")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Therapeutic potential of BLT1 antagonist for COPD: involvement of inducing autophagy and ameliorating inflammation
Authors Zhang L , Huang J , Dong R, Feng Y , Zhou M
Received 12 May 2019
Accepted for publication 10 August 2019
Published 4 September 2019 Volume 2019:13 Pages 3105—3116
DOI https://doi.org/10.2147/DDDT.S215433
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Li Zhang1,2, Jingwen Huang1,2, Ran Dong3, Yun Feng1,2, Min Zhou1,2
1Department of Respiratory and Critical Care Medicine, Shanghai Institute of Respiratory Disease, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 2Institute of Respiratory Diseases, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 3Department of Respiratory Medicine, Tongji Hospital, Tongji University School of Medicine, Shanghai, People’s Republic of China
Correspondence: Min Zhou
Department of Pulmonary and Critical Care Medicine, Shanghai Institute of Respiratory Disease, Ruijin Hospital Affiliated to Shanghai Jiaotong University School of Medicine, No.197 Ruijin Er Road, Shanghai 200025, People’s Republic of China
Tel +86 216 437 0045
Fax +86 216 467 4301
Email [email protected]
Purpose: Leukotriene B4 (LTB4) is a major pro-inflammatory mediator that leads to the persistence of chronic inflammation in chronic obstructive pulmonary disease (COPD). The purpose of this study was to evaluate therapeutic potential of BLT1 antagonist for cigarette smoke (CS)-induced COPD and to explore the underlying mechanism.
Materials and methods: In vitro, autophagy proteins were determined by Western blotting in RAW264.7 macrophages treated with U75302 (BLT1 antagonist) or autophagy inhibitor in cigarette smoke extract-induced inflammation. In vivo, C57BL/6J mice were randomly divided into three groups: Control group, CS group and CS+U75302 group. After 12-week exposure, histological analysis and lung function tests were performed to evaluate the inflammatory infiltration and emphysema. The expression of inflammatory cytokines was measured by real-time PCR and enzyme-linked immunosorbent assay. Immunohistochemical analysis and Western blotting detected the expression of autophagy-related proteins. Transmission electron microscopy (TEM) showed the alterations of autophagosomes and lysosomes.
Results: Lower levels of inflammatory factors and autophagy markers were detected in U75302-treated cells and mice after CS exposure than control. In vitro, LC3 mRNA expression was elevated when treated with U75302. Autophagy inhibition resulted in augmented inflammatory response and autophagy proteins even with U75302 treatment. Furthermore, BLT1 antagonist decreased the number of lysosomes and autophagosomes in alveolar macrophages of mice and potentially enhanced the expression of transcriptional activation of transcription factor-EB (TFEB) in vitro and vivo.
Conclusion: Insufficient autophagy of macrophages was associated with LTB4-mediated inflammation in CS-exposure models. BLT1 antagonist ameliorated inflammatory response through inducing autophagy.
Keywords: cigarette smoke, inflammation, autophagy, BLT1 antagonist, COPD
Introduction
Cigarette smoke (CS) is the primary risk factor for chronic obstructive pulmonary disease (COPD). Although preventable, it is a life-threatening lung condition characterized by persistent airflow obstruction, progressive decline in lung function, and excessive mucus secretion.1,2 COPD affects approximately 400 million people worldwide and is the third leading cause of death.3 It imposes a substantial economic burden on the society and reduces the quality of life.1 The management of CS‐induced COPD remains a challenge.
The detrimental effects of CS exposure are multifaceted, as it impacts cellular homeostasis, including cell growth and metabolism, autophagy, and host immune defense.4–7 CS exposure has been shown to induce an abnormal airway inflammatory response mediated by the activation of innate immune cells, such as macrophages, causing airway remodeling and pulmonary emphysema.8 Chronic exposure to CS-induced oxidative stress and inflammation results in impaired autophagy that in turn, accelerates the pathogenesis of COPD-emphysema.9,10 Insufficient autophagy could not completely remove the aged or defective mitochondria, misfolded or damaged protein aggregates, autophagosomes, via the lysosomes, further damaging the cellular clearance processes, which drives COPD initiation and progression.11–13 P62 (P62/SQSTMI), an adaptor protein, can directly bind ubiquitinated proteins and LC3 and then is delivered to autophagosome. Subsequently, the cargo protein and p62 protein are degraded by autophagy-lysosome pathway.14 Hence, autophagy deficiency resulted in the accumulation of p62, ubiquitinated proteins and LC3, which may exert a pro-inflammatory effect in COPD.15 Therefore, elimination of inflammation and induction of autophagy may be the main strategies for COPD treatment.
Leukotriene B4 (LTB4) is a key chemokine that effectuates the accumulation of granulocytes and macrophages at the sites of inflammation.16,17 BLT1, a highly specific receptor for LTB4, was expressed on the surface of inflammatory and immune cells.18 Presently, inhibition of LTB4/BLT1 signaling pathway using BLT1 antagonists has been used to alleviate a range of inflammatory conditions and mitochondrial dysfunction in sepsis, asthma and arthritis.16,19,20 Evidence suggested that BLT1 blockade with its specific antagonist U75302 can prevent the later development of tissue fibrosis.21 Patients with COPD exhibited an increased number of BLT1-positive alveolar macrophages and cells in the alveolar walls.22 We have shown that LTB4-mediated inflammation played a critical role in COPD patients.23 However, whether BLT1 antagonist can affect autophagy, thus improving airway inflammation lacks sufficient experimental evidence in CS-induced COPD.
This study aimed to evaluate the therapeutic potential of BLT1 antagonist (U75302) for COPD and explored its relative molecular mechanism.
Materials and methods
Reagents
BLT1 antagonist U75302 was purchased from Cayman Chemical. Autophagy inhibitor chloroquine (CQ) was obtained from Sigma-Aldrich. Antibodies against LC3B (1:1000; Cell Signaling Technology), p62 (1:2000; Abcam), TFEB (1:2000; Benthyl Laboratories), LAMP1 (1:2000; Santa Cruz Biotechnology) and GAPDH (1:2000; Cell Signaling Technology) were used. ELISA kits for mouse-LTB4 and mouse-IL-6 were purchased from R&D system.
