth factor beta. Covaccination with three weekly oral doses of these two types of engineered bacteria significantly reduced the rate of development of diabetes in NOD mice, improving the response to glucose challenge, preserving pancreatic beta cell mass, and reducing the severity of insulitis when compared with controls and autoantigen administration alone. This vaccination protocol also increased circulating levels of IL-10 for up to 4 weeks post-vaccination and IL2 for 12 weeks post-vaccination, but had no effect on production of proinflammatory cytokines, ie, IL6, IL12 (p70), IL17, and interferon gamma. Although highly successful in responding mice, the nonresponding animals showed a significant increase in IL12, which increases the cytotoxic potential of CD8+ cells and merits further investigation.35
Use of DNA plasmids is another strategy for modulation of antigen expression. This approach has been used in a Phase I clinical trial in which 80 patients with T1DM received weekly intramuscular doses of plasmid encoding proinsulin (BHT-3021) in four distinct concentrations. BHT-3021 was well tolerated, and after the first administration, C-peptide levels were improved for up to 6 months; however, the response was not homogeneous across all individuals, and this could be linked to individual autoantibody levels, HLA genotype, or time since diagnosis. BHT-3021 was also able to selectively decrease CD8+ proinsulin reactive cells but not other T-cells, but unfortunately this effect was only observed during treatment and reverted to basal levels 3 months after the end of administration.36 Although encouraging, there is a need to optimize this particular strategy in order to improve the results in terms of achieving a more effective and durable response37 (Table 1).
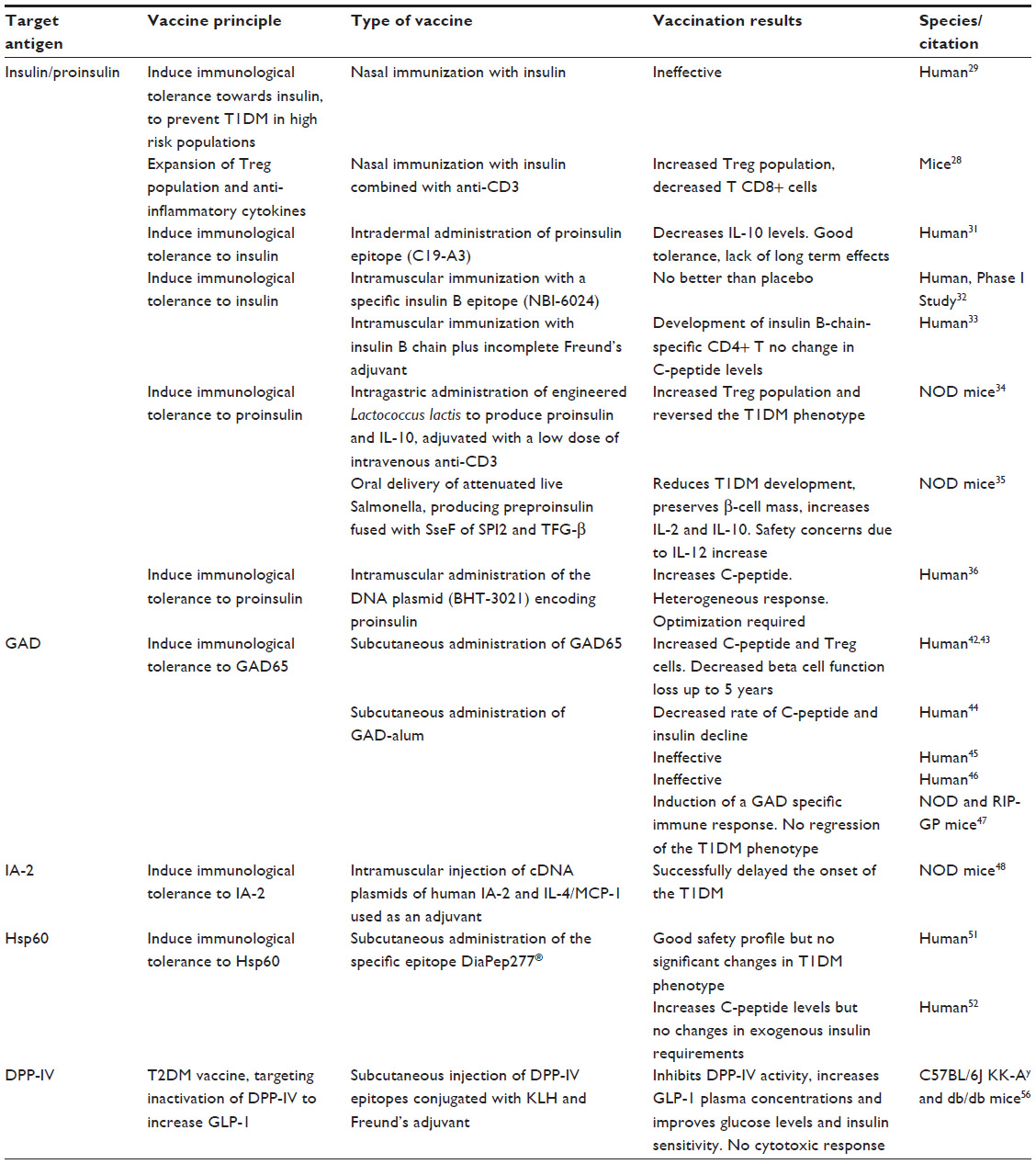 | Table 1 Vaccination approaches for diabetes |
Vaccines targeting glutamic acid decarboxylase
Glutamic acid decarboxylase (GAD) or glutamate decarboxylase, the enzyme that catalyzes the decarboxylation of glutamate to γ-aminobutyric acid, and more specifically GAD65, the only isoform of the enzyme present in pancreatic beta cells, is another target antigen that has been widely used in the field of diabetes vaccination.38 Although the role of this enzyme in the pancreas is still unclear, it has been postulated that γ-aminobutyric acid may regulate the first phase of glucose-dependent insulin release.39 GAD is a promising antigen, since anti GAD65 autoantibodies usually precede development of T1DM and latent autoimmune diabetes of adults, while immune tolerance or antibody suppression has been shown to be very effective in preventing the T1DM phenotype in animal models.40,41 A recent Phase II clinical trial used an alum-formulated human recombinant GAD65 to test the effects of this immunomodulation approach. Forty-seven patients with latent autoimmune diabetes of adults were subcutaneously administered two antigen doses 4 weeks apart, with four rising doses (4, 20, 100, and 500 μg).42 At one of the tested concentrations, fasting C-peptide levels and the ratio of CD4+CD25+ cells to CD4+CD25− cells increased significantly up to 24 weeks42 and there was no decrease in beta cell function during 5 years of follow up, suggesting that this approach could be a promising one for developing a diabetes vaccine.43 Unfortunately, subsequent clinical trials with GAD-alum did not yield the same positive results. A trial designed to test the ability to reverse the T1DM phenotype enrolled 70 patients with recently diagnosed T1DM (ie, less than 18 months since the initial diagnosis) aged 11–18 years. Two subcutaneous injections of GAD-alum on days 1 and 30 elicited a specific immune response and were able to significantly decrease the rate of decline of C-peptide and insulin in the first 6 months, although these effects were lost over a period of 30 months.44 Unfortunately, these results could not be replicated in subsequent larger studies. In a trial involving 334 patients aged 10–20 years, the patients were randomized into three groups to receive either four doses of GAD-alum, two doses of GAD-alum followed by two of placebo, or four doses of placebo, with no differences in C-peptide decline or clinical improvement over 15 months.45 In another study conducted in 145 T1DM patients who were less than 100 days from diagnosis, this vaccine was unable to show any differences in C-peptide, glycated hemoglobin, or insulin requirements across the three groups for up to one year.46 Furthermore, a recent study performed in diabetic mice, NOD, and transgenic rat insulin promoter-glycoprotein (RIP-GP) mice, showed that the dose of GAD-alum used in human trials induces a GAD-alum Th2-specific response but fails to revert the T1DM phenotype47 (Table 1).
Vaccines targeting other antigens
Other antigen targets suggested for the development of T1DM vaccines include tyrosine phosphatase-related islet antigen 2 (IA-2) and heat shock protein 60 (HSP 60), which seems to be a far more promising antigen. To test the ability of the IA-2 vaccine to prevent the development of T1DM, a vaccine consisting of cDNA of human IA-2 and a plasmid expressing IL-4/monocyte chemoattractant protein-1 used as an adjuvant, these were intramuscularly coadministered to NOD mice, demonstrating that both the IA-2 plasmid alone or adjuvant combined, managed to delay the onset of the disease,48 although vaccines targeting IA-2 have not yet been reported in humans.
HSP60, or p277, is an immunomodulatory molecule49 that has been demonstrated to prevent low-dose streptozotocin-induced diabetes in mice.50 DiaPep277® is a 24-amino acid peptide derived from the 37–460 sequence of human HSP60, has been developed for subcutaneous administration, and is being tested in several Phase II clinical trials and Phase III studies, which are either underway or nearing completion. DiaPep277 has been tested in 40 adults and 40 children recently diagnosed with T1DM, who were given one of three doses of the peptide at four different time points. The safety profile of this vaccine was very good, with no adverse reactions, but there were no significant results with regard to protection against disease progression or insulin requirements.51 In another study that enrolled 35 males, DiaPep277 fared better, demonstrating a significant ability to maintain C-peptide levels for up to 6 months after year-long treatment and to reduce the need for exogenous insulin, although no changes in glycated hemoglobin were reported.52 Notwithstanding the lack of adverse effects, such conflicting results are troublesome since marginal increases in C-peptide that are not accompanied by a reduction in the exogenous insulin requirement are far from being a success (Table 1).
