")
Back to Journals » Drug Design, Development and Therapy » Volume 14
The Traditional Chinese Medicine Compound, GRS, Alleviates Blood–Brain Barrier Dysfunction
Authors Zhang Y, Hu Y, Li M, Wang J, Guo G, Li F, Yu B, Kou J
Received 30 August 2019
Accepted for publication 10 February 2020
Published 28 February 2020 Volume 2020:14 Pages 933—947
DOI https://doi.org/10.2147/DDDT.S229302
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Yan Zhu
Yuanyuan Zhang, Yang Hu, Min Li, Jieman Wang, Gengshuo Guo, Fang Li, Boyang Yu, Junping Kou
Jiangsu Key Laboratory of TCM Evaluation and Translational Research, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing, People’s Republic of China
Correspondence: Boyang Yu; Junping Kou
Jiangsu Key Laboratory of TCM Evaluation and Translational Research, China Pharmaceutical University, Nanjing 211198, People’s Republic of China
Tel +86 25-86185157
Fax +86 25-86185158
Email [email protected]; [email protected]
Introduction: Traditional Chinese medicine (TCM) provides unique advantages for treatment of ischemic stroke, an aging-related vascular disease. Shengmai powder (GRS) is composed of three active components, specifically, ginsenoside Rb1, ruscogenin and schisandrin A, at a ratio of 6:0.75:6. The main objective of this study was to evaluate the effects of GRS on blood–brain barrier (BBB) dysfunction under conditions of middle cerebral artery occlusion/reperfusion (MCAO/R).
Methods: C57BL/6J mice subjected to MCAO/R were used as a model to assess the protective effects of varying doses of GRS (6.4, 12.8, and 19.2 mg/kg) on BBB dysfunction.
Results: GRS reduced cerebral infarct volume and degree of brain tissue damage, improved behavioral scores, decreased water content and BBB permeability, and restored cerebral blood flow. Moreover, GRS promoted expression of zona occludens-1 (ZO-1) and claudin-5 while inhibiting matrix metalloproteinase 2/9 (MMP-2/9) expression and myosin light chain (MLC) phosphorylation. In vitro, GRS (1, 10, and 100 ng/mL) enhanced the viability of bEnd.3 cells subjected to oxygen glucose deprivation/reoxygenation (OGD/R) and decreased sodium fluorescein permeability.
Conclusion: Consistent with in vivo findings, ZO-1 and claudin-5 were significantly upregulated by GRS in bEnd.3 cells under OGD/R and MMP-2/9 levels and MLC phosphorylation reduced through the Rho-associated coil-forming protein kinase (ROCK)/cofilin signaling pathway. Based on the collective findings, we propose that the TCM compound, GRS, plays a protective role against I/R-induced BBB dysfunction.
Keywords: GRS, ischemic stroke, blood–brain barrier, endothelial, tight junctions, ROCK/cofilin
Introduction
Stroke is a leading cause of death worldwide and a major contributory factor to permanent disability.1,2 Blood–brain barrier (BBB) injury is a significant physiological and pathological consequence of ischemic stroke, contributing to early death, severe long-term cognitive impairment, and poor clinical prognosis.3,4 Evaluation of BBB permeability in early cerebral ischemia and development of effective strategies to protect BBB function and prevent secondary brain injury after stroke, therefore remains an urgent medical requirement.5
BBB function depends on tight junctions (TJ) between adjacent endothelial cells, which are composed of transmembrane molecules (claudins, occludins, junctional adhesion molecules, and others), scaffold proteins (zona occludens (ZOs), afadin, and membrane-associated guanylate kinase), and connexins (cingulin and myosin).6 Matrix metalloproteinases (MMP), a superfamily of calcium-dependent zinc endopeptidases, act to remodel and degrade the extracellular matrix (ECM), which is critical for BBB function and signal transduction in the neurovascular unit.7 Stroke triggers alterations in MMP levels (including increased expression of MMP-2/9), promoting ECM degradation, TJ destruction, and consequently, BBB permeability.
TJs are connected via connexins to the actin cytoskeleton, which maintains the endothelial cell structure and vascular permeability.8,9 Early BBB destruction is associated with endothelial cytoskeleton rearrangement. Stress fiber contraction transfers tension to cell junctions anchored to the actin cytoskeleton, resulting in damage to the junction complex, destabilization of endothelial cell junctions, and increased BBB permeability.10–12 Rho-related coiled helix-forming protein kinases (ROCK1 and ROCK2) are molecular switches that regulate cytoskeletal rearrangement. ROCK1/2 activation increases phosphorylation of myosin light chains (MLC) and cofilin, regulates actin kinetics, enhances stress fiber formation, and alters cell contractility13,14.
GRS is a traditional Chinese medicine (TCM) comprising three pharmacologically active components (ginsenoside Rb1, ruscogenin, and schisandrin A). Ginsenoside Rb1 has been shown to exert protective effects against central nervous system (CNS) diseases.15 Ruscogenin, the main active component of Ophiopogon japonicus, protects against BBB disruption in ischemia/reperfusion (I/R) injury by inhibiting inflammation and mitogen-activated protein kinase signaling.16 Schisandrin A exerts protective effects against D-galactose-induced oxidative brain damage and improves cognitive impairment in rats.17 Previous findings suggest that GRS mitigates myocardial I/R injury by suppressing mitochondria-mediated apoptosis and modulating AMP-activated protein kinase-mediated mitochondrial fission.18 However, the effects of GRS on ischemic stroke are yet to be characterized.
The main objective of the current study was to evaluate the effects of GRS on BBB permeability in ischemic stroke with the aid of a middle cerebral artery (MCA) occlusion/reperfusion (MCAO/R) C57BL/6J mouse model. The mechanisms underlying the protective effects of GRS against I/R were further investigated using an oxygen glucose deprivation/reoxygenation (OGD/R)-induced endothelial barrier permeability model of bEnd.3 cells. Our collective findings indicate that GRS exerts a significant protective effect on the bEnd.3 endothelial barrier, preventing OGD/R-induced damage.
Methods
Reagents
GRS isolated by our group was prepared for use by dissolving in ethanol and subsequently diluting to 5% ethanol using normal saline (chemical structures of the three components of GRS are depicted in Figure 1A). Fluorescein isothiocyanate-conjugated goat anti-rabbit immunoglobulin G and horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse immunoglobulin G were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA). All chemicals were of the highest available purity.
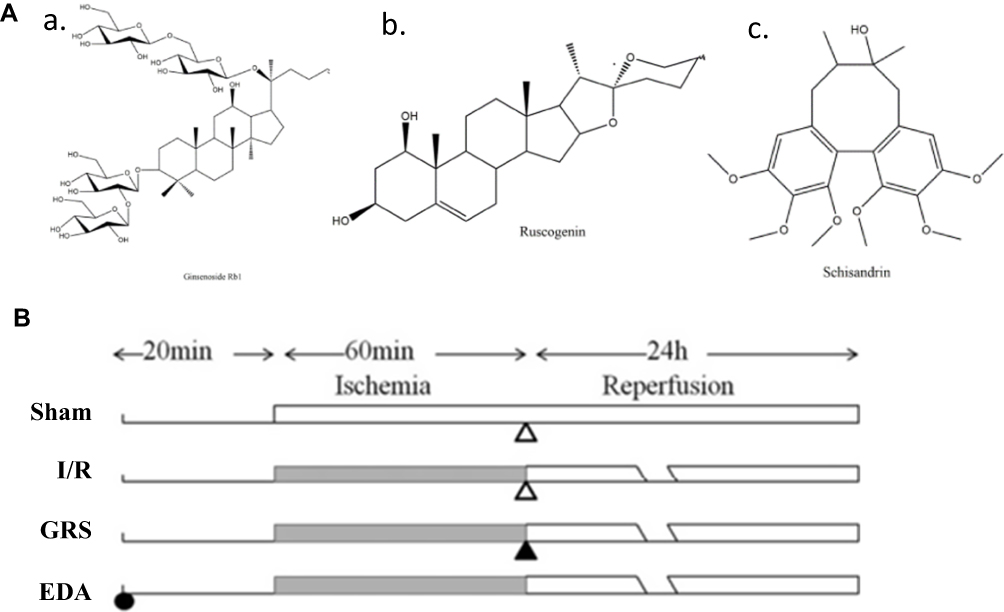 |
Figure 1 Chemical structures of the three components of GRS. (A) Ginsenoside Rb1 (a), ruscogenin (b) and schisandrin (c). (B) Vehicle (open triangle) and GRS were administered i.p. 1 h after ischemia. EDA was administered i.p. 20 min before middle cerebral artery occlusion. |
Cell Culture
The bEnd.3 cells, which were purchased from the KeyGen BioTECH (Nanjing, China), were cultured in RPMI1640 complete medium containing 15% fetal bovine serum. After reaching 80–90% confluence, the medium was removed and cells washed with 2 mL phosphate-buffered saline (PBS). Next, 1 mL trypsin was added for ~2 mins to detach cells. Once the cells were detached, trypsin was removed and 2 mL complete culture medium added. Cells were centrifuged at 800 rpm for 5 mins and supernatant discarded, followed by the addition of fresh medium. The suspension was blown gently. Cells were subsequently plated on 60 mm culture dishes at a density of 2 × 105 cells/mL (4 mL total) and incubated at 37°C in a 5% CO2 for 2–3 days.