Experimental protocols
Male C57BL/6J mice (8 weeks old) were obtained from the Centre for Experimental Animals of Ruijin Hospital (Shanghai Jiaotong University School of Medicine, China). The mice (n=10) were exposed to chronic cigarette smoke (12 weeks, 7 days/week) for burning 10 sticks of 3R4F research cigarettes (Tobacco Research Institute, University of Kentucky, Lexington, Ky) two times per day in a sealed 80 L chamber. Control group mice (n=10) were exposed to room-air, while in the treated group (n=10), CS-exposed mice were also intraperitoneally disposed with 5 μg of U75302 diluted in 200 μl saline, one time per week, before CS.21 Twenty-four hours after the last exposure, the mice were anesthetized with a sodium pentobarbital intraperitoneal injection dissolved in saline. All experimental procedures were approved by and conducted in accordance with the Ethics Committee and Animal Care Committee of Ruijin Hospital (Shanghai Jiaotong University School of Medicine, China).
Lung function measurement
Mice were placed in a sealed whole-body plethysmograph after intubation and connected to a computer-controlled ventilator controlled by AniRes2005 software (Peking Biolab Tech Co, Beijing, China). Breathing, expiratory resistance (Re), dynamic compliance (Cdyn), and peak expiratory flow (PEF) were assessed with an average breathing frequency of 90 breaths/min. Under pressure control of 30 cm H2O, the forced vital capacity (FVC), forced expiratory volume at 100 ms (FEV0.1s) and the FEV0.1s/FVC ratio were calculated.
Bronchoalveolar lavage and histopathology
Bronchoalveolar lavage fluid (BALF) was obtained to detect the levels of inflammatory factors after infusing the lung three times with 0.8 mL sterile saline. Histologic examination was performed on the tissue samples, which were fixed in 4% buffered formalin and embedded in paraffin. The deparaffinized sections (5 µm) were stained with hematoxylin and eosin (H&E). The peribronchiolar and perivascular inflammation was evaluated on a subjective scale of 0 to 3 for histological assessment (200×) as described elsewhere.24 A value of 0 was adjudged when no inflammation was detectable, a value of 1 for occasional cuffing with inflammatory cells, a value of 2 for most bronchi or vessels surrounded by thin layer (one to five cells) of inflammatory cells, and a value of 3 when most bronchi or vessels were surrounded by a thick layer (>5 cells) of inflammatory cells. A total of 20 randomly selected fields from two H&E-stained paraffin sections per tissue sample were captured at 200× magnification using the mean linear intercept technique to assess the alveolar diameter and emphysema.25 The first 10 images that did not contain airways and/or blood vessels were overlaid with an 11–horizontal line template. Intercepts of alveolar walls with lines were enumerated and the alveolar diameter calculated by dividing the total length of the 11 lines by the average number of intercepts per lung section.
Immunohistochemical analysis
Sections obtained from formalin-fixed, paraffin-embedded lung tissue were used for immunohistological staining. Endogenous peroxidases were inhibited with 0.5% hydrogen peroxide in methanol for 10 min, following, the slides were immunostained overnight at 4 °C with rabbit antibody against LC3B. Immunodetection was performed as reported in our previous study.23 Quantitative analysis was performed to calculate integral optical density using Image-Pro Plus software (Media Cybernetics, Rockville, MD, USA). Six random fields of two sections at 200× magnification in each tissue sample were evaluated.
Transmission electron microscopy (TEM)
Extracted lung tissue was fixed overnight with 2% glutaradehyde in PBS. The samples were washed in PBS, then fixed in 1% osmium tetroxide for 2 h and stained with 3% uranyl acetate. After dehydration, the samples were embedded in embedding medium and ultrathin sections were stained with lead citrate. Images were taken using a CM-120 transmission electron microscope (PHILIP, Holland). In order to quantify the alteration of the number of the lysosomes (Lys), the area of the cell cytoplasm was measured by using Image-Pro Plus 6.0. The data were represented as Lys per 100 μm2.
Cigarette smoke extract (CSE)
CS was collected in a chamber filled with 500 mL Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher Scientifc, Waltham, MA, USA) connected to a vacuum by continuous suction. The smoke from ten cigarettes was drawn into the DMEM for 5 mins. The resulting solution was sterile filtered (0.2 μm) to remove large particles and used as 100% CSE stock solution. The stock solution was diluted to obtain a 25% CSE solution for cell culture.
Cell culture and treatment
The RAW264.7 mouse macrophage cell line was obtained from the Cell Collection and Research Center of the Chinese Academy of Science and maintained in DMEM with 10% fetal bovine serum (FBS), 50 U/mL penicillin, and 50 U/mL streptomycin. The most appropriate condition was determined using a combination of different concentrations of CSE stimulation at different exposure times of 3, 6, 12, 24, and 48 hrs. Basically, 35% CSE was the maximal dilution that achieved positive growth without excessive cell mortality after 24 hrs exposure. For U75302 and CQ treatment, RAW264.7 cells were seeded onto 6-well or 12-well plates overnight and treated with 25% CSE, or 10 nmol/mL U75302, or 50 μmol/mL CQ, before total RNA and protein extraction.
RNA extraction and real-time PCR
Total RNA was extracted using the TRIzol reagent (Invitrogen), according to the manufacturer’s instructions. Reverse transcription was performed with RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, Waltham, MA, USA). The expression of mRNA transcripts was measured by a 7500 Fast Real-Time PCR System (Thermo Fisher Scientific) using FastStart Universal SYBR Green (Hoffman-La Roche Ltd, Basel, Switzerland) after cDNA synthesis. The sequences of primers used were listed in Table 1. The fold change in gene expression was calculated by 2−ΔΔCt relative to the internal reference gene (glyceraldehyde-3-phosphate dehydrogenase, GAPDH).
 |
Table 1 Primer sequences for real-time PCR |
Western blotting
Cell and lung homogenates were lysed in RIPA buffer (Sigma) to detect relative protein expression. Protein concentration was quantified using the bicinchoninic acid (BCA) protein assay reagent. The samples (30 μg/lane) were loaded and blotted with primary antibodies, then incubated overnight at 4 °C with secondary antibody. The signal was detected and quantified using a chemiluminescence system (ImageQuant LAS-4000 MINI).
ELISA
The concentrations of LTB4 and IL6 in culture supernatants and BALF were determined with ELISA kits following the manufacturer’s protocol.
