")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Ulinastatin Inhibits the Proliferation, Invasion and Phenotypic Switching of PDGF-BB-Induced VSMCs via Akt/eNOS/NO/cGMP Signaling Pathway
Authors Huang C, Huang W, Wang R , He Y
Received 4 August 2020
Accepted for publication 4 October 2020
Published 14 December 2020 Volume 2020:14 Pages 5505—5514
DOI https://doi.org/10.2147/DDDT.S275488
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Tin Wui Wong
Cheng Huang, Weihui Huang, Rui Wang, Yongli He
Department of Cardiology, Guangdong Cardiovascular Institute, Guangdong Provincial Key Laboratory of Coronary Heart Disease Prevention, Guangdong Academy of Medical Sciences, Guangzhou, Guangdong 510055, People’s Republic of China
Correspondence: Cheng Huang
Guangdong Cardiovascular Institute, Guangdong Provincial Key Laboratory of Coronary Heart Disease Prevention, Guangdong Academy of Medical Sciences, Dongchuan Road, 96#, Guangzhou, Guangdong 510055, People’s Republic of China
Email [email protected]
Background: Atherosclerosis is a chronic inflammatory disease responsible for thrombosis, blood supply disorders, myocardial infarction and strokes, eventually leading to increased deaths and reduced quality of life. As inflammation plays a vital role in the development of this disease, the present study aims to investigate whether urinary trypsin inhibitor (UTI) with anti-inflammatory property can inhibit the proliferation, invasion and phenotypic switching of PDGF-BB-induced vascular smooth muscle cells (VSMCs) and probe its potential mechanism.
Methods: Western blot was used to detect the expressions of the proteins related to the Akt/eNOS/NO/cGMP signaling pathway, phenotypic switching and proliferation. CCK-8 assay and EdU staining were used to detect cell proliferation of VSMCs. Transwell and wound healing assays were respectively conducted to measure the invasion and migration of VSMCs. The concentration of NO was evaluated by NO detection kit. ELISA assay analyzed the expression of cyclic GMP (cGMP).
Results: The expressions of p-Akt and p-eNOS were elevated by UTI treatment. Furthermore, UTI inhibited the proliferation, migration and invasion of VSMCs. UTI also increased the expressions of proteins related to phenotypic switching. The amount of NO and expression of cGMP were both elevated under UTI treatment.
Conclusion: UTI inhibits the proliferation, invasion and phenotypic switching of PDGF-BB-induced VSMCs via Akt/eNOS/NO/cGMP signaling pathway, which might provide a theoretical basis for the UTI treatment of atherosclerosis.
Keywords: ulinastatin, atherosclerosis, proliferation, invasion, phenotypic switching, Akt/eNOS/NO/cGMP signaling pathway
Introduction
As a chronic inflammatory disease, atherosclerosis is accountable for other cardiovascular diseases such as thrombosis, blood supply disorders, myocardial infarction and strokes.1,2 Characterized by lipid accumulation in the vessel wall, atherosclerosis brings about high morbidity and lethality, leading to a large amount of deaths and loss of life quality among patients and their families.3,4 Despite great expenditure and decades of research, the underlying mechanisms triggering the occurrence, progression and the clinical events at the end stage, such as plaque rupture and myocardial infarction, still puzzle numerous researchers. Thus, it is urgent to take measures to understand the complex mechanism in this disease.5
Urinary trypsin inhibitor (UTI) is also called as “ulinastatin”, a kind of glycoprotein and a nonspecific wide-spectrum protease inhibitor.6,7 It could reduce pro-inflammatory cytokines and increase anti-inflammatory cytokines to protect the organs of the patients and alleviate tissue injury.8–10 What is more, existing evidence has illustrated that the inhibitory impacts of UTI on inflammation and oxidative stress could attenuate ischemia-reperfusion injury and lower the generation of oxygen free radicals. UTI has been extensively used to treat various diseases, such as pancreatitis, sepsis, and hemorrhagic shock.11 Considering that the contribution of inflammatory cells to the occurrence and development of atherosclerosis have been emphasized by numerous studies, drugs with anti-inflammatory property may be an excellent option for its treatment.12
In mature and normal blood vessels, vascular smooth muscle cells (VSMCs) present contractile phenotype, with elevated levels of contractile markers such as α-smooth muscle actin (α-SMA) and smooth muscle 22 alpha (SM22α).13 However, when VSMCs are stimulated by cytokines like platelet-derived growth factor BB (PDGF-BB), they will convert to synthetic phenotype, along with enhanced dedifferentiation, proliferation and migration, evoking the occurrence of atherosclerosis.14 Since proliferation and migration of vascular smooth muscle cells (VSMCs) are the crucial factors for atherosclerosis, and phenotypic switching is believed to be a common vascular pathological condition in atherosclerosis, we intended to test the influences of UTI on the proliferation, invasion and phenotypic switching of VSMCs.15 PDGF family is composed of five proteins, PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC and PDGF-DD contained.16 Among them, PDGF-BB, one of the most effective stimulants for VSMC proliferation and migration, is chosen to induce VSMCs proliferation and migration as per a previous report.17
In the present study, we aimed to investigate whether UTI could suppress the proliferation, invasion and phenotypic switching of PDGF-BB-induced VSMCs and its potential mechanism.
Materials and Methods
Cell Culture
VSMC cell line T/G HA-VSMC cells (RRID: CVCL_4009) from human were obtained from ATCC (Manassas, USA). These cells were cultured with the Dulbecco’s modified Eagle’s medium (DMEM; Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA). These cells were placed in a humid atmosphere at 37°C with 5% CO2. PDGF-BB (20 ng/mL; TL644l; Shanghai Xitang Biotechnology Co., LTD., China) was applied to treat the cells for 24 h.