Animals
C57BL/6J mice weighing 18–22 g with 6–8 weeks were provided by the Reference Animal Research Centre of Yangzhou University (Yangzhou, China). All procedures were approved by the Animal Ethics Committee of the School of Chinese Materia Medica, China Pharmaceutical University (Nanjing, China). Experiments were performed in accordance with the National Institutes of Health Guide for the Care and use of Laboratory Animals (National Institutes of Health Publication No 80–23, revised in 1996). All animals were randomly assigned according to weight to experimental groups and operators blinded to the study procedures.
Focal Cerebral I/R
MCAO was induced in C57BL/6J mice, followed by reperfusion, as reported previously (Figure 1B).19 Mice were anesthetized with 4% chloral hydrate (0.1 mL per 10 g body weight, i.p.). Neck vessels were exposed by a midline incision, and the branches of the right external carotid artery carefully isolated and cauterized. A 6–0 nylon monofilament suture, blunted at the tip and coated with 1% poly-L-lysine, was advanced 9–10 mm into the internal carotid artery for MCAO. Body temperature was maintained at 37.0 ± 0.5°C using a heating pad (ALCBIO, Shanghai, China) during surgery and ischemia. Sham animals underwent the same procedure, but the suture was not advanced into the internal carotid artery. A laser Doppler flow meter (FLPI2; Moor Instruments Ltd., Axminster, UK) was used to confirm decreased MCA blood flow. Mice with 30% blood flow relative to pre-ischemia levels were used for further experiments.
Evaluation of Infarct Volume, Neurological Deficits, and Brain Water Content
The protective effects of GRS were examined in mice randomly divided into six groups (n = 6 per group): sham, I/R, GRS after 1 h MCAO, GRS (i.p.), and 0.9% sodium chloride (i.p.). After reperfusion for 24 h, mice were anesthetized with 4% choral hydrate and sacrificed. Mouse brains were removed, dissected, and sectioned. Five slices (1.5 mm thickness) were incubated with 1% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich Co., St Louis, MO, USA) for 30 min in the dark, followed by fixing with 4% paraformaldehyde (PFA). Lesions areas that were not stained red were quantitatively analyzed with Quantity One analysis software (Bio-Rad Laboratories Inc., Hercules, CA, USA). Infarct volume was calculated based on slice thickness and measured lesion areas. Data are expressed as a percentage of infarct volume, compared to total hemisphere volume. Behavioral assessment was performed 24 h after reperfusion. Neurological deficits were graded on an 18-point scale as described previously. Spontaneous activity, symmetry of movement, symmetry of forelimbs, climbing, reaction to touch, and response to vibrissae touch were evaluated.20 Each test value was scored on a four-point scale (0–3). The total score was the sum of the individual test scores, with a maximum score of 18 for healthy animals. Brain water content was determined 24 h after reperfusion with the wet–dry method using the following equation: ((wet weight – dry weight)/wet weight) × 100%.
Cerebral Blood Flow Measurement
Laser Doppler flowmetry was applied for cerebral blood flow (CBF) assessment. A computer-controlled optical scanner directed a low-power laser beam over the exposed cortex. The scanner head was positioned parallel to the cortex at a distance of ~20 cm. A color-coded image indicating relative perfusion levels was displayed on a video monitor. Images were acquired at ischemia onset and during reperfusion (n = 6 per group).
Hematoxylin and Eosin Staining
Histological analysis was performed using hematoxylin and eosin (H&E) staining. Animal brains were immediately removed 24 h after reperfusion under deep anesthesia with chloral hydrate, fixed in 10% phosphate-buffered formalin for 24 h, and embedded in paraffin wax. A series of adjacent brain sections (5 µm thick) were cut in the coronal plane, followed by staining with H&E. Histological analysis was performed by a pathologist blinded to the treatment groups.
Evaluation of BBB Permeability
We evaluated leakage of Evans blue (EB) dye into the brain for the BBB permeability test following tail vein injection.21 Two hours before sacrifice, 0.1 mL/10 g body weight of 2% EB in normal saline was injected into each animal. Animals were anesthetized and perfused with normal saline. For quantitative measurement of EB leakage, the ipsilateral hemisphere was removed and homogenized in 1 mL trichloroacetic acid, followed by centrifugation at 12,000 × g for 20 min. The EB concentration in the supernatant was determined at 620 nm using a spectrophotometer, quantified using a standard curve, and calculated as micrograms EB per gram of tissue.
Western Blot
Proteins extracted from ischemic cortices and corresponding cortices of sham-operated mice were analyzed via Western blot. Membranes were incubated overnight at 4°C with primary antibodies: ZO-1 (1:100; ab59720, Abcam, Cambridge, UK), claudin-5 (1:500; Abcam, Cambridge, UK), MMP-2 (1:500; 1:500; Abcam, Cambridge, UK); MMP-9 (1:500, millipore, USA); p-MLC (1:500; CST, USA); MLC (1:1000, CST, USA); ROCK1 (1:1000; Santa Cruz, USA); p-cofilin (1:500; Abcam, UK); cofilin (1:1000, Santa Cruz, USA) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 1:8000; Kangchen Bio-tech Inc., Shanghai, China). After washing, membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1.5 h. Immunoreactive bands were detected using the chemiluminescence system (ECL Plus; Amersham Life Science, Arlington Heights, IL, USA) and analyzed with Quantity One analysis software (Bio-Rad Laboratories Inc.). Each ischemic sample was normalized against GAPDH as well as the sham sample.
Immunofluorescence Staining
Mice were anesthetized i.p. with 4% chloral hydrate (0.1 mL/10 g body weight) and transcardially perfused with 100 mL normal saline, followed by 100 mL of 0.1 M PBS containing 4% PFA (pH 7.4). Brains were post-fixed in 0.1 M PBS + 4% PFA overnight and subsequently dehydrated in 40% sucrose solution. Coronal brain sections (20 µm thick) were obtained using a cryostat (CM1950; Leica Microsystems, Wetzlar, Germany) and blocked for 1.5 h in 5% bovine serum albumin (BSA) in PBS with 0.1% Triton X-100. Sections were incubated overnight at 4°C in 3% BSA in PBS containing 0.1% Triton X-100 with an anti-ZO-1 antibody (1:50; ab59720, Abcam). After rinsing three times with PBS, sections were incubated for 2 h with a fluorochrome-coupled secondary antibody. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Beyotime Institute of Biotechnology, Shanghai, China). After rinsing with PBS, sections were visualized under a fluorescence microscope (Leica Microsystems).
Statistical Analysis
All results are expressed as means ± SD. Student’s two-tailed t-test was used for comparisons between two groups and one-way analysis of variance followed by Dunnett’s test for three or more groups. Data were considered statistically significant at P < 0.05. All analyses were performed using GraphPad Prism software (Version 5.01; San Diego, CA, USA).
Results
GRS Reduces Infarct Volume and Brain Water Content and Improves Behavioral Outcomes in I/R Mice
Infarct volume was evaluated in I/R mouse brains using 2,3,5-triphenyltetrazolium chloride (TTC) staining and visualized with ImageJ software (National Institutes of Health, Bethesda, MD, USA). Representative images of TTC-stained brain sections are presented in Figure 2A, with corresponding infarct volumes and statistical data summarized in Figure 2B. As shown in Figure 2A and B, ischemic hemispheres of the model group showed severe infarction (P<0.01), compared to the sham-operated group. I/R mice additionally exhibited lower neurobehavioral scores (P<0.01; Figure 2C). All three doses of GRS reduced cerebral infarct volume and improved neurological function. The efficacy of GRS at high doses was similar to that of edaravone (EDA), which was used as the positive control, indicative of GRS-induced improvement of ischemic brain injury in mice. As shown in Figure 2D, brain water content in the model group was significantly higher than that in the sham operation group (P < 0.01). Treatment with medium and high GRS doses significantly reduced brain water content and edema in I/R mice, compared to their sham-operated counterparts. High-dose GRS (19.2 mg/kg) induced protective effects to a similar extent as EDA (P < 0.01).