Statistical analysis
All data were processed using SPSS 23.0 software (Chicago, IL, USA) and results were presented as mean ± standard error of the mean (Mean ± SEM) or median with range (minimum – maximum). Two-group comparisons were analyzed using Student’s t-test, and multi-sample comparisons were analyzed using one-way ANOVA followed by post hoc multiple comparisons if data satisfied normal distribution criteria. Otherwise, the non-parametric tests were applied as appropriate. Wilcoxon rank sum test was used to compare the results between two groups and Kruskal Wallis test determined the difference among groups. P<0.05 was considered statistically significant.
Results
Expression of autophagy markers in CSE-induced RAW264.7 cells
The expression of LC3 mRNA was upregulated at 6 h and reduced at 12 h, 24 h with CSE treatment (Figure 1A). When treated with U75302 at 24 h, LC3 mRNA expression was significantly increased (Figure 1B). Western blotting results exhibited that LC3-II and p62 protein expression was induced at 24 h in CSE-treated cells. Because of U75302 treatment, the protein expression was lower relative only to CSE treatment (Figure 1C). Furthermore, TFEB protein expression (an autophagy regulator) was inhibited with CSE treatment and partially restored by U75302 treatment (Figure 1D).
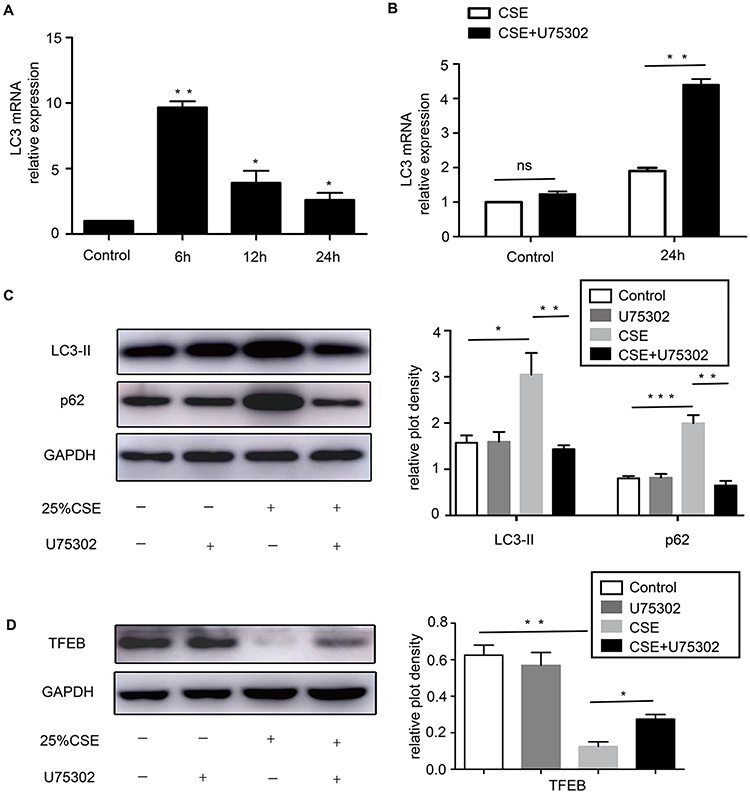 |
Figure 1 Expression of autophagy markers in CSE-induced RAW264.7 cells. (A) The relative expression of LC3 mRNA was normalized to GAPDH in CSE-treated cells by real-time PCR. (B) The effect of U75302 on LC3 mRNA at 24 h. (C) LC3 and p62 protein expression was measured by Western blotting in CSE-treated cells with or without U75302 at 24 h. Densitometry analysis calculated the ratio normalized to GAPDH. (D) The protein expression of TFEB with treatment for 24 h and corresponding quantification normalized to GAPDH. The data were represented as means ± SEM from three independent experiments, *P<0.05, **P<0.01, ***P<0.001. Statistical analysis was performed by two-tailed unpaired Student’s t-test and one-way ANOVA followed by post hoc multiple comparisons. Abbreviations: CSE, cigarette smoke extract; LC3, microtubule-associated protein light chain 3; p62, Sequestosome1/p62; TFEB, transcription factor-EB; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CQ, chloroquine; ns, not significant. |
Effect of autophagy inhibitor on LTB4-mediated inflammation
CQ addition blocking the fusion of autophagosomes with lysosomes caused an accumulation of LC3-II and p62 in “no-exposure” control and under CSE treatment for 24 h, LC3-II and p62 expression was not impacted by BLT1 antagonist in CQ-treated lanes (Figure 2A). The level of IL-6 was markedly increased when treated with CQ before U75302 treatment, but no significant difference was observed in the level of LTB4 with the treatment of U75302 or CQ (Figure 2B–C).