Vaccines for type 2 diabetes
T2DM is typically not an autoimmune disease, and has been connected primarily with lifestyle factors, such as obesity, and genetics.53 Glucagon-like peptide 1 (GLP-1), a peptide that plays an important role in glucose homeostasis, increasing insulin release and insulin sensitivity, is decreased in patients with T2DM.54 Since the half-life of GLP-1 is very short due to degradation by the enzyme dipeptidyl peptidase (DPP)-4, inhibition of this enzyme has become an important pharmacological target in T2DM therapy.55
More recently, a vaccine aimed at developing an immune response against DPP-4, thus raising endogenous GLP-1 levels, has been tested in diabetic animal models. This study showed that the vaccine was able to inhibit DPP-4 activity and significantly increase plasma GLP-1 levels, while improving both postprandial glucose and insulin sensitivity. Furthermore, in diabetic KK-Ay and db/db mice strains subjected to a high-fat diet, anti-DPP-4 vaccination was able to significantly improve insulin secretion and sensitivity without activating a cytotoxic Th1-type response from T-cells, thus increasing the safety of this method56 (Table 1).
Although progress has been made in the field of vaccination for diabetes, there is still a long way for research to go in order to achieve relevant clinical efficacy, given that success in animal models does not always translate well into humans and some promising methods may fail in larger clinical trials.57 Careful planning and rigorous thought must be applied when trying to extrapolate data from animals to humans. In addition, difficulties in modulating the immune system are highlighted by the fact that, for every successful trial, there is a similar trial that was unsuccessful.
Vaccines for dyslipidemia and cholesterol
Most of the cholesterol in plasma is transported by three lipoprotein classes, ie, very low-density lipoprotein (VLDL) cholesterol, low-density lipoprotein (LDL) cholesterol, and HDL cholesterol. There is good evidence that both VLDL and LDL are associated with promotion of atherogenesis, while HDL cholesterol is able to prevent this pathological process,58 which has a predominant role in the development of cardiovascular disease.
In order to prevent and treat cardiovascular disease, the search for a therapeutic vaccine to manage cholesterol has been the focus of several studies over the years, and has been encouraged by the finding of natural protective autoantibodies anti-oxidized LDL in human plasma.59,60 These vaccines have been mainly developed to target lipoproteins, cholesterol itself, or molecules involved in the metabolism of cholesterol.61–65
Vaccines targeting lipoproteins
The first report of a vaccine with positive results in atherosclerotic disease was published by Gero et al, who demonstrated that immunization for beta-lipoproteins in rabbits and cocks fed with a high-cholesterol diet decreased the progression of atherosclerotic plaques in the aorta.59,66 However, other studies using similar protocols, in spite of observing improvement in the lipoprotein profile, failed to show a significant decrease in the rate of development of atherosclerotic plaques in the aorta.65
Demonstration of the role of oxidized LDL in the pathology of atherosclerosis and the finding that oxidized LDL is a target for the immune system, since circulating autoantibodies that recognize several forms of oxidized LDL, in particular one of the dominant antigen malondialdehyde-modified lysine in oxidized LDL, are quite prevalent in humans and other species,60,67 scientific interest in development of an anti-oxidized LDL vaccine has increased. Immunization with malondialdehyde-modified LDL to stimulate production of antibodies anti-oxidized LDL, in LDL receptor-deficient rabbits and mice resulted in high antibody titers and reduction of atherosclerotic plaques.68,69 Ameli et al confirmed the results of previous studies by demonstrating that low-level immunization of hypercholesterolemic rabbits with homologous LDL and copper-oxidized LDL reduced the formation of early atherosclerotic lesions in response to a high cholesterol diet.70 These studies have also contributed to our understanding of the regulation of atherogenesis by adaptive and innate immunity.71 The mechanisms leading to LDL modification into oxidized forms via myeloperoxidase and its oxidant product, hypochlorite, have also been used as an alternative target to decrease oxidized LDL. In a recent study, passive immunization of LDLr–/– mice with the monoclonal antibody MoabA7S8 directed against hypochlorite-oxidized LDL resulted in induction of specific immunoglobulin M and G titers against oxidized LDL, and decreased cholesterol levels and atherosclerosis.72
After oxidized LDL, other molecular targets for atheroprotective immune responses were identified, namely apolipoprotein B-100 peptide sequences,73,74 that are the primary apolipoprotein component found in LDL cholesterol particles and required for their formation. Fredrikson et al confirmed that immunization with apolipoprotein B-100 peptides (p2, p143, and p210) using albumin as a carrier and aluminum hydroxide (alum) as the adjuvant resulted in a significant reduction in size and inflammation of atherosclerotic lesions in apolipoprotein E-null mice, which have increased plasma cholesterol levels and develop spontaneous atherosclerotic plaques by 2–3 months of age75 (Table 2).
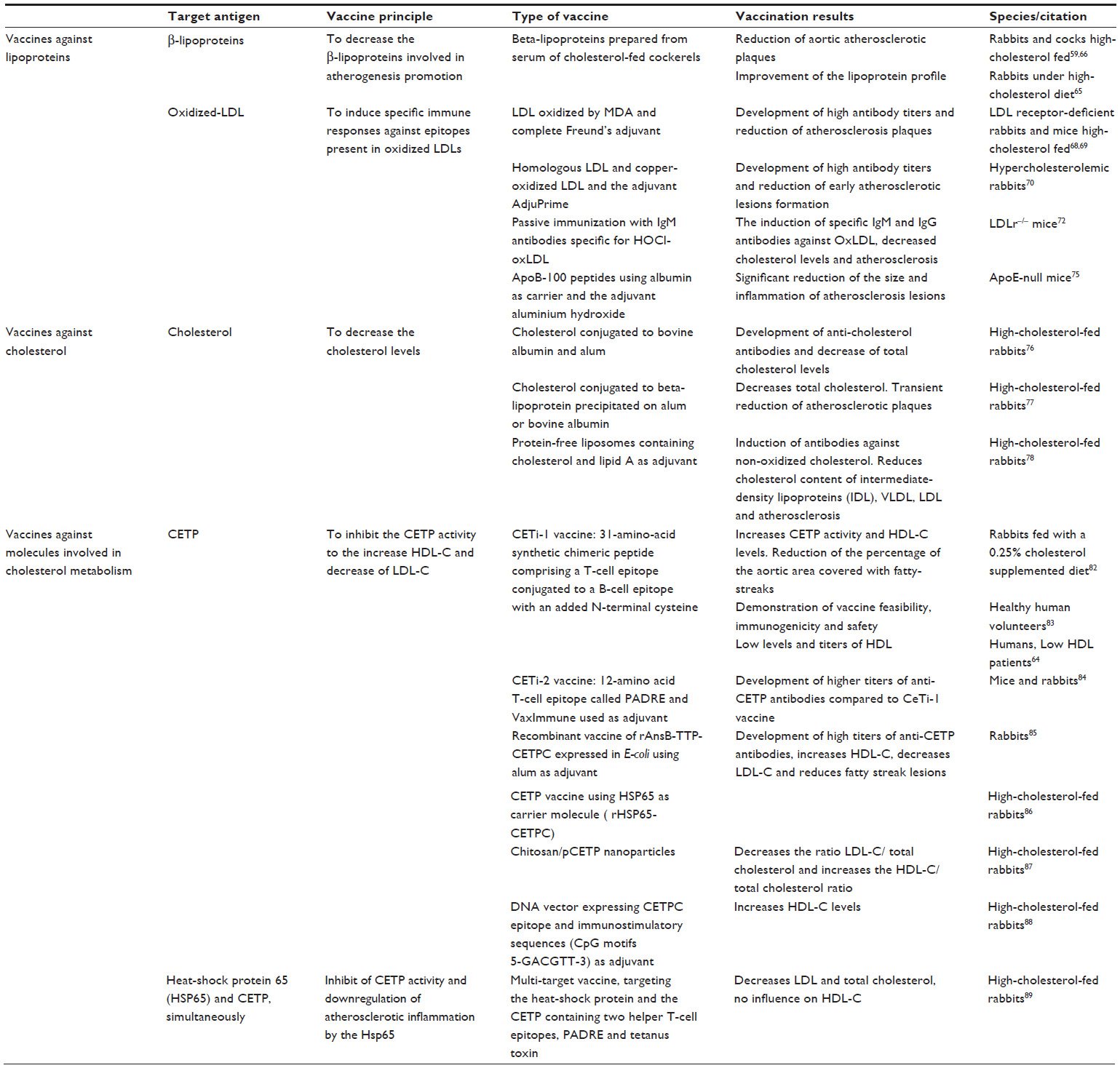 | Table 2 Vaccination approaches for dyslipidemia |
Vaccines targeting cholesterol
The first reports of attempts to develop a cholesterol vaccine to reduce progression of atherosclerosis date back to the early 1960s.76 Bailey et al showed increased anticholesterol antibodies and decreased total cholesterol levels in cholesterol-fed rabbits inoculated with a vaccine containing cholesterol conjugated to bovine albumin and alum.76 Years later, using similar vaccines, the same group showed that immunized rabbits fed a high-cholesterol diet had a reduction in atherosclerotic plaques; however, this effect was not sustained, with progression of the atherosclerotic plaques in almost all of the inoculated rabbits after 9 months of the high-cholesterol diet.77
Another study demonstrated that rabbits fed a high-cholesterol diet and immunized with protein-free liposomes containing 71% cholesterol and lipid A as the adjuvant developed antibodies against nonoxidized cholesterol, leading to a reduction of the plasma cholesterol present in intermediate-density lipoprotein, VLDL, and LDL cholesterol and also a reduction in atherosclerosis78 (Table 2).