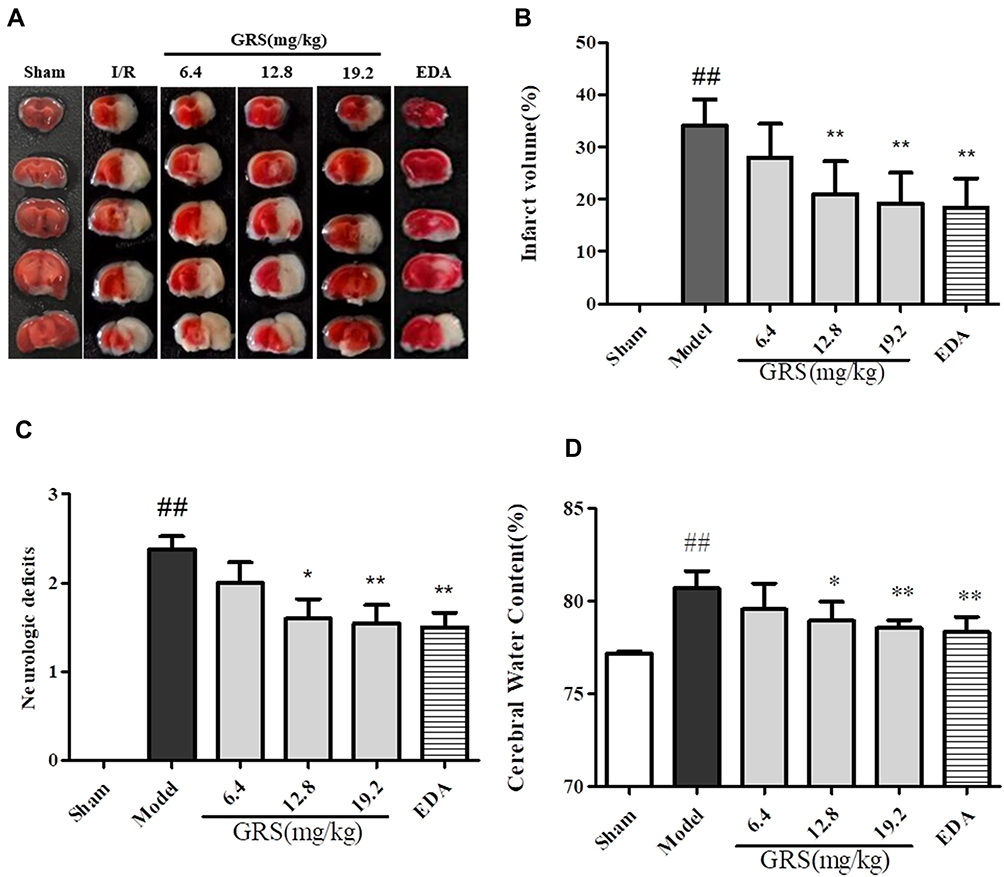 |
Figure 2 GRS reduces infarct volume and brain water content and improves behavioral outcomes in I/R mice. Mice were subjected to reperfusion for 24 h after 1 h of ischemia. GRS (6.4, 12.8, 19.2 mg/kg) and EDA (3 mg/kg) were administered immediately before reperfusion. (A) Protective effects of GRS on I/R-induced infarct volume. (B) Evaluation of brain infarct volume after transient MCAO/R. (C) Evaluation of mouse neurobehavioral scores after transient MCAO/R. (D) Protective effects of GRS on I/R-induced brain edema. Data are presented as means ± SD, n = 6. ##P < 0.01 vs sham group, **P < 0.01 vs I/R mice, *P < 0.05 vs I/R mice. |
Effects of GRS on BBB Permeability in I/R Mice
All three GRS doses induced varying degrees of CBF in the ischemic hemisphere of I/R mice (Figure 3A), with the highest dose exerting a similar effect as EDA (P < 0.01). I/R led to increased vacuolation, nuclear pyknosis and decreased numbers of nerve cells in the infarct area. Treatment with GRS (19.2 mg/kg) significantly reduced vacuolation and cell damage (P<0.01), indicative of a protective effect against I/R-induced brain damage (Figure 3B). At 24 h after reperfusion, EB leakage was greater in the I/R group, compared with that in the sham group (Figure 3C and D; P < 0.01). GRS (12.8 and 19.2 mg/kg) significantly inhibited I/R-induced EB leakage to an equivalent level as EDA at the highest dose (P < 0.01). Our findings clearly indicate that GRS effectively decreases I/R-induced BBB permeability.
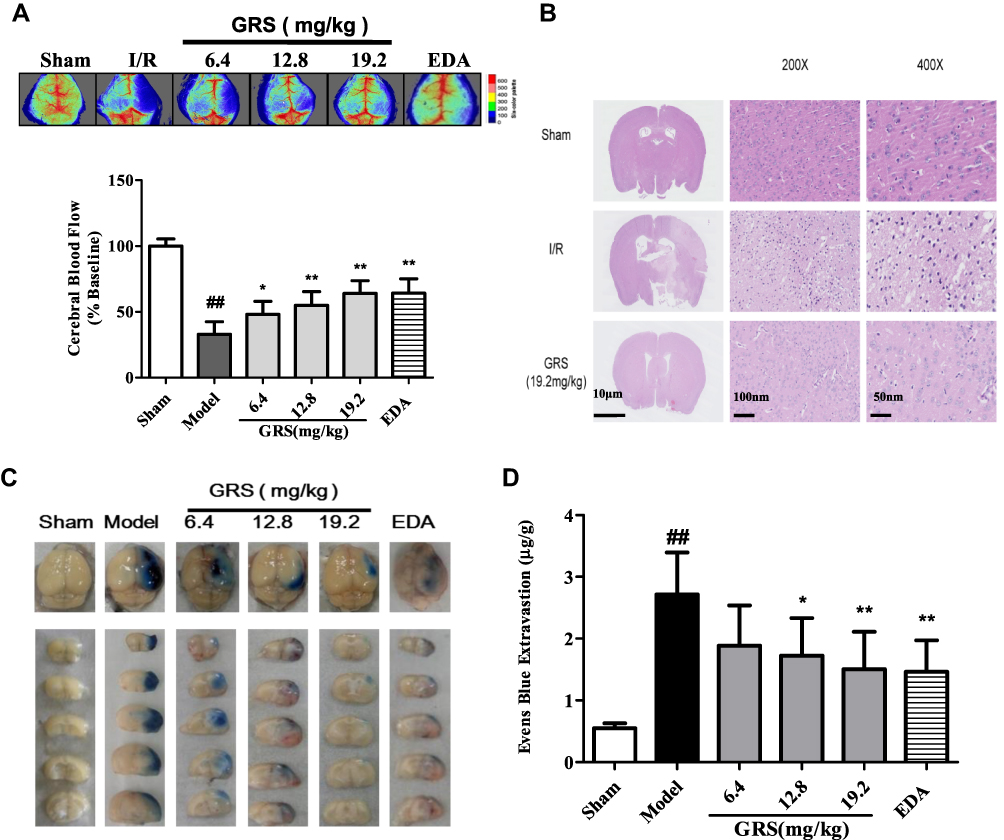 |
Figure 3 GRS reduces BBB permeability in I/R mice. Following MCAO, CBF was measured using laser Doppler flowmetry and histological analysis performed with H&E staining. At 2 h before euthanization, 0.1 mL per 10 g body weight of 2% EB in normal saline was injected into each animal. Animals were anesthetized and perfused with normal saline. (A) Protective effect of GRS against I/R-induced decrease in CBF in ischemic brain regions. (B) Effects of GRS on I/R-induced histological features of brain slices from ischemic brain regions. (C) Effects of GRS on MCAO/R-induced EB leakage. Use images of EB-stained brain sections detected in different groups following 24 h of reperfusion after MCAO to show. (D) Amount and statistical result of EB extravasation. Data are presented as means ± SD, n = 6. ##P < 0.01 vs sham group, **P < 0.01 vs I/R mice, *P < 0.05 vs I/R mice. |
GRS Enhances ZO-1 and Claudin-5 Levels in I/R Mice
ZO-1 protein levels were lower in brain tissue subjected to I/R, compared to the sham operation group, as shown in Figure 4A and B. Treatment with medium and high doses of GRS induced a significant increase in ZO-1 levels (P<0.01). Consistently, immunofluorescence data showed markedly enhanced ZO-1 expression and distribution in brain microvascular endothelial cells (BMEC) of I/R mice treated with 19.2 mg/kg GRS.