Western Blot
Protein extracts were prepared from cells using RIPA lysis buffer (Sigma-Aldrich, St. Louis, MO). Protein concentration was measured using the bicinchoninic acid method. Equal amounts of proteins were separated by 10% SDS-PAGE and next transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% nonfat milk for 1 h, followed by incubation with the following primary antibodies: CDK2 (1:1000, Abcam, cat. no. ab206038), PCNA (1:1000, Abcam, cat. no. ab92552), p-Akt Ser473 (1:1000, Abcam, cat. no. ab18206), Akt (1:1000, Abcam, cat. no. ab8805), p-eNOS Ser1177 (1:1000, Cell Signaling Technology, Danvers, MA; cat. no. 9571), eNOS (1:1000, Abcam, cat. no. ab199956), MMP-2 (1:1000, Abcam, cat. no. ab37150), MMP-9 (1:1000, Abcam, cat. no. ab38898), α-SMA (1:1000, Abcam, cat. no. ab204573), and SM22α (1:1000, Arigo, cat. no. ARG63416). The membranes were subsequently cultured with horseradish peroxidase-conjugated goat anti-rabbit IgG. The protein was detected using an enhanced chemiluminescence kit (Pierce Biotechnology, Rockford, IL) and the band intensity was quantified with Image-Pro Plus 4.5 software. GAPDH served as the internal control.
Cell Counting Kit-8 (CCK-8) Assay and EdU Assay
VSMCs were seeded into a 96-well plate at 3×103 cells/well in DMEM containing 10% FBS. PDGF-BB treatment (20ng/mL) was used for 24 h after starvation of the cells in serum-free medium for 24 h. 10 µL of CCK-8 reagent (Dojindo, Japan) was added into per well for 3 h. Absorbance at 450 nm was determined by a spectrophotometer (Model 680; Bio-Rad, USA). For EdU analysis, the nuclear DNA was counterstained using Hoechst 33,342, and the EdU-positive images were captured by a fluorescence microscopy (80i, Nikon, Japan), as previously performed.18
RT-qPCR
Total cellular RNA was extracted from cells using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the instruction. SYBR Green PCR Master Mix (Applied Biosystems; Thermo Fisher Scientific) was used to detect mRNA expression level. GAPDH was used as an internal control. The primer sequences are as follows: CDK2: forward 5′- GCGAATTCCCCAGCCCTAATCTCA-3′, reverse: 5ʹ‐GCCTCGAGAACCCTCTTCAGCAATAA‐3ʹ; PCNA forward: 5ʹ-TCCCAGTTCACCATCCATGTC-3ʹ, reverse: 5ʹ-GGTCCCCTAAGGCCCATTCCT-3ʹ; MMP2: forward: 5ʹ-CAACGGTCGGGAATACAGCAG-3ʹ, reverse: 5ʹ-CCAGGAAAGTGAAGGGGAAGA-3ʹ; MMP9: forward: 5ʹ-CGTGTCTGGAGATTCGACTTGA-3ʹ, reverse: 5ʹ-TGGAAGATGTCGTGTGAGTTCC-3ʹ;, whereas those for the internal control, GAPDH: forward: 5′-ACCACAGTCCATGCCATCAC-3′, reverse: 5′-TCCACC ACCCTGTTGCTGTA-3′.
Wound Healing Assay
Cells of each group were collected and seeded into 24-well plates with a density of 5×103/mL. When the cells were completely covered, a vertical line was drawn with a 10μL tip on the well. Cells were washed with PBS thrice and visualized under an inverted microscope. The images were labeled as 0 h as a reference. Then, serum-free DMEM was added and cells were continuously cultured for 24 h. The images were taken under an inverted microscope and labeled as 24 h. The relative cell migration was analyzed by Image J software.
Transwell Assay
The invasion of cells was evaluated using a Matrigel-based assay in 24-well Transwell chambers. 2×104 cells were plated in the upper chambers precoated with Matrigel (BD Biosciences) membrane, and DMEM (600 μl) containing 10% FBS was added to the lower chamber. Following incubation for 24 h, the cells on the upper surface of the membrane were removed by a sterile cotton swab. The invading cells were stained with hematoxylin and eosin (H&E, Sigma-Aldrich) and then observed under a microscope (Nikon E100; Nikon, Tokyo, Japan). Thereafter, the number of invading cells was counted.
Immunofluorescence Staining
After being fixed with 4% paraformaldehyde for 30 min, the stimulated VSMCs were permeabilized with 0.1% Triton X-100 in PBS for 15 min. Cells were blocked with 5% BSA for 1 h at room temperature and then incubated with indicated primary antibodies at 4°C overnight. After three times of wash with PBS, cells were detected with goat anti-rabbit IgG H&L Alexa Fluor® 488. Finally, the nuclei were stained with 4ʹ,6-diamidino-2-phenylindole (DAPI) for 10 min. Images were photographed by a fluorescence microscope (80i, Nikon, Tokyo, Japan).
Detection of NO
NO production was measured using a Nitric Oxide Colorimetric Assay Kit (BioVision, Mountain View, CA, U.S.), and OD values were measured at 540 nm.
Enzyme-Linked Immunosorbent Assay (ELISA)
Cell supernatant was collected by centrifugation, and then the concentrations of the inflammatory cytokines were measured as per the instructions of the corresponding ELISA kits (Multi Science, Hangzhou, China). All the experiments were repeated in triplicate.