Vaccines targeting other molecules involved in cholesterol metabolism
The most promising target for anticholesterol vaccines is cholesteryl ester transfer protein (CETP), a molecule that mediates the transport of cholesterol esters from HDLs to triglyceride-rich lipoproteins and to LDLs.79 Mutations that cause congenital CETP deficiency are associated with elevated HDL cholesterol.79,80 Therefore, pharmacological inhibition of CETP has been pursued as a treatment approach for management of cholesterol.79,81
In 2000, Rittershaus et al inoculated New Zealand White rabbits fed a 0.25% cholesterol diet with CETi-1 vaccine, which consists of a 31-amino acid synthetic chimeric peptide comprising a T-cell epitope conjugated to a B-cell epitope with an added N-terminal cysteine.64,82 Compared with the control group, inoculated rabbits showed an increase in CETP activity and HDL cholesterol levels, together with a reduction of the percentage of the aortic area covered with fatty streaks.82 This CETi-1 vaccine went into a Phase I clinical trial and was tested in 48 adult healthy volunteers with an HDL <60 mg/dL. This trial has demonstrated the feasibility, immunogenicity, and safety of a single and booster dose for induction of anti-CETP antibodies.83 In a Phase II clinical trial, the CETi-1 vaccine was administered to 203 patients with low HDL cholesterol levels (<40 mg/dL for men and <50 mg/dL for women), who showed an increase in HDL levels that correlated with antibody titers, although lower levels and titers than expected were reached.64 In order to render this vaccination strategy more immunogenic, the same research group has created the CETi-2 vaccine, in which the 14 amino acids comprising the T-cell epitope from tetanus toxin were replaced by a 12-amino acid T-cell epitope called PADRE, with Vax-Immune was added as an adjuvant. This vaccine has been shown to be able to induce higher anti-CETP antibody titers in mice and rabbits than those observed after inoculation with the CeTi-1 vaccine.84
Gaofu et al developed a recombinant vaccine of rAnsB-TTP-CETPC expressed in Escherichia coli using alum as an adjuvant. Inoculated rabbits developed high titers of anti-CETP antibodies, followed by increased HDL cholesterol, decreased LDL cholesterol, and fewer fatty streak lesions.85 This same group also developed the rHSP65-CETPC vaccine, which uses HSP65 as a carrier molecule, and achieved similar results when tested in cholesterol-fed rabbits.86 Recombinant vaccines expressed in E. coli represent a more suitable process and are less expensive for large-scale production than the vaccines produced using synthetic methods.85,86
DNA vaccines directed against CETP were also trialed as an approach to control in vivo cholesterol levels.87–89 Mao et al using a vaccine with DNA vector expressing CETPC epitope and containing immunostimulatory sequences which included CpG motifs 5-GACGTT-3 as adjuvant were intramuscularly inoculated to high cholesterol-fed rabbits showing a significant elevation of plasma HDL cholesterol.88 Mucosal immunization with chitosan/plasmid pCR-X8-HBc-CETP nanoparticles also achieved a decrease in the ratio of LDL cholesterol to total cholesterol and an increase in the HDL cholesterol to total cholesterol ratio, demonstrating that noninvasive mucosal immunization can be an alternative route to deliver DNA vaccines targeting CETP.87 More recently, Jun et al used a multitarget vaccine targeting HSP65 and CETP simultaneously for cholesterol management. When administered to cholesterol-fed rabbits, this intranasal vaccine decreased LDL and total cholesterol, but had no influence on HDL cholesterol levels when compared with the control group89 (Table 2).
In summary, most animal studies of vaccine approaches for cholesterol management have yielded significant results with regard to modulation of the immune system for prevention and treatment of atherosclerosis. However, further studies are needed in order to assess the feasibility and safety of these vaccines in the clinical setting, as well as comparison with other well established cholesterol treatments.
Vaccines for hypertension
Hypertension is defined by the World Health Organization90 as a systolic blood pressure (BP) ≥140 mmHg and/or a diastolic BP ≥90 mmHg. Hypertension is a major risk factor for cardiovascular disease,90 and has been recognized by the Global Burden of Diseases, Injuries, and Risk Factors Study in 2010 as the leading cause of death worldwide.91 In 2000, 26.4% of the adult population was hypertensive, and this number is projected to increase to 29.2% by 2025.92 Although there are several effective therapeutic approaches for hypertension, including lifestyle modification by increasing physical activity and decreasing salt intake, as well as pharmacological treatment, this condition requires chronic therapy. Furthermore, only a small percentage of hypertensive patients are able to adequately control their hypertension, which results in an enormous economic burden directly related to hypertension or its comorbid complications.93
Vascular resistance has a key role in determining BP and is largely regulated by the renin–angiotensin system, a hormonal cascade that begins with the synthesis of renin in the juxtaglomerular complex of the kidneys. Renin cleaves circulating angiotensinogen produced by the liver to form angiotensin I, which in turn is cleaved by angiotensin-converting enzyme to form angiotensin II,94 the physiologically active component of the system that acts through binding to AT1 and AT2 receptors. AT1 receptors are present in vascular tissue, and it is believed that angiotensin II exerts its actions on the cardiovascular system via these receptors.94 Since its discovery, this system has been the most attractive target for various therapeutic approaches to hypertension, starting with angiotensin-converting enzyme inhibitors that block the cleavage of angiotensin I into angiotensin II,95 followed by angiotensin II receptor blockers that prevent the signaling of angiotensin II through the AT1 receptor,96 and finally renin inhibitors that block production of angiotensin I.97 Every pharmacological approach has proven effective for the treatment of hypertension; however, they have limitations concerning undesirable side effects, poor patient compliance due to the asymptomatic nature of the disease, and the need for chronic therapy, rendering this pathological condition an attractive target for a therapeutic vaccine. Therefore, active vaccination against components of the renin–angiotensin system has been suggested as a good therapeutic strategy and several vaccines using different approaches have already been proposed.97–99
Vaccines targeting renin
The first attempt to develop an antihypertension vaccine consisted of active immunization against renin, and was performed in 1951100,101 when Goldblatt et al immunized eight hypertensive patients with renin isolated from hog kidneys. This first attempt, despite inducing production of antirenin antibodies, had no effect on BP, which was attributed to the lack of cross-reactivity between human and hog renin.101 Later, Dheodhar et al observed that the antibodies produced in response to immunization with acetylated dog renin were capable of blocking not only acetylated renin but also untreated renin; this was accompanied by a decrease in BP to the prehypertensive level, once again demonstrating the importance of renin blockade as a therapeutic approach for hypertension.100 Michel et al further investigated this immunization approach in normotensive marmosets using human renin with Freund’s adjuvant that, although not approved for human use, allowed production of high titers of antirenin antibodies which were able to block human and marmoset renin in vitro and significantly decrease BP in vivo, but unfortunately caused sickness and death due to autoimmune kidney disease.102 The same research team was able to immunize spontaneous hypertensive and normotensive rats against murine renin and to induce production of specific antirenin antibodies, which were higher in hypertensive rats than in normotensive rats, with consequent blockade of renin activity and a decrease in BP in both groups, although the rats developed autoimmune kidney disease as well.103 Understandably, for safety reasons, this approach has been abandoned until recently, when six different peptides, representing potential epitopes of rat and human renin, coupled with keyhole limpet hemocyanin, were used to immunize normotensive and hypertensive rats.104 This study showed that all peptides elicited strong antibody responses and all antisera could bind to renin in vitro; in addition, human and rat epitope R32 vaccines also decreased systolic BP in spontaneous hypertensive rats significantly and by up to 15 mmHg, without significant signs of immune-mediated damage in vaccinated animals104 (Table 3).