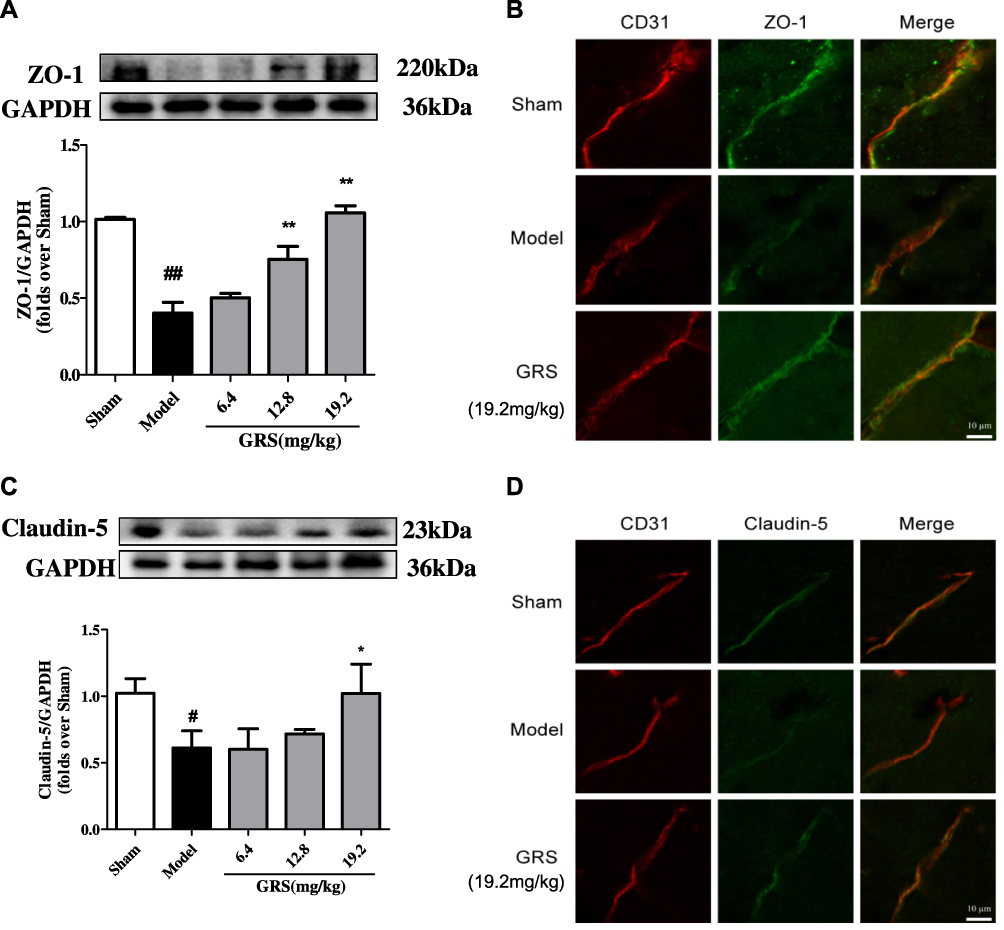 |
Figure 4 Effects of GRS on TJ protein expression in I/R mice. Mice were subjected to 1 h of ischemia, followed by reperfusion for 24 h. GRS (6.4, 12.8, or 19.2 mg/kg) was administered immediately before reperfusion. Proteins extracted from the cortex of the ischemic hemisphere in operated mice and corresponding areas in sham-operated mice were examined via Western blot analysis. (A) Effects of GRS on I/R-induced ZO-1 expression. (B) Confocal immunofluorescent images of ZO-1 (green) and CD31, a marker of endothelial cells (red), in ischemic areas. (C) Expression of claudin-5. (D) Confocal immunofluorescent images of claudin-5 (green) and CD31 (red) in the ischemic area. Data are presented as mean ± SD, n = 3. ##P < 0.01 vs sham group; #P < 0.05 vs sham group; **P < 0.01 vs model group, *P < 0.05 vs I/R mice. |
As shown in Figure 4C and D, claudin-5 protein levels were lower in brain tissue under I/R conditions, compared to the sham operation group. GRS (19.2 mg/kg) induced a significant increase in claudin-5 expression (P<0.05). In immunofluorescence analyses, 19.2 mg/kg GRS significantly enhanced claudin-5 expression and distribution in BMECs in I/R mice.
GRS Suppresses MMPs and p-MLC Levels in I/R Mice
MMP-2/9 levels were significantly increased in brain tissue following I/R. The I/R-induced increase was mitigated by GRS (Figure 5A and B). The protective effect of GRS on BBB may thus be attributable to inhibition of I/R-induced production of MMP-2/9. In addition, GRS significantly inhibited I/R-induced phosphorylation of MLC, suggesting that protective effects on BBB are mediated by regulating stress fiber formation in endothelial cells (Figure 5C and D).
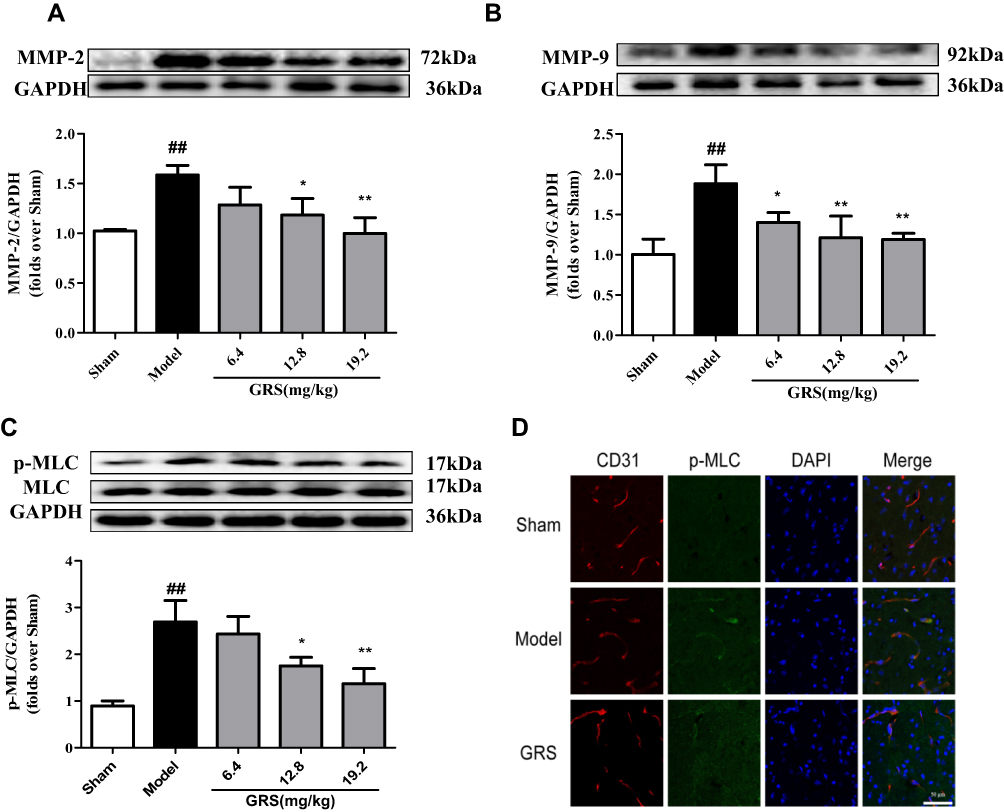 |
Figure 5 Effects of GRS on MMP-2/9 and p-MLC in I/R mice. Mice were subjected to 1 h of ischemia, followed by reperfusion for 24 h. GRS (6.4, 12.8, or 19.2 mg/kg) was administered immediately before reperfusion. MMP-2 (A), MMP-9 (B) and p-MLC (C) levels were quantitatively analyzed via Western blot. Data are presented as means ± SD, n =3. ##P < 0.01 vs sham group; *P < 0.05 vs model group; **P < 0.01 vs model group. (D) Confocal immunofluorescent images of p-MLC (green) and the endothelial marker CD31 (red) in the ischemic hemisphere. Scale bar = 100 µm. |
GRS Attenuates Endothelial Cell Permeability Disruption in OGD/R-Injured bEnd.3 Cells
bEnd.3 cells exposed to OGD/R were treated with GRS (1, 10, and 100 ng/mL) using EDA as a positive control. GRS (1–100 ng/mL) reduced OGD/R-induced sodium fluorescein leakage to a similar extent as EDA (Figure 6A), signifying effective protection against OGD/R-induced bEnd.3 cell barrier permeability. As shown in Figure 6B and C, all doses of GRS mitigated downregulation of ZO-1 protein in bEnd.3 cells exposed to OGD/R. ZO-1 continuously distributed in the control group. In contrast, ZO-1 protein expression was decreased and not continuously distributed in OGD/R cells. High-dose GRS (100 ng/mL) enhanced the expression and distribution of ZO-1 in bEnd.3 cells subjected to OGD/R to a significant extent. GRS (1–100 ng/mL) additionally inhibited OGD/R-induced downregulation of claudin-5 protein (Figure 6D). Claudin-5 protein was continuously distributed in the control group but decreased in the OGD/R group, with fragmented distribution. High-dose GRS (100 ng/mL) induced significant upregulation of claudin-5 protein and improved its distribution in bEnd.3 cells subjected to OGD/R. In view of these results, we propose that GRS reduces OGD/R-induced bEnd.3 cell barrier permeability by regulating the expression of TJ proteins ZO-1 and claudin-5.