Statistical Analysis
All results were defined as mean ± standard deviation (SD). Comparisons between two groups were made by Student’s t-test. For comparisons among multiple groups, statistical analysis was performed by ANOVA followed by Tukey’s test. The criterion for statistical significance was set at p < 0.05.
Results
UTI Activates the Expression of Akt/eNOS Signal in PDGF-BB-Induced VSMCs
Akt is a crucial signaling pathway regulating eNOS in the endothelium.19 The expressions of Akt and eNOS were evaluated by Western blot. As shown in Figure 1, compared with the control group, the PDGF-BB group exhibited significantly lower expressions of p-Akt and p-eNOS, while the addition of UTI dose-dependently increased their expressions. Herein, UTI at doses of 500 μmol/L and 1000 μmol/L was chosen for the next experiments.
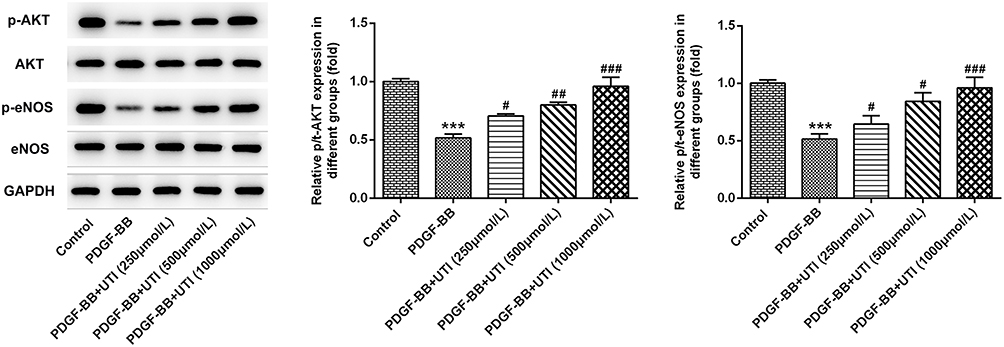 |
Figure 1 UTI activates the expression of Akt/eNOS signal in PDGF-BB-induced VSMCs. Western blot was used to detect the Akt/eNOS signaling-related proteins. ***P<0.001 vs Control; #P<0.05, ##P<0.01, ###P<0.001 vs PDGF-BB. |
UTI Inhibits the Proliferation of PDGF-BB-Stimulated VSMCs
To further investigate the role of UTI in the proliferation of PDGF-BB-induced VSMCs, CCK-8 assay, EdU staining, RT-qPCR and Western blot were conducted. The results of CCK-8 showed that PDGF-BB dramatically elevated the cell viability of VSMCs; however, the addition of UTI inhibited the cell viability dose-dependently, which could be further reversed by AKT inhibitor or eNOS inhibitor (L-NAME) (Figure 2A). Western blot (Figure 2B) and RT-qPCR (Figure 2C) discovered that the elevated levels of proliferation-related proteins CDK2 and PCNA in VSMCs induced by PDGF-BB were decreased after UTI treatment, and the effects were restored when AKT inhibitor or eNOS inhibitor were added. Furthermore, the result of EdU was consistent with that of CCK8 experiment (Figure 2D and E). These results suggest that UTI represses the proliferation of PDGF-BB-treated VSMCs.
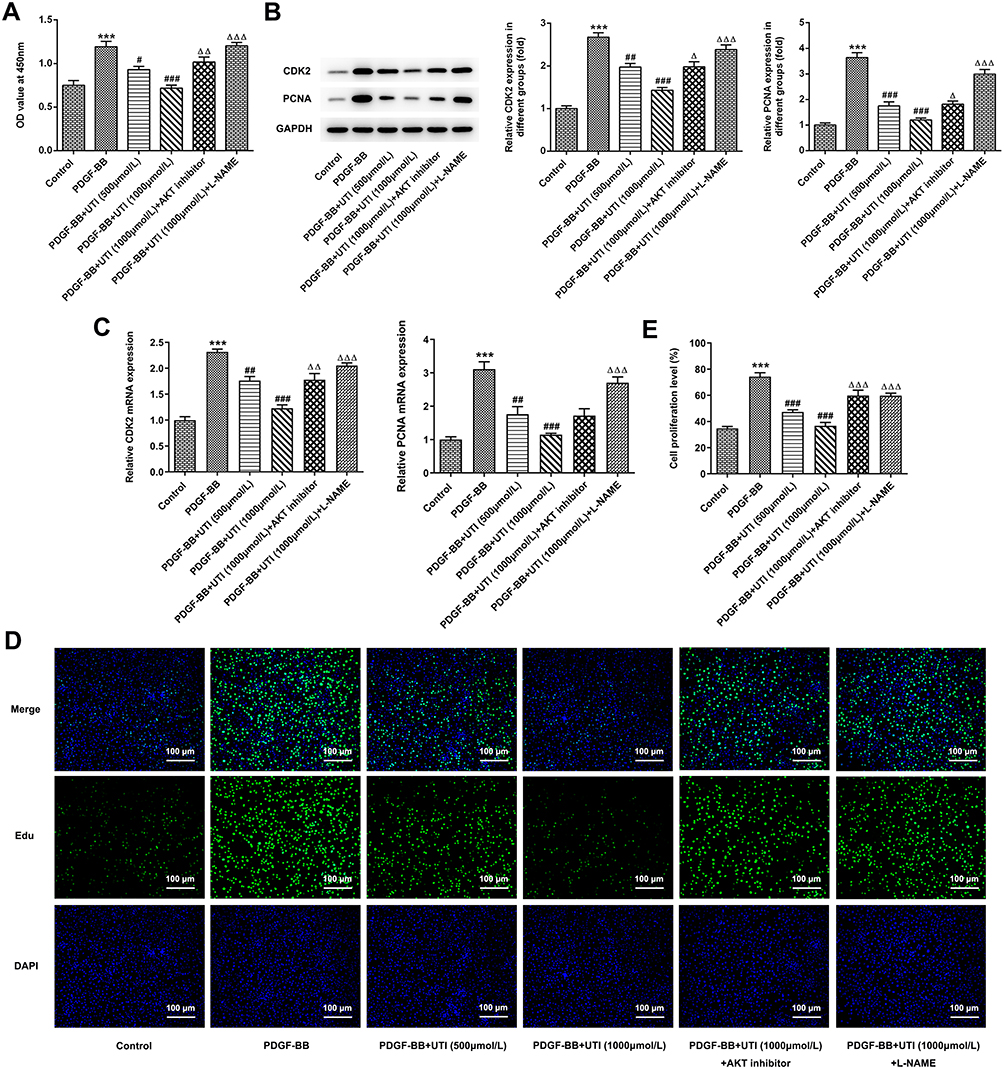 |