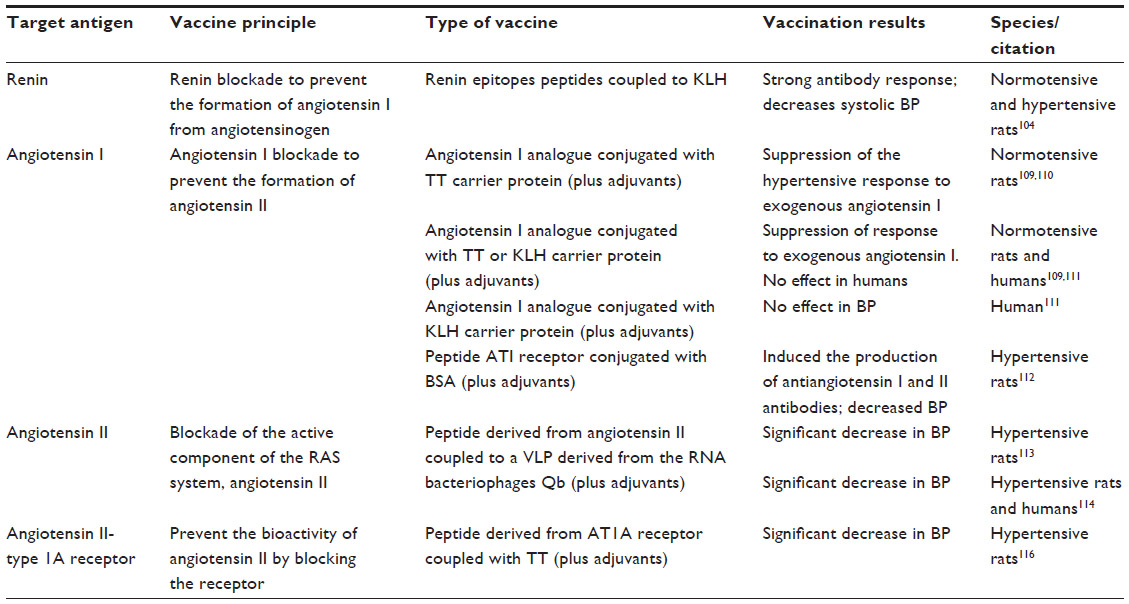 | Table 3 Vaccination approaches for hypertension |
Vaccines targeting angiotensin
The angiotensins have similarly been a target of several active immunization attempts for treatment of hypertension, although most of them were not able to significantly affect BP.105–108
Antiangiotensin I conjugated with tetanus toxoid (PMD 2850)109,110 or a 12-amino acid analog of angiotensin I covalently coupled to keyhole limpet hemocyanin109,111 (PMD 3117) vaccines have been proposed, with more promising results than the previous approaches. Both vaccines were able to elicit an antibody response after only the second immunization. The patients were challenged with angiotensin I to address the effect on BP, and although there were no significant changes in this regard, the higher the antibody titer, the more angiotensin I was needed to increase BP, indicating a biological effect. PMD 3117 vaccine has been tested in 27 hypertensive patients who were subcutaneously injected with either the vaccine or aluminum hydroxide (Alhydrogel™) four times over a 6-week period. The antibody titers increased from the second inoculation onwards, peaking at day 64 and having a half-life of approximately 100 days. This vaccination strategy did not influence BP, but significantly blunted the fall in plasma renin following withdrawal of the hypertension medication. In addition, in hypertensive patients, PMD3117 generated a prolonged antibody response to angiotensin I, although higher titers seem to be required in order to achieve a significant decrease in BP. More recently, Hong et al reported a new vaccine for hypertension based on peptide AngI-R conjugated with bovine serum albumin and adjuvanted with aluminum hydroxide. This vaccine proved capable of inducing production of both angiotensin I and angiotensin II antibodies and successfully decreased BP.112
In contrast, Cyt006-AngQb, an antiangiotensin II vaccine consisting of a peptide derived from angiotensin II chemically linked to a virus-like particle from RNA bacteriophage Qb, has been shown to be effective at producing specific antibodies in both spontaneous hypertensive rats113 and humans.113,114 Hypertensive patients (n=72) were injected with either 100 μg of Cyt006-AngQb (n=24), 300 μg of Cyt006-AngQb (n=24), or placebo (n=24), at weeks 0, 4, and 12. The vaccine was well tolerated and the higher vaccine concentration was able to decrease BP and the early morning BP surge when compared with placebo. Unfortunately, in subsequent studies, Cyt006 failed to reproduce this BP reduction, despite shorter dosing intervals and higher antibody titers115 (Table 3).
Vaccines targeting the angiotensin receptor
The AT1 receptor has also been tested as a possible hypertension vaccine target in spontaneously hypertensive rats by repeated subcutaneous injections of a peptide-tetanus-toxoid complex in combination with Freund’s adjuvant over 64 weeks. This resulted in induction of specific antibodies, a decrease in BP and cardiac hypertrophy, and attenuation of kidney injury, with no signs of autoimmune heart or kidney disease116 (Table 3).
In brief, due to the alarming prevalence of hypertension and the chronicity of the disease, alternative and definitive or long-lasting therapeutic approaches to ensure better BP control and patient compliance are welcomed. The proposed hypertension vaccines could be a good alternative, although future research is required before these can reach the clinical setting.
Vaccines for obesity
Obesity and overweight rates are increasing worldwide and are now one of the most serious public health problems of the century.117,118 Obesity is a medical condition characterized by accumulation of excess body fat associated with an increased risk of adverse health effects, which is more often the result of a positive energy balance due to a combination of excessive food intake and lack of physical activity in genetically predisposed individuals.118 Only a limited number of obesity cases are secondary to monogenetic diseases, endocrine disorders, or use of drugs that cause weight gain.119
Obesity is a risk factor for many chronic metabolic conditions, including T2DM, hypertension, dyslipidemia, cardiovascular disease, and metabolic syndrome.119 In addition, obesity is a major cause of decreased life expectancy, and to an extent similar to that observed in smokers.120 Since weight loss is able to improve or resolve a number of comorbidities,121 obesity is currently considered the leading cause of preventable death worldwide.122–124 Since there is a high likelihood of weight regain after weight loss, obesity requires chronic treatment and a long-term approach, for which pharmacological options remain quite limited.125 One of the most exciting strategies on the horizon is the possibility of targeting molecular factors associated with energy homeostasis regulation using vaccines as a possible means to control this major global disease.
Vaccines targeting adipose tissue antigens
Given that obesity is associated with a chronic inflammatory response, one of the first antiobesity vaccination approaches has focused on inducing immune tolerance against adipose tissue-derived antigens, in an attempt to modulate the inflammatory response. Oral administration of adipose tissue-pooled antigens has been demonstrated to be safe, able to reduce waist and thigh circumferences, and improve the lipid profile by decreasing triglycerides and increasing HDL cholesterol levels.126 However, this simple and attractive obesity vaccine failed to have a significant effect on body weight and lacked specificity, and could benefit from a more focused approach after identification of the appropriate molecular targets (Table 4).
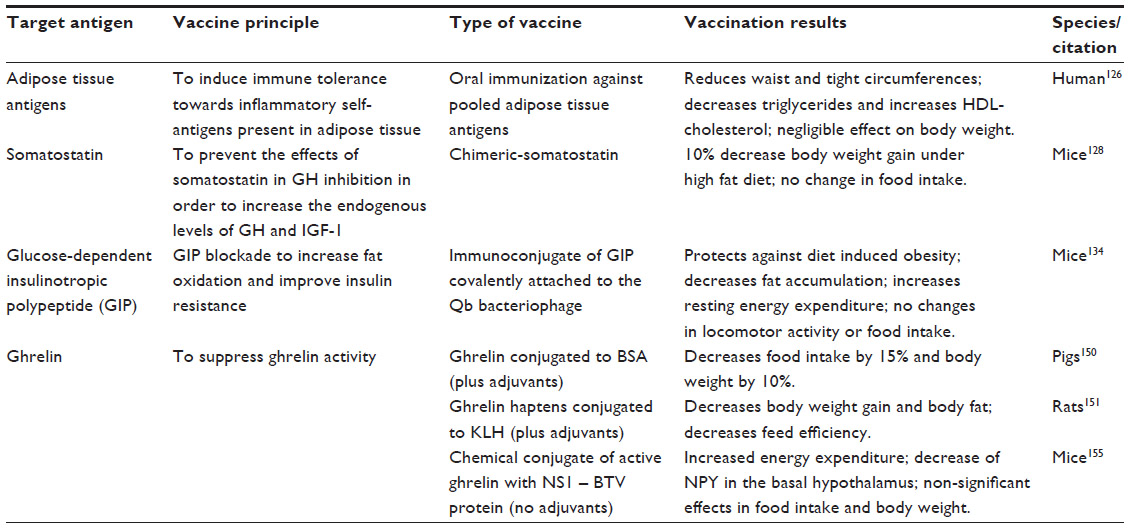 | Table 4 Vaccination approaches for obesity |
Vaccines targeting somatostatin
Obesity is characterized by a decrease in basal secretion of growth hormone, which has been demonstrated to reduce fat and increase lean mass.127 Somatostatin is a peptide hormone produced in the hypothalamus and gastrointestinal system, among other tissues, and inhibits secretion of growth hormone and insulin-like growth factor 1. Therefore, blocking the physiological effects of somatostatin increases endogenous levels of growth hormone and insulin-like growth factor 1, without the need to use anabolic hormones. Antisomatostatin vaccination in mice has shown to decrease body weight gain by 10% despite continuous feeding with a high-fat diet.128 This vaccination approach, despite the benefit of having a specific molecular target, is capable of upregulating growth hormone with the advantage of decreasing fat mass, but has the potential shortcomings of impairing glucose metabolism secondary to growth hormone excess and promoting neuroendocrine cell proliferation that is inhibited by somatostatin (Table 4).
Vaccines targeting glucose-dependent insulinotropic polypeptide
Glucose-dependent insulinotropic polypeptide (GIP) is a gastrointestinal hormone released after carbohydrate ingestion by neuroenteric K-cells and stimulates glucose-dependent insulin secretion and release.129 Besides the pancreas, the GIP receptor is widely distributed in peripheral organs, and in adipose tissue has a key role in fat deposition.130
GIP-R knockout mice exposed to a high-fat diet show decreased weight gain and increased uncoupling protein-1 expression in brown adipose tissue that is associated with increased energy expenditure.131 GIP antagonists have also been shown to prevent or reverse obesity in rodent models.132 Although GIP does not participate in regulation of food intake, GIP blockade has been shown to deplete adipose deposits and improve insulin resistance, so has been considered a potential drug target for treatment of obesity-related diabetes.133 Based on these findings, an antiobesity vaccination targeting GIP has been attempted, using an immunoconjugate of GIP covalently attached to the Qb bacteriophage to induce production of anti-GIP neutralizing antibodies. This anti-GIP vaccine has been shown to protect against diet-induced weight gain due to decreased fat accumulation and increased energy expenditure, similar to the effects observed after chemical blockade of the receptor134 (Table 4).
Vaccines targeting ghrelin
Ghrelin is the only peripheral hormone known to stimulate food intake and therefore is considered the most promising target for obesity treatment.135 Neutralization of the biologic effects of ghrelin on energy homeostasis, achieved in a number of experimental models, has already been used in order to prove this concept, and includes genetic deletion of ghrelin or the ghrelin receptor, antagonism of the ghrelin receptor, and inhibition of ghrelin O-acyltransferase (GOAT), the enzyme responsible for ghrelin acylation with subsequent activation.
