")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Use of levosimendan in the treatment of cerebral vascular vasospasm: a case study
Authors Onichimowski D, Nosek K , Goraj R , Jalali R , Wińska A, Pawlos A, Tuyakov B
Received 27 November 2017
Accepted for publication 14 March 2018
Published 20 June 2018 Volume 2018:12 Pages 1777—1783
DOI https://doi.org/10.2147/DDDT.S158237
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Dariusz Onichimowski,1 Krzysztof Nosek,2 Radosław Goraj,1 Rakesh Jalali,3 Aleksandra Wińska,2 Aleksandra Pawlos,4 Bułat Tuyakov1
1Department of Anesthesiology and Intensive Care, School of Medicine, Collegium Medicum, University of Warmia and Mazury, Olsztyn, Poland; 2Department of Neurology, School of Medicine, Collegium Medicum, University of Warmia and Mazury, Olsztyn, Poland; 3Department of Emergency Medicine, School of Medicine, Collegium Medicum, University of Warmia and Mazury, Olsztyn, Poland; 4Department of Pharmacology and Toxicology, Center for Experimental Medicine, Faculty of Medical Sciences, University of Warmia and Mazury, Olsztyn, Poland
Abstract: Despite the progress in the management of cerebral arterial aneurysms, subarachnoid hemorrhage (SAH) remains the major cause of neurological disability. While SAH-related deaths usually occur as a result of brain impairment due to hemorrhage, permanent neurological deficits are caused by cerebral ischemia due to edema and spasm of cerebral arteries. Additionally, ~20%–30% of patients with SAH develop secondary cardiomyopathy; this phenomenon is known as neurogenic stress cardiomyopathy (NSC), which is associated with increased mortality and poor long-term prognosis. Levosimendan is a new inotropic drug that causes calcium sensitization of troponin C, thus increasing contraction force of myofilaments. The drug also causes opening of ATP-dependent potassium channels in vascular smooth muscles, which results in dilatation of veins and arteries, including cerebral arteries. To date, there have been several reports of levosimendan application in patients with SAH and neurogenic stress cardiomyopathy, and the effect of the drug on vasospasm has been previously advocated. This paper presents a case report of a 57-year-old patient with massive SAH, where levosimendan was used for reducing vasospasm.
Keywords: cerebral arterial aneurysm, subarachnoid hemorrhage, cerebral vasospasm, levosimendan
Introduction
Subarachnoid hemorrhage (SAH) is one of the most common causes of neurological disability in patients with intracranial pathology. Bleeding from an aneurysm of a cerebral vessel (aneurysmal SAH [aSAH]) is not only a major risk factor leading to death, but may also cause long-term complications. The long-term effects primarily include neurological disability; the cardiovascular system can also get affected resulting in myocardial ischemia or infarction with acute circulatory failure. According to data from the Brain Aneurysm Foundation,1 the incidence of an unruptured aneurysm of a cerebral vessel is 1 per 50 persons and that of a ruptured aneurysm equals 8–10 cases per 100,000, which accounts for 30,000 cases per year in the USA alone. The majority of deaths due to SAH are the result of a fast-progressing, massive, and difficult or impossible to treat cerebral impairment due to hemorrhage. Permanent neurological impairment is caused by ischemic and hypoxic damage in the brain due to hypoperfusion as a result of cerebral edema and cerebral vasospasm.1
According to some authors, symptomatic cerebral vasospasm in SAH usually occurs between days 4 and 14,2 while according to others, it occurs between day 3 and week 2–4 of the disease.3 The peak incidence of vasospasm is at 7–8 days after hemorrhage, while <4% of symptoms occur after day 13.4 Angiography performed in the second week after SAH is reported to induce vasospasm in 67% of patients.5 The angiographic constriction is caused by prolonged, but reversible spasm of the smooth muscles. Vessel perfusion is inversely proportional to its radius raised to the fourth power. Slight changes in a vessel diameter may lead to profound disturbances of blood flow, and decrease in diameter below the critical level may cause cerebral tissue ischemia. Ischemic edema may compromise local perfusion despite normal systemic blood pressure. This results from compromised tissue perfusion in areas supplied by vasoconstricted arteries. Thus, prevention and treatment of vasospasm and consequent cerebral tissue ischemia are the critical issues in the management of SAH patients.4
It is believed that the underlying cause of a vasospasm is extravasation of hemoglobin, and more precisely, its nonprotein component heme. It causes constriction of arteries by not fully explained mechanisms. These mechanisms may be associated with neuronal apoptosis,6 reduced amount of nitric oxide (NO),7 increased endothelin-1 level,8 direct effect of oxidative stress on smooth muscle cells,9 production of free radicals, lipid peroxidation in cell membranes,10 and modification of potassium and calcium channels.11