Figure 2 UTI inhibits the proliferation of PDGF-BB-induced VSMCs. (A) CCK-8 assay was used to detect the cell viability of VSMCs. (B) Western blot detected the expressions of proliferation-related proteins. (C) RT-qPCR detected the expressions of proliferation-related proteins. (D) EdU staining was employed to detect the proliferation of VSMCs. (E) Statistical graph of D. ***P<0.001 vs Control; #P<0.05, ##P<0.01, ###P<0.001 vs PDGF-BB; ΔP<0.05, ΔΔP<0.01, ΔΔΔP<0.001 vs PDGF-BB+UTI (1000μmol/L). |
UTI Hinders the Invasion and Migration of PDGF-BB-Treated VSMCs
The invasion and migration of PDGF-BB-treated VSMCs were evaluated by transwell and wound healing assays, respectively. Compared with control cells, PDGF-BB induced VSMCs presented enhanced migration (Figure 3A and C) and invasion (Figure 3B and D). Moreover, this effect was inhibited by UTI treatment. In addition, AKT inhibitor and eNOS inhibitor recovered the UTI-inhibited invasion and migration of PDGF-BB-activated VSMCs. Also, compared with control cells, the expressions of MMP-2 and MMP-9 in PDGF-BB-activated VSMCs were upregulated, which was markedly downregulated upon UTI treatment (Figure 3E and F). Furthermore, AKT inhibitor and eNOS inhibitor reversed the suppressive effects of UTI on MMP-2 and MMP-9 in PDGF-BB-induced VSMCs. The expression of MMP-2 and MMP-9 promoted by RT-qPCR was consistent with that of Western blot (Figure 3G). Thus, these results demonstrate that UTI hampers the invasion and migration of PDGF-BB-induced VSMCs.
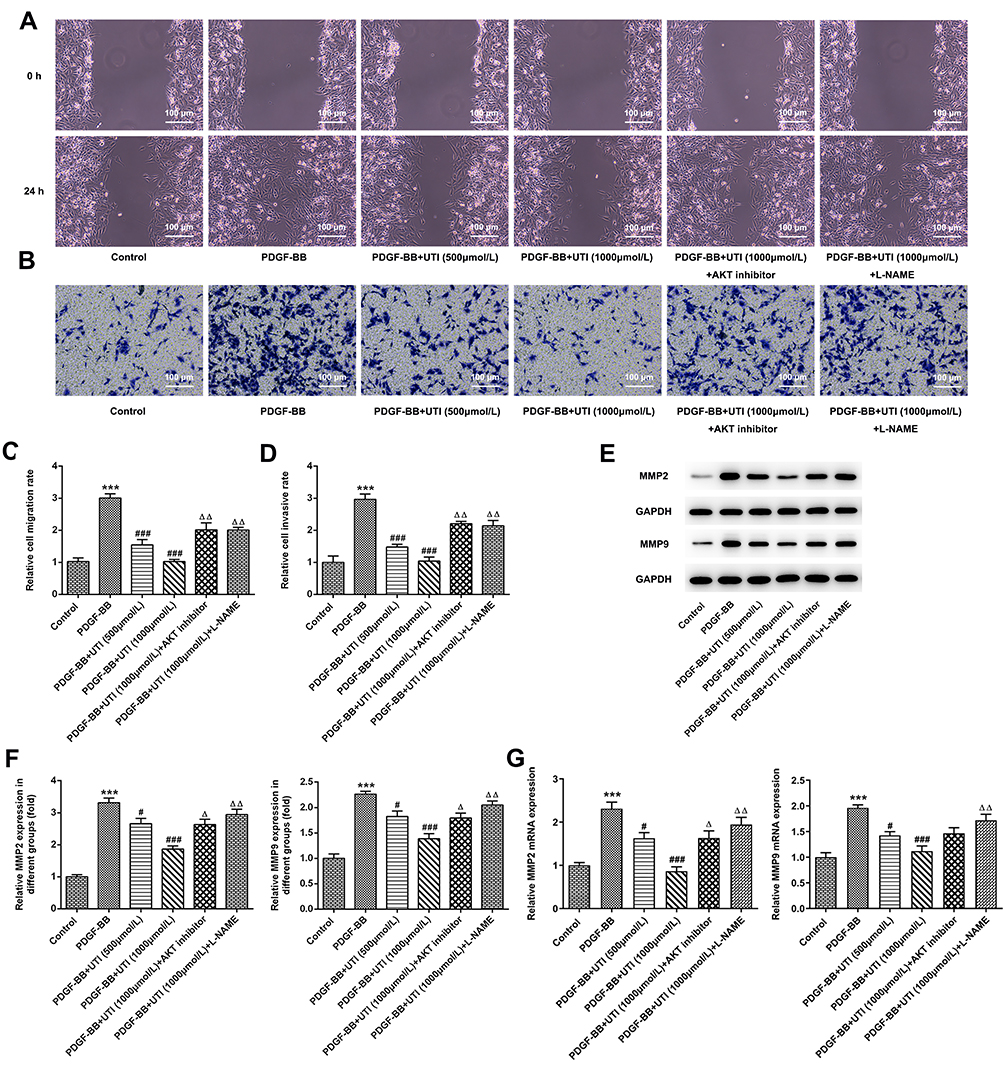 |
Figure 3 UTI impedes the invasion and migration of PDGF-BB-treated VSMCs. (A and C) Transwell and (B and D) wound healing were utilized to analyze the invasion and migration of VSMCs. (E) Western blot was used to detect the expressions of proliferation-related proteins. (F) Statistical graph of E. (G) RT-qPCR was used to detect the expressions of proliferation-related proteins. ***P<0.001 vs Control; #P<0.05, ###P<0.001 vs PDGF-BB; ΔP<0.05, ΔΔP<0.01 vs PDGF-BB+UTI (1000μmol/L). |
UTI Resists the Phenotypic Switching of PDGF-BB-Stimulated VSMCs