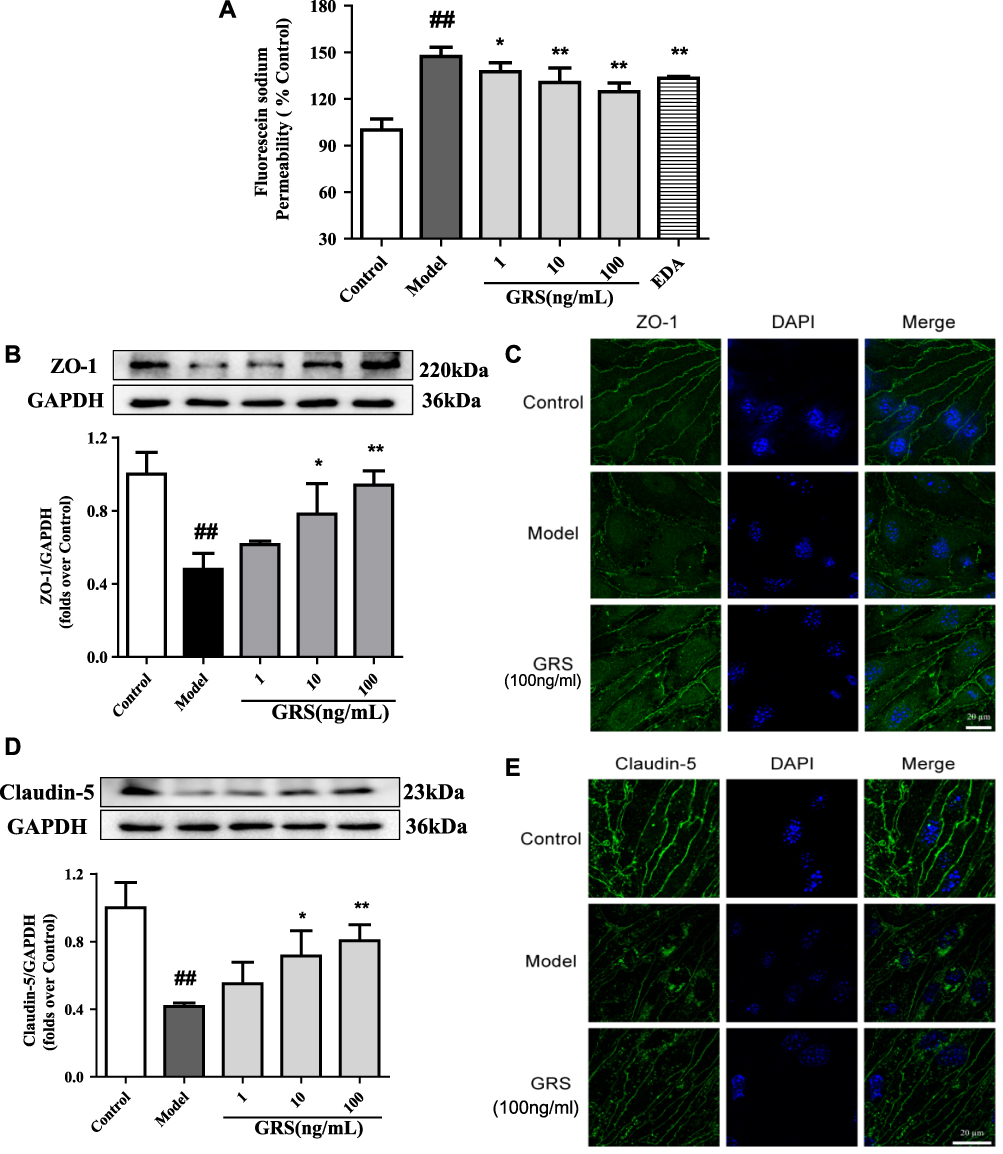 |
Figure 6 Effects of GRS on TJ protein expression in OGD/R-treated bEnd.3 cells. Cells were pretreated with GRS (1-100 ng/mL) and EDA (100 μmol/L), and subjected to OGD for 6 h followed by reperfusion for 18 h. The barrier function protection effect of GRS was evaluated with sodium fluorescein assay. (A) Protective effect of GRS against OGD/R-induced bEnd.3 cell injury. Results are expressed as means ± SD, n = 5. ##P < 0.01 vs Control; *P < 0.05 vs Model; **P < 0.01 vs Model. (B) ZO-1 protein levels were quantitatively analyzed via Western blot. (C) Representative confocal immunofluorescent images of ZO-1 (green) and DAPI-stained nuclei (blue). (D) Western blot analysis of claudin-5 protein expression. (E). Confocal immunofluorescent images of claudin-5 (green) and DAPI-stained nuclei (blue). Results are presented as means ± SD, n = 3. ##P < 0.01 vs Control; * P < 0.05 vs Model; ** P < 0.01 vs Model. |
Effects of GRS on MMP-2/9 and p-MLC Levels in OGD/R-Injured bEnd.3 Cells
Enhanced MMP-2/9 expression under OGD/R conditions was inhibited upon treatment of bEnd.3 cells with GRS (1–100 ng/mL) (Figure 7A and B). The results suggest that GRS reduces OGD/R-induced permeability of the bEnd.3 cell barrier via inhibiting MMP-2/9 release. Furthermore, GRS significantly suppressed OGD/R-induced MLC phosphorylation in bEnd.3 cells (Figure 7C and D), supporting GRS-mediated prevention of formation of bEnd.3 stress fibers under OGD/R.