Vasospasm was first visualized angiographically by Ecker and Riemenschneider in 1951. Since 1982, it has been possible to assess and monitor vasospasm by means of Transcranial Doppler (TCD) ultrasonography, and since 1990 Transcranial Color-Coded Duplex (TCCD) has been used for this purpose.12
Cardiovascular system disorders in SAH are well-recognized complications that occur in 14%–25% of patients. These disorders result in significantly worse outcomes despite the fact that myocardial impairment is often of transient nature. It is associated with release of large amounts of endogenous catecholamines, which causes shrinking of the vascular bed, including coronary vessels, resulting in myocardial ischemia with changes of ST-T segment and myocardial infarction, acute contractility disorders, myocardial stunning syndrome, or pulmonary edema. SAH may therefore be complicated by left ventricular dysfunction and cardiogenic shock as well as increased risk of brain ischemia. The suggested mechanisms that cause myocardial dysfunction include shrinking and thrombosis of coronary arteries, impaired balance of oxygen supply and utilization, and tachycardia, due to the combination of neuronal and hormonal mechanisms. It is also thought that rise of intracranial pressure (ICP) due to SAH causes sympathetic activation resulting in enhanced contraction of myocytes, which leads to myocardial impairment.13,14 Another mechanism that may play a role in cardiovascular instability is fever of central origin.15,16
The diagnostic criterion for neurogenic stress cardiomyopathy (NSC) is increased level of at least one marker of myocardial impairment. Independent risk factors for NSC in SAH are degree of neurological damage, higher grade on Hunt–Hess scale on presentation, degree of elevation of troponin, CK-MB, cerebral natriuretic peptide levels, and smoking. Arterial hypertension may be protective against NSC.17 Rise in biomarkers levels of myocardial injury (CK-MB and troponin) tends to be minimal in NSC, and it is disproportionately low in relation to degree of severity of contractility abnormalities. NSC generates particularly high risk of potentially fatal complications, such as ventricular dysrhythmias or exacerbation of vasospasm.18
Considering the fact that one of the mechanisms underlying the development of neurogenic stunned myocardium is excessive sympathetic activation, use of traditional positive inotropic drugs (catecholamines and phosphodiesterase inhibitors) may be ineffective. These drugs by causing a rise in calcium ion concentration may lead to increased oxygen consumption in the myocardium and thus to deepening of oxygen deficit, which in consequence may result in reduced cardiac output (CO) and cerebral blood flow. In recent years, several reports on the use of levosimendan in the management of SAH have been published. The drug acts by increasing myocardial contractility without increasing oxygen consumption. The long-term result is decreased mortality when compared with the results of using dobutamine or milrinone. Levosimendan binds to troponin C and increases its sensitivity to calcium ions, acting as the so-called calcium sensitizer. This increases the power of myofilament contraction without energy-consuming increase in calcium ion concentration in cells. Additionally, the drug causes opening of ATP-dependent potassium (KATP) channels in vascular smooth muscles, which results in the dilatation of venous and arterial vessels, including cerebral vessels. The third mechanism of action of the drug is opening of KATP channels in mitochondria. This action allows better energy utilization in a cell, which increases its resistance to hypoxia. The effect was observed in myocardial and brain cells. Improved utilization of energy in cells contributes to reduced production of free radicals and their increased disposal. This in turn reduces cell damage and stimulation of production of inflammatory response mediators.19 Use of levosimendan results in increase in CO and decrease in systemic vascular resistance (SVR). The beneficial changes in hemodynamic profile allow for alleviation or resolution of symptoms of acute decompensated heart failure, leading to increased organ perfusion. The drug also shows neuro- and cardioprotective properties.20
The drug’s mode of action suggests that its use may be beneficial in patients with aSAH, particularly in those who develop vasospasm followed by NSC. To date, only a few cases of using levosimendan in patients with aSAH have been reported.21–23 In all cases, both improvement and hemodynamic stability have been obtained. Nevertheless, the drug’s effect on vasospasm was not assessed. So far, there has been no report published on using levosimendan with the intention of reducing vasospasm without accompanying NSC. This report presents a case of the beneficial effect of using levosimendan in aSAH with vasospasm.