Genetic deletion of ghrelin or GHS-R1a, the receptor that mediates the stimulatory effects of ghrelin on growth hormone and food intake, protects mice from diet-induced obesity by increasing energy expenditure and locomotor activity, without a significant change in food intake or impairment of growth hormone levels.136–138,139 In addition, patients harboring missense mutations of GHS-R are characterized by short stature and obesity.140
Ghrelin receptor antagonists have been demonstrated to decrease food intake and body weight and to improve glucose tolerance in diet-induced obese mice and in a mouse model of postmenopausal obesity, confirming the potential of blocking ghrelin as a treatment for obesity and T2DM.141,142 In addition, ghrelin neutralizing molecules, such as RNA Spiegelmer, a non-natural nucleic acid with specific binding activity towards a given molecule, has also been shown to decrease food intake, promote weight loss, and decrease food efficiency.143 Antibody-mediated GOAT inactivation increases metabolic rate and suppresses refeeding after food deprivation,144,145 while a GOAT-specific inhibitor, a bisubstrate analog GO-CoA-Tat composed of ghrelin, octanoyl Co-A, and a Tat sequence that allows the analog to penetrate within the cell cytoplasm where acylation of ghrelin occurs, has been shown to decrease serum levels of active ghrelin, prevent body weight gain, increase insulin secretion, and improve glucose tolerance.146
The first attempts at an antighrelin vaccination strategy consisted of passive antibody transfer. As a proof-of-concept, intracerebroventricular administration of polyclonal antighrelin antibodies has demonstrated to dose-dependently inhibit fast-induced feeding and suppress dark phase food intake.147 In addition, monoclonal antiacylated ghrelin antibodies were shown to inhibit acute ghrelin-mediated orexigenic effects without modifying long-term food intake in mice,148 while more recently, a mixture of monoclonal antibodies targeting different ghrelin haptens demonstrated increase energy expenditure during fasting and deprivation-induced food intake, and reduced overall food intake upon refeeding.149 Although representative as a proof-of-concept, passive immunizations have the limitation of lacking long-term effectiveness, due to the reduced half-lives of the antibodies and a need for periodic administration, as well as the possibility of activation of compensatory pathways of ghrelin production, as may occur with other ghrelin inactivation procedures.
Active immunization strategies have been developed more recently with the rationale of inducing an immune response to suppress endogenous ghrelin bioactivity. The first ghrelin immunization attempts included vaccines using bovine serum albumin150 and keyhole limpet hemocyanin as carrier proteins and immunogenic substances.151 Ghrelin-bovine serum albumin conjugates with Freund’s incomplete adjuvant and diethylaminoethyl dextran decreased voluntary food intake by 15% and body weight by 10% in vaccinated pigs, with no evidence of interference in the somatotropic axis.150 Zorrilla et al used different ghrelin haptens conjugated to keyhole limpet hemocyanin and Ribi emulsion or alum as adjuvants in order to induce production of antighrelin antibodies, which decreased body weight gain, leptin levels, body fat, and feed efficiency in immunized rats.151 In contrast, vaccination with ghrelin-PADRE, a synthetic Pan-DR helper T-cell epitope that binds with high affinity to HLA-DR molecules, was able to induce a strong immunoglobulin G immune response to various antigens coupled with PADRE, but failed to show any effect on body weight or food intake despite inducing the development of high antighrelin antibody titers.152 The main limitations of these antighrelin vaccination strategies is the need to use adjuvants in order to achieve an appropriate antibody response, which may be associated with an inflammatory response, a risk of an exacerbated immune response, or have restricted use in humans (Table 4).
An alternative antighrelin vaccination approach has been active immunization using virus-like particles. Virus-like proteins are viral proteins without genetic material, so have no pathogenic phenotype. These structures have a highly repetitive nature that has the advantage of allowing B-cell receptor cross-linking due to the ordered presentation of epitopes on the molecule surface, which induces efficient B-cell activation. The high immunogenicity allows use of a small number of inoculations and a low quantity of vaccine in order to obtain an effective immunization.153 Compared with classic immunization strategies, virus-like protein vaccines are usually more safe, efficient, and cost-effective,154 so are currently used as carrier molecules for several recombinant vaccines used for hepatitis B and human papillomavirus. The virus-like protein-based antighrelin vaccine is a chemical conjugate of active ghrelin with NS1 protein tubules of the Bluetongue virus, developed with the aim of obtaining a safe and more effective vaccine that could be used to treat humans.155 Although NS1 protein is not part of the viral capsid, it has the same immunogenic characteristics as classical virus-like proteins,156,157 with the advantage of having been used previously as a carrier protein for several prophylactic vaccines against common human infectious diseases, including foot-and-mouth disease and influenza A virus.157,158
The ability of this immunoconjugate to trigger an immune response without adjuvants has been demonstrated in normal weight and diet-induced obese male mice that developed specific antighrelin antibodies,155 although in low titers when compared with the titers reached after common infectious diseases. However, this antibody profile is further reassuring in terms of safety concerns, because complete ghrelin neutralization is not desirable as a strategy for obesity treatment, since ghrelin also intervenes in the regulation of growth hormone secretion and has functions in the gastrointestinal and nervous systems.135 Vaccinated animals also show increased energy expenditure, providing additional evidence of the effectiveness of ghrelin neutralization, since ghrelin is known to suppress energy metabolism.136,159 In addition, a significant decrease of NPY gene expression in the basal hypothalamus, the most potent orexigenic signal in the central nervous system, was also observed in vaccinated diet-induced obese mice.160 In comparison, in vaccinated normal weight mice, there was no significant difference in genetic expression of the NPY gene. The fact that this finding was only observed in diet-induced obese mice suggests the likelihood of a compensatory mechanism from the periphery in order to prevent a reduction in the feeding threshold of normal weight mice.155 Vaccinated mice also showed a nonsignificant decrease in long-term food intake and weight gain, which could be explained by the activation of compensatory mechanisms of energy homeostasis pathways; since the signaling pathways that control energy homeostasis and appetite are highly redundant,161 it is possible that ghrelin blockade could lead to activation of compensatory mechanisms similar to what occurs in ghrelin knockout mice.136,138
As ghrelin is the only orexigenic peripheral hormone identified, has attracted particular attention for the development of different neutralization approaches for obesity treatment, which has shown to be an effective means of decreasing food intake and increasing energy expenditure, both important contributions to establish a negative energy balance and promote weight loss. However, the available data suggest that the utility of ghrelin antagonism as a broad antiobesity agent is questionable. The role of ghrelin in regulation of food intake seems to act predominantly in response to conditions of low energy intake, driving hunger rather than regulating basal food intake or appetite. In addition, since most obese patients have low ghrelin levels, it is not expected that a vaccine would be effective in the absence of a diet-induced ghrelin rise, so obese patients who could benefit from these therapeutic approaches are more likely to be individuals enrolling in a diet and exercise program as adjuvant therapy for weight loss in an effort to prevent weight regain. Furthermore, since food intake is the result of a highly regulated and redundant network, it is unlikely that inhibition of the ghrelin pathway alone or any of the other pathways explored so far as a means to interfere with body weight regulation will be effective for obesity treatment. Combination therapies targeting multiple pathways will most likely be needed.
Conclusion
Chronic metabolic disorders are a major public health problem in western societies.2 Treatment of diabetes, hypertension, dyslipidemia, and obesity in order to prevent cardiovascular disease requires a chronic treatment approach that, in addition to lifestyle modification, often requires use of multiple drugs and can be associated with considerable costs, side effects, and a low rate of therapeutic compliance due to the asymptomatic nature of these conditions in most patients.162 Vaccines targeting the molecular risk factors associated with chronic noncommunicable diseases are an attractive option for controlling a major global health problem such as cardiovascular disease. Therefore, an expansion of the vaccination goals from infectious disease prevention to the therapeutic perspective is underway. Nevertheless, in the context of cardiovascular disease, advances have been slow, mostly due to the multifactorial pathogenesis of the disease with many intervening elements. Nonetheless, successes with vaccines for diabetes, cholesterol, hypertension, and obesity have highlighted the significant potential of such drug targets, despite the fact that before these vaccines can be used in the clinical field for the prevention and treatment of chronic metabolic diseases, there are several technical challenges that need to be met. These include identification of the most efficient molecular targets for each risk factor, determining the antibody titers and affinities that need to be reached in order to achieve clinical efficacy, and assessment of clinical safety.
Acknowledgments
UMIB is funded by grants from the Foundation for Science and Technology POCTI/FEDER Fcomp-01-0124-FEDER-015896. The authors’ research concerning the development of an antighrelin vaccine is funded by FEDER through the Competitiveness Operational Programme and national funds through the Foundation for Science and Technology under grant FCOMP-01-0124-FEDER-027 651 and project grant EXPL/BIM-MET/0618/2012.
Disclosure
The authors report no conflicts of interest in this work.