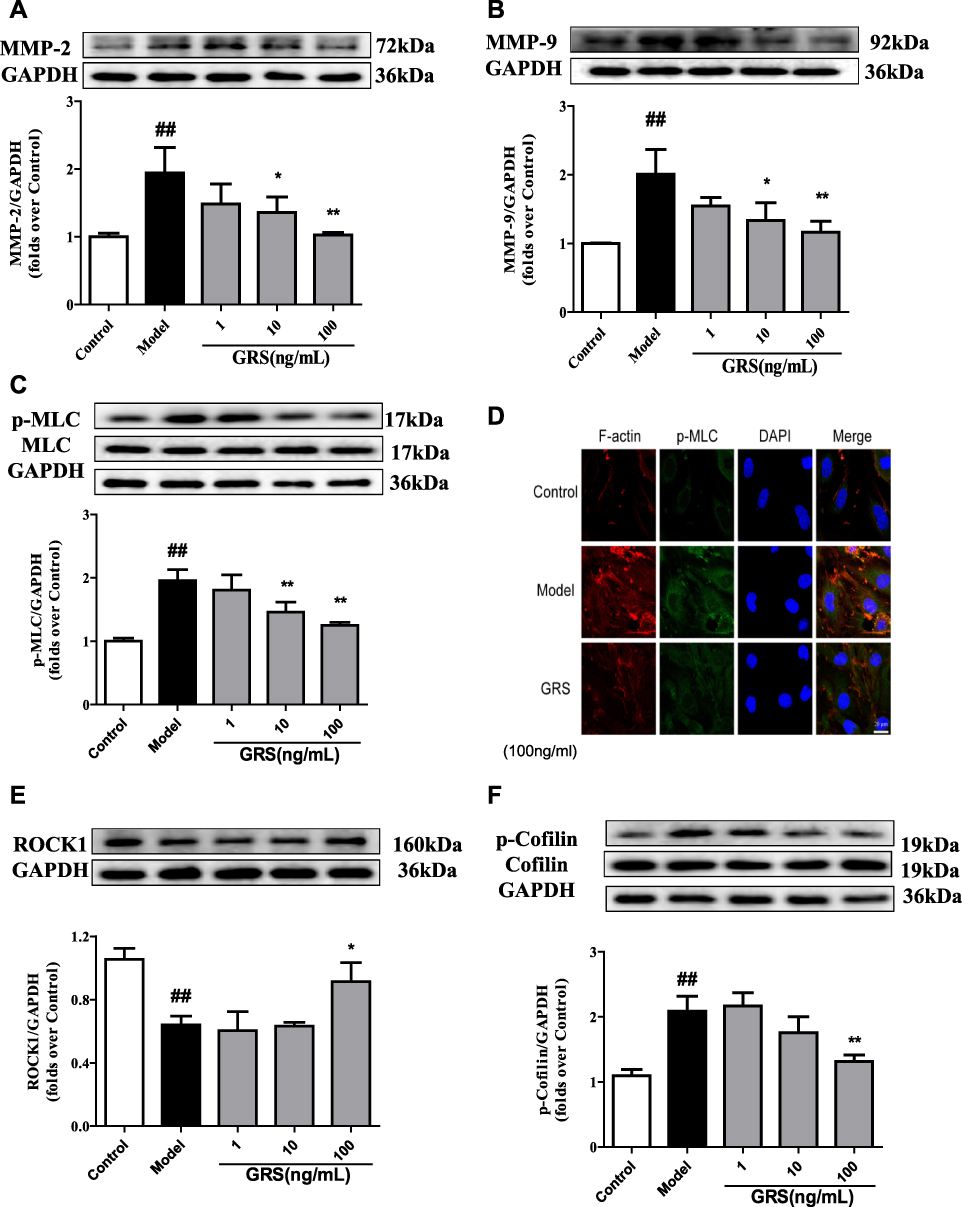 |
Figure 7 Effects of GRS on MMP-2/9, MLC, and ROCK/cofilin levels in OGD/R- injured bEnd.3 cells. Cells were treated with GRS (1–100 ng/mL) prior to 6 h of OGD and 18 h of reoxygenation. (A) Quantitative analysis of MMP-2 and (B) MMP-9 levels via Western blot. Band intensities were assessed via scanning densitometry and data normalized against GAPDH. Cells were treated with GRS (1–100 ng/mL) before 6 h of OGD in a hypoxic chamber and subjected to reoxygenation for 1 h. (C) Western blot analysis of phosphorylated MLC. (D) Confocal immunofluorescent images of F-actin (red), p-MLC (green) and nuclei (blue) in bEnd.3 cells. Scale bar = 20 µm. ROCK1 (E) and p-cofilin (F) levels were quantitatively analyzed via Western blot. Data are expressed as means ± SD, n = 3. ##P < 0.01 vs Control; *P < 0.05 vs Model; **P < 0.01 vs. Model. |
Effect of GRS on ROCK/Cofilin Signaling in OGD/R-Injured bEnd.3 Cells
ROCK1 was sheared and protein expression decreased in response to OGD/R. GRS at all three doses significantly inhibited OGD/R-induced shearing and enhanced expression and activation of ROCK1 (Figure 7E). In contrast, ROCK2 expression was not affected by GRS at any of the doses examined (data not shown). Additionally, OGD/R significantly enhanced cofilin phosphorylation (Ser-3), which was inhibited by GRS (10 and 100 ng/mL) (Figure 7F). Our results collectively demonstrate that GRS protects the bEnd.3 cell barrier against OGD/R-induced damage through the ROCK/cofilin signaling pathway.
Discussion
Data from the current study showed that GRS (6.4–19.2 mg/kg) significantly improves the neurobehavioral score and CBF, reduces cerebral infarct volume, brain water content, and edema, and mitigates brain pathological changes in MCAO/R mice. GRS additionally reduced BBB permeability, as evident from decreased leakage of EB, degradation of barrier-related TJ proteins ZO-1 and claudin-5, expression of MMP-2/9 and phosphorylation of MLC. Our collective results clearly indicate that GRS, the active component group of Shengmai powder, effectively protects against ischemia-reperfusion-induced BBB injury. In vitro, GRS reduced OGD/R-induced bEnd.3 cell death and fluorescein sodium leakage, confirming improvement of the barrier function of bEnd.3 cells. Furthermore, GRS mitigated OGD/R-induced decrease in ZO-1 and claudin-5 expression in bEnd.3 cells and increase in MMP-2/9 expression and phosphorylation of MLC through the ROCK/cofilin signaling pathway. Clearly, GRS exerts significant protective effects against OGD/R-induced endothelial barrier damage in bEnd.3 cells.
MCA and its branches are the most commonly affected cerebral vessels in human ischemic stroke, accounting for about 70% infarctions. Therefore, an MCAO animal model is the best experimental representation of human ischemic stroke.20–22 Neurobehavioral score and cerebral infarction volume are the main outcomes used to evaluate the degree of ischemic brain damage and efficacy of therapeutic drugs. Longa quintile is the most commonly used method for comprehensive neurobehavioral scoring in mice. TTC is a rapid and reliable indicator of hypoxic brain tissue, facilitating the determination of infarct volume and penumbra area.
A common characteristic of clinical cerebral ischemia is brain edema, which occurs as a result of fluid leakage into the brain across the BBB.22 Evaluation of CBF and HE staining are typical strategies for assessing ischemic brain injury. Detection of EB, an azo dye that binds to plasma albumin and only crosses the BBB upon disruption or damage, staining regions of the brain that are inaccessible under normal physiological conditions in brain tissue, reflects the degree of BBB damage.21 To confirm the effects of GRS on cerebral ischemia-reperfusion injury, three doses were administered intraperitoneally 1 h after MCAO induction in mice. GRS administration resulted in asignificant reduction in infarct volume and structural damage to ischemic tissue and brain edema, along with improved CBF. Our finding that GRS decreases I/R-induced brain injury in mice supports its therapeutic potential for cerebrovascular diseases (Figures 2 and 3).
BBB permeability is a characteristic feature of the early stages of ischemic stroke that contributes to stroke-induced brain injury.23 BBB is a crucial selective barrier for restricting transportation of substances between blood and CNS24 that helps maintain a healthy brain microenvironment and is essential for brain homeostasis. The function of BBB depends primarily on BMECs.25 These CNS endothelial cells have unique properties that ensure strict control of movement of ions and molecules between the blood and brain. Adjacent endothelial cells are connected by TJs, providing a physical barrier for transfer from blood to brain.26 Claudins are a family of more than 25 proteins with four transmembrane domains. Two of the domains connect with claudins of adjacent endothelial cells, forming the main barrier of TJs. The two inner rings are bound by carboxyl ends to the PDZ domain of ZO protein for stabilization of the cell structure. Claudin-1, -2, -3, -5, -11, and -12 are primarily expressed in endothelial cells of BBB. In particular, claudin-5 is highly expressed in BMECs and participates in regulating the paracellular permeability of small molecules through the BBB. Consequently, decreased claudin-5 expression results in augmentation of BBB permeability.27 We evaluated the TJ proteins, ZO-1 and claudin-5, as indicators of BBB integrity. Under conditions of MCAO/R, ZO-1 and claudin-5 levels were significantly reduced. These changes were attenuated by GRS, confirming protective effects against I/R-induced BBB damage (Figure 4).
MMPs are members of a calcium-dependent zinc endopeptidase superfamily that remodel and degrade ECM, with important roles in inflammation, tissue transformation, and the innate immune response.7 The ECM provides a scaffold that allows BBB to participate in signal transduction within the neurovascular unit. Therefore, MMPs that degrade the neurovascular matrix have become a major research target for prevention and treatment of CNS diseases.28 Stroke can trigger aberrant MMP expression, leading to ECM degradation and TJ damage due to type IV collagen loss, resulting in increased BBB permeability.29,30 Previous studies have reported that inhibition of MMP-9 activity and deletion of the MMP-9 gene can reduce I/R-induced BBB damage in mice.31,32 Our experiments clearly showed inhibition of I/R-induced increase in MMP-2/9 expression and prevention of BBB damage by GRS (Figure 5A and B).