Case study
A 57-year-old woman with massive SAH of grade V (according to the Hunt and Hess scale), impaired consciousness, and respiratory insufficiency was admitted to the Intensive Care Unit (ICU) of the Voivodal Specialist Hospital in Olsztyn. In the history, the patient, with no prior history of chronic disease, suddenly experienced severe headache and a loss of consciousness (6 points in the Glasgow Coma Scale [GCS]), was intubated and mechanically ventilated at home, and transported to the Regional Specialist Hospital in Olsztyn. Computed tomography (CT) angiography revealed extensive fresh SAH of grade IV in the Fisher scale, involving the ventricular system (Figure 1), and a 2 mm aneurysmal protrusion on the vascular wall was visualized in M2 segment of the left middle central artery (MCAsin; Figure 1). On admission to the ICU the patient was in critical condition, unconscious with GCS 3 points without analgosedation, with cough reflex, ciliary and corneal reflexes, and pupillary reactivity to light preserved, hemodynamically stable with BP 130/70 mmHg, regular sinus rhythm of 80/min, spontaneous diuresis, and intestinal peristalsis.
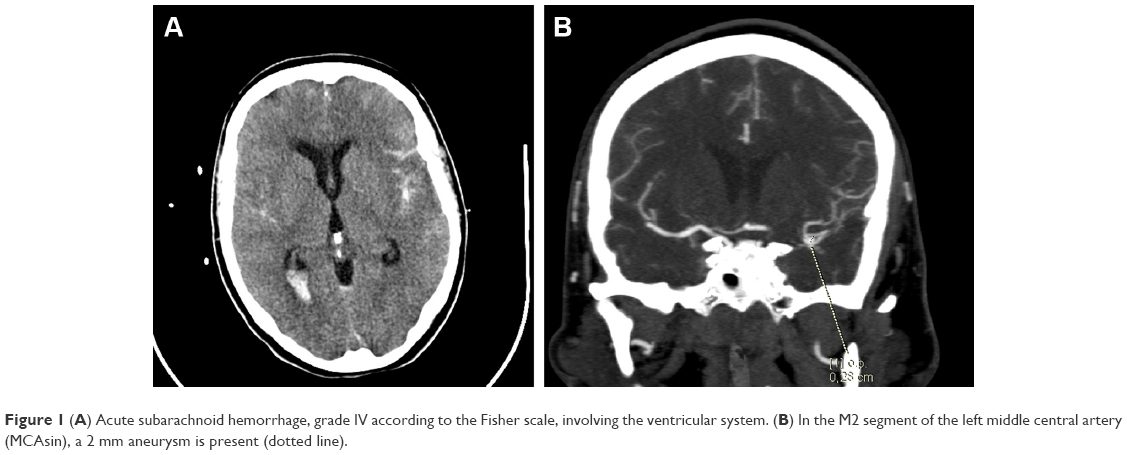 | Figure 1 (A) Acute subarachnoid hemorrhage, grade IV according to the Fisher scale, involving the ventricular system. (B) In the M2 segment of the left middle central artery (MCAsin), a 2 mm aneurysm is present (dotted line). |
After ICU admission, the following treatment was implemented: analgosedation, mechanical ventilation, mannitol and hypertonic saline infusion, nimodipine, tranexamic acid (SAH with no possibility of immediate direct management of the aneurysm due to patient’s critical condition on presentation), and bronchodilators. In order to ensure optimal cerebral perfusion norepinephrine infusion of 0.1 mcg/kg/min was started. Hemodynamic parameters were monitored with transpulmonary thermodilution (Edwards LifeSciences EV1000 Clinical Platform; the values are presented in Figure 2). Over the next few days, the patient’s neurological condition did not improve. Due to hydrocephalus in the control CT scans, on day 4, external ventricular drainage was implemented. The patient qualified for the surgical clipping of the aneurysm, and the surgery was carried out on the 7th day of hospitalization. Over the first few days of hospitalization (days 1–9) norepinephrine infusion was used in small doses (0.1–0.025 mc/kg/min) to control cerebral perfusion pressure (CPP). No beta-receptor agonists were administered.