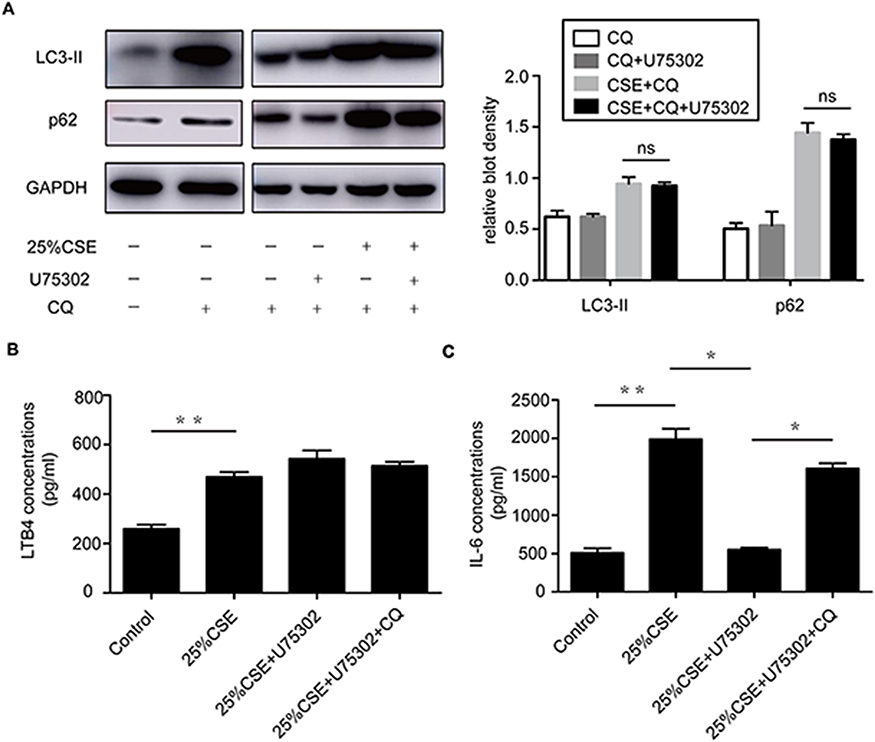 |
Figure 2 Effect of autophagy inhibitor on LTB4-mediated inflammation. (A) Western blotting analyses of LC3 and p62 protein expression with U75302 or CQ treatment by CSE at 24 h in RAW264.7 cells. CQ addition was determined at 2 h before CSE or U75302 treatment. (B, C) The levels of LTB4 and IL-6 in the culture supernatants were detected by ELISA at 24 h. The data were represented as means ± SEM from three independent experiments, *P<0.05, **P<0.01. Statistical analysis was performed by two-tailed unpaired Student’s t-test and one-way ANOVA followed by post hoc multiple comparisons. Abbreviations: CSE, cigarette smoke extract; LC3, microtubule-associated protein light chain 3; p62, Sequestosome1/p62; ns, not significant. |
Histopathological findings and lung function of mice
Following 12 weeks exposure, H&E staining of lung tissues was performed. As shown in Figure 3A, the structure of alveoli was normal in the control group. Inflammatory cell infiltration and alveolar space was notably reduced in CS+U75302 group relative to CS group. The inflammation scores and alveolar diameter showed that, in the CS group, scores were much higher accompanied by increased alveolar diameter compared to controls; U75302 treatment could decrease the airway inflammation and alveolar space enlargement (Figure 3B and C). CS exposure induced a decreased lung function, which could be improved by U75302 treatment. However, no statistically significant difference was observed in cdyn (dynamic compliance) among the three groups (Table 2).
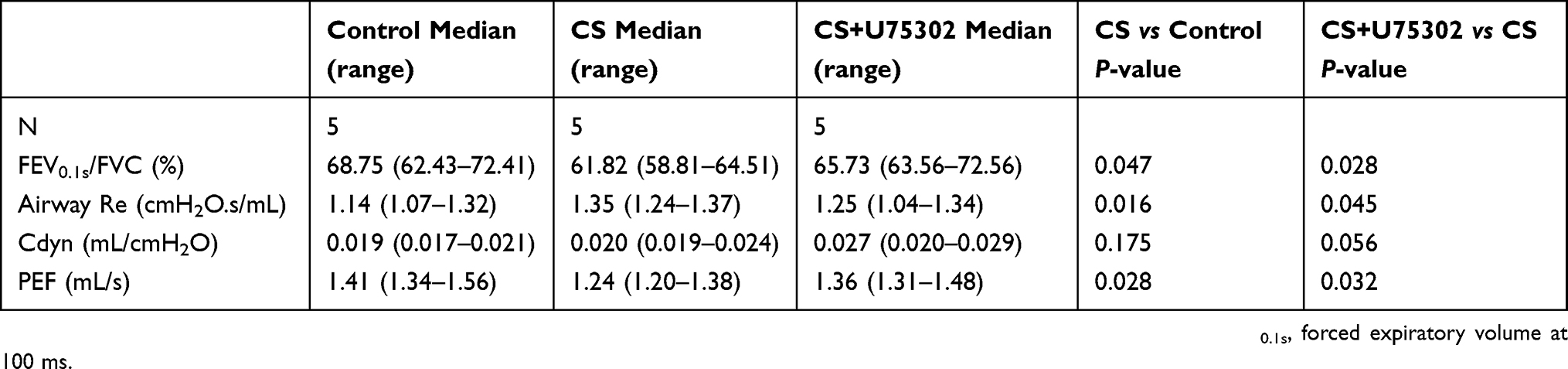 |
Table 2 Parameters comparation of lung function measurements among the three groups |
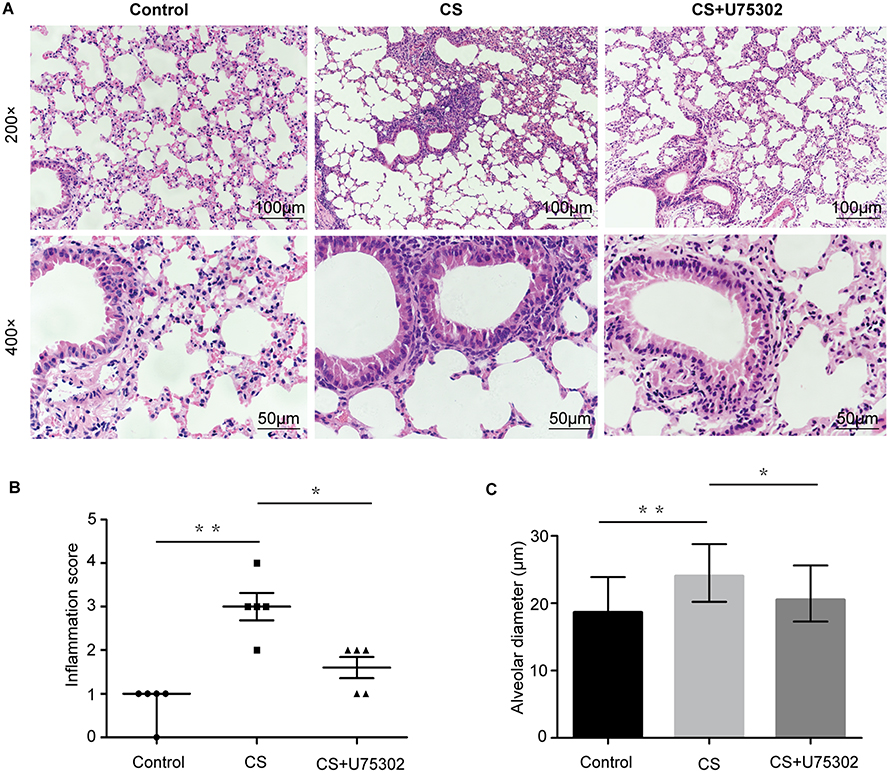 |
Figure 3 Histopathological examination and assessment of lung tissues stained with H&E. (A) Wild-type (WT)s C57BL/6J mice were exposed to cigarette smoke with or without U75302 for 12 weeks. Histopathological images of lung tissues stained with H&E. The images were observed at 200× or 400× magnification. (B) Semiquantified inflammation scores with H&E staining, the results were presented as median with range (10 images per section, n=5 mice/group). (C) Alveolar diameter values were detected at 200× magnification and the bars show median with range (10 images each section, n=5 mice/group). Statistical analysis was performed by non-parametric tests of Kruskal Wallis test and Wilcoxon rank sum test. *P<0.05, **P<0.01. Abbreviations: H&E, hematoxylin and eosin; CS, cigarette smoke. |
Inflammatory factors in lung tissue and BALF
Treatment with U75302 decreased the mRNA expression of TNF-α, IL-6, IL-1β, Muc5ac in CS-induced lung tissues of mice compared to the control group (Figure 4A). The levels of LTB4 and IL-6 in BALF were also reduced in treated group relative to CS group (Figure 4B).
