")
Back to Journals » Drug Design, Development and Therapy » Volume 12
The total alkaloids from Coptis chinensis Franch improve cognitive deficits in type 2 diabetic rats
Authors Li JC, Shen XF, Shao JA, Tao MM, Gu J, Li J, Huang N
Received 13 April 2018
Accepted for publication 2 July 2018
Published 31 August 2018 Volume 2018:12 Pages 2695—2706
DOI https://doi.org/10.2147/DDDT.S171025
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Jia-Chuan Li,1,2 Xiao-Fei Shen,3 Jun-Ao Shao,2 Meng-Min Tao,2 Jian Gu,2 Jingyu Li,1 Ning Huang1
1Department of Pathophysiology, West China College of Basic and Forensic Medicine, Sichuan University, Chengdu, China; 2Department of Traditional Chinese Medicine, College of Pharmacy, Southwest Minzu University, Chengdu, China; 3Key Laboratory of Birth Defects and Related Diseases of Women and Children (Ministry of Education), West China Second University Hospital, Sichuan University, Chengdu, China
Background: Coptis chinensis Franch is extensively used in traditional Chinese medicine to treat diabetes and dementia. Alkaloids are the main active ingredients of C. chinensis.
Purpose: This study was designed to probe the effects and possible mechanisms of the total alkaloids from C. chinensis (TAC) on cognitive deficits in type 2 diabetic rats.
Methods: Cognitive deficits were induced in rats by streptozotocin and high glucose/high fat diet. After treatment with TAC (80, 120, and 180 mg/kg) for 24 weeks, the behavioral parameters of each rat were assessed by Morris water maze and Y-maze tests. The indexes of glucose and lipid metabolism, pathological changes of brain tissue, and the phosphorylation levels of insulin signaling related proteins were also evaluated.
Results: The type 2 diabetic rats showed significantly elevated levels of fasting blood glucose, glycosylated hemoglobin and glycosylated serum protein, as well as apolipoprotein B, free fatty acid, triglyceride and total cholesterol but decreased the content of apolipoprotein A1, and TAC treatment dose-dependently reversed these abnormal changes. Furthermore, the behavioral results showed that TAC alleviated the cognitive deficits in type 2 diabetic rats. Moreover, immunohistochemical and histopathologic examinations indicated that the diabetic rats showed significant Aβ deposition, and neuronal damage and loss, which can be reversed by TAC treatment. The western blot results showed that TAC treatment markedly increased the phosphorylation of IRS, PI3K, and Akt, and inhibited the overactivation of GSK3β in the brain of type 2 diabetic rats.
Conclusion: These findings conclude that TAC prevents diabetic cognitive deficits, most likely by ameliorating the disorder of glucose and lipid metabolism, attenuating Aβ deposition, and enhancing insulin signaling.
Keywords: cognitive deficits, type 2 diabetes, Coptis chinensis Franch, total alkaloids, insulin signaling
Introduction
Cognitive deficits, one of the most common complications of diabetes, are strongly correlated with the degeneration and dysfunction of the central nervous system.1,2 The incidence rate of diabetes and related cognitive deficits increases steadily together with the changes of lifestyle and population aging.3,4 Thus, identification of treatment strategies for diabetes-induced cognitive deficits has been an important research goal. Cognitive dysfunction in diabetics appears to be caused by various factors.5 Increasing studies have revealed that chronic hyperglycemia and hyperlipidemia is a major trigger factor for the initiation and development of diabetes-induced cognitive deficits.6,7 In addition, disorder of carbohydrate, lipid, and protein metabolism under diabetic circumstances can cause amyloid β (Aβ) deposition, abnormal insulin signaling, and severe inflammatory response, which also contribute to diabetes-induced neuronal damage and cognitive deficits.8–10 Therefore, amelioration of hyperglycemia and hyperlipidemia and alternation of abnormal insulin signaling may be beneficial for the treatment of cognitive deficits induced by diabetes.