References
Isomaa B, Almgren P, Tuomi T, et al. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24(4):683–689. | |
Zimmet P, Magliano D, Matsuzawa Y, Alberti G, Shaw J. The metabolic syndrome: a global public health problem and a new definition. J Atheroscler Thromb. 2005;12(6):295–300. | |
Bachmann MF, Jennings GT. Therapeutic vaccines for chronic diseases: successes and technical challenges. Philos Trans R Soc Lond B Biol Sci. 2011;366(1579):2815–2822. | |
Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640–1645. | |
Buscemi S, Sprini D, Grosso G, et al. Impact of lifestyle on metabolic syndrome in apparently healthy people. Eat Weight Disord. 2014;19(2):225–232. | |
Gallagher EJ, LeRoith D. Epidemiology and molecular mechanisms tying obesity, diabetes, and the metabolic syndrome with cancer. Diabetes Care. 2013;36 Suppl 2:S233–S239. | |
Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356–359. | |
Ervin RB. Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race and ethnicity, and body mass index: United States, 2003–2006. Natl Health Stat Report. 2009;13:1–7. | |
Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S1–S45. | |
Zimmet P, Cowie C, Ekoe J-M, Shaw J. Classification of diabetes mellitus and other categories of glucose intolerance. In: Alberti KGMM, DeFronzo RA, Keen H, Zimmet P, editors. International Textbook of Diabetes Mellitus. Chichester, UK: John Wiley & Sons, Ltd; 2003. | |
International Diabetes Federation. Diabetes Atlas. 6th ed. Brussels, Belgium: International Diabetes Federation; 2013. Available from: http://www.idf.org/diabetesatlas. Accessed July 26, 2014. | |
Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365(9467):1333–1346. | |
Canivell S, Gomis R. Diagnosis and classification of autoimmune diabetes mellitus. Autoimmun Rev. 2014;13(4–5):403–407. | |
Cnop M, Welsh N, Jonas J-C, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54 Suppl 2:S97–S107. | |
Yoon JW, Jun HS. Autoimmune destruction of pancreatic beta cells. Am J Ther. 2005;12(6):580–591. | |
Gianani R, Campbell-Thompson M, Sarkar SA, et al. Dimorphic histopathology of long-standing childhood-onset diabetes. Diabetologia. 2010;53(4):690–698. | |
Moraes-Vasconcelos D, Costa-Carvalho BT, Torgerson TR, Ochs HD. Primary immune deficiency disorders presenting as autoimmune diseases: IPEX and APECED. J Clin Immunol. 2008;28 Suppl 1:S11–S19. | |
Garg G, Tyler JR, Yang JH, et al. Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol. 2012;188(9):4644–4653. | |
Anjos S, Polychronakos C. Mechanisms of genetic susceptibility to type I diabetes: beyond HLA. Mol Genet Metab. 2004;81(3):187–195. | |
Todd JA. Etiology of type 1 diabetes. Immunity. 2010;32(4):457–467. | |
Gan MJ, Albanese-O’Neill A, Haller MJ. Type 1 diabetes: current concepts in epidemiology, pathophysiology, clinical care, and research. Curr Probl Pediatr Adolesc Health Care. 2012;42(10):269–291. | |
Cambuli VM, Incani M, Cossu E, et al. Prevalence of type 1 diabetes autoantibodies (GADA, IA2, and IAA) in overweight and obese children. Diabetes Care. 2010;33(4):820–822. | |
Wong FS. Insulin – a primary autoantigen in type 1 diabetes? Trends Mol Med. 2005;11(10):445–448. | |
Larche M. Peptide immunotherapy for allergic diseases. Allergy. 2007;62(3):325–331. | |
Larche M. Peptide immunotherapy. Immunol Allergy Clin North Am. 2006;26(2):321–332, viii. | |
Peakman M. Can we vaccinate against type 1 diabetes? F1000 Biol Rep. 2012;4:19. | |
Tree TI, Lawson J, Edwards H, et al. Naturally arising human CD4 T-cells that recognize islet autoantigens and secrete interleukin-10 regulate proinflammatory T-cell responses via linked suppression. Diabetes. 2010;59(6):1451–1460. | |
Bresson D, Togher L, Rodrigo E, et al. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006;116(5):1371–1381. | |
Nanto-Salonen K, Kupila A, Simell S, et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet. 2008;372(9651):1746–1755. | |
Peakman M, von Herrath M. Antigen-specific immunotherapy for type 1 diabetes: maximizing the potential. Diabetes. 2010;59(9):2087–2093. | |
Thrower SL, James L, Hall W, et al. Proinsulin peptide immunotherapy in type 1 diabetes: report of a first-in-man Phase I safety study. Clin Exp Immunol. 2009;155(2):156–165. | |
Walter M, Philotheou A, Bonnici F, Ziegler AG, Jimenez R. No effect of the altered peptide ligand NBI-6024 on beta-cell residual function and insulin needs in new-onset type 1 diabetes. Diabetes Care. 2009;32(11):2036–2040. | |
Orban T, Farkas K, Jalahej H, et al. Autoantigen-specific regulatory T cells induced in patients with type 1 diabetes mellitus by insulin B-chain immunotherapy. J Autoimmun. 2010;34(4):408–415. | |
Takiishi T, Korf H, Van Belle TL, et al. Reversal of autoimmune diabetes by restoration of antigen-specific tolerance using genetically modified Lactococcus lactis in mice. J Clin Invest. 2012;122(5):1717–1725. | |
Husseiny MI, Rawson J, Kaye A, et al. An oral vaccine for type 1 diabetes based on live attenuated Salmonella. Vaccine. 2014;32(20):2300–2307. | |
Roep BO, Solvason N, Gottlieb PA, et al. Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8+ T cells in type 1 diabetes. Sci Transl Med. 2013;5(191):191ra182. | |
Gottlieb P, Utz PJ, Robinson W, Steinman L. Clinical optimization of antigen specific modulation of type 1 diabetes with the plasmid DNA platform. Clin Immunol. 2013;149(3):297–306. | |
Hagopian WA, Michelsen B, Karlsen AE, et al. Autoantibodies in IDDM primarily recognize the 65,000-M(r) rather than the 67,000-M(r) isoform of glutamic acid decarboxylase. Diabetes. 1993;42(4):631–636. | |
Shi Y, Kanaani J, Menard-Rose V, et al. Increased expression of GAD65 and GABA in pancreatic beta-cells impairs first-phase insulin secretion. Am J Physiol Endocrinol Metab. 2000;279(3):E684–E694. | |
Tian J, Atkinson MA, Clare-Salzler M, et al. Nasal administration of glutamate decarboxylase (GAD65) peptides induces Th2 responses and prevents murine insulin-dependent diabetes. J Exp Med. 1996;183(4):1561–1567. | |
Yoon J-W, Yoon C-S, Lim H-W, et al. Control of autoimmune diabetes in NOD mice by GAD expression or suppression in β cells. Science. 1999;284(5417):1183–1187. | |
Agardh CD, Cilio CM, Lethagen A, et al. Clinical evidence for the safety of GAD65 immunomodulation in adult-onset autoimmune diabetes. J Diabetes Complications. 2005;19(4):238–246. | |
Agardh CD, Lynch KF, Palmer M, Link K, Lernmark A. GAD65 vaccination: 5 years of follow-up in a randomised dose-escalating study in adult-onset autoimmune diabetes. Diabetologia. 2009;52(7):1363–1368. | |
Ludvigsson J, Faresjo M, Hjorth M, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008;359(18):1909–1920. | |
Ludvigsson J, Krisky D, Casas R, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med. 2012;366(5):433–442. | |
Wherrett DK, Bundy B, Becker DJ, et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet. 2011;378(9788):319–327. | |
Boettler T, Pagni PP, Jaffe R, Cheng Y, Zerhouni P, von Herrath M. The clinical and immunological significance of GAD-specific autoantibody and T-cell responses in type 1 diabetes. J Autoimmun. 2013;44:40–48. | |
Guan Y, Zhang M, Li Y, Cao W, Ji M, Liu Y. Vaccination with IA-2 autoantigen can prevent late prediabetic nonobese diabetic mice from developing diabetes mellitus. Diabetes Res Clin Pract. 2012;95(1):93–97. | |
Quintana FJ, Cohen IR. The HSP60 immune system network. Trends Immunol. 2011;32(2):89–95. | |
Elias D, Cohen IR. The hsp60 peptide p277 arrests the autoimmune diabetes induced by the toxin streptozotocin. Diabetes. 1996;45(9):1168–1172. | |
Schloot NC, Meierhoff G, Lengyel C, et al. Effect of heat shock protein peptide DiaPep277 on β-cell function in paediatric and adult patients with recent-onset diabetes mellitus type 1: two prospective, randomized, double-blind phase II trials. Diabetes Metab Res Rev. 2007;23(4):276–285. | |
Raz I, Avron A, Tamir M, et al. Treatment of new-onset type 1 diabetes with peptide DiaPep277® is safe and associated with preserved beta-cell function: extension of a randomized, double-blind, phase II trial. Diabetes Metab Res Rev. 2007;23(4):292–298. | |
Ashcroft Frances M, Rorsman P. Diabetes mellitus and the β cell: the last ten years. Cell. 2012;148(6):1160–1171. | |
Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87(4):1409–1439. | |
Pratley RE, Salsali A. Inhibition of DPP-4: a new therapeutic approach for the treatment of type 2 diabetes. Curr Med Res Opin. 2007;23(4):919–931. | |
Pang Z, Nakagami H, Osako MK, et al. Therapeutic vaccine against DPP4 improves glucose metabolism in mice. Proc Natl Acad Sci U S A. 2014;111(13):E1256–E1263. | |
Herold KC, Vignali DA, Cooke A, Bluestone JA. Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol. 2013;13(4):243–256. | |