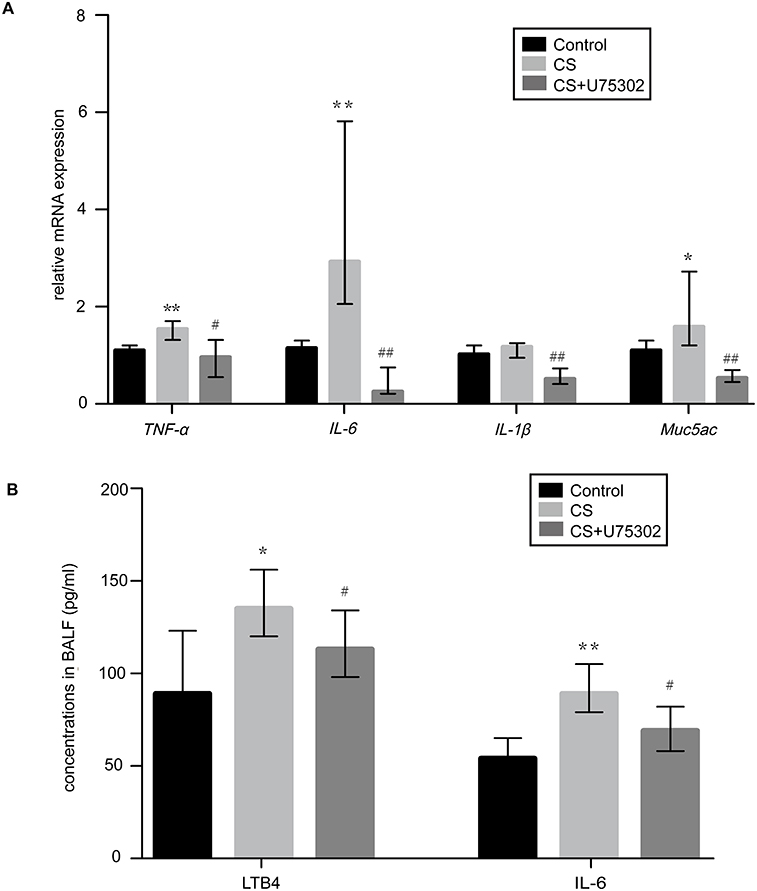 |
Figure 4 Expression of inflammatory factors in lung tissues and BALF. (A) The relative mRNA expression of inflammatory genes (TNF-α, IL-6, IL-1β, and Muc5ac) was normalized to GAPDH by real-time PCR in the three groups. (B) The levels of LTB4 and IL-6 in BALF by ELISA. Data were presented as median with range (n=5 mice/group). *P<0.05, **P<0.01, vs Control group, #P<0.05, ##P<0.01, vs CS group. Statistical analysis was performed by non-parametric tests of Kruskal Wallis test and Wilcoxon rank sum test. Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; IL-1β, interleukin-1β; Muc5ac, Mucin-5 subtype ac; LTB4, Leukotriene B4; BALF, bronchoalveolar lavage fluid. |
Effect of U75302 on autophagy in CS-induced mice
Immunohistological analysis showed that LC3 protein expression in lung tissues was significantly attenuated by treatment with a BLT1 antagonist (Figure 5A). In Figure 5B, we observed a higher expression of p62 and LAMP1 in the CS group compared to the control group and a lower expression in the treated group. The expression of TFEB protein was also elevated in the treated group (Figure 5B). Also, TEM showed that the number of autophagosomes and lysosomes in alveolar macrophages were increased in the CS group and decreased in the treated group (Figure 5C).