Cytoskeletal rearrangement in BMECs is the initiating factor for BBB damage during the early stage of I/R. Inhibition of early endothelial cell cytoskeletal rearrangement suppressed stroke-induced BBB damage and secondary brain injury, thereby reducing long-term neurological deficits.33 Stress fibers are actinomyosin bundles that participate in cytoskeleton formation. Excessive production of stress fibers disrupts homeostasis, resulting in cell shrinkage and enlargement of intercellular gaps.34 Actinomyosin filament polarity determines the degree to which stress filaments induce cell shrinkage.35 Previous studies have demonstrated that myosin II activation is mediated by MLC phosphorylation (Thr-18/Ser-19), which is regulated by MLC kinase, MLC phosphatase, and ROCK.36,37 Excessive phosphorylation of MLC results in increased Mg2+-ATPase activity, promoting myosin aggregation with actin to form stress fibers and consequent cytoskeletal rearrangement.14 In our experiments, GRS significantly inhibited I/R-induced phosphorylation of MLC, suggestive of protective effects against stress fiber-induced cytoskeletal rearrangement (Figure 5C and D).
In vitro evaluation of BBB damage is typically performed by subjecting primary or immortalized BMECs to OGD/R.38 Using cell culture to evaluate BBB damage reduces the risk of interindividual differences among animals and facilitates evaluation of single cells39 and experiments with immortalized cell lines should minimize the number of animals for testing. Based on the finding that in ischemic stroke, damage to BMECs aggravates injury to the neurovascular unit leading to poor clinical outcomes,25 BMECs are frequently used as an in vitro model for evaluation of BBB function. ZO-1, claudin-5, and other TJ proteins are more abundant in bEnd.3 cells than other cell lines, which exhibit several BBB characteristics.40 Accordingly, bEnd.3 cells are the most commonly used in vitro BBB model.41 The OGD/R model simulates pathological changes that occur during I/R, including low blood oxygen concentration and secondary injury after reperfusion. Previous studies have shown that OGD for 4 h, followed by reperfusion, leads to increased bEnd.3 cell permeability and TJ protein degradation.42 In the current study, the bEnd.3 cell model was applied to simulate the effects of I/R in vitro.
Microvascular endothelial cell permeability is a key index reflecting endothelial barrier integrity.43 Changes in cell barrier permeability in vitro can be evaluated by measuring the exudation rate of sodium fluorescein. bEnd.3 cells cultured for 7 days formed a dense monolayer with high transepithelial electrical resistance, resulting in extensive barrier function and low exudation of sodium fluorescein.38 In our study, GRS (1–100 ng/mL) significantly reduced sodium fluorescein leakage to a similar extent as the positive control, indicating effective reduction in OGD/R-induced bEnd.3 permeability (Figure 6A).
The TJ protein complex comprises a series of transmembrane and cytoplasmic attachment proteins critical for maintaining the structure and function of the BMEC barrier. Changes in TJ protein expression and distribution in response to OGD/R lead to increased endothelial barrier permeability. Therefore, TJ status is an important indicator of endothelial barrier permeability.44 Our experiments showed that GRS reduces OGD/R-induced permeability of the bEnd.3 cell barrier by regulating the expression of the TJ proteins, ZO-1 and claudin-5 (Figure 6).
OGD/R is known to induce endothelial cell injury and MMP-2/9 expression and activity, along with the degradation of ECM and TJ proteins.33 As shown in Figure 7, GRS (1-100 ng/mL) inhibited MMP-2/9 expression and MLC phosphorylation in bEnd.3 cells, clearly suggesting a role in the maintenance of BBB integrity.
Rho/ROCK is a molecular switch for several signal transduction pathways and the main regulator of the actin cytoskeleton.45 ROCK is the most important effector protein downstream of Rho. Activation of ROCK promotes cytoskeletal rearrangement through phosphorylation of cofilin and MLC. Cofilin is a critical regulator of actin that cleaves and depolymerizes F-actins, produces free actin monomers, promotes actin recombination, and regulates cytoskeleton dynamics.46 Phosphorylation of cofilin at Ser-3 inhibits the depolymerization of F-actin.47 Phosphorylated MLC activates ATPases to promote myosin contraction, resulting in increased interactions between myosin and actin, along with the formation of stress fibers. The contractile force produced by stress fibers promotes the formation of intercellular microspaces.48 Our results suggest that GRS protects against OGD/R-induced bEnd.3 barrier damage, thus, GRS could inhibit the expression of the trough ROCK1 and p-cofilin. (Figure 7).
Ischemic stroke recurrence and mortality rates are increasing due to the lack of effective treatments. Shengmai, a TCM compound used for prevention and treatment of Qi and Yin deficiency, is composed of ginseng, Ophiopogon japonicus, and Schisandra chinensis. Shengmai invigorates Qi, generates Jin, nourishes Yin, and relieves sweat. The active ingredient group is obtained as Shengmai powder originating from Gufang Shengmai powder, which has been screened repeatedly using orthogonal design quantitative pharmacology based on a myocardial ischemia model (ginsenoside Rb1: Rus: schisandrin A= 6:0.75:6). Several reports have demonstrated that Shengmai preparation, administered via injection or orally, and its active ingredients, Rb1 and ruscogenin, exert obvious protective effects against inflammation and BBB damage induced by ischemic brain injury.16,19,49 However, the beneficial activity of GRS against ischemic stroke and associated mechanisms have not been established until now.
Conclusion
The pathogenesis of ischemic cerebral stroke is complex. BBB disruption results in secondary brain injury and poor clinical prognosis. In this study, a C57BL/6J mouse model under MCAO/R was employed to investigate the effects of GRS on BBB permeability in ischemic stroke. The protective effects of Shengmai active ingredients on OGD/R-induced endothelial cell barrier permeability and associated mechanisms were examined in bEnd.3 cells. The results show that the ROCK/cofilin pathway plays a major role in the protective activity of GRS in vitro experiment. Since GRS is a compound preparation that potentially affects multiple signaling pathways, further studies are required to evaluate whether other pathways additionally contribute to these effects.