Kwiterovich PO Jr. The metabolic pathways of high-density lipoprotein, low-density lipoprotein, and triglycerides: a current review. Am J Cardiol. 2000;86(12A):5L–10L. | |
Gero S. Some data on the influence of cholesterol atherosclerosis by immunological means. Rev Atheroscler (Paris). 1967;9 Suppl 1:194–198. | |
Bergmark C, Wu R, de Faire U, Lefvert AK, Swedenborg J. Patients with early-onset peripheral vascular disease have increased levels of autoantibodies against oxidized LDL. Arterioscler Thromb Vasc Biol. 1995;15(4):441–445. | |
de Carvalho JF, Pereira RM, Shoenfeld Y. Vaccination for atherosclerosis. Clin Rev Allergy Immunol. 2010;38(2–3):135–140. | |
Rittershaus CW. Vaccines for cholesterol management. World J Surg. 2007;31(4):690–694. | |
Nilsson J, Wigren M, Shah PK. Vaccines against atherosclerosis. Expert Rev Vaccines. 2013;12(3):311–321. | |
Ryan US, Rittershaus CW. Vaccines for the prevention of cardiovascular disease. Vasc Pharmacol. 2006;45(5):253–257. | |
Bailey JM, Tomar R. Lipoprotein immunization and induced atherosclerosis in rabbits. 1. Cockerel beta-lipoproteins as antigens. J Atheroscler Res. 1965;5(2):203–214. | |
Gero S, Gergely J, Jakab L, et al. Inhibition of cholesterol atherosclerosis by immunisation with beta-lipoprotein. Lancet. 1959;2(7088):6–7. | |
Yla-Herttuala S, Palinski W, Butler SW, Picard S, Steinberg D, Witztum JL. Rabbit and human atherosclerotic lesions contain IgG that recognizes epitopes of oxidized LDL. Arterioscler Thromb. 1994;14(1):32–40. | |
Palinski W, Miller E, Witztum JL. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc Natl Acad Sci U S A. 1995;92(3):821–825. | |
Palinski W, Tangirala RK, Miller E, Young SG, Witztum JL. Increased autoantibody titers against epitopes of oxidized LDL in LDL receptor-deficient mice with increased atherosclerosis. Arterioscler Thromb Vasc Biol. 1995;15(10):1569–1576. | |
Ameli S, Hultgardh-Nilsson A, Regnstrom J, et al. Effect of immunization with homologous LDL and oxidized LDL on early atherosclerosis in hypercholesterolemic rabbits. Arterioscler Thromb Vasc Biol. 1996;16(8):1074–1079. | |
Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6(7):508–519. | |
van Leeuwen M, Kemna MJ, de Winther MP, et al. Passive immunization with hypochlorite-oxLDL specific antibodies reduces plaque volume in LDL receptor-deficient mice. PLoS One. 2013; 8(7):e68039. | |
Fredrikson GN, Andersson L, Soderberg I, et al. Atheroprotective immunization with MDA-modified apo B-100 peptide sequences is associated with activation of Th2 specific antibody expression. Autoimmunity. 2005;38(2):171–179. | |
Fredrikson GN, Hedblad B, Berglund G, et al. Identification of immune responses against aldehyde-modified peptide sequences in apoB associated with cardiovascular disease. Arterioscler Thromb Vasc Biol. 2003;23(5):872–878. | |
Fredrikson GN, Soderberg I, Lindholm M, et al. Inhibition of atherosclerosis in apoE-null mice by immunization with apoB-100 peptide sequences. Arterioscler Thromb Vasc Biol. 2003;23(5):879–884. | |
Bailey JM, Bright R, Tomar R. Immunization with a synthetic cholesterol-ester antigen and induced atherosclerosis in rabbits. Nature. 1964;201:407–408. | |
Bailey JM, Bright R, Tomar R, Butler J. Anti-atherogenic effects of cholesterol vaccination. Biochem Soc Trans. 1994;22(4):433S. | |
Alving CR, Swartz GM Jr, Wassef NM, et al. Immunization with cholesterol-rich liposomes induces anti-cholesterol antibodies and reduces diet-induced hypercholesterolemia and plaque formation. J Lab Clin Med. 1996;127(1):40–49. | |
Barter PJ, Brewer HB Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23(2):160–167. | |
Inazu A, Brown ML, Hesler CB, et al. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N Engl J Med. 1990;323(18):1234–1238. | |
Barter PJ, Kastelein JJ. Targeting cholesteryl ester transfer protein for the prevention and management of cardiovascular disease. J Am Coll Cardiol. 2006;47(3):492–499. | |
Rittershaus CW, Miller DP, Thomas LJ, et al. Vaccine-induced antibodies inhibit CETP activity in vivo and reduce aortic lesions in a rabbit model of atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20(9):2106–2112. | |
Davidson MH, Maki K, Umporowicz D, Wheeler A, Rittershaus C, Ryan U. The safety and immunogenicity of a CETP vaccine in healthy adults. Atherosclerosis. 2003;169(1):113–120. | |
Thomas LJ, Hammond RA, Forsberg EM, et al. Co-administration of a CpG adjuvant (VaxImmune, CPG 7909) with CETP vaccines increased immunogenicity in rabbits and mice. Hum Vaccin. 2009;5(2):79–84. | |
Gaofu Q, Jun L, Xiuyun Z, Wentao L, Jie W, Jingjing L. Antibody against cholesteryl ester transfer protein (CETP) elicited by a recombinant chimeric enzyme vaccine attenuated atherosclerosis in a rabbit model. Life Sci. 2005;77(21):2690–2702. | |
Gaofu Q, Jun L, Xin Y, et al. Vaccinating rabbits with a cholesteryl ester transfer protein (CETP) B-Cell epitope carried by heat shock protein-65 (HSP65) for inducing anti-CETP antibodies and reducing aortic lesions in vivo. J Cardiovasc Pharmacol. 2005;45(6):591–598. | |
Yuan X, Yang X, Cai D, et al. Intranasal immunization with chitosan/pCETP nanoparticles inhibits atherosclerosis in a rabbit model of atherosclerosis. Vaccine. 2008;26(29–30):3727–3734. | |
Mao D, Kai G, Gaofu Q, et al. Intramuscular immunization with a DNA vaccine encoding a 26-amino acid CETP epitope displayed by HBc protein and containing CpG DNA inhibits atherosclerosis in a rabbit model of atherosclerosis. Vaccine. 2006;24(23):4942–4950. | |
Jun L, Jie L, Dongping Y, et al. Effects of nasal immunization of multi-target preventive vaccines on atherosclerosis. Vaccine. 2012;30(6):1029–1037. | |
World Health Organization. A global brief on hypertension. Silent killer, global public health crisis. Geneva, Switzerland. Available from: http://www.who.int/cardiovascular_diseases/publications/global_brief_hypertension/en/. Accessed July 26, 2014. | |
Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2224–2260. | |
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365(9455):217–223. | |
Wolf-Maier K, Cooper RS, Kramer H, et al. Hypertension treatment and control in five European countries, Canada, and the United States. Hypertension. 2004;43(1):10–17. | |
Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev. 2003;24(3):261–271. | |
Brown NJ, Vaughan DE. Angiotensin-converting enzyme inhibitors. Circulation. 1998;97(14):1411–1420. | |
Burnier M, Brunner HR. Angiotensin II receptor antagonists. Lancet. 2000;355(9204):637–645. | |
Lam S, Choy M. Aliskiren: an oral renin inhibitor for the treatment of hypertension. Cardiol Rev. 2007;15(6):316–323. | |
Nakagami H, Koriyama H, Morishita R. Therapeutic vaccines for hypertension and dyslipidemia. Int Heart J. 2014;55(2):96–100. | |
Pandey R, Quan WY, Hong F, Jie SL. Vaccine for hypertension: modulating the renin-angiotensin system. Int J Cardiol. 2009;134(2):160–168. | |
Deodhar SD, Haas E, Goldblatt H. Production of antirenin to homologous renin and its effect of experimental renal hypertension. J Exp Med. 1964;119:425–432. | |
Goldblatt H, Haas E, Lamfrom H. Antirenin in man and animals. Trans Assoc Am Physicians. 1951;64:122–125. | |
Michel JB, Guettier C, Philippe M, Galen FX, Corvol P, Menard J. Active immunization against renin in normotensive marmoset. Proc Natl Scad Sci U S A. 1987;84(12):4346–4350. | |
Michel JB, Sayah S, Guettier C, et al. Physiological and immunopathological consequences of active immunization of spontaneously hypertensive and normotensive rats against murine renin. Circulation. 1990;81(6):1899–1910. | |
Qiu Z, Chen X, Zhou Y, et al. Therapeutic vaccines against human and rat renin in spontaneously hypertensive rats. PLoS One. 2013;8(6):e66420. | |
Johnston CI, Hutchinson JS, Mendelsohn FA. Biological significance of renin angiotensin immunization. Circ Res. 1970;27:Suppl 2:215+. | |
MacDonald GJ, Louis WJ, Renzini V, Boyd GW, Peart WS. Renal-clip hypertension in rabbits immunized against angiotensin II. Circ Res. 1970;27(2):197–211. | |
Oates HF, Stokes GS, Storey BG, Glover RG, Snow BF. Renal hypertension in rats immunized against angiotensin I and angiotensin II. J Exp Med. 1974;139(2):239–248. | |
Reade R, Michel JB, Carelli C, Huang H, Baussant T, Corvol P. [Immunisation of spontaneously hypertensive rats against angiotensin I]. Arch Mal Coeur Vaiss. 1989;82(7):1323–1328. French. | |
Downham MR, Auton TR, Rosul A, et al. Evaluation of two carrier protein-angiotensin I conjugate vaccines to assess their future potential to control high blood pressure (hypertension) in man. Br J Clin Pharmacol. 2003;56(5):505–512. | |