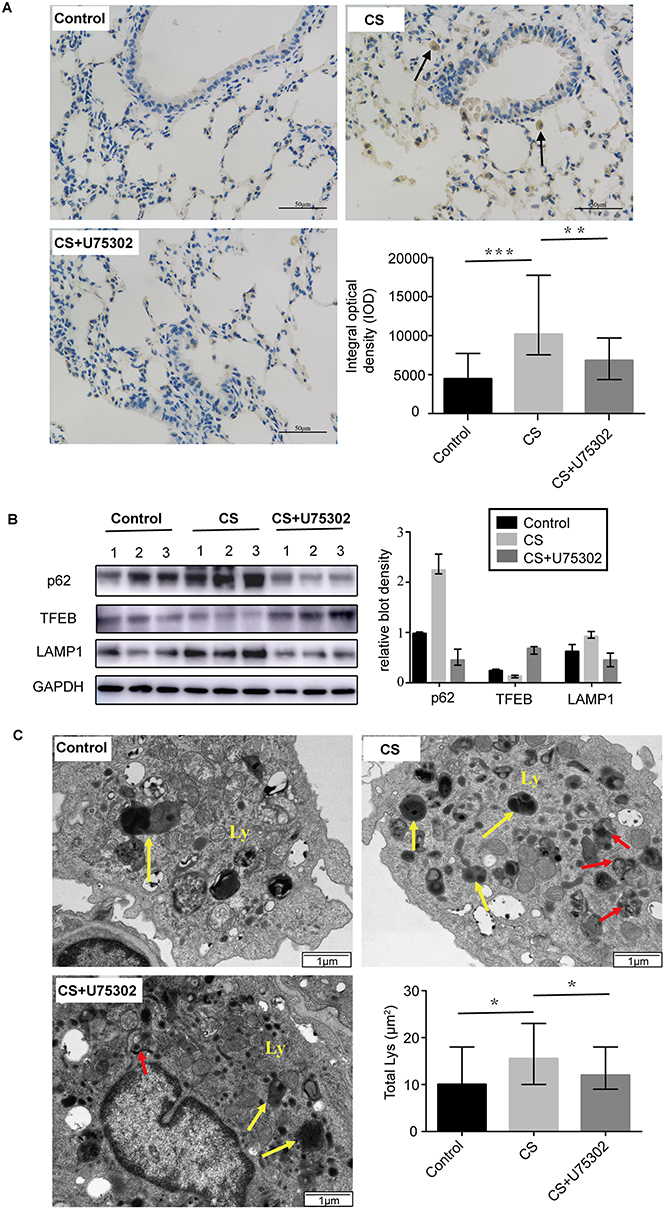 |
Figure 5 Effect of U75302 on autophagy in CS-induced mice. (A) Immunohistochemistry with antibodies against LC3 in lung tissues (brown staining) and the result of quantificational analysis (6 images for each section, n=5 mice/group). Black arrows, alveolar macrophages. (B) Western blotting detected p62, TFEB and LAMP1 protein expression and corresponding quantification normalized to GAPDH (n=3 mice/group). (C) TEM showed the alterations of autophagosomes and lysosomes. Ly, lysosomes (yellow arrows); red arrows, autophagosomes. Semiquantified number of lysosomes (Lys) in TEM images was represented as per 100 µm2 (6 images for each section, n=5 mice/group). The bars represent median with range. *P<0.05, **P<0.01, ***P<0.001. Statistical analysis was performed by non-parametric tests of Kruskal Wallis test and Wilcoxon rank sum test. Abbreviations: CS, cigarette smoke; LC3, microtubule-associated protein light chain 3; p62, Sequestosome1/p62; TFEB, transcription factor-EB; LAMP1, lysosomal-associated membrane protein1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. |
Discussion
Based on this current study, selective BLT1 antagonist (U75302) exerted beneficial effects on the CS-exposure models and cell lines. Using the common markers of autophagy progression (LC3 and p62) and autophagy regulator (TFEB), we showed that autophagy regulation was associated with LTB4-mediated inflammation in CS-induced COPD. Upregulated LC3 mRNA and reduced autophagy proteins expression after U75302 treatment indicated an insufficient autophagy in CSE-treated macrophages. In the treated-mice model of COPD, TEM showed a decreased autophagosomes and lysosomes in alveolar macrophages. The results suggested that BLT1 antagonist could ameliorate inflammatory response through promoting autophagy-lysosomal pathway (Figure 6).
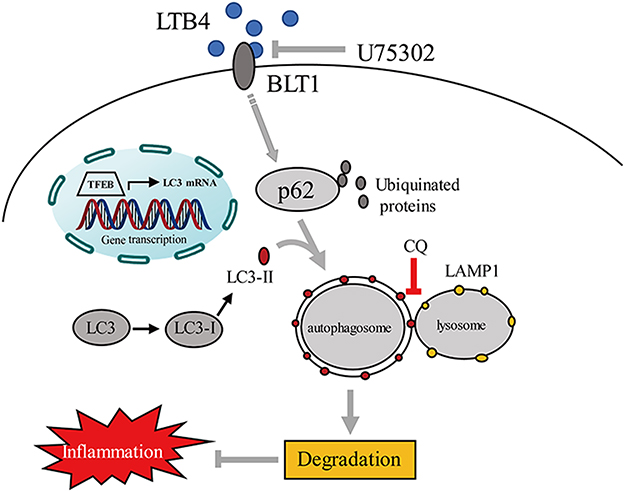 |
Figure 6 A schematic diagram of therapeutic effects of BLT1 antagonist (U75302) in CS-induced COPD. Abbreviations: LC3, microtubule-associated protein light chain 3; p62, Sequestosome1/p62; TFEB, transcription factor-EB; LAMP1, lysosomal-associated membrane protein 1. |
Autophagy is a known lysosome-degradation pathway for the removal and subsequent recycling of dysfunctional intracellular components. A recent study reported that 14,15-epoxyeicosatrienoic acid from metabolic products of free arachidonic acid suppresses the CS-induced inflammation by inhibiting autophagy in lung epithelial cells.26 Autophagy was found to be essential for particulate matter (PM)-induced airway epithelial injury, inhibition of airway inflammation, and mucus hyperproduction.27,28 Various studies have revealed that CS can induce autophagy impairment and deficiency, resulting in inflammatory responses.4,9,11,15,29 Accordingly, it is reasonable to hypothesize that insufficient autophagy degradation may be involved in LTB4-mediated inflammation in COPD. Our results showed that LC3 mRNA expression was upregulated with U75302 treatment in CSE-treated macrophages whereas ameliorated autophagy proteins expression by BLT1 antagonist were detected in vivo and vitro, including LC3-II and p62. Decreases in LC3-II may imply the induction of autophagy degradation. Furthermore, autophagy inhibitor resulted in autophagy markers accumulation even with BLT1 antagonist treatment. The findings indicated that BLT1 antagonist reduced autophagy levels due to enhance autophagic process not because of inhibiting autophagy. Increased autophagy proteins by CS exposure may be accounted for insufficient autophagic clearance in macrophages. Autophagy inhibition with CQ treatment exaggerated inflammatory response, suggesting that the potential for autophagy inducing associated with reduced inflammation by BLT1 antagonist. Therefore, U75302 may be capable of improving autophagy-lysosomal pathway.