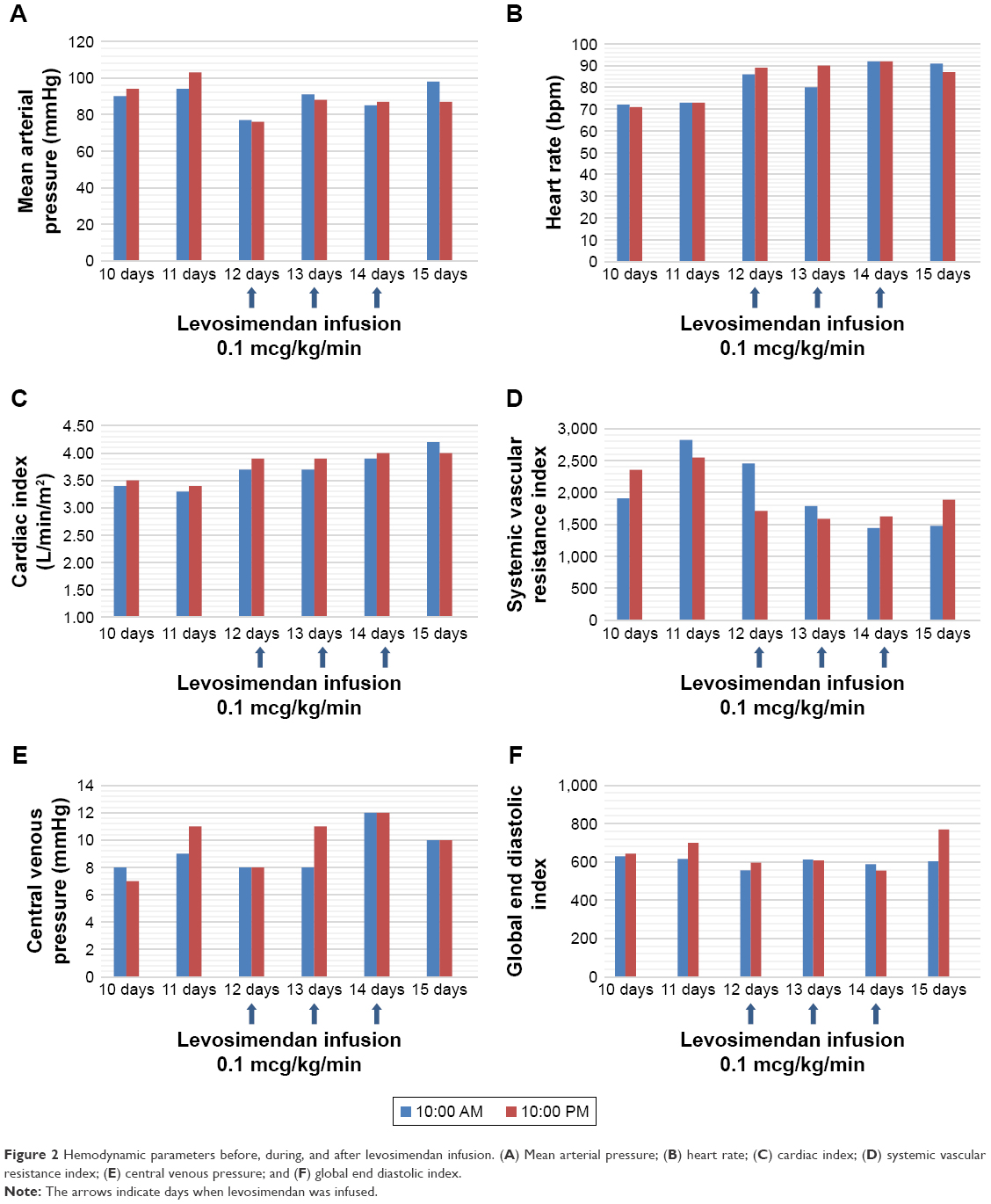 | Figure 2 Hemodynamic parameters before, during, and after levosimendan infusion. (A) Mean arterial pressure; (B) heart rate; (C) cardiac index; (D) systemic vascular resistance index; (E) central venous pressure; and (F) global end diastolic index. |
The TCD assessment of cerebral vessels on the 10th day revealed developing vasospasm, which increased over the following days (the values of blood flow velocities on the consecutive days are shown in Table 1). Significant acceleration of blood flow velocities was detected in the middle cerebral arteries (MCAs) bilaterally and in the left anterior cerebral artery (ACA), with a slight acceleration of flow in the posterior cerebral arteries and the right ACA. Flow velocities were normal in the vertebral, common, internal and external carotid arteries (the values are shown in Table 1). The mean values of the blood flow velocities in the intracranial and internal carotid were used to calculate Lindegaard index that led to the diagnosis of vasospasm affecting MCAs in M1 segment and spasm of the left ACA. The maximum values of blood flow velocities were observed on day 12 of hospitalization (Table 1).