Gardiner SM, Auton TR, Downham MR, et al. Active immunization with angiotensin I peptide analogue vaccines selectively reduces the pressor effects of exogenous angiotensin I in conscious rats. Br J Pharmacol. 2000;129(6):1178–1182. | |
Brown MJ, Coltart J, Gunewardena K, Ritter JM, Auton TR, Glover JF. Randomized double-blind placebo-controlled study of an angiotensin immunotherapeutic vaccine (PMD3117) in hypertensive subjects. Clin Sci (Lond). 2004;107(2):167–173. | |
Hong F, Quan WY, Pandey R, et al. A vaccine for hypertension based on peptide AngI-R: a pilot study. Int J Cardiol. 2011;148(1):76–84. | |
Ambuhl PM, Tissot AC, Fulurija A, et al. A vaccine for hypertension based on virus-like particles: preclinical efficacy and phase I safety and immunogenicity. J Hypertens. 2007;25(1):63–72. | |
Tissot AC, Maurer P, Nussberger J, et al. Effect of immunisation against angiotensin II with CYT006-AngQb on ambulatory blood pressure: a double-blind, randomised, placebo-controlled phase IIa study. Lancet. 2008;371(9615):821–827. | |
Unger T, Paulis L, Sica DA. Therapeutic perspectives in hypertension: novel means for renin-angiotensin-aldosterone system modulation and emerging device-based approaches. Eur Heart J. 2011;32(22):2739–2747. | |
Zhu F, Liao YH, Li LD, et al. Target organ protection from a novel angiotensin II receptor (AT1) vaccine ATR12181 in spontaneously hypertensive rats. Cell Mol Immunol. 2006;3(2):107–114. | |
do Carmo I, Dos Santos O, Camolas J, et al. Overweight and obesity in Portugal: national prevalence in 2003–2005. Obes Rev. 2008;9(1):11–19. | |
World Health Organization. Obesity and overweight fact sheet No 311. Available from: http://www.who.int/mediacentre/factsheets/fs311/en/print.html. Acessed July 3, 2009. | |
Pi-Sunyer FX. The obesity epidemic: pathophysiology and consequences of obesity. Obes Res. 2002;10 Suppl 2:97S–104S. | |
Peeters A, Barendregt JJ, Willekens F, Mackenbach JP, Al Mamun A, Bonneux L. Obesity in adulthood and its consequences for life expectancy: a life-table analysis. Ann Intern Med. 2003;138(1):24–32. | |
Christou NV, Sampalis JS, Liberman M, et al. Surgery decreases long-term mortality, morbidity, and health care use in morbidly obese patients. Ann Surg. 2004;240(3):416–423. | |
Moayyedi P. The epidemiology of obesity and gastrointestinal and other diseases: an overview. Dig Dis Sci. 2008;53(9):2293–2299. | |
Puska P, Nishida C, Porter D. World Health Organization. Obesity and overweight. Available from: http://www.who.int/dietphysicalactivity/publications/facts/obesity/en/print.html. Accessed July 3, 2009. | |
World Health Organization. Diet, nutrition and the prevention of chronic diseases, 2003. Available from: http://www.who.int/mediacentre/news/releases/2003/pr20/en/. Accessed July 3, 2009. | |
Bray GA. Lifestyle and pharmacological approaches to weight loss: efficacy and safety. J Clin Endocrinol Metab. 2008;93(11 Suppl 1):S81–S88. | |
Bourinbaiar AS, Jirathitikal V. Effect of oral immunization with pooled antigens derived from adipose tissue on atherosclerosis and obesity indices. Vaccine. 2010;28(15):2763–2768. | |
Johansen T, Malmlof K. Treatment of obesity using GH. Metab Syndr Relat Disord. 2006;4(1):57–69. | |
Haffer KN. Effects of novel vaccines on weight loss in diet-induced-obese (DIO) mice. J Anim Sci Biotechnol. 2012;3(1):1–7. | |
Calanna S, Christensen M, Holst JJ, et al. Secretion of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes: systematic review and meta-analysis of clinical studies. Diabetes Care. 2013;36(10):3346–3352. | |
Yip RG, Boylan MO, Kieffer TJ, Wolfe MM. Functional GIP receptors are present on adipocytes. Endocrinology. 1998;139(9):4004–4007. | |
Hansotia T, Maida A, Flock G, et al. Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest. 2007;117(1):143–152. | |
Gault VA, McClean PL, Cassidy RS, Irwin N, Flatt PR. Chemical gastric inhibitory polypeptide receptor antagonism protects against obesity, insulin resistance, glucose intolerance and associated disturbances in mice fed high-fat and cafeteria diets. Diabetologia. 2007;50(8):1752–1762. | |
Irwin N, Flatt PR. Evidence for beneficial effects of compromised gastric inhibitory polypeptide action in obesity-related diabetes and possible therapeutic implications. Diabetologia. 2009;52(9):1724–1731. | |
Fulurija A, Lutz TA, Sladko K, et al. Vaccination against GIP for the treatment of obesity. PLoS One. 2008;3(9):0003163. | |
Kojima M, Kangawa K. Ghrelin: structure and function. Physiol Rev. 2005;85(2):495–522. | |
Wortley KE, Anderson KD, Garcia K, et al. Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc Natl Acad Sci U S A. 2004;101(21):8227–8232. | |
Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci U S A. 2004;101(13):4679–4684. | |
De Smet B, Depoortere I, Moechars D, et al. Energy homeostasis and gastric emptying in ghrelin knockout mice. J Pharmacol Exp Ther. 2006;316(1):431–439. | |
Wortley KE, del Rincon JP, Murray JD, et al. Absence of ghrelin protects against early-onset obesity. J Clin Invest. 2005;115(12):3573–3578. | |
Pantel J, Legendre M, Cabrol S, et al. Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J Clin Invest. 2006;116(3):760–768. | |
Esler WP, Rudolph J, Claus TH, et al. Small-molecule ghrelin receptor antagonists improve glucose tolerance, suppress appetite, and promote weight loss. Endocrinology. 2007;148(11):5175–5185. | |
Maletinska L, Matyskova R, Maixnerova J, et al. The peptidic GHS-R antagonist [D-Lys(3)] GHRP-6 markedly improves adiposity and related metabolic abnormalities in a mouse model of postmenopausal obesity. Mol Cell Endocrinol. 2011;343(1–2):55–62. | |
Shearman LP, Wang SP, Helmling S, et al. Ghrelin neutralization by a ribonucleic acid-SPM ameliorates obesity in diet-induced obese mice. Endocrinology. 2006;147(3):1517–1526. | |
Yang J, Zhao TJ, Goldstein JL, Brown MS. Inhibition of ghrelin O-acyltransferase (GOAT) by octanoylated pentapeptides. Proc Natl Acad Sci U S A. 2008;105(31):10750–10755. | |
Mayorov AV, Amara N, Chang JY, et al. Catalytic antibody degradation of ghrelin increases whole-body metabolic rate and reduces refeeding in fasting mice. Proc Natl Acad Sci U S A. 2008;105(45):17487–17492. | |
Barnett BP, Hwang Y, Taylor MS, et al. Glucose and weight control in mice with a designed ghrelin O-acyltransferase inhibitor. Science. 2010;330(6011):1689–1692. | |
Nakazato M, Murakami N, Date Y, et al. A role for ghrelin in the central regulation of feeding. Nature. 2001;409(6817):194–198. | |
Lu SC, Xu J, Chinookoswong N, et al. An acyl-ghrelin-specific neutralizing antibody inhibits the acute ghrelin-mediated orexigenic effects in mice. Mol Pharmacol. 2009;75(4):901–907. | |
Zakhari JS, Zorrilla EP, Zhou B, Mayorov AV, Janda KD. Oligoclonal antibody targeting ghrelin increases energy expenditure and reduces food intake in fasted mice. Mol Pharm. 2012;9(2):281–289. | |
Vizcarra JA, Kirby JD, Kim SK, Galyean ML. Active immunization against ghrelin decreases weight gain and alters plasma concentrations of growth hormone in growing pigs. Domest Anim Endocrinol. 2007;33(2):176–189. | |
Zorrilla EP, Iwasaki S, Moss JA, et al. Vaccination against weight gain. Proc Natl Acad Sci U S A. 2006;103(35):13226–13231. | |
Kellokoski E, Kummu O, Serpi R, et al. Ghrelin vaccination decreases plasma MCP-1 level in LDLR(–/–)-mice. Peptides. 2009;30(12):2292–2300. | |
Noad R, Roy P. Virus-like particles as immunogens. Trends Microbiol. 2003;11(9):438–444. | |
Lechner F, Jegerlehner A, Tissot AC, et al. Virus-like particles as a modular system for novel vaccines. Intervirology. 2002;45(4–6):212–217. | |
Andrade S, Pinho F, Ribeiro AM, et al. Immunization against active ghrelin using virus-like particles for obesity treatment. Curr Pharm Des. 2013;19(36):6551–6558. | |
Hewat EA, Booth TF, Wade RH, Roy P. 3-D reconstruction of bluetongue virus tubules using cryoelectron microscopy. J Struct Biol. 1992;108(1):35–48. | |
Ghosh MK, Borca MV, Roy P. Virus-derived tubular structure displaying foreign sequences on the surface elicit CD4+ Th cell and protective humoral responses. Virology. 2002;302(2):383–392. | |
Mikhailov M, Monastyrskaya K, Bakker T, Roy P. A new form of particulate single and multiple immunogen delivery system based on recombinant bluetongue virus-derived tubules. Virology. 1996;217(1):323–331. | |
Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407(6806):908–913. | |
Kalra SP, Dube MG, Sahu A, Phelps CP, Kalra PS. Neuropeptide Y secretion increases in the paraventricular nucleus in association with increased appetite for food. Proc Nat Acad Sci U S A. 1991;88(23):10931–10935. | |
Cone RD, Cowley MA, Butler AA, Fan W, Marks DL, Low MJ. The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int J Obes Relat Metab Disord. 2001;25 Suppl 5:S63–S67. | |
Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.