The role of autophagy in human diseases is complex that has been shown to be both protective and harmful in different models. Emerging data have improved our understanding of the autophagy process in the development of COPD emphysema. In response to CS exposure, activated autophagy in epithelial cells appears to aggravate airway inflammation and mucociliary clearance dysfunction.30,31 Consistently, genetic depletion of autophagy mediators LC3B and Beclin1 suppressed CSE-induced cell death and LC3B-deficient mice exhibited a reduced emphysema airspace enlargement.27 Conversely, other studies showed a protective role for autophagy that loss of autophagy enhanced cell senescence and oxidative stress in CSE-treated epithelial cells.15,32 Previous study has observed a deficit in the alveolar macrophages of smokers due to the impairment of delivery of bacteria to lysosomes by CS exposure.11 LC3 knockout alveolar macrophages also exhibited increased inflammation.33 Recent study reported that CS-induced autophagy impairment via TFEB pathway resulted in phagocytosis defect in murine macrophages.34 Autophagy process in macrophages was consistently defective in this study, which can be improved by BLT1 antagonist.
A series of autophagy regulators have been identified in the pathogenesis of COPD, among which, TFEB is a master regulator of autophagic and lysosomal genes, with a critical role in neurodegenerative and cardiovascular diseases.35–37 A recent study revealed that TFEB upregulation by autophagy-inducing drugs (Gemfibrozil/Fisetin) treatment could control the CS-induced airway inflammation and autophagy-impairment, which alleviated the pathogenesis of COPD-emphysema.31 The overexpression of TFEB in endothelial cells could inhibit the inflammation and atherosclerosis development due to reduced oxidative-antioxidant imbalance.36 TFEB has been identified as a potent activator of the autophagy-lysosomal pathway, and TFEB induction-enhanced cellular clearance via regulation of lysosomal exocytosis, a process mediated by the activation of lysosomal Ca2+ channel mucolipin1 (MCOLN1).38 Lysosomes are cellular organelles involved in the degradation and recycling processes. In this study, the expression of TFEB was upregulated by BLT1 antagonist treatment in vivo and vitro. Moreover, the autophagosomes and lysosomes in alveolar macrophages were accumulated in the CS group and decreased by U75302 treatment. The observations further confirmed our hypothesis that autophagy-lysosome pathway involved in the regulation of LTB4-mediated inflammation. Inhibition of LTB4/BLT1 pathway may promote the ability of autolysosomes formation and degradation in response to CS exposure. Studies have identified the impairment of autophagy in macrophages from smokers and murine mediated by TFEB expression,11,34 which is consistent with our results. The anti-inflammatory effects of BLT1 antagonist involved in autophagy regulation and promotion of the cellular degradation pathway.
As for the construction of CS-exposure model, different exposure protocols were designed in the vitro and vivo study, which is compatible with previous studies.9,28,39 Indeed, the cell lines with CSE exposure somewhat represent acute process relative to the vivo study of COPD model and such single-factor approaches are actually not representative of the complex pathology that occurs in vivo events. The vitro study was designed to observe the alterations in cell lines, which contributes to the further exploration in the vivo events. Nevertheless, the current different exposure protocols were generally valuable for evaluating the autophagy and inflammation responses and our results indicated similar outcomes as anticipated. The difference was observed that LTB4 levels can be reduced in the treated models but not in the cell lines. The pro-inflammatory mediator LTB4, combined with BLT1 receptor, can lead to the release of inflammatory factors via activating the downstream signaling pathway. Therefore, BLT1 specific antagonist U-75302 blocking the LTB4/BLT1 pathway can reduce the levels of IL-6 both in vitro and vivo. However, LTB4 synthesis is generated from arachidonic acid that is released from membrane phospholipids and signaling via BLT1 is determined by the inflammatory and immune cells. Therefore, it is possible that the levels of LTB4 was not affected by the treatment of BLT1 antagonist in vitro study and can be reduced following the decrease of innate immune cells in the treated group of animal models.
Of note, due to the limitation of equipment and room space, a small size of animal models was performed, and statistical analysis was not evaluated for specific experiments in this study. Given the optimal concentration administration and confounding effects of intraperitoneal injection of CQ and U75302, we did not evaluate the groups of controls with autophagy and BLT1 antagonist. On the other hand, relative negative/positive controls in cell lines was not determined perfectly, which will be an alert for the future work. For further translational research, application in large groups and humans will be needed to verify the beneficial effect of BLT1 antagonist in the subsequent studies.
Conclusion
Taken together, BLT1 antagonist exhibits the anti-inflammatory property through inducing autophagy, and promoting lysosome pathway. This finding indicated that BLT1 antagonist might provide potential therapeutic intervention for improving the management of CS-exposure COPD.
Ethics approval
All the animal experiments were strictly conducted in accordance with the protocols approved by the Ethics Committee and Animal Care Committee of Ruijin Hospital (Shanghai Jiaotong University School of Medicine, China).
Acknowledgment
The study was supported by National Key R&D Program of China (granted number 2017YFC1309701 and 2017YFC1309700), National Natural Science Foundation of China (granted number 81570029), Shanghai Key Discipline for Respiratory Diseases (granted number 2017ZZ02014) and Innovative research team of high-level local universities in Shanghai. We thank the laboratory of Shanghai Ninth People’s Hospital (China) for providing professional assistances and reagent supplement.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rosenberg SR, Kalhan R, Mannino DM. Epidemiology of chronic obstructive pulmonary disease: prevalence, morbidity, mortality, and risk factors. Semin Respir Crit Care Med. 2015;36(4):457–469. doi:10.1055/s-0035-1555607
2. Macnee W. Pathogenesis of chronic obstructive pulmonary disease. Clin Chest Med. 2007;28(3):479–513. doi:10.1016/j.ccm.2007.06.008
3. Davies A, Stephen C, Chinwei L, et al. Global and regional estimates of COPD prevalence: systematic review and meta–analysis. J Glob Health. 2015;5(2):020415. doi:10.7189/jogh.05.020415
4. Bodas M, Vij N. Augmenting autophagy for prognosis based intervention of COPD-pathophysiology. Respir Res. 2017;18(1):83. doi:10.1186/s12931-017-0560-7
5. Hassett DJ, Borchers MT, Panos RJ. Chronic obstructive pulmonary disease (COPD): evaluation from clinical, immunological and bacterial pathogenesis perspectives. J Microbiol. 2014;52(3):211–226. doi:10.1007/s12275-014-4068-2
6. Min T, Bodas M, Mazur S, Vij N. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J Mol Med. 2011;89(6):577–593. doi:10.1007/s00109-011-0732-8
7. Harvey CJ, Thimmulappa RK, Sethi S, et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med. 2011;3(78):78ra32. doi:10.1126/scitranslmed.3002042
8. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
9. Vij N, Chandramani P, Westphal CV, Hole R, Bodas M. Cigarette smoke induced autophagy-impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am J Physiol Cell Physiol. 2016;314(1):C73–C87.