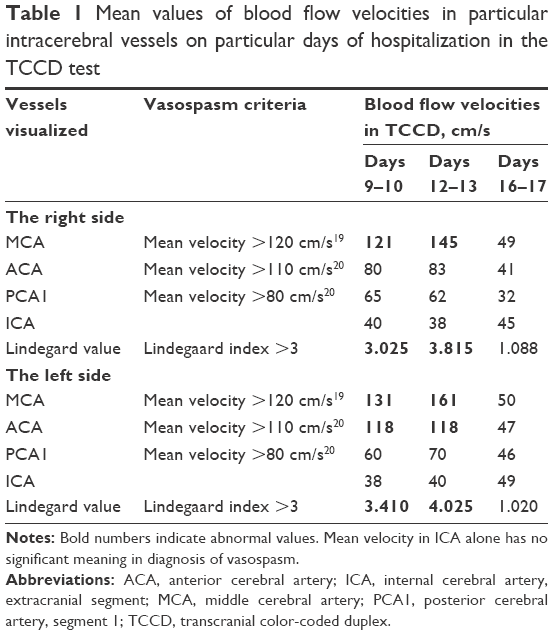 | Table 1 Mean values of blood flow velocities in particular intracerebral vessels on particular days of hospitalization in the TCCD test |
On day 12 of hospitalization, levosimendan was started with a dose of 0.1 mcg/kg/min (without an initial bolus) and was continued for 3 days due to its vasodilatory effect on cerebral arteries. The patient was unconscious at the time of the study and so the patient’s written informed consent was impossible to obtain; for this reason, it was obtained from the family. Following this treatment, gradual improvement was observed with reduction of blood flow velocities, which finally normalized (Figure 2 and Table 1). On day 16, intraventricular drainage of cerebrospinal fluid was removed with no evidence of hydrocephalus progression in control CT scans. Sedation was discontinued, the patient gradually regained consciousness, spontaneous breathing, and full hemodynamic stability and oral feeding was started. After 27 days in ICU, the patient was discharged to the Department of Neurosurgery for further treatment and physiotherapy.
On ICU discharge the patient was stable, conscious, spontaneously opening her eyes and fixating eyes periodically, and spontaneously moving her left upper limb; she did not follow commands.
Due to paresis, verbal informed consent has been provided by the patient and written informed consent from the patient’s legal guardian, for the case details and accompanying images to be published.