To further clarify the role of UTI in phenotypic switching of PDGF-BB-stimulated VSMCs, we examined the protein expressions of contraction phenotype makers, α-SMA and SM22α. Compared with control, VSMCs treated with PDGF-BB exhibited lower expressions of α-SMA and SM22α, which were elevated by the treatment of UTI in a dose-dependent manner (Figure 4A). However, addition of AKT inhibitor and eNOS inhibitor downregulated their expressions. Similar results are found in Figure 4B and C, which confirmed the changes of α-SMA expression. Thus, the above results hint that UTI resists the phenotypic switching of PDGF-BB-induced VSMCs.
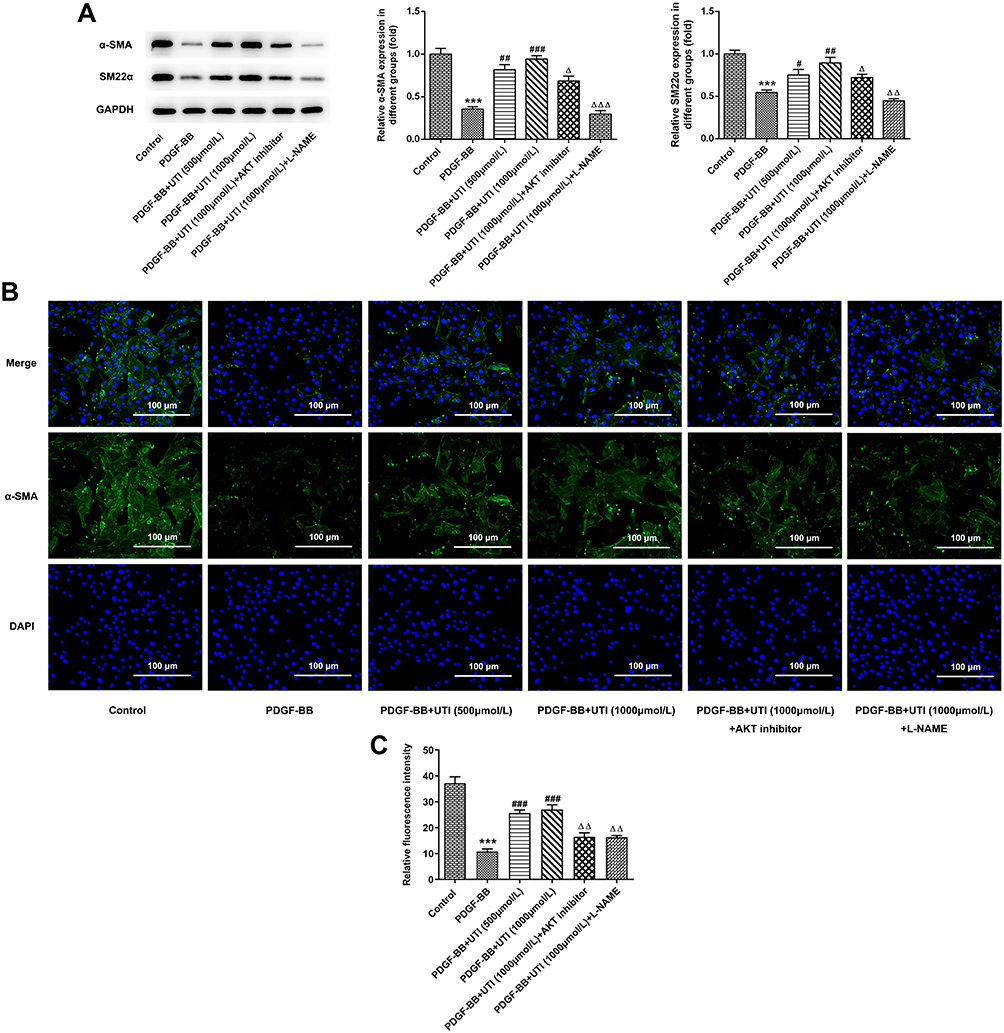 |
Figure 4 UTI resists the phenotypic switching of PDGF-BB-treated VSMCs. (A) Western blot was applied to detect the expressions of α-SMA and SM22α. (B) Immunofluorescence staining analysis of the expression of α-SMA. (C) Statistical graph of B. ***P<0.001 vs Control; #P<0.05, ##P<0.01, ###P<0.001 vs PDGF-BB; ΔP<0.05, ΔΔP<0.01, ΔΔΔP<0.001 vs PDGF-BB +UTI (1000μmol/L). |
UTI Triggers the Activation of Akt/eNOS/NO/cGMP Signaling Pathway
The results from Western blot showed that compared with control, the expression levels of p-Akt and p-eNOS in VSMCs were decreased by PDGF-BB (Figure 5A). What is more, their levels were elevated when PDGF-BB-induced VSMCs were treated with UTI, and the additional treatment of AKT inhibitor and eNOS inhibitor reversed these effects. Next, we detected the concentration of NO, a critical endothelium-derived relaxing factor produced by eNOS.20 The results elucidated that compared with control, the concentration of NO was decreased in PDGF-BB-induced VSMCs but increased under UTI treatment (Figure 5B). Furthermore, AKT inhibitor and eNOS inhibitor decreased the increase of NO concentration caused by UTI in PDGF-BB-treated VSMCs. The result from ELISA assay illustrated the similar trend of cGMP expression with the changes in NO concentration (Figure 5B). Therefore, the results indicate that UTI triggers the activation of Akt/eNOS/NO/cGMP signaling pathway.