10. Ahmad T, Sundar IK, Lerner CA, et al. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. Faseb J. 2015;29(7):2912. doi:10.1096/fj.14-268276
11. Monick MM, Powers LS, Walters K, et al. Identification of an autophagy defect in smokers’ alveolar macrophages. J Immunol. 2010;185(9):5425. doi:10.4049/jimmunol.1001603
12. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222.
13. Aravamudan B, Kiel A, Freeman M, et al. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2014;306(9):840–854. doi:10.1152/ajplung.00155.2013
14. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. doi:10.1038/nature09782
15. Fujii S, Hara H, Araya J, et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology. 2012;1(5):630–641. doi:10.1016/j.ccm.2007.06.008
16. Miyahara N, Miyahara S, Takeda K, Gelfand EW. Role of the LTB4/BLT1 pathway in allergen-induced airway hyperresponsiveness and inflammation. Allergol Int. 2006;55(2):91–97. doi:10.2332/allergolint.55.91
17. Goodarzi K, Goodarzi M, Tager AM, Luster AD, Andrian UHV. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat Immunol. 2003;4(10):965–973. doi:10.1038/ni972
18. Tager AM, Luster AD. BLT1 and BLT2: the leukotriene B(4) receptors. Prostag Leukot Essent Fatty Acids. 2003;69(2–3):123. doi:10.1016/S0952-3278(03)00073-5
19. Sun M, Wang R, Han Q. Inhibition of leukotriene B4 receptor 1 attenuates lipopolysaccharide-induced cardiac dysfunction: role of AMPK-regulated mitochondrial function. Sci Rep. 2017;7:44352. doi:10.1038/srep44352
20. Chou RC, Kim ND, Sadik CD, et al. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity. 2010;33(2):266–278. doi:10.1016/j.immuni.2010.07.018
21. Lv J, Xiong Y, Li W, et al. BLT1 mediates bleomycin-induced lung fibrosis independently of neutrophils and CD4+ T cells. J Immunol. 2017;198(4):1673. doi:10.4049/jimmunol.1600465
22. Marian E, Baraldo S, Visentin A, et al. Up-regulated membrane and nuclear leukotriene B4 receptors in COPD. Chest. 2006;129(6):1523–1530. doi:10.1378/chest.129.6.1523
23. Dong R, Xie L, Zhao K, et al. Cigarette smoke-induced lung inflammation in COPD mediated via LTB4/BLT1/SOCS1 pathway. Int J Chron Obstruct Pulmon Dis. 2016;11(default):31–41. doi:10.2147/COPD.S96412
24. Lee KS, Lee HK, Hayflick JS, Yong CL, Puri KD. Inhibition of phosphoinositide 3-kinase δ attenuates allergic airway inflammation and hyperresponsiveness in murine asthma model. FASEB J. 2006;20(3):455–465. doi:10.1096/fj.05-5045com
25. Horvat JC, Starkey MR, Kim RY, et al. Early-life chlamydial lung infection enhances allergic airways disease through age-dependent differences in immunopathology. J Allergy Clin Immunol. 2010;125(3):
26. Li Y, Yu G, Yuan S, et al. 14,15-Epoxyeicosatrienoic acid suppresses cigarette smoke condensate-induced inflammation in lung epithelial cells by inhibiting autophagy. Am J Physiol Lung Cell Mol Physiol. 2016;311(5):L970–L980. doi:10.1152/ajplung.00161.2016
27. Chen ZH, Lam HC, Jin Y, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Autophagy. 2010;107(44):18880–18885.
28. Zhou JS, Zhao Y, Zhou HB, et al. Autophagy plays an essential role in cigarette smoke-induced expression of MUC5AC in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2016;310(11):L1042–L1052. ajplung.00418.02015. doi:10.1152/ajplung.00418.2015
29. Netea-Maier RT, Plantinga TS, Fl VDV, Smit JW, Netea MG. Modulation of inflammation by autophagy: consequences for human disease. Autophagy. 2016;12(2):245.
30. Lam HC, Cloonan SM, Bhashyam AR, et al. Histone deacetylase 6–mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2013;123(12):5212–5230. doi:10.1172/JCI69636
31. Bodas M, Patel N, Silverberg D, Walworth K, Vij N. Master autophagy regulator transcription factor EB regulates cigarette smoke-induced autophagy impairment and chronic obstructive pulmonary disease-emphysema pathogenesis. Antioxid Redox Signal. 2017;27(3):150. doi:10.1089/ars.2016.6842
32. Ito S, Araya J, Kurita Y, et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. 2015;11(3):547–559. doi:10.1080/15548627.2015.1017190
33. Wen Z, Fan L, Li Y, et al. Neutrophils counteract autophagy-mediated anti-inflammatory mechanisms in alveolar macrophage: role in post-hemorrhagic shock acute lung inflammation. J Immunol. 2014;193(9):4623–4633. doi:10.4049/jimmunol.1400899
34. Pehote G, Bodas M, Brucia K, Vij N. Cigarette smoke exposure inhibits bacterial killing via TFEB-mediated autophagy impairment and resulting phagocytosis defect. Mediat Inflamm. 2017;2017(2):3028082.
35. Kinghorn KJ, Asghari AM, Castilloquan JI. The emerging role of autophagic-lysosomal dysfunction in Gaucher disease and Parkinson’s disease. Neural Regen Res. 2017;12(3):380–384. doi:10.4103/1673-5374.202934
36. Lu H, Fan Y, Qiao C, et al. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Sci Signal. 2017;10(464):eaah4214. doi:10.1126/scisignal.aah4214
37. Settembre C, Di MC, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Autophagy. 2011;332(11):1429–1433.
38. Medina DL, Fraldi A, Bouche V, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21(3):421–430. doi:10.1016/j.devcel.2011.07.016
39. Tran I, Ji C, Ni I, et al. Role of cigarette smoke-induced aggresome formation in chronic obstructive pulmonary disease-emphysema pathogenesis. Am J Respir Cell Mol Biol. 2015;53(2):159–173. doi:10.1165/rcmb.2014-0107OC
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.