Discussion
Rupture of a cerebral aneurysm leading to SAH may cause brain damage, which develops within 72 h following blood extravasation. It is not only the rise of ICP associated with a decrease of CPP that is responsible for this damage, but also complex pathophysiological mechanisms such as blood–brain barrier (BBB) dysfunction, microcirculation disorders, neuronal apoptosis, or cerebral vasospasm. In SAH, vasospasm occurs in up to 67% of patients. It may develop after 2 days to 4 weeks of SAH. Moreover, 20%–30% of patients with aSAH may develop NSC with reduced CO and increase in SVR, which may reduce both CPP and cerebral blood perfusion.24 Additional reflex increase of sympathetic stimulation may further exacerbate vasospasm and impair autoregulation in cerebral microcirculation, leading to cerebral ischemia. The intracranial arteries have rich sympathetic innervation; hence they readily respond to sympathetic stimulation by developing vasospasm. Exogenously administered catecholamines may intensify vasospasm or may cause persistent spasm. Additionally, catecholamines enhance activation of endothelin, a vasoconstrictor, which also has an important role in generating vasospasm in SAH. KATP channels, found both in large cerebral arteries and in microcirculation, are involved in the regulation of vascular tone and are mediators of vasodilation via nitric oxide (NO).25 In patients with SAH, KATP channels dysfunction leads to reduction in NO production and response to vasodilators released from the endothelium. This mechanism may also enhance vasospasm and enlarge the area of ischemia. In SAH, active oxygen forms are released by lipid peroxidation and hemoglobin autooxidation; they induce oxidative stress and contribute to early and delayed ischemia. Reactive oxygen species cause smooth muscles and endothelium damage as well as BBB dysfunction. Moreover, oxidative stress leads to release of mediators of apoptosis, such as cytochrome C.26
In the management of aSAH with NSC, quick restoration of decreased CO may prevent further damage of brain tissue by maintenance of adequate cerebral perfusion and reversing vasospasm. Additional use of catecholamines may not only fail to provide improvement in CO and cerebral perfusion, but even enhance vasospasm. Similarly, hypervolemia may exacerbate symptoms of cardiomyopathy and due to pulmonary congestion lead to decreased cerebral oxygen supply and enhanced brain edema. Use of levosimendan seems to be a beneficial therapeutic option in the management of aSAH with NSC. This drug increases CO without a rise in calcium concentration in myocardial cells and, unlike epinephrine and norepinephrine, has a vasodilatory effect. Taccone et al23 reported a case of a patient with aSAH and stunned heart syndrome, where standard treatment (dobutamine and milrinone) was not effective. After implementing levosimendan, significant improvement in circulatory hemodynamic parameters was observed; however, the patient died due to central nervous system damage. No assessment of perfusion in the cerebral arteries was conducted.23 Two other cases of a beneficial use of levosimendan in aSAH were published by Papanikolaou et al.22 Apart from the improvement in hemodynamic parameters and organ perfusion, the authors observed stabilization of CPP after administration of the drug. However, in those cases, cerebral perfusion was also not assessed.22 Authors of those reports did not use the loading dose of levosimendan to avoid hypotension with potentially disastrous effect on CPP.
So far, the authors have presented the beneficial effect of levosimendan on improving hemodynamic parameters in patients with cardiomyopathy, which in turn may have an advantageous effect on cerebral circulation in patients with vasospasm. In order to determine a group of aSAH patients with developing vasospasm who could benefit from levosimendan, it is essential to determine whether this drug may have a beneficial effect on cerebral circulation itself or only through improvement within the systemic circulation. Levosimendan, apart from its influence on myocardium and coronary arteries, may also act favorably on some pathophysiological processes underlying permanent brain damage following vasospasm in SAH.27
In the majority of patients with SAH treated with levosimendan, cardiac failure was observed.21–23 This report presents the use of levosimendan and its effect on cerebral circulation in a patient with SAH without NSC. Hemodynamic parameters were assessed by means of transpulmonary thermodilution and cerebral perfusion with TCD. Vasospasm is a dynamic phenomenon with different blood flow velocities on particular days. On the basis of tests aiming to compare mean values of blood flow in angiography versus TCCD, it is assumed that the mean velocity in MCA >100 cm/s is associated with possible vasospasm with 88% specificity, the mean velocity >110 cm/s with probable vasospasm with 94% specificity, and the mean velocity >130 cm/s with definite (pronounced) vasospasm with 96% specificity. In the case of ACA, spasm is recognized when the mean value exceeds 110 cm/s in patients >55 years of age or 140 cm/s in patients <55 years of age; in the case of the posterior cerebral artery, spasm is recognized when the mean value exceeds 80 cm/s in patients >55 years of age or 110 cm/s in patients <55 years of age, and in the case of the vertebral arteries over 55 cm/s in patients >55 years of age or over 70 cm/s in those <55 years of age.
An additional criterion of vasospasm is the Lindegaard index, which is the ratio between the mean velocity in the MCA and the mean velocity in the internal carotid artery. A result >3 correlates with the evidence of spasm in MCA.12 In this case, after the administration of levosimendan, CO increased by several percent; however, it was primarily due to increased heart rate. Systemic vascular resistance index decreased by ~30%. No significant decrease in mean arterial pressure was noted. TCD tests revealed developing vasospasm starting on day 9–10 with maximum values of perfusion velocity on day 12, when the mean value of blood velocity in the right MCA reached 145 cm/s with a Lindegaard index of 3.815, and in the left MCA, 161 cm/s with a Lindegaard index of 4.025. The velocity in the left ACA was 118 cm/s. After starting levosimendan (measurements conducted on day 16 of hospitalization), the mean velocity in the right MCA of 49 cm/s was obtained, with a Lindegaard index of 1.088, while in the left MCA the mean velocity value was 50 cm/s with a Lindegaard index of 1.020. The mean value of blood velocity in the left ACA on day 16 of hospitalization was 47 cm/s. As the result of treatment, the patient’s neurological condition improved (from GCS 3 to 8), and the patient regained respiratory and circulatory stability. It seems that in our case the main mechanism of reducing vasospasm was the vasodilatory effect in the cerebral circulation and not improvement of CO. This means that the drug may provide benefits not only for patients with heart insufficiency, but also for those without it.
A similar decrease in blood velocity values in cerebral arteries following implementation of levosimendan was also reported by Papanikolaou et al in patients with SAH and underlying trauma, in the presentation of two clinical cases.28
The beneficial effect of levosimendan on CO and coronary perfusion has already been described in many experimental and clinical trials. The drug unquestionably improves hemodynamic parameters in cardiomyopathy of various etiologies. In contrast, its effect on the cerebral circulation and neuronal physiology has only been the subject of studies in experimental trials. Moreover, levosimendan caused significant and dose-dependent dilatation of vessels in the cerebral circulation via the prostaglandin system.29 Levosimendan has antiinflammatory properties, which may reduce neuronal damage caused by vasospasm. In studies on brain impairment associated with ischemia and reperfusion, levosimendan reduced inflammatory response and TNF-α expression 24 h after reperfusion.27,30
The presented case seems to confirm the beneficial pharmacodynamic properties of the drug in clinical conditions, with regard to the cerebral circulation and neurons, which have been already described in experimental trials.31 Defining the range of use of levosimendan in standard SAH therapy requires further clinical research.
Conclusion
SAH followed by vasospasm still remains the cause of severe neurological impairment and high mortality. Considerable advances in the surgical management of both ruptured and unruptured aneurysms of cerebral arteries have been achieved over recent years. Nevertheless, there is no major progress in the treatment and prevention of SAH consequences, especially when SAH is accompanied by NSC. Owing to the beneficial mechanisms of action, levosimendan seems to be the drug offering a chance for improved outcome and prognosis in patients with SAH and vasospasm. Determining indications and use of levosimendan in therapy of SAH with NSC requires further clinical trials.
Disclosure
The authors report no conflicts of interest in this work.
References
Brain Aneurysm Foundation. Brain aneurysm statistics and facts. Available from: https://www.bafound.org/about-brain-aneurysms/. Accessed November 1, 2017. | ||
Bradley WG, Daroff RB, Fenichel GM, Jankovic J. Neurologia w Praktyce Klinicznej. Lublin: Czelej; 2006:1133–1134. | ||
Stępień A. Neurologia. Warszawa: Medical Tribune Polska; 2014:230–243. | ||
Macdonald RL. Management of Cerebral Vasospasm. Section of Neurosurgery, MC3026, Chicago, IL, USA: University of Chicago Medical Center; 2005. | ||
Macdonald RL, Weir B. Cerebral Vasospasm. San Diego: Academic; 2001. | ||
Cahill J, Calvert JW, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26(11):1341–1353. | ||
Pluta RM. Delayed cerebral vasospasm and nitric oxide: review, new hypothesis, and proposed treatment. Pharmacol Ther. 2005;105(1):23–56. | ||
Seifert V, Loffler B, Zimmermann M, Roux S, Stolke D. Endothelin concentrations in patients with aneurysmal subarachnoid hemorrhage. Correlation with cerebral vasospasm, delayed ischemic neurological deficits and volume of hematoma. J Neurosurg. 1995;82(1):55–62. | ||
Ostrowski RP, Colohan AR, Zhang JH. Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28(4):399–414. | ||
Sehba FA, Bederson JB. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28(4):381–398. | ||
Zuccarello M, Bonasso CL, Lewis AI, Sperelakis N, Rapoport RM. Relaxation of subarachnoid hemorrhage-induced spasm of rabbit basilar artery by the K+ channel activator cromakalim. Stroke. 1996;27(2):311–316. | ||
Kaźmierski R. Podręcznik Diagnostyki Ultrasonograficznej w Neurologii [Textbook of ultrasonographic diagnostics in neurology]. Lublin: Czelej; 2011. Polish. | ||
Bybee KA, Prasad A. Stress-related cardiomyopathy syndromes. Circulation. 2008;118(4):397–409. | ||
Ahmadian A, Mizzi A, Banasiak M, et al. Cardiac manifestations of subarachnoid hemorrhage. HSR Proc Intensive Care Cardiovasc Anesth. 2013;5(1):1–11. | ||
Zawadzka M, Szmuda M, Mazurkiewicz-Bełdzińska M. Thermoregulation disorders of central origin – how to diagnose and treat. Anaesthesiol Intensive Ther. 2017;49(3):227–234. | ||
Tunali Y, Akcil EF, Dilmen O. Fever treatment with a catheter-based heat exchange system in the neurointensive care unit. Anaesthesiol Intensive Ther. 2016;48(3):208–210. | ||
Malik AN, Gross BA, Rosalind Lai PM, Moses ZB, Du R. Neurogenic stress cardiomyopathy after aneurysmal subarachnoid hemorrhage. World Neurosurg. 2015;83(6):880–885. | ||
Mayer SA, Lin J, Homma S, et al. Myocardial injury and left ventricular performance after subarachnoid hemorrhage. Stroke. 1999;30(4):780–786. | ||
Perrone SV, Kaplinsky EJ. Calcium sensitizer agents: a new class of inotropic agents in the treatment of decompensated heart failure. Int J Cardiol. 2005;103(3):248–255. | ||
Papp Z, Edes I, Fruhwald S, et al. Levosimendan: molecular mechanisms and clinical implications: consensus of experts on the mechanisms of action of levosimendan. Int J Cardiol. 2012;159(2):82–87. | ||
Busani S, Rinaldi L, Severino C, Cobelli M, Pasetto A, Girardis M. Levosimendan in cardiac failure after subarachnoid hemorrhage. J Trauma. 2010;68(5):E108–E110. | ||
Papanikolaou J, Tsolaki V, Makris D, Zakynthinos E. Early levosimendan administration may improve outcome in patients with subarachnoid hemorrhage complicated by acute heart failure. Int J Cardiol. 2014;176(3):1435–1437. | ||
Taccone FS, Brasseur A, Vincent JL, De Backer D. Levosimendan for the treatment of subarachnoid hemorrhage-related cardiogenic shock. Intensive Care Med. 2013;39(8):1497–1498. | ||
Naidech AM, Kreiter KT, Janjua N, et al. Cardiac troponin elevation, cardiovascular morbidity, and outcome after subarachnoid hemorrhage. Circulation. 2005;112(18):2851–2856. | ||
Sobey CG, Faraci FM. Subarachnoid haemorrhage: what happens to the cerebral arteries? Clin Exp Pharmacol Physiol. 1998;25(11):867–876. | ||
Rondeau N, Cinotti R, Rozec B, et al. Dobutamine-induced high cardiac index did not prevent vasospasm in subarachnoid hemorrhage patients: a randomized controlled pilot study. Neurocrit Care. 2012;17(2):183–190. | ||
Varvarousi G, Xantos T, Sarafidou P, et al. Role of levosimendan in the management of subarachnoid hemorrhage. Am J Emerg Med. 2016;34(2):298–306. | ||
Papanikolaou J, Spathoulas K, Makris D, Koukoubani T, Zakynthinos E. Hemodynamic challenges in traumatic subarachnoid hemorrhage complicated by cerebral vasospasm. Am J Emerg Med. 2016;34(5):904–906. | ||
Konczalla J, Wanderer S, Mrosek J, et al. Levosimendan, a new therapeutic approach to prevent delayed cerebral vasospasm after subarachnoid hemorrhage? Acta Neurochir (Wien). 2016;158(11):2075–2083. | ||
Cengiz SL, Erdi MF, Tosun M, et al. Beneficial effects of levosimendan on cerebral vasospasm induced by subarachnoid haemorrhage: an experimental study. Brain Inj. 2010;24(6):877–885. | ||
Kivikko M, Kuoppamäki M, Soinne L, et al. Oral levosimendan increases cerebral blood flow velocities in patients with a history of stroke or transient ischemic attack: a Pilot Safety Study. Curr Ther Res Clin Exp. 2015;77:46–51. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.