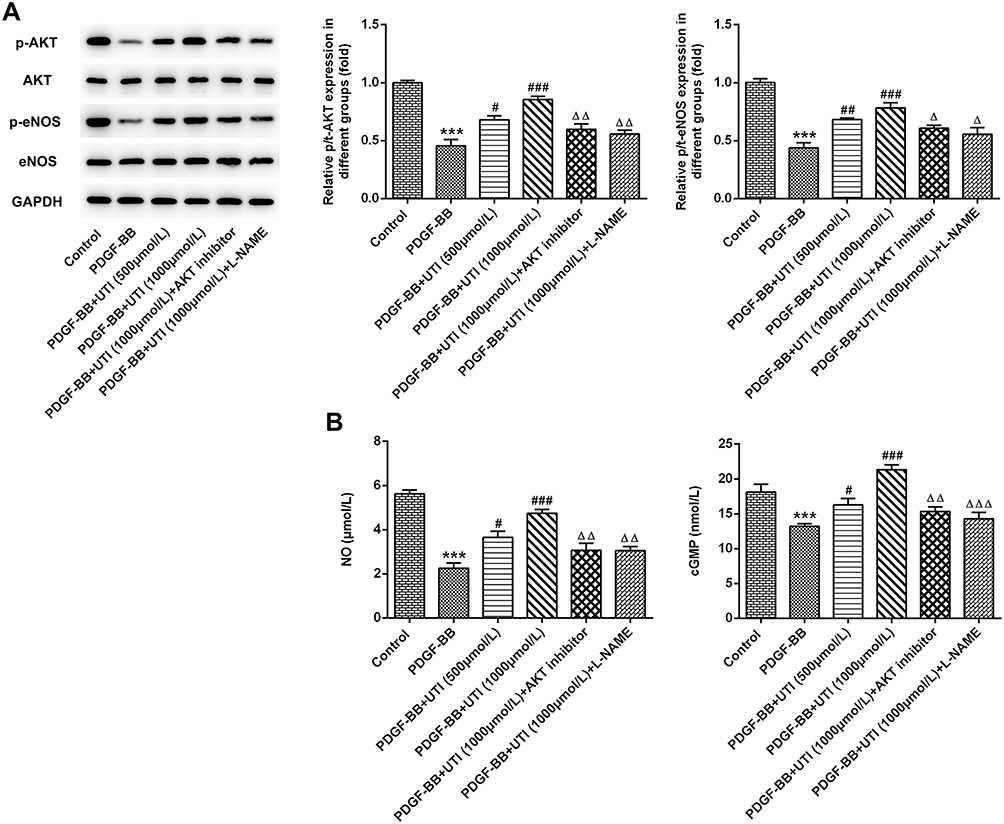 |
Figure 5 UTI triggers the activation of Akt/eNOS/NO/cGMP signaling pathway. (A) Western blot examined the Akt/eNOS/NO/cGMP signaling-related proteins. (B) The amount of NO and the expression of cGMP were measured by corresponding commercial kits. ***P<0.001 vs Control; #P<0.05, ##P<0.01, ###P<0.001 vs PDGF-BB; ΔP<0.05, ΔΔP<0.01, ΔΔΔP<0.001 vs PDGF-BB +UTI (1000μmol/L). |
Discussion
UTI is broadly known as a serine protease inhibitor that can suppress several pro-inflammatory proteases, inhibit neutrophil elastase, and decrease the release of inflammatory cytokines.21,22 Based on its physiological characteristics, UTI has been extensively applied in the treatment of many acute inflammatory disorders.23,24 Studies have demonstrated that the serum levels of UTI are decreased in patients with sepsis, and genetically modified mice without genes for synthesis of UTI possess relatively higher risk of sepsis.25,26 Some researchers also found the vital role of UTI in preserving the functions of myocardial mitochondria to prevent against hemorrhagic shock.27 One animal study indicated that UTI is crucial for the reduction of key inflammatory mediators in the vascular endothelium resulting in atherosclerosis.28 But there was no evidence exploring its role in the proliferation, invasion and phenotypic switching of VSMCs during the development of atherosclerosis.