Ethical Approval
All experimental protocols were carried out according to the National Institutes of Health (NIH) guidelines. The research was approved by the Institutional Animal Care and Use Committee of Animal Ethics of the School of Traditional Chinese Pharmacy, China Pharmaceutical University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fisher M, Saver JL. Future directions of acute ischaemic stroke therapy. Lancet Neurol. 2015;14(7):758–767. doi:10.1016/S1474-4422(15)00054-X
2. Correction to: Heart Disease and Stroke Statistics-2017. Update: a report from the American Heart Association. Circulation. 2017;136(10):e196. doi:10.1161/CIRCULATIONAHA.117.027612
3. Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67(2):181–198. doi:10.1016/j.neuron.2010.07.002
4. Rodrigo R, Fernandez-Gajardo R, Gutierrez R, et al. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013;12(5):698–714. doi:10.2174/1871527311312050015
5. Khatri R, McKinney AM, Swenson B, et al. Blood-brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology. 2012;79(13 Suppl 1):S52–S57. doi:10.1212/WNL.0b013e3182697e70
6. Van Itallie CM, Anderson JM. Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol. 2014;36:157–165. doi:10.1016/j.semcdb.2014.08.011
7. Ricci S, Grandgirard D, Wenzel M, et al. Inhibition of matrix metalloproteinases attenuates brain damage in experimental meningococcal meningitis. BMC Infect Dis. 2014;14:726. doi:10.1186/s12879-014-0726-6
8. Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta. 2008;1778(3):794–809. doi:10.1016/j.bbamem.2007.09.003
9. Endres M, Fink K, Zhu J, et al. Neuroprotective effects of gelsolin during murine stroke. J Clin Invest. 1999;103(3):347–354. doi:10.1172/JCI4953
10. Shi Y, Zhang L, Pu H, et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat Commun. 2016;7:10523. doi:10.1038/ncomms10523
11. Chapin LM, Blankman E, Smith MA, et al. Lateral communication between stress fiber sarcomeres facilitates a local remodeling response. Biophys J. 2012;103(10):2082–2092. doi:10.1016/j.bpj.2012.09.038
12. Pellegrin S, Mellor H. Actin stress fibres. J Cell Sci. 2007;120(Pt 20):3491–3499. doi:10.1242/jcs.018473
13. Suurna MV, Ashworth SL, Hosford M, et al. Cofilin mediates ATP depletion-induced endothelial cell actin alterations. Am J Physiol Renal Physiol. 2006;290(6):F1398–F1407. doi:10.1152/ajprenal.00194.2005
14. Ruiz-Loredo AY, Lopez E, Lopez-Colome AM. Thrombin promotes actin stress fiber formation in RPE through Rho/ROCK-mediated MLC phosphorylation. J Cell Physiol. 2011;226(2):414–423. doi:10.1002/jcp.v226.2
15. Ahmed T, Raza SH, Maryam A, et al. Ginsenoside Rb1 as a neuroprotective agent: a review. Brain Res Bull. 2016;125:30–43. doi:10.1016/j.brainresbull.2016.04.002
16. Cao G, Jiang N, Hu Y, et al. Ruscogenin attenuates cerebral ischemia-induced blood-brain barrier dysfunction by suppressing TXNIP/NLRP3 inflammasome activation and the MAPK pathway. Int J Mol Sci. 2016;17(9):1418. doi:10.3390/ijms17091418
17. Wang CP, Li GC, Shi YW, et al. Neuroprotective effect of schizandrin A on oxygen and glucose deprivation/reperfusion-induced cell injury in primary culture of rat cortical neurons. J Physiol Biochem. 2014;70(3):735–747. doi:10.1007/s13105-014-0342-3
18. Li F, Fan X, Zhang Y, et al. Cardioprotection by combination of three compounds from ShengMai preparations in mice with myocardial ischemia/reperfusion injury through AMPK activation-mediated mitochondrial fission. Sci Rep. 2016;6:37114. doi:10.1038/srep37114
19. Cao G, Ye X, Xu Y, et al. YiQiFuMai powder injection ameliorates blood-brain barrier dysfunction and brain edema after focal cerebral ischemia-reperfusion injury in mice. Drug Des Devel Ther. 2016;10:315–325. doi:10.2147/DDDT.S96818
20. Yonemori F, Yamaguchi T, Yamada H, et al. Evaluation of a motor deficit after chronic focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1998;18(10):1099–1106. doi:10.1097/00004647-199810000-00006
21. Zhang T, Fang S, Wan C, et al. Excess salt exacerbates blood-brain barrier disruption via a p38/MAPK/SGK1-dependent pathway in permanent cerebral ischemia. Sci Rep. 2015;5:16548. doi:10.1038/srep16548
22. Yao X, Derugin N, Manley GT, et al. Reduced brain edema and infarct volume in aquaporin-4 deficient mice after transient focal cerebral ischemia. Neurosci Lett. 2015;584:368–372. doi:10.1016/j.neulet.2014.10.040
23. Gliem M, Krammes K, Liaw L, et al. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia. 2015;63(12):2198–2207. doi:10.1002/glia.v63.12
24. Zhu JC, Si MY, Li YZ, et al. Circulating tight junction proteins mirror blood-brain barrier integrity in leukaemia central nervous system metastasis. Hematol Oncol. 2017;35(3):365–373. doi:10.1002/hon.2289
25. Krueger M, Bechmann I, Immig K, et al. Blood-brain barrier breakdown involves four distinct stages of vascular damage in various models of experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 2015;35(2):292–303. doi:10.1038/jcbfm.2014.199
26. Coomber BL, Stewart PA. Morphometric analysis of CNS microvascular endothelium. Microvasc Res. 1985;30(1):99–115. doi:10.1016/0026-2862(85)90042-1
27. Liu WY, Wang ZB, Zhang LC, et al. Tight junction in blood-brain barrier: an overview of structure, regulation, and regulator substances. CNS Neurosci Ther. 2012;18(8):609–615. doi:10.1111/cns.2012.18.issue-8
28. Seo JH, Guo S, Lok J, et al. Neurovascular matrix metalloproteinases and the blood-brain barrier. Curr Pharm Des. 2012;18(25):3645–3648. doi:10.2174/138161212802002742
29. Abbott NJ, Patabendige AA, Dolman DE, et al. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25. doi:10.1016/j.nbd.2009.07.030
30. Copin JC, Goodyear MC, Gidday JM, et al. Role of matrix metalloproteinases in apoptosis after transient focal cerebral ischemia in rats and mice. Eur J Neurosci. 2005;22(7):1597–1608. doi:10.1111/j.1460-9568.2005.04367.x
31. Ren C, Li N, Wang B, et al. Limb ischemic perconditioning attenuates blood-brain barrier disruption by inhibiting activity of MMP-9 and occludin degradation after focal cerebral ischemia. Aging Dis. 2015;6(6):406–417. doi:10.14336/AD.2015.0812
32. Turner RJ, Sharp FR. Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front Cell Neurosci. 2016;10:56. doi:10.3389/fncel.2016.00056
33. Heo JH, Lucero J, Abumiya T, et al. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19(6):624–633. doi:10.1097/00004647-199906000-00005
34. Tojkander S, Gateva G, Lappalainen P. Actin stress fibers–assembly, dynamics and biological roles. J Cell Sci. 2012;125(Pt 8):1855–1864. doi:10.1242/jcs.098087
35. Kassianidou E, Kumar S. A biomechanical perspective on stress fiber structure and function. Biochim Biophys Acta. 2015;1853(11 Pt B):3065–3074. doi:10.1016/j.bbamcr.2015.04.006
36. Chen G, Hou Z, Gulbranson DR, et al. Actin-myosin contractility is responsible for the reduced viability of dissociated human embryonic stem cells. Cell Stem Cell. 2010;7(2):240–248. doi:10.1016/j.stem.2010.06.017
37. Kimura K, Ito M, Amano M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 1996;273(5272):245–248. doi:10.1126/science.273.5272.245
38. Li G, Simon MJ, Cancel LM, et al. Permeability of endothelial and astrocyte cocultures: in vitro blood-brain barrier models for drug delivery studies. Ann Biomed Eng. 2010;38(8):2499–2511. doi:10.1007/s10439-010-0023-5
39. Naik P, Cucullo L. In vitro blood-brain barrier models: current and perspective technologies. J Pharm Sci. 2012;101(4):1337–1354. doi:10.1002/jps.23022
40. Watanabe T, Dohgu S, Takata F, et al. Paracellular barrier and tight junction protein expression in the immortalized brain endothelial cell lines bEND.3, bEND.5 and mouse brain endothelial cell 4. Biol Pharm Bull. 2013;36(3):492–495. doi:10.1248/bpb.b12-00915
41. Lee HT, Chang YC, Tu YF, et al. CREB activation mediates VEGF-A’s protection of neurons and cerebral vascular endothelial cells. J Neurochem. 2010;113(1):79–91. doi:10.1111/j.1471-4159.2010.06584.x
42. Ku JM, Taher M, Chin KY, et al. Characterisation of a mouse cerebral microvascular endothelial cell line (bEnd.3) after oxygen glucose deprivation and reoxygenation. Clin Exp Pharmacol Physiol. 2016;43(8):777–786. doi:10.1111/cep.2016.43.issue-8
43. Deli MA, Abraham CS, Kataoka Y, et al. Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol. 2005;25(1):59–127. doi:10.1007/s10571-004-1377-8
44. Tietz S, Engelhardt B. Brain barriers: crosstalk between complex tight junctions and adherens junctions. J Cell Biol. 2015;209(4):493–506. doi:10.1083/jcb.201412147
45. Pritchard CA, Hayes L, Wojnowski L, et al. B-Raf acts via the ROCKII/LIMK/cofilin pathway to maintain actin stress fibers in fibroblasts. Mol Cell Biol. 2004;24(13):5937–5952. doi:10.1128/MCB.24.13.5937-5952.2004
46. Ohashi K. Roles of cofilin in development and its mechanisms of regulation. Dev Growth Differ. 2015;57(4):275–290. doi:10.1111/dgd.12213
47. Heemskerk N, Schimmel L, Oort C, et al. F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat Commun. 2016;7:10493. doi:10.1038/ncomms10493
48. Lee SR, Xu YN, Jo YJ, et al. The Rho-GTPase effector ROCK regulates meiotic maturation of the bovine oocyte via myosin light chain phosphorylation and cofilin phosphorylation. Mol Reprod Dev. 2015;82(11):849–858. doi:10.1002/mrd.22524
49. Chen W, Guo Y, Yang W, et al. Protective effect of ginsenoside Rb1 on integrity of blood-brain barrier following cerebral ischemia. Exp Brain Res. 2015;233(10):59–127. doi:10.1007/s00221-015-4352-3
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.