")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Ursolic acid sensitizes cisplatin-resistant HepG2/DDP cells to cisplatin via inhibiting Nrf2/ARE pathway
Received 13 April 2016
Accepted for publication 12 July 2016
Published 25 October 2016 Volume 2016:10 Pages 3471—3481
DOI https://doi.org/10.2147/DDDT.S110505
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Shouhai Wu,1,2 Tianpeng Zhang,1 Jingsheng Du3
1School of Life Sciences, Sun Yat-sen University, 2Center for Regenerative and Translational Medicine, 3Department of Pharmacy, The Second Affiliated Hospital, Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, People’s Republic of China
Background: Combinations of adjuvant sensitizers with anticancer drugs is a promising new strategy to reverse chemoresistance. Ursolic acid (UA) is one of the natural pentacyclic triterpene compounds known to have many pharmacological characteristics such as anti-inflammatory and anticancer properties. This study investigates whether UA can sensitize hepatocellular carcinoma cells to cisplatin.
Materials and methods: Cells were transfected with nuclear factor erythroid-2-related factor 2 (Nrf2) small interfering RNA and Nrf2 complementary DNA by using Lipofectin 2000. The cytotoxicity of cells was investigated by Cell Counting Kit 8 assay. Cell apoptosis, cell cycle, reactive oxygen species, and mitochondrial membrane potential were detected by flow cytometry fluorescence-activated cell sorting. The protein level of Nrf2, NAD(P)H quinone oxidoreductase 1 (NQO1), glutathione S-transferase (GST), and heme oxygenase-1 (HO-1) was detected by Western blot analysis.
Results: The results showed that the reverse index was 2.9- and 9.69-fold by UA of 1.125 µg/mL and 2.25 µg/mL, respectively, for cisplatin to HepG2/DDP cells. UA–cisplatin combination induced cell apoptosis and reactive oxygen species, blocked the cell cycle in G0/G1 phase, and reduced the mitochondrial membrane potential. Mechanistically, UA–cisplatin dramatically decreased the expression of Nrf2 and its downstream genes. The sensibilization of UA–cisplatin combination was diminished in Nrf2 small interfering RNA-transfected HepG2/DDP cells, as well as in Nrf2 complementary DNA-transfected HepG2/DDP cells.
Conclusion: The results confirmed the sensibilization of UA on HepG2/DDP cells to cisplatin, which was possibly mediated via the Nrf2/antioxidant response element pathway.
Keywords: ursolic acid, chemoresistance, cisplatin, liver cancer, Nrf2/ARE
Introduction
Hepatocellular carcinoma is one of the most common malignant tumors worldwide.1 Chemotherapy is one of the important methods in the comprehensive treatment of liver cancer, and the effect of drugs on the treatment of liver cancer is particularly weighty.1 The combined chemotherapy based on cisplatin recommended by international cancer organizations has become a line of liver cancer standard chemotherapy regimens.1 With the widespread application of platinum drugs, the tumor cell has inevitably developed resistance to platinum, which significantly reduces the effect of chemotherapy.2 Earlier studies reported that the drug resistance of recurrent liver cancer increased significantly, while the response rate of chemotherapy drugs was <30%.2 Thus, there is an emergent need to develop a new drug sensitizer that can increase the efficacy of cisplatin-based chemotherapy and overcome drug resistance.
Nuclear factor erythroid-2-related factor 2 (Nrf2) is known as the “primary supervisor” of the antioxidant response through the antioxidant response elements (AREs), regulating the expression of numerous genes including heme oxygenase-1 (HO-1), NAD(P)H quinone oxidoreductase 1 (NQO1), and glutathione S-transferase (GST) and several adenosine triphosphate-dependent drug efflux pumps (multi-drug resistance proteins).3–5 Therefore, many studies have demonstrated that Nrf2 is a transcription factor that can regulate various cytoprotective genes. Recently, Nrf2 has been reported to be a pharmacological target to overcome drug resistance. Overexpression of Nrf2 increases chemoresistance; on the contrary, knockdown of Nrf2 sensitizes various cancer cells including liver,6 leukemia,7 neuroblastoma,8 lung,9 breast,10 and pancreatic11 cells to chemotherapeutic drugs. Hence, screening Nrf2 inhibitors as an adjuvant sensitizer to overcome drug resistance is a desirable treatment strategy.
Earlier studies reported that one of the potential drug targets of several pentacyclic triterpene compounds, such as maslinic acid,12 boswellic acid,13 and oleanolic acid,14 was Nrf2. This finding make us to assess whether other pentacyclic triterpene compounds can inhibit Nrf2/ARE pathway to reverse resistance to chemotherapy drugs. Ursolic acid (UA), a natural pentacyclic triterpene compound (Figure 1), which is widely found in medicinal plants such as holy basil, food, and other plants, exhibits anticancer potential effect in vitro and in vivo.15 It inhibited proliferation and caused apoptosis in cells of numerous cancers, including breast cancer,16 colon cancer,17 non-small cell lung cancer,18 cervical cancer,19 multiple myeloma,20 pancreatic cancer,21 melanoma,22 and prostate cancer.23 UA has been shown to prevent CCl4-induced hepatotoxicity and fibrosis via Nrf2/ARE pathway.24 In this study, we tested the effect of UA to sensitize cisplatin-resistant human hepatocarcinoma HepG2/DDP cells to cisplatin-induced cytotoxicity and identified the underlying mechanism of its action. The results demonstrated that the combination of UA with low dose of cisplatin exhibited significantly higher cytotoxic response in HepG2/DDP cells. Mechanistically, UA–cisplatin combination significantly decreased the level of Nrf2 and its downstream genes, leading to a reversal of cisplatin-resistant phenotype in HepG2/DDP cells. Thus, UA is considered to be a promising adjuvant sensitizer to overcome chemoresistance.
 | Figure 1 Chemical structure of UA. |
Materials and methods
Regents
HepG2 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). HepG2/DDP cells were obtained from the Cell Bank, Chinese Academy of Sciences, Shanghai, People’s Republic of China. Cisplatin, UA, and dichloro-dihydro-fluorescein diacetate were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Cell Counting Kit 8 (CCK8), JC-1 dye, and BCA Protein Assay Kit were obtained from Beyotime (Nantong, People’s Republic of China). Annexin V Fluorescein Isothiocyanate Apoptosis KGA107 Detection Kit was purchased from KeyGene (Nanjing, People’s Republic of China). Dulbecco’s Modified Eagle’s Medium and fetal bovine serum were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Polyvinylidene difluoride (PVDF) membrane was obtained from EMD Millipore (Billerica, MA, USA). Anti-Nrf2, anti-HO-1, anti-NQO1, anti-GST, anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody, and horseradish-peroxidase-conjugated secondary antibody were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA).