Atherosclerosis is mainly triggered by the excessive proliferation and migration of VSMCs, and phenotypic switching is the key element to control the proliferation and migration of VSMCs.15,29 Phenotypic switching has long been thought as a major force for the atherosclerosis development, generating a pro-atherogenic VSMC phenotype.15 VSMCs express a series of “SMC markers” in the normal arterial media, including SM22α and α-SMA.15 However, under the pathological conditions of atherosclerosis, the expressions of these markers are reduced in VSMCs, giving rise to enhanced proliferation, migration and secretion of multiple extracellular matrix proteins and cytokines.30 In this study, PDGF-BB significantly reduced expressions of SM22α and α-SMA in VSMCs, in agreement with the observed phenomena in the previous study.19 It was also proved that UTI could inhibit the phenotypic switching induced by PDGF-BB in VSMCs, which might be beneficial to the treatment of advanced atherosclerosis.31
Studies have indicated that eNOS is one of the downstream substrates of Akt that can induce eNOS phosphorylation.32 In the present study, UTI promoted the phosphorylation of Akt and activated its downstream effector eNOS in PDGF-BB-treated VSMCs. Furthermore, AKT inhibitor and eNOS inhibitor enhanced the proliferation, migration and invasion that were suppressed by UTI in PDGF-BB-induced VSMCs. Decreased concentration of NO, a characteristic of endothelial dysfunction, is a significant mediator in close association with cardiovascular diseases.33 High amount of NO protects the vasculature through hindering platelet aggregation, monocyte adhesion, and smooth muscle cell proliferation.34 Herein, UTI treatment abolished the inhibitory effect of PDGF-BB on the concentration of NO, suggesting that the endothelial dysfunction of VSMCs was alleviated by UTI. The activation of eNOS can bring about rapid vasodilatation. Specifically, NO release can promote endothelial cells to proliferate into VSMCs and activate soluble guanylyl cyclase (sGC), thereby producing cyclic GMP (cGMP).35,36 In the present study, the activation of p-Akt and p-eNOS by UTI gave rise to higher production of NO and expression of cGMP, demonstrating that UTI could activate the whole Akt/eNOS/NO/cGMP signaling pathway.
Conclusion
Taken together, this study demonstrates that UTI inhibits the proliferation, invasion and phenotypic switching of PDGF-BB-induced VSMCs via Akt/eNOS/NO/cGMP signaling pathway, which might provide a theoretical basis for the UTI treatment of atherosclerosis.
Data Sharing Statement
The analyzed data sets generated during the present study are available from the corresponding author on reasonable request.
Funding
National Natural Science Foundation of China (81300230). Medical Research Fund of Guangdong Province (2019612114114896).
Disclosure
The authors declare that they have no competing interests.
References
1. Huang Y, Li T, Gao W, et al. Platelet-derived nanomotor coated balloon for atherosclerosis combination therapy. J Mater Chem B. 2020.
2. Kyaw T, Peter K, Li Y, Tipping P, Toh BH, Bobik A. Cytotoxic lymphocytes and atherosclerosis: significance, mechanisms and therapeutic challenges. Br J Pharmacol. 2017;174(22):3956–3972.
3. Zhou L, Long J, Sun Y, Chen W, Qiu R, Yuan D. Resveratrol ameliorates atherosclerosis induced by high-fat diet and LPS in ApoE(-/-) mice and inhibits the activation of CD4(+) T cells. Nutr Metab (Lond). 2020;17(1):41. doi:10.1186/s12986-020-00461-z
4. Steven S, Frenis K, Oelze M, et al. Vascular inflammation and oxidative stress: major triggers for cardiovascular disease. Oxid Med Cell Longev. 2019;2019:7092151. doi:10.1155/2019/7092151
5. Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20(5):1262–1275. doi:10.1161/01.ATV.20.5.1262
6. Pugia MJ, Lott JA. Pathophysiology and diagnostic value of urinary trypsin inhibitors. Clin Chem Lab Med. 2005;43(1):1–16. doi:10.1515/CCLM.2005.001
7. Pugia MJ, Valdes R
8. He S, Lin K, Ma R, Xu R, Xiao Y. Effect of the urinary trypsin inhibitor ulinastatin on cardiopulmonary bypass-related inflammatory response and clinical outcomes: a meta-analysis of randomized controlled trials. Clin Ther. 2015;37(3):643–653. doi:10.1016/j.clinthera.2014.12.015
9. He QL, Zhong F, Ye F, et al. Does intraoperative ulinastatin improve postoperative clinical outcomes in patients undergoing cardiac surgery: a meta-analysis of randomized controlled trials. Biomed Res Int. 2014;2014:630835. doi:10.1155/2014/630835
10. Chen HM, Huang HS, Ruan L, He YB, Li XJ. Ulinastatin attenuates cerebral ischemia-reperfusion injury in rats. Int J Clin Exp Med. 2014;7(5):1483–1489.
11. Fan H, Zhao Y, Zhu JH, Ye JH, Yao XQ. Ulinastatin and thymosin a1 therapy in adult patients with severe sepsis: a meta-analysis with trial sequential analysis of randomized controlled trials. Iran J Public Health. 2016;45(9):1234–1235.
12. von Vietinghoff S, Koltsova EK. Inflammation in atherosclerosis: a key role for cytokines. Cytokine. 2019;122:154819. doi:10.1016/j.cyto.2019.154819
13. Cao T, Zhang L, Yao LL, et al. S100B promotes injury-induced vascular remodeling through modulating smooth muscle phenotype. Biochim Biophys Acta. 2017;1863(11):2772–2782. doi:10.1016/j.bbadis.2017.07.002
14. Zhu LH, Huang L, Zhang X, et al. Mindin regulates vascular smooth muscle cell phenotype and prevents neointima formation. Clin Sci (Lond). 2015;129(2):129–145. doi:10.1042/CS20140679
15. Chistiakov DA, Orekhov AN, Bobryshev YV. Vascular smooth muscle cell in atherosclerosis. Acta Physiol (Oxf). 2015;214(1):33–50.
16. Ha JM, Yun SJ, Kim YW, et al. Platelet-derived growth factor regulates vascular smooth muscle phenotype via mammalian target of rapamycin complex 1. Biochem Biophys Res Commun. 2015;464(1):57–62. doi:10.1016/j.bbrc.2015.05.097
17. Shawky NM, Segar L. Sulforaphane inhibits platelet-derived growth factor-induced vascular smooth muscle cell proliferation by targeting mTOR/p70S6kinase signaling independent of Nrf2 activation. Pharmacol Res. 2017;119:251–264. doi:10.1016/j.phrs.2017.02.010
18. Lu QB, Wan MY, Wang PY, et al. Chicoric acid prevents PDGF-BB-induced VSMC dedifferentiation, proliferation and migration by suppressing ROS/NFkappaB/mTOR/P70S6K signaling cascade. Redox Biol. 2018;14:656–668. doi:10.1016/j.redox.2017.11.012
19. Zhang N, Zhang Y, You S, et al. Septin4 prevents PDGF-BB-induced HAVSMC phenotypic transformation, proliferation and migration by promoting SIRT1-STAT3 deacetylation and dephosphorylation. Int J Biol Sci. 2020;16(4):708–718. doi:10.7150/ijbs.39843
20. Hamilton CA, Brosnan MJ, McIntyre M, Graham D, Dominiczak AF. Superoxide excess in hypertension and aging: a common cause of endothelial dysfunction. Hypertension. 2001;37(2 Pt 2):529–534. doi:10.1161/01.HYP.37.2.529
21. Hirose J, Ozawa T, Miura T, et al. Human neutrophil elastase degrades inter-alpha-trypsin inhibitor to liberate urinary trypsin inhibitor related proteins. Biol Pharm Bull. 1998;21(7):651–656. doi:10.1248/bpb.21.651
22. Karnad DR, Bhadade R, Verma PK, et al. Intravenous administration of ulinastatin (human urinary trypsin inhibitor) in severe sepsis: a multicenter randomized controlled study. Intensive Care Med. 2014;40(6):830–838. doi:10.1007/s00134-014-3278-8
23. Inamo Y, Okubo T, Wada M, et al. Intravenous ulinastatin therapy for Stevens-Johnson syndrome and toxic epidermal necrolysis in pediatric patients. Three case reports. Int Arch Allergy Immunol. 2002;127(1):89–94. doi:10.1159/000048174
24. Ribeiro JK, Cunha DD, Fook JM, Sales MP. New properties of the soybean trypsin inhibitor: inhibition of human neutrophil elastase and its effect on acute pulmonary injury. Eur J Pharmacol. 2010;644(1–3):238–244. doi:10.1016/j.ejphar.2010.06.067
25. Lim YP, Bendelja K, Opal SM, Siryaporn E, Hixson DC, Palardy JE. Correlation between mortality and the levels of inter-alpha inhibitors in the plasma of patients with severe sepsis. J Infect Dis. 2003;188(6):919–926. doi:10.1086/377642
26. Shigetomi H, Onogi A, Kajiwara H, et al. Anti-inflammatory actions of serine protease inhibitors containing the kunitz domain. Inflamm Res. 2010;59(9):679–687. doi:10.1007/s00011-010-0205-5
27. Masuda T, Sato K, Noda C, et al. Protective effect of urinary trypsin inhibitor on myocardial mitochondria during hemorrhagic shock and reperfusion. Crit Care Med. 2003;31(7):1987–1992. doi:10.1097/01.CCM.0000057037.44171.BA
28. Deng Y, Kong J. Urinary trypsin inhibitor reduced inflammation response induced by hyperlipidemia. J Cardiovasc Pharmacol Ther. 2015;20(6):572–578.
29. Liao XH, Wang N, Zhao DW, et al. STAT3 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem. 2015;290(32):19641–19652. doi:10.1074/jbc.M114.630111
30. Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74(1):13–40. doi:10.1146/annurev-physiol-012110-142315
31. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801.
32. Jiang F, Wang H, Bao S, et al. Thyrotropin regulates eNOS expression in the endothelium by PGRN through Akt pathway. Front Endocrinol (Lausanne). 2018;9:353. doi:10.3389/fendo.2018.00353
33. Diamant M, Tushuizen ME. The metabolic syndrome and endothelial dysfunction: common highway to type 2 diabetes and CVD. Curr Diab Rep. 2006;6(4):279–286. doi:10.1007/s11892-006-0061-4
34. Chenou F, Albuquerque DM, Leonardo DP, et al. Endothelial nitric oxide synthase (eNOS) gene polymorphisms and markers of hemolysis, inflammation and endothelial dysfunction in brazilian sickle cell anemia patients. Biochem Genet. 2020;58(4):580–594. doi:10.1007/s10528-020-09959-w
35. Hilgers RH, Kundumani-Sridharan V, Subramani J, et al. Thioredoxin reverses age-related hypertension by chronically improving vascular redox and restoring eNOS function. Sci Transl Med. 2017;9(376):eaaf6094. doi:10.1126/scitranslmed.aaf6094
36. Lundberg JO, Gladwin MT, Weitzberg E. Strategies to increase nitric oxide signalling in cardiovascular disease. Nat Rev Drug Discov. 2015;14(9):623–641.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.