Herbal medicines have been used in the treatment of diseases since the earliest days of human kind. In China, and some other oriental countries, there are plenty of medicinal plants that are used in the treatment of cognitive deficits. For example, Acorus tatarinowii and its main active components α-asarone and β-asarone have been demonstrated to be effective in experimental models of cognitive deficits.11–13 Coptis chinensis Franch, a famous Chinese herbal medicine, has been used for thousand years to treat diabetes and related complications in China, Japan, and Korea.14 Our previous results showed that the extract from C. chinensis exhibited significant antidiabetic effect in streptozotocin (STZ)- and high-glucose/high-fat diet-induced diabetic rats through antioxidant and pancreatic β-cell protection.14 Furthermore, the total alkaloids from C. chinensis (TAC) including berberine have also been demonstrated as potential anti-dementia agents.15 However, the underlying mechanisms and activity of the TAC on diabetic cognitive deficits have not been fully understood. In the present study, for the first time, we assessed the possible mechanisms and effect of TAC on cognitive deficits in STZ- and high-glucose/high-fat diet-induced type 2 diabetic rats.
Materials and methods
Preparation of the TAC
C. chinensis Franch was purchased from Derentang Pharmacy (Chengdu, China), and identified by Prof. Jian Gu (College of Pharmacy, Southwest Minzu University). The rhizomes of C. chinensis Franch (83.5 g) was soaked in 835 mL of 70% ethanol for 1 hour at room temperature and thereafter reflux-extracted three times (2 hours each time). Total alkaloids were enriched and purified using D101 macroporous resin and then concentrated in vacuo and freeze-dried (yield 13.40%). The extract was suspended in 0.5% sodium carboxymethyl cellulose before administration to animals during the experiment. The quality of TAC was checked using HPLC, and the results are shown in Figure S1.
Chemicals and drugs
STZ was purchased from Sigma-Aldrich (St. Louis, MO, USA). Metformin was provided by Beijing Coway Pharmaceutical Co., Ltd. (Beijing, China). OneTouch Ultra™ glucometer and test paper were purchased from Johnson & Johnson (New Brunswick, NJ, USA). All other chemicals were of analytical grade.
Experimental animals
All protocols involving animal experiments were conducted strictly adhering to and in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No 85-23, revised 1996), and approved by Medical Ethics Committee of Sichuan University. Male Wistar rats (220–240 g) were obtained from Laboratory Animal Institute, Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital (Chengdu, China). All animals were allowed to acclimatize to the laboratory environment for 7 days before the experiments and were group-housed under a normal 12-hour light/dark cycle in plastic cages with food and water available ad libitum.
Induction of type 2 diabetes and treatment
Induction of type 2 diabetes was performed as per our previous method.14 In brief, diabetes was induced in the rats by intraperitoneal injection of a single dose of 40 mg/kg STZ freshly dissolved in citrate buffer. Rats with fasting plasma glucose levels >11.1 mmol/L were defined as diabetic and selected for further study. Then, the diabetic and nondiabetic rats were divided into six groups, each comprising ten rats, as follows: normal control rats; diabetic rats without any drug treatment; diabetic rats treated with TAC at a dose of 80, 120, and 180 mg/kg; and diabetic rats treated with metformin at a dose of 250 mg/kg (as a positive control). The rats were intragastrically administrated continuously for 24 weeks once a day. In addition, the diabetic rats were allowed free access to the high-glucose/high-fat diet for 24 weeks to induce dyslipidemia. Subcutaneous injection of D-galactose was started 20 weeks after the STZ injection and continued for 4 weeks to aggravate cognitive deficits.
Morris water maze test
Spatial memory ability of rats was evaluated by Morris water maze test. The test was carried out in a black circular pool (diameter: 120 cm). The temperature of water was kept at 25°C ± 2°C. A black round platform (diameter: 9 cm) was placed 1 cm below the water surface in a middle fixed position of the pool. The pool was divided into four equal-spaced quadrants. The rats were subjected to three consecutive daily training trials for 4 days (two times per day), with each trial having a time limit of 90 seconds and with an intertrial interval of 30 minutes. For each trial, the rat swam until it climbed onto the submerged platform. After climbing onto the platform, the rat was allowed to remain on the platform for 20 seconds. If a rat failed to find the platform within a maximum of 90 seconds, it was gently placed on the platform and allowed to remain there for an equivalent amount of time. The escape latency to find the platform was measured in each trial and averaged over three trials for each rat.