Cell culture
HepG2 cells were maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in a humidified 5% CO2 atmosphere. HepG2/DDP cells were maintained in the same medium and then treated with 0.03 μM cisplatin for at least 4 weeks before the experiment.25
Cell proliferation assay
Cell proliferation was detected using the CCK8 according to the manufacturer’s protocol. The cells were seeded at 1×103 per well in a 96-well plate and treated as experiment design. Then, 10 μL of CCK8 reagent was added to each well and incubated for 4 hours at 37°C. VMax® microplate spectrophotometer (Molecular Devices LLC, Sunnyvale, CA, USA) was used to measure the absorbance at 490 nm.
Cell apoptosis assay
The Annexin-V Fluorescein Isothiocyanate Apoptosis KGA107 Detection Kit for flow cytometry fluorescence-activated cell sorting (FACS; BD Biosciences, San Jose, CA, USA) was used. In this protocol, 5×105 cells were seeded and washed twice with phosphate-buffered saline, and then mixed with 500 μL binding buffer to form a cell suspension. Annexin-V Fluorescein Isothiocyanate 5 μL was mixed with 5 μL of propidium iodide. The cells were incubated for 10 minutes at room temperature in the dark and analyzed immediately using an FACS.
Cell cycle analysis
The cells were plated at 2×105 per well in a six-well plate. After 24 hours, cells were harvested and fixed with ice cold 70% ethanol at −4°C overnight. Then, the cells were incubated with 10 mg/mL RNase A, 400 mg/mL propidium iodide, and 0.1% Triton-X in phosphate-buffered saline at room temperature for 30 minutes and analyzed immediately using an FACS.
Measurement of reactive oxygen species
According to manufactures’ protocol, the level of reactive oxygen species (ROS) was measured by dichloro-dihydro-fluorescein diacetate. Briefly, cells were incubated with dichloro-dihydro-fluorescein diacetate (final concentration of 10 μM) at room temperature in the dark for 30 minutes, then washed with cold Hank’s balanced salt solution (pH 7.2), and then analyzed immediately using a FACS.
Measurement of mitochondrial membrane potential
According to the manufacturer’s instruction, mitochondrial membrane potential (MMP) was determined using JC-1 dye. Briefly, the cells (5×104 per well) were plated in six-well plates. After treatment, cells were collected and mixed with 500 mL JC-1 working solution at room temperature in the dark for 15–20 minutes. Then, the cells were resuspended in 500 μL 1× incubation buffer, and analyzed immediately using a FACS.
Western blotting
The cell lysates were harvested in lysis buffer. Total protein content was analyzed using the BCA Protein Assay Kit. Samples were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrotransferred to the PVDF membrane. The PVDF membrane was incubated with 10% nonfat dried milk in Tris-buffered saline (TBS; 100 mM NaCl, 10 mM Tris–HCl, pH 7.4) containing 0.1% Tween 20 (TBST) for 2 hours. Overnight incubation was performed with anti-Nrf2 (diluted 1:2,000), anti-HO-1 (diluted 1:2,000), anti-NQO1 (1:2,000), anti-GST (diluted 1:2,000), or anti-GAPDH antibody (diluted 1:10,000) at 4°C, followed by incubation with horseradish-peroxidase-conjugated secondary antibodies. After rinsing in Tris-buffered saline containing 0.1% Tween 20, the PVDF membrane was exposed to an X-ray film using Western blot detection reagents (Thermo Fisher Scientific). Gel band density was scanned using the Gel Doc 2000® system and analyzed by Quantity One® image software, version 5.2.1 (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Transient transfection with Nrf2 small interfering RNA or plasmids
The small interfering RNA (siRNA) targeting human Nrf2 (5′-GAGUUACAGUGUCUUAAUA-3′), the nontargeting negative control siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′), the plasmid pGL3-Nrf2, and empty vector DNA were obtained from Shanghai GenePharma (Shanghai, People’s Republic of China). Cells were plated at a density of 1.5×105 cells/well in six-well plates. After 24 hours, cells were transfected with siRNA or plasmid mixed with Lipofectamine 2000 (Thermo Fisher Scientific).
Statistical analysis
All the data were expressed as mean ± standard deviation. The statistical analysis was performed using one-way analysis of variance or two-tailed Student’s t-test for multiple comparisons. The differences between comparisons were considered to be statistically significant at P<0.05. SPSS software version 18.0 (SPSS Inc., Chicago, IL, USA) was used for data analysis. All experiments were performed in triplicate.
Results
Nrf2 was overexpressed in cisplatin-resistant human hepatocellular carcinoma HepG2/DDP cells
Cell survival rates were detected by CCK8 assay to observe the cisplatin resistance phenotype of cells exposed to cisplatin. HepG2 cells were treated with series concentration of cisplatin (0.1–25.6 μg/mL) for 48 hours (Figure 2A), and HepG2/DDP cells were treated with series concentration of cisplatin (2–512 μg/mL) for 48 hours (Figure 2B). Then, the inhibition rate of the cells was calculated. The results revealed that the 50% inhibitory concentration (IC50) of HepG2/DDP cells was higher than that of HepG2 cells. To evaluate whether Nrf2 and its substrates were different between HepG2 and HepG2/DDP cells, Western blot assay was performed. The level of Nrf2 and its downstream target genes HO-1, NQO1, and GST in HepG2/DDP cells was significantly higher than that in HepG2 cells (Figure 2C).
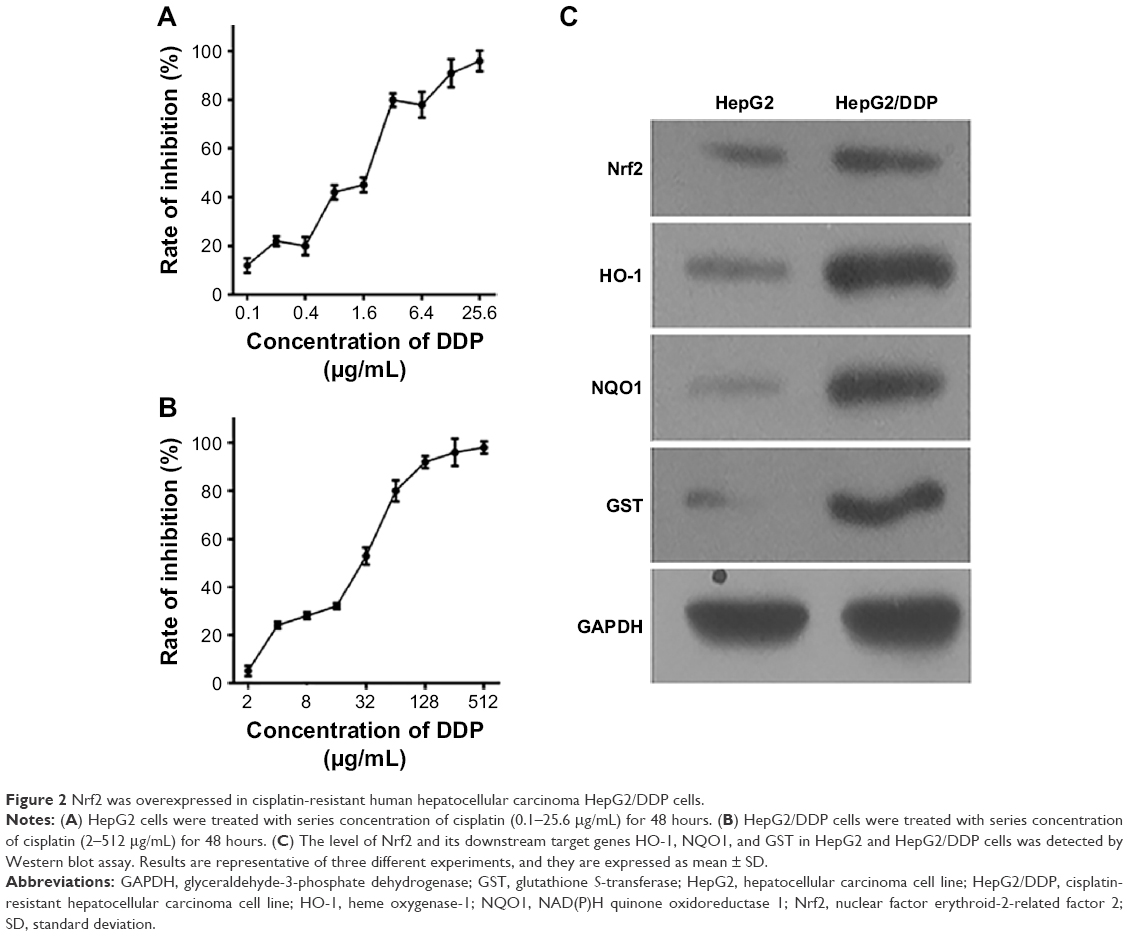 | Figure 2 Nrf2 was overexpressed in cisplatin-resistant human hepatocellular carcinoma HepG2/DDP cells. |
UA potentiates cisplatin-induced growth inhibition
As shown in Figure 3A, HepG2 and HepG2/DDP cells were treated with increasing concentrations of UA (1.125–288 μg/mL) for 48 hours. Then, the inhibition rate of the cells was calculated. It was found that the proliferation of HepG2 and HepG2/DDP cells was inhibited by UA after 48-hour incubation. We chose 1.125 μg/mL and 2.25 μg/mL of UA for the subsequent study as low cytotoxicity occurred at these two concentrations, thus excluding the anti-proliferation effect of high-dose UA in cancer cells.
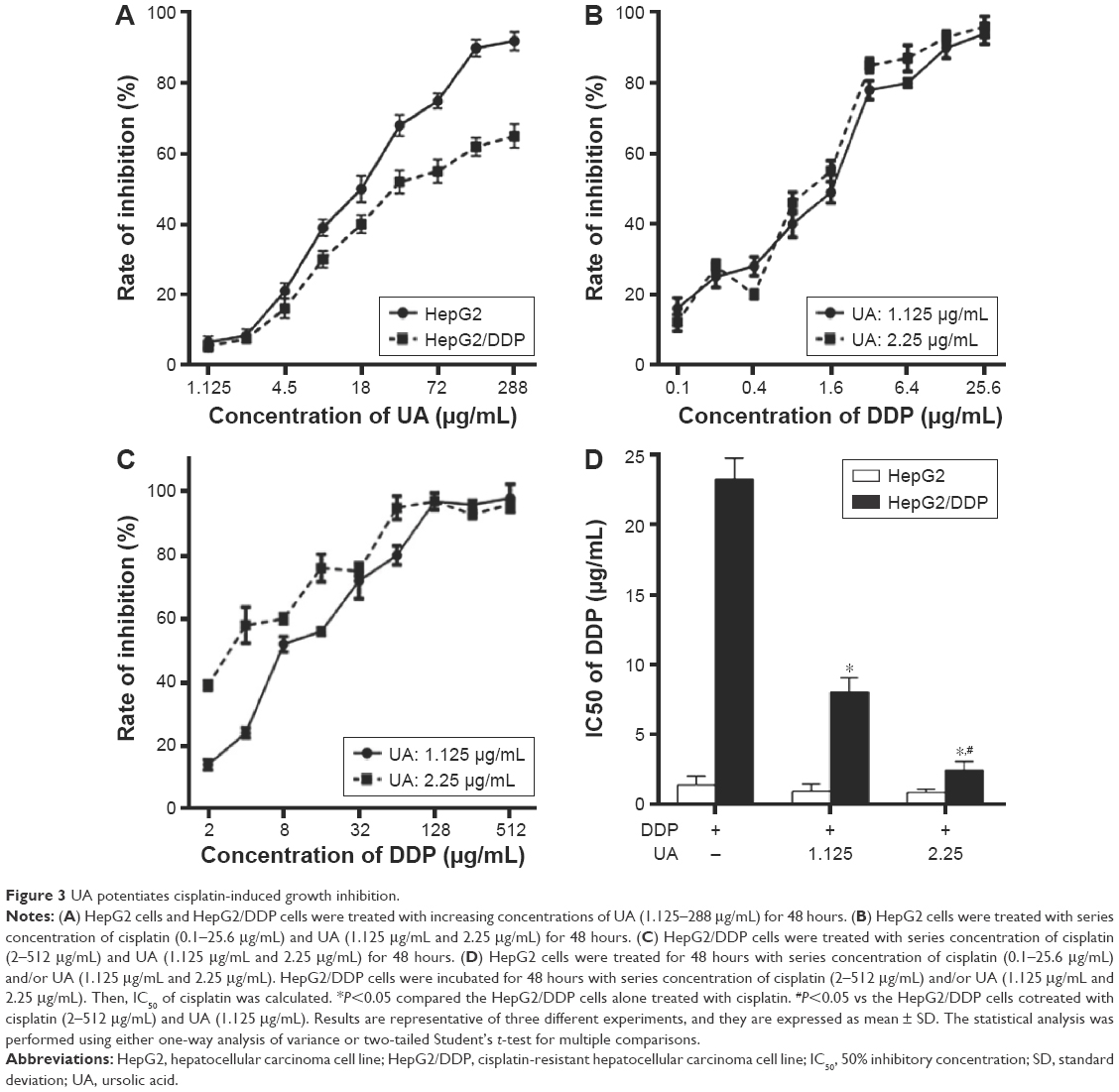 | Figure 3 UA potentiates cisplatin-induced growth inhibition. |
To investigate whether UA can increase the sensitivity of HepG2/DDP cells to cisplatin, HepG2 cells were treated with series concentration of cisplatin (0.1–25.6 μg/mL) and/or UA (1.125 μg/mL and 2.25 μg/mL) for 48 hours (Figure 3B), and HepG2/DDP cells were treated with series concentration of cisplatin (2–512 μg/mL) and/or UA (1.125 μg/mL and 2.25 μg/mL) for 48 hours (Figure 3C). Then, IC50 was calculated. The results showed that the reversal index was 2.9- and 9.69-fold by UA of 1.125 μg/mL and 2.25 μg/mL, respectively, for cisplatin (Figure 3D).
UA–cisplatin combination enhances low-dose cisplatin-induced apoptosis and causes G0/G1 arrest in resistant cells
HepG2/DDP cells were treated with dimethyl sulfoxide as control group (final concentration of 0.1%). HepG2/DDP cells were treated with 8.92 μg/mL cisplatin (30% inhibitory concentration [IC30] of cisplatin for HepG2/DDP cells) and/or UA (2.25 μg/mL) for 48 hours. The results showed that the proportion of apoptotic cells in the groups treated with UA and low-dose cisplatin combination was increased when compared to the groups treated with low-dose cisplatin (P<0.01; Figure 4A). As shown in Figure 4B, the groups treated with UA and low-dose cisplatin combination enhanced the cells in G0/G1 phase, and reduced the number of cells in G2/M phases (P<0.05). These results demonstrate that UA increased the sensitivity of HepG2/DDP cells to low-dose cisplatin by retaining cells in G0/G1 phase and promoting cell apoptosis.
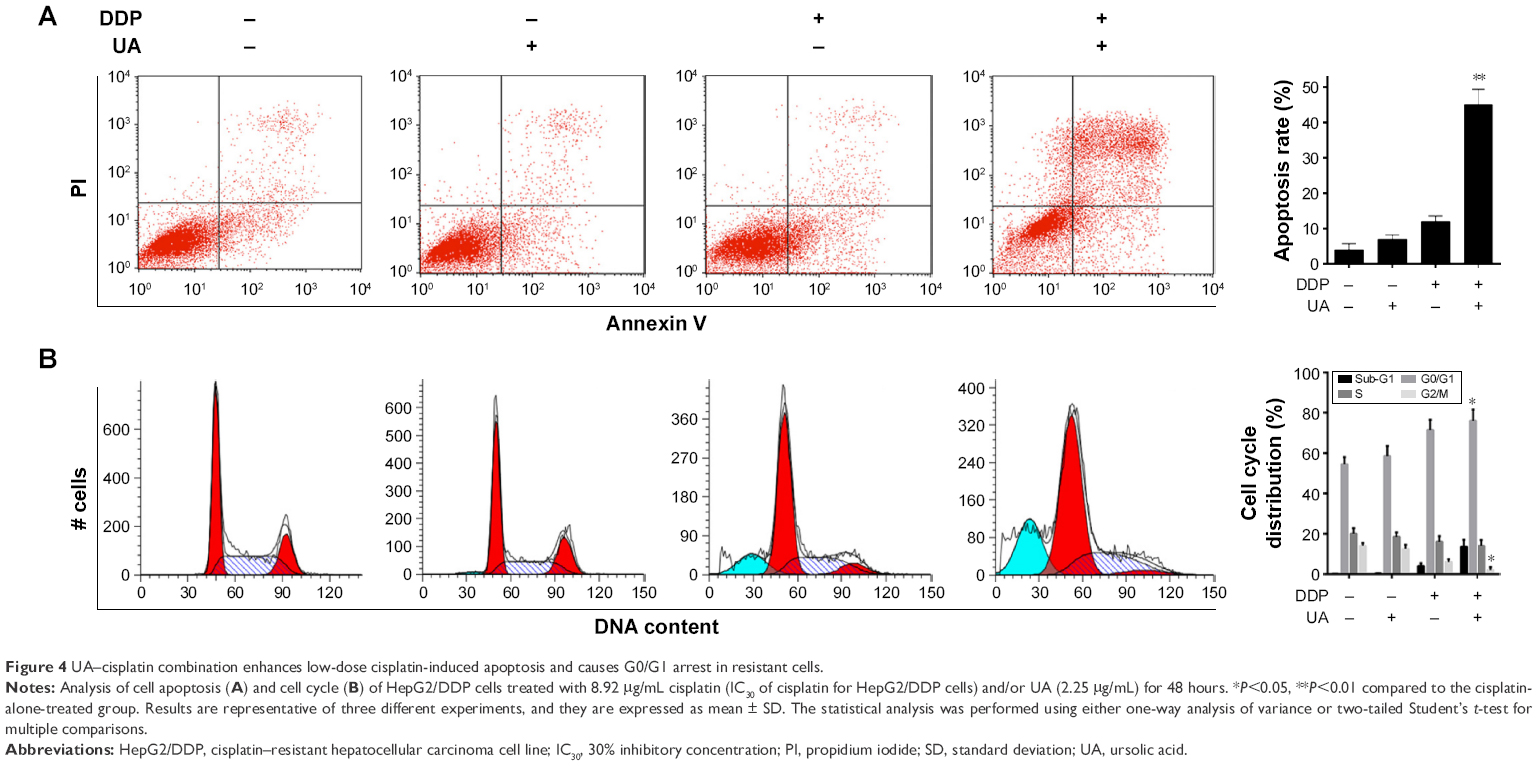 | Figure 4 UA–cisplatin combination enhances low-dose cisplatin-induced apoptosis and causes G0/G1 arrest in resistant cells. |
UA–cisplatin combination increases low-dose cisplatin-induced mitochondrial oxidative stress in resistant cells
To explore the sensibilization of UA on HepG2/DDP cells to low-dose cisplatin, the level of ROS and MMP was detected. The results showed that the level of ROS in the groups treated with UA and low-dose cisplatin combination was increased when compared to the groups treated with cisplatin (P<0.01; Figure 5A). In addition, we used JC-1 dye and the flow cytometry system to analyze the MMP (Figure 5B). The results showed that cells with low MMP in the groups of HepG2/DDP cells treated with UA and low-dose cisplatin combination increased markedly compared to the groups treated with low-dose cisplatin (P<0.01).
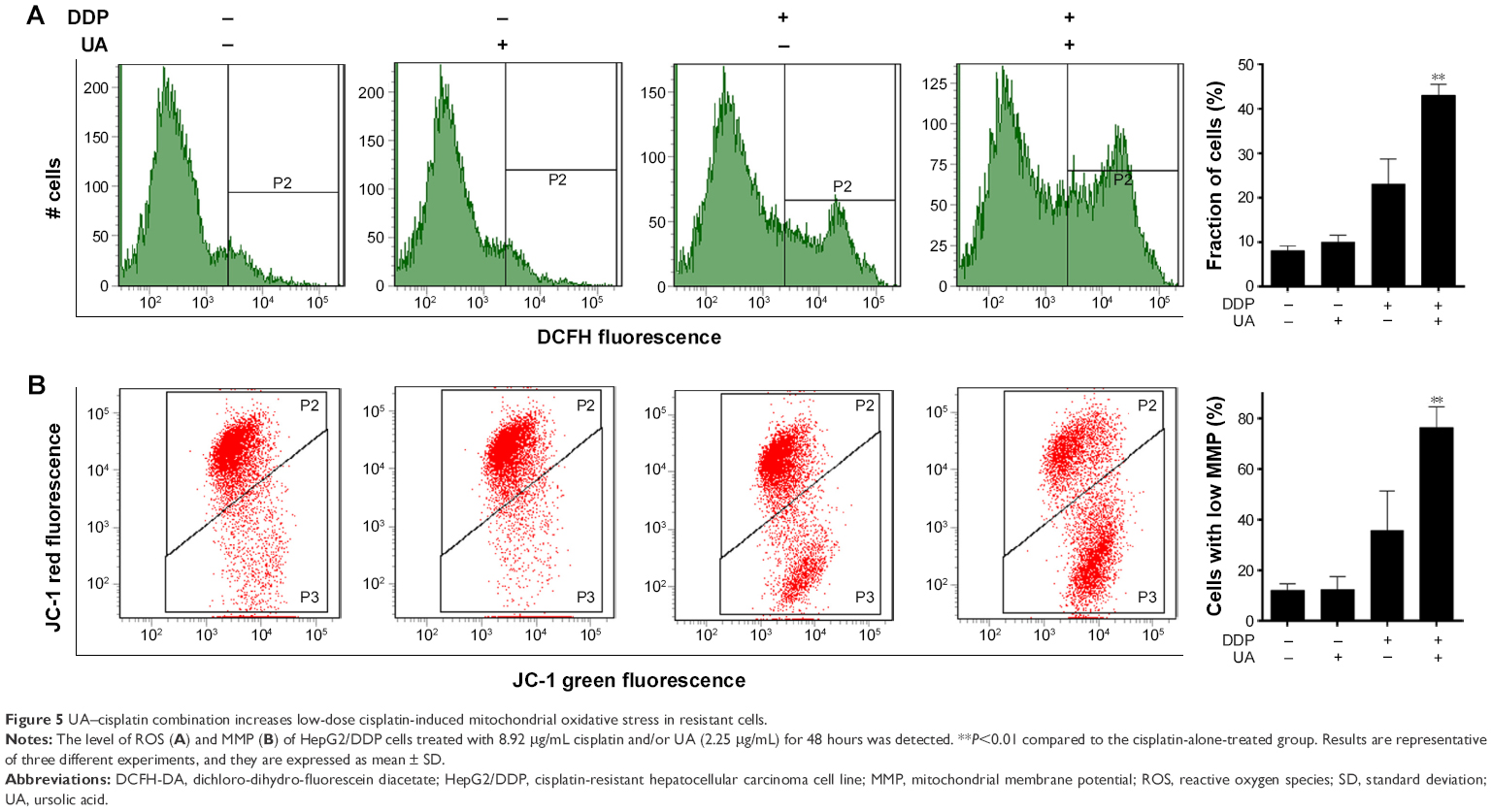 | Figure 5 UA–cisplatin combination increases low-dose cisplatin-induced mitochondrial oxidative stress in resistant cells. |
UA–cisplatin combination downregulates Nrf2 and its substrates
To investigate the regulation mechanism of the sensibilization of UA on HepG2/DDP cells to low-dose cisplatin, the protein expression levels of Nrf2, HO-1, NQO1, and GST were detected by Western blot analysis. As shown in Figure 6, the expression levels of Nrf2, HO-1, NQO1, and GST were decreased in the groups treated with UA and low-dose cisplatin combination compared with the groups treated with low-dose cisplatin.
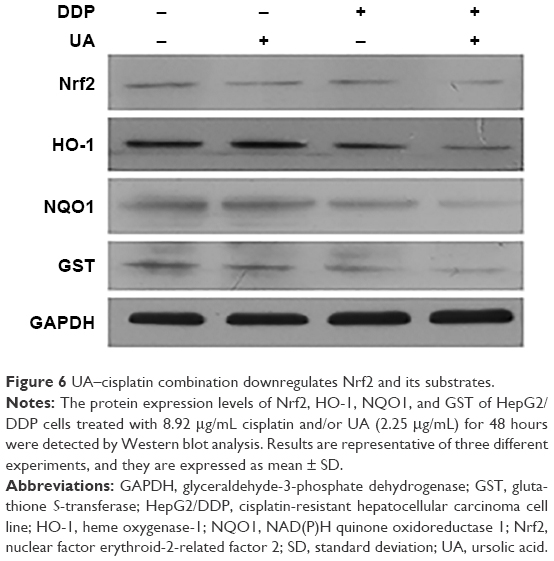 | Figure 6 UA–cisplatin combination downregulates Nrf2 and its substrates. |
UA sensitizes HepG2/DDP cells to low-dose cisplatin via inhibition of Nrf2/ARE signaling pathway
To investigate the potential involvement of Nrf2/ARE signaling pathway, HepG2/DDP cells were transfected with Nrf2 siRNA (si-Nrf2) or Nrf2 complementary DNA (Nrf2 cDNA) and then treated with 8.92 μg/mL cisplatin (IC30 of cisplatin for HepG2/DDP cells) and/or UA (2.25 μg/mL) for 48 hours. The level of Nrf2, HO-1, NQO1, and GST was detected by Western blot analysis. As shown in Figure 7A, when compared to negative control (si-Con) transfected groups, the protein level of Nrf2, HO-1, NQO1, and GST was successfully downregulated in si-Nrf2 transfected groups. Furthermore, UA and low-dose cisplatin combination did not significantly influence the level of Nrf2, HO-1, NQO1, and GST in si-Nrf2-transfected HepG2/DDP cells. As shown in Figure 7C, when compared to empty vector (vector) transfected groups, the protein level of Nrf2, HO-1, NQO1, and GST was successfully upregulated in Nrf2 cDNA-transfected groups. Furthermore, UA and low-dose cisplatin combination significantly decreased the protein expression of Nrf2, HO-1, NQO1, and GST in Nrf2 cDNA-transfected HepG2/DDP cells.
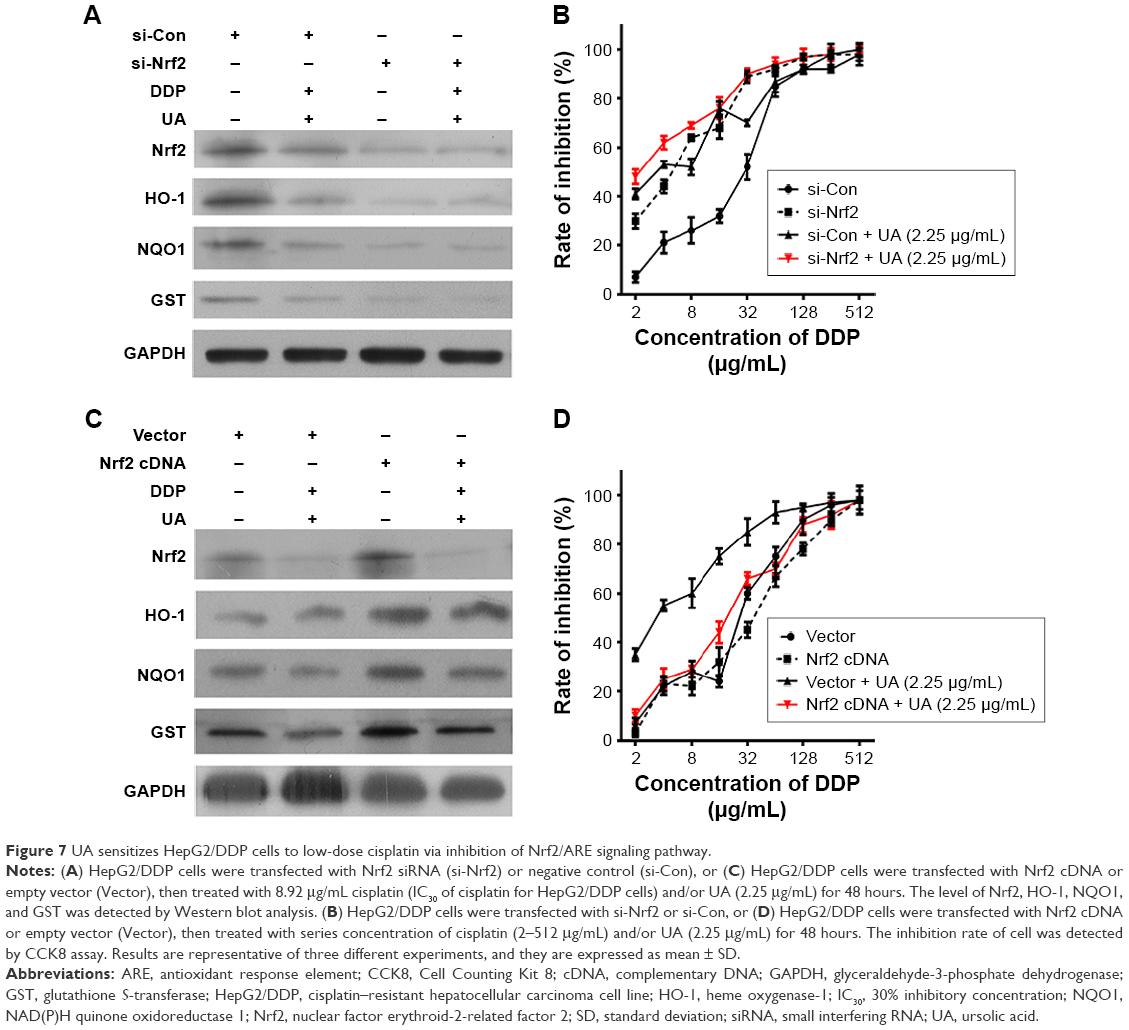 | Figure 7 UA sensitizes HepG2/DDP cells to low-dose cisplatin via inhibition of Nrf2/ARE signaling pathway. |
Then, we observed the sensibilization of UA to cisplatin in HepG2/DDP cells with Nrf2 knockdown or overexpression. HepG2/DDP cells were transfected with si-Nrf2 or Nrf2 cDNA, and then treated with series concentration of cisplatin (2–512 μg/mL) and/or UA (2.25 μg/mL) for 48 hours. Transfection with si-Nrf2 in HepG2/DDP cells was much more sensitive to cisplatin than in HepG2/DDP cells (Figure 7B). Figure 7D shows that Nrf2 cDNA transfection did not significantly affect the chemoresistance of HepG2/DDP cells. However, the sensibilization of UA–cisplatin combination was diminished in si-Nrf2-transfected HepG2/DDP cells (Figure 7B), as well as in Nrf2 cDNA-transfected HepG2/DDP cells (Figure 7D).
Discussion
Drug resistance is one of the main hurdles to effective therapy of numerous tumors, which is taken into account during chemotherapy.2 Even a small increase in chemotherapy drug can cause severe toxicity to dose-limiting normal tissue.2 Thus, to accomplish higher curability with least toxicity during chemotherapy, strategies using dual agents rather than using single agents represent the most useful alternative. Various natural products have been considered as potential sources of chemosensitizer. Gao et al26 reported that “apigenin sensitized doxorubicin-resistance BEL-7402/ADM cells to doxorubicin”. Chian et al27 discovered that “luteolin sensitized non-small cell lung cancer cell lines and colorectal cancer cells to oxaliplatin, bleomycin, and doxorubicin”. Hou et al9 found that “3′,4′,5′,5,7-pentamethoxyflavone sensitized cisplatin-resistant lung carcinoma A549 cells to cisplatin”. UA is one of the natural pentacyclic triterpene compounds. It has numerous pharmacological activities including antioxidant, anti-inflammatory, antirheumatic, antiviral, and anticancer properties. In this study, we investigated the potential of UA to sensitize the cisplatin-resistant human hepatocellular carcinoma cells to cisplatin-induced cytotoxicity. We demonstrated that UA reduces the recalcitrance of HepG2/DDP cells to low-dose cisplatin-induced cytotoxicity via downregulation of Nrf2/ARE signaling pathway.
The anticancer effect of cisplatin is:
binding to DNA and causing the DNA strands to crosslink, which ultimately triggers cells to die in a programmed way including the suppression of DNA synthesis, the restraint of RNA transcription, the blocking of cell cycle, and cell apoptosis.28
Antiproliferative activity of UA which related to cell apoptosis and cell cycle arrest has been exhibited in several cancers. Mahmoudi et al29 reported that:
UA significantly suppressed cell proliferation after 24 and 48 h in the presence of UA, and increased apoptosis by enhancing the Annexin-V-positive population of isolated human melanoma cells.
Yang et al30 reported that “UA significantly arrested cycle in G0/G1 phase of hepatocellular carcinoma cells”. However, the underlying mechanism of synergized antiproliferative effects of UA–cisplatin on cancer cells is not completely understood. We observed an increase in low-dose cisplatin-induced apoptosis and an accumulation of G0/G1 cell population in UA–cisplatin-treated HepG2/DDP cisplatin-resistant human hepatocellular carcinoma cells. These results imply that the combined treatment of low-dose UA and cisplatin manifests as the synergetic growth inhibitory effect on cisplatin-resistant cells.
In the cytoplasm, cisplatin interacts with glutathione, metallothioneins, or mitochondrial proteins such as the voltage-dependent anion channel, resulting in the generation of ROS; ROS can directly trigger membrane permeabilization, thereby triggering cisplatin-induced cell death; quenching proapoptotic ROS is known to be responsible for cisplatin resistance.28 Recent research has suggested that:
UA enhances radiation effects by increasing ROS, downregulating Ki-67 level, and improving apoptosis in BGC-823 human adenocarcinoma gastric cancer cells.31
Kim et al32 reported that UA induced apoptosis by decreasing MMP as shown with JC-1 staining in breast cancer cells. In our study, we observed that UA–cisplatin combination increased low-dose cisplatin-induced mitochondrial oxidative stress in resistant cells. Our findings suggested that an increase in ROS and a decrease in MMP are responsible for, at least in part, cisplatin resistance reduced by UA.
Nrf2 is known to be a pharmacological target to overcome drug resistance.33 In our study, the expression level of Nrf2 and its downstream target genes HO-1, NQO1, and GST in HepG2/DDP cells was significantly higher than that in HepG2 cells. The expression level of Nrf2-mediated pathway showed a decrease in Nrf2 siRNA-transfected HepG2/DDP cells, which led to slight recovery of sensitivity to treatment with cisplatin. Our findings suggest a crucial role of Nrf2/ARE signaling pathway in the chemoresistance of human hepatocellular carcinoma cells. Therefore, the identification of potent small-molecule inhibitors of Nrf2 is desirable. Nrf2 is the potential drug target of several pentacyclic triterpene compounds, including UA. The study by Ma et al24 suggested that:
UA protected liver from carbon tetrachloride (CCL4)-induced hepatotoxicity and liver fibrosis through Nrf2/ARE pathway in an experimental mice model.
Moreover, earlier studies showed that:
UA relieved cytotoxicity and the stimulation of external deleterious factors, reverted the intracellular redox balance in a cigarette-smoke-extract-induced human bronchial epithelial cell model; UA reduced cigarette-smoke-extract-induced DNA damage via the Nrf2 pathway.34
In this study, we confirmed UA as an available Nrf2 inhibitor. UA–cisplatin treatment sensitized HepG2/DDP cells to low-dose cisplatin and inhibited the level of Nrf2 and its substrates. However, in Nrf2 siRNA- or Nrf2 cDNA-transfected HepG2/DDP cells, the sensibilization of UA–cisplatin combination was diminished, indicating that suppression of Nrf2/ARE signaling pathway was one of the main mechanisms by which UA can weaken chemoresistance.
Many articles have reported that other signaling pathways also regulate the Nrf2/ARE pathway, including various transcription factors such as peroxisome proliferator-activated receptor, estrogen receptor α, and activating transcription factor 3.35 Therefore, agents that downregulate Nrf2/ARE pathway might activate these transcription factors directly in cancer cells. Whether the regulatory mechanism of Nrf2/ARE pathway by UA–cisplatin combination is related to these transcription factors has yet to be studied.
Conclusion
Our data experimentally showed the chemosensitization of UA on hepatocellular carcinoma cisplatin-resistant HepG2/DDP cells to low-dose cisplatin via Nrf2/ARE pathway and suggested that UA as a possible natural adjuvant sensitizer might have clinical significance with therapeutic capability on overcoming cisplatin-resistant hepatocellular carcinoma cells. However, different hepatocellular carcinoma cells, or even more other cancer cells, should be used for further study to see whether it is a universal phenomenon, or it occurs only in this cell line. Recently, many researches have suggested that the overcoming cisplatin resistance by using different adjuvant molecules and nanoparticle technology,36,37 the comparison between these new drugs and UA–cisplatin combination should be studied in the future. Further safety and efficacy investigations in animal experiment and clinical trials are essential before UA can be used as a chemosensitizer in the treatment of liver cancer.
Acknowledgment
The authors are thankful to the members of our laboratories for their insight and technical support.
Disclosure
The authors report no conflicts of interest in this work.
References
Buendia MA, Neuveut C. Hepatocellular carcinoma. Cold Spring Harb Perspect Med. 2015;5(2):a021444. | ||
Gordon RR, Nelson PS. Cellular senescence and cancer chemotherapy resistance. Drug Resist Updat. 2012;15(1–2):123–131. | ||
Xu W, Shao L, Zhou C, Wang H, Guo J. Upregulation of Nrf2 expression in non-alcoholic fatty liver and steatohepatitis. Hepatogastroenterology. 2011;58(112):2077–2080. | ||
Liu J, Wu KC, Lu YF, Ekuase E, Klaassen CD. Nrf2 protection against liver injury produced by various hepatotoxicants. Oxid Med Cell Longev. 2013;2013:305861. | ||
Wu KC, Liu JJ, Klaassen CD. Nrf2 activation prevents cadmium-induced acute liver injury. Toxicol Appl Pharmacol. 2012;263(1):14–20. | ||
Guan L, Zhang L, Gong Z, et al. FoxO3 inactivation promotes human cholangiocarcinoma tumorigenesis and chemoresistance through Keap1-Nrf2 signaling. Hepatology. 2016;63(6):1914–1927. | ||
Yao J, Wei X, Lu Y. Chaetominine reduces MRP1-mediated drug resistance via inhibiting PI3K/Akt/Nrf2 signaling pathway in K562/Adr human leukemia cells. Biochem Biophys Res Commun. 2016;473(4):867–873. | ||
Furfaro AL, Piras S, Domenicotti C, et al. Role of Nrf2, HO-1 and GSH in neuroblastoma cell resistance to bortezomib. PLoS One. 2016;11(3):e0152465. | ||
Hou X, Bai X, Gou X, et al. 3′,4′,5′,5,7-pentamethoxyflavone sensitizes Cisplatin-resistant A549 cells to Cisplatin by inhibition of Nrf2 pathway. Mol Cells. 2015;38(5):396–401. | ||
Syu JP, Chi JT, Kung HN. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget. 2016;7(12):14659–14672. | ||
Hong YB, Kang HJ, Kwon SY, et al. Nuclear factor (erythroid-derived 2)-like 2 regulates drug resistance in pancreatic cancer cells. Pancreas. 2010;39(4):463–472. | ||
Qin X, Qiu C, Zhao L. Maslinic acid protects vascular smooth muscle cells from oxidative stress through Akt/Nrf2/HO-1 pathway. Mol Cell Biochem. 2014;390(1–2):61–67. | ||
Ding Y, Chen M, Wang M, et al. Neuroprotection by acetyl-11-keto-beta-Boswellic acid, in ischemic brain injury involves the Nrf2/HO-1 defense pathway. Sci Rep. 2014;4:7002. | ||
Chen P, Zeng H, Wang Y, et al. Low dose of oleanolic acid protects against lithocholic acid-induced cholestasis in mice: potential involvement of nuclear factor-E2-related factor 2-mediated upregulation of multidrug resistance-associated proteins. Drug Metab Dispos. 2014;42(5):844–852. | ||
Wozniak L, Skapska S, Marszalek K. Ursolic acid – a pentacyclic triterpenoid with a wide spectrum of pharmacological activities. Molecules. 2015;20(11):20614–20641. | ||
Zhao C, Yin S, Dong Y, et al. Autophagy-dependent EIF2AK3 activation compromises ursolic acid-induced apoptosis through upregulation of MCL1 in MCF-7 human breast cancer cells. Autophagy. 2013;9(2):196–207. | ||
Kim JH, Kim YH, Song GY, et al. Ursolic acid and its natural derivative corosolic acid suppress the proliferation of APC-mutated colon cancer cells through promotion of beta-catenin degradation. Food Chem Toxicol. 2014;67:87–95. | ||
Wu J, Zhao S, Tang Q, et al. Activation of SAPK/JNK mediated the inhibition and reciprocal interaction of DNA methyltransferase 1 and EZH2 by ursolic acid in human lung cancer cells. J Exp Clin Cancer Res. 2015;34:99. | ||
Leng S, Hao Y, Du D, et al. Ursolic acid promotes cancer cell death by inducing Atg5-dependent autophagy. Int J Cancer. 2013;133(12):2781–2790. | ||
Pathak AK, Bhutani M, Nair AS, et al. Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Mol Cancer Res. 2007;5(9):943–955. | ||
Leal AS, Wang R, Salvador JA, Jing Y. Synthesis of novel ursolic acid heterocyclic derivatives with improved abilities of antiproliferation and induction of p53, p21waf1 and NOXA in pancreatic cancer cells. Bioorg Med Chem. 2012;20(19):5774–5786. | ||
Junco JJ, Mancha-Ramirez A, Malik G, et al. Ursolic acid and resveratrol synergize with chloroquine to reduce melanoma cell viability. Melanoma Res. 2015;25(2):103–112. | ||
Meng Y, Lin ZM, Ge N, Zhang DL, Huang J, Kong F. Ursolic acid induces apoptosis of prostate cancer cells via the PI3K/Akt/mTOR pathway. Am J Chin Med. 2015;43(7):1471–1486. | ||
Ma JQ, Ding J, Zhang L, Liu CM. Protective effects of ursolic acid in an experimental model of liver fibrosis through Nrf2/ARE pathway. Clin Res Hepatol Gastroenterol. 2015;39(2):188–197. | ||
Wang XB, Wang SS, Zhang QF, et al. Inhibition of tetramethylpyrazine on P-gp, MRP2, MRP3 and MRP5 in multidrug resistant human hepatocellular carcinoma cells. Oncol Rep. 2010;23(1):211–215. | ||
Gao AM, Ke ZP, Wang JN, Yang JY, Chen SY, Chen H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis. 2013;34(8):1806–1814. | ||
Chian S, Li YY, Wang XJ, Tang XW. Luteolin sensitizes two oxaliplatin-resistant colorectal cancer cell lines to chemotherapeutic drugs via inhibition of the Nrf2 pathway. Asian Pac J Cancer Prev. 2014;15(6):2911–2916. | ||
Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–1883. | ||
Mahmoudi M, Rabe SZ, Balali-Mood M, Karimi G, Tabasi N, Riahi-Zanjani B. Ursolic acid induced apoptotic cell death following activation of caspases in isolated human melanoma cells. Cell Biol Int. 2015;39(2):230–236. | ||
Yang LJ, Tang Q, Wu J, et al. Inter-regulation of IGFBP1 and FOXO3a unveils novel mechanism in ursolic acid-inhibited growth of hepatocellular carcinoma cells. J Exp Clin Cancer Res. 2016;35(1):59. | ||
Yang Y, Jiang M, Hu J, et al. Enhancement of radiation effects by ursolic acid in BGC-823 human adenocarcinoma gastric cancer cell line. PLoS One. 2015;10(7):e0133169. | ||
Kim KH, Seo HS, Choi HS, Choi I, Shin YC, Ko SG. Induction of apoptotic cell death by ursolic acid through mitochondrial death pathway and extrinsic death receptor pathway in MDA-MB-231 cells. Arch Pharm Res. 2011;34(8):1363–1372. | ||
Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16(2):123–140. | ||
Liu W, Tan X, Shu L, et al. Ursolic acid inhibits cigarette smoke extract-induced human bronchial epithelial cell injury and prevents development of lung cancer. Molecules. 2012;17(8):9104–9115. | ||
Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34(4):176–188. | ||
Johnstone TC, Suntharalingam K, Lippard SJ. The next generation of platinum drugs: targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) Prodrugs. Chem Rev. 2016;116(5):3436–3486. | ||
Pathak RK, Dhar S. A nanoparticle cocktail: temporal release of predefined drug combinations. J Am Chem Soc. 2015;137(26):8324–8327. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.