Y-maze test
The Y-maze also served as an exteroceptive model to evaluate the acquisition of spatial memory in rats. A Y-maze with three identical Plexiglas arms (100×20×20 cm, named as A, B, and C arm, respectively, 120° apart) was placed at the center of a room under dim lighting conditions. The walls of each arm had a distinct design to provide visual cues. The A arm was kept in dark and B arm under light. The rats were subjected to five consecutive daily training trials for 4 days (10 times per day). At the beginning of the test, each rat was placed at the end of C arm facing the center and allowed to explore the maze. Successful response was defined as arriving at the end of B arm within 15 seconds. A total of 10 attempts were allowed for each rat. Percent successful response was calculated according to the following formula: Successful response (%) = 100% × (Number of successful responses)/(Total number of attempts).
Biochemical analysis
At the end of the experimental period, all rats were fasted for 8 hours. Then, rats were anesthetized with chloral hydrate (360 mg/kg), and blood specimens were collected from femoral artery with or without EDTA for plasma or serum separation, respectively. The biochemical indexes including fasting blood glucose (FBG), glycosylated hemoglobin (GHb), glycosylated serum protein (GSP), total cholesterol (TC), total glyceride (TG), free fatty acid, apolipoprotein A1 (ApoA1), and apolipoprotein B (ApoB) were tested using commercial diagnostic kits (Nanjing Jiancheng Bioengineering Research Institute, Nanjing, China).
Western blot assay
The brain tissues were removed carefully and quickly to ice-cold saline (0.9% sodium chloride) and weighed. The hippocampus was homogenized in an ice-cold lysis buffer. Then, the homogenate was centrifuged at 15,000 × g for 5 minutes at 4°C prior to collecting the supernatants. Protein concentrations were measured using the bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Samples (50 μg protein) were subjected to SDS-PAGE. The proteins were then blotted onto nitrocellulose membranes. Western blotting analyses were performed with the primary antibodies, followed by horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:1,000, #A0208 and #A0216; Beyotime Institute of Biotechnology, Haimen, China). Signals were detected by enhanced chemiluminescence reagent (Beyotime Institute of Biotechnology). Primary antibodies against phospho-IRS1 (#2384), IRS1 (#2382), phospho-PI3K (#4228), PI3K (#4249), phospho-Akt (#13038), Akt (#9272), phospho-GSK3β (#9322), and GSK3β (#5676) with 1:1,000 dilution were obtained from Cell Signaling Technologies (Danvers, MA, USA). GAPDH (1:1,000, #AF0006) was purchased from Beyotime Institute of Biotechnology, which was used as the reference.
H&E staining and toluidine blue Nissl staining
After behavioral evaluation and biochemical analysis, remaining rats were used for histopathological examination. Briefly, the remaining rats were anesthetized using 10% chloral hydrate, transcardially perfused with 150 mL cold 0.1 M PBS (pH 7.4), and treated with 150 mL cold 4% paraformaldehyde for 30 minutes. The brain tissues were taken and immersed in 10% formaldehyde solution. The tissues were dehydrated in series concentrations of ethanol, cleared in xylene, and embedded in paraffin wax, and then cut into 7 μm thick sections. Deparaffinization was performed by heating the sections for 25 minutes at 60°C. After gradual hydration through graded alcohols, the sections were rinsed in distilled water and stained by H&E or toluidine blue (1%; Sigma-Aldrich). Finally, the sections were viewed under a light microscope to observe the pathological changes of brain tissues.
Immunohistochemical analysis
The paraffin sections were dewaxed with xylene and hydrated with gradient ethanol. Then, the sections were treated in a microwave oven at low power for 10 minutes in 0.01 M sodium citrate buffer (pH 6.0) and blocked with 10% goat serum at room temperature for 2 hours. Subsequently, the sections were stained with anti-Aβ42 (1:100; Abcam, Cambridge, UK) rabbit polyclonal antibody at 4°C overnight. The sections were then washed with tris-buffered saline/Tween 20 (TBST), and endogenous peroxidase was inactivated using 3% hydrogen peroxide for 15 minutes. Next, the sections were incubated with biotin-labeled goat anti-rabbit secondary antibody and horseradish peroxidase-labeled streptomycin ovalbumin (1:1,000; Zhongshan Golden Bridge Biotechnology, Beijing, China) for 1 hour at room temperature, and antibody-binding sites were visualized by diaminobenzidine (DAB) kit (Zhongshan Golden Bridge Biotechnology). Thereafter, the sections were stained with hematin for 10 minutes and dehydrated and cleared using gradient ethanol and xylene, respectively. Finally, the sections were sealed with neutral balsam on slides and observed under a light microscope.
Statistical analysis
Data were expressed as mean values ± SD. Statistical analyses were performed with GraphPad Prism 5.0 software (GraphPad, La Jolla, CA, USA). The data were compared by one-way ANOVA followed by Dunnett’s post hoc test. The differences were considered statistically significant at P<0.05.
Results
TAC improves the disorder of glucose and lipid metabolism in type 2 diabetic rats
To test the effects of TAC on disorder of glucose and lipid metabolism, type 2 diabetic rats were established by STZ and high-glucose/high-fat diet. After STZ injection and administration of high-glucose/high-fat diet, disorder of glucose and lipid metabolism was induced. TAC at the doses of 80, 120, and 180 mg/kg ameliorated STZ- and high-glucose/high-fat diet-induced hyperglycemia, which manifested as decreased FBG, GHb, and GSP levels (Figure 1). Moreover, elevated levels of lipid components including nonesterified fatty acid (NEFA), TC, and TG (Figure 2), as well as reduced level of ApoA1 and elevated level of ApoB (Figure 2), were significantly adjusted by TAC treatment. These results suggest that TAC could improve the disorder of glucose and lipid metabolism in type 2 diabetic rats.
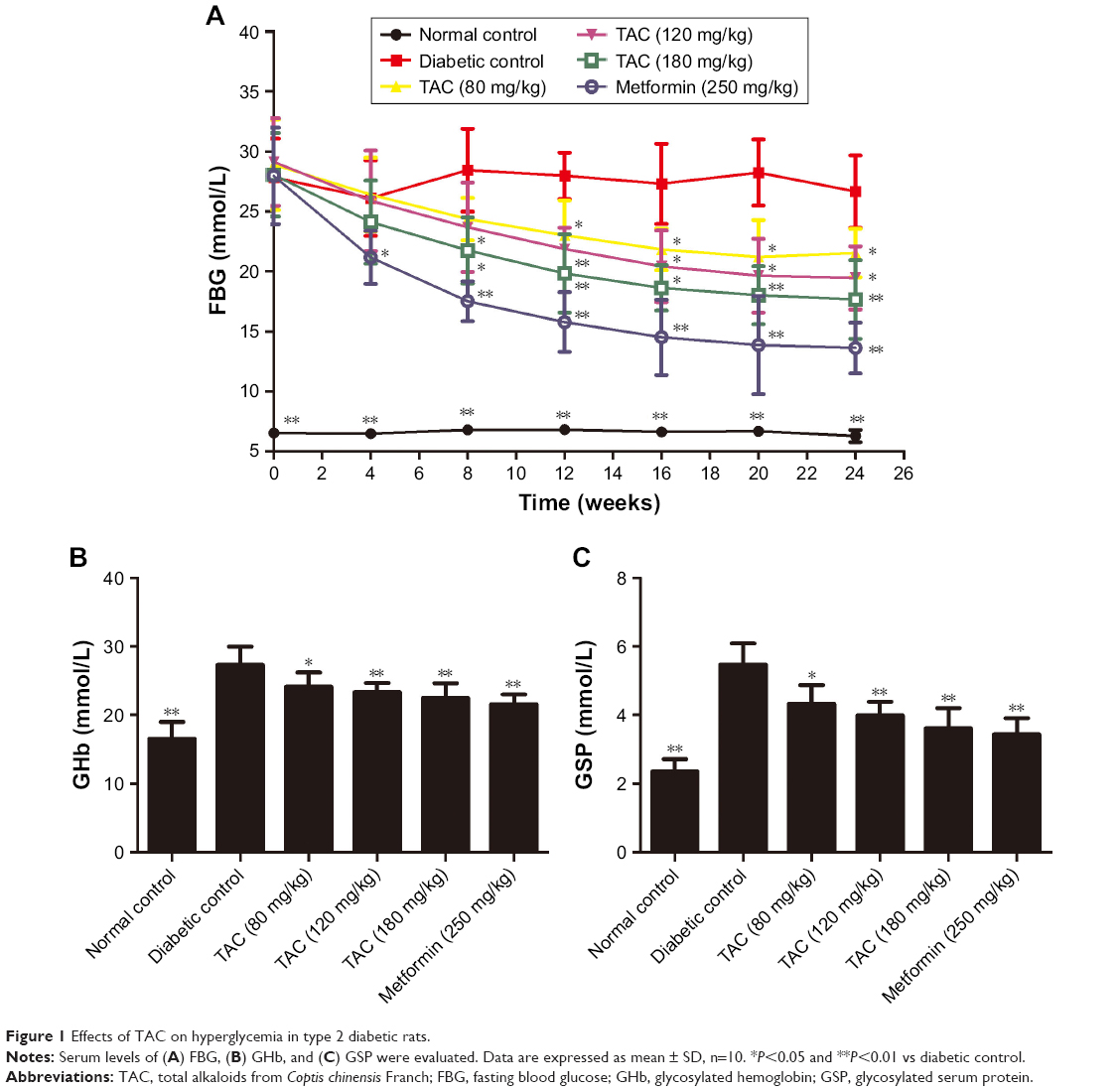 | Figure 1 Effects of TAC on hyperglycemia in type 2 diabetic rats. |
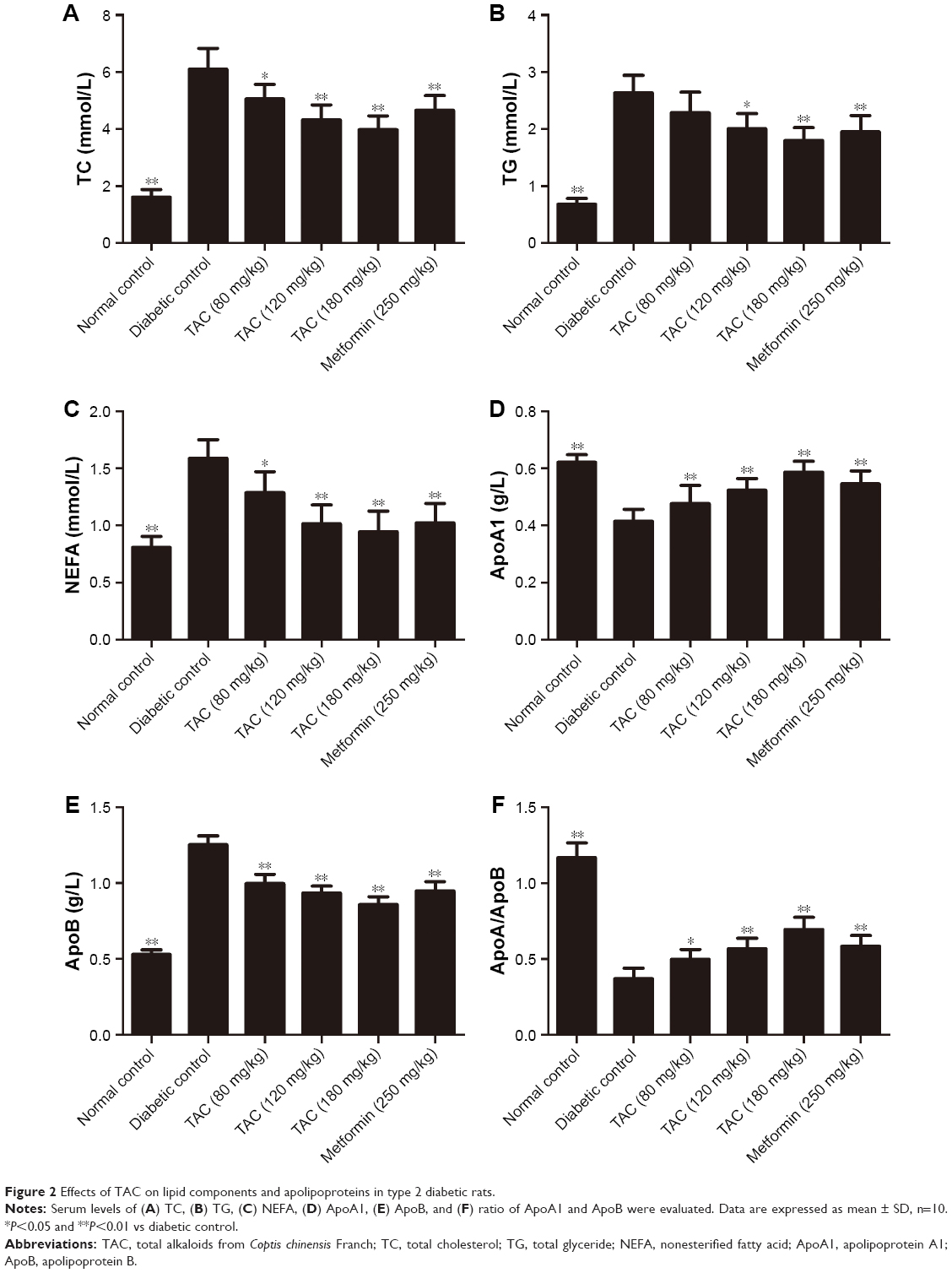 | Figure 2 Effects of TAC on lipid components and apolipoproteins in type 2 diabetic rats. |
TAC treatment enhances the cognitive ability in type 2 diabetic rats
Type 2 diabetes is a strong risk factor for cognitive deficits. To assess whether TAC treatment affects the cognitive function, we performed Morris water maze test and Y-maze test in the present study. During the tests, type 2 diabetic rats showed severe learning deficit in the Morris water maze test (Figure 3A) and Y-maze test (Figure 3B), including prolonged escape latency and increased error responses, respectively. After 24 weeks of TAC treatment (120 and 180 mg/kg), diabetic rats showed decreased escape latency in Morris water maze test and elevated successful response in Y-maze test (Figure 3A and B). The data suggested that TAC is beneficial in improving the cognitive deficits of rats with type 2 diabetes.
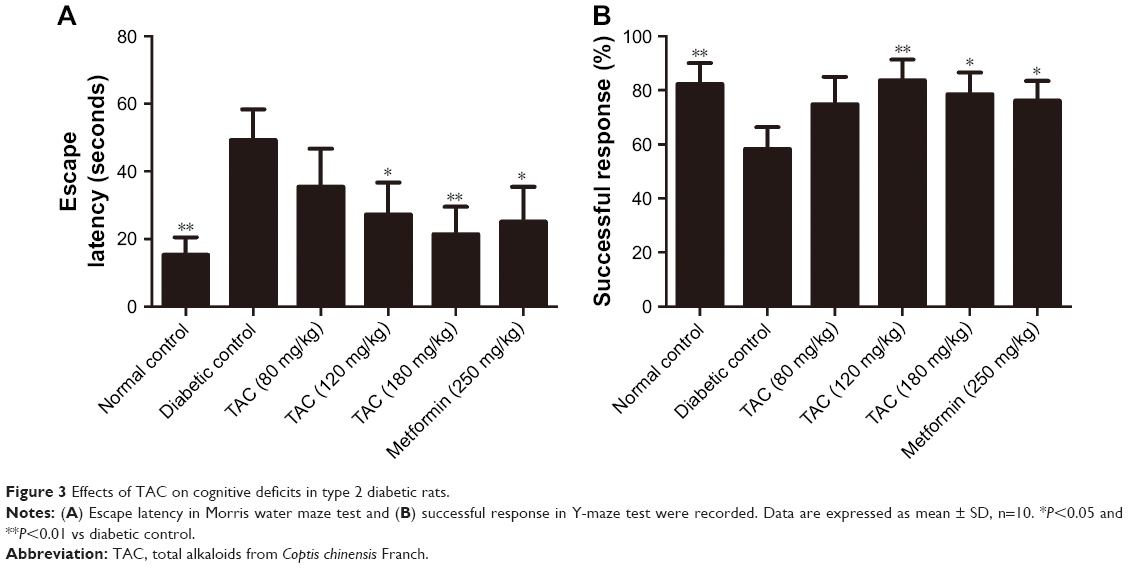 | Figure 3 Effects of TAC on cognitive deficits in type 2 diabetic rats. |
TAC attenuates the pathological changes of brain tissue in type 2 diabetic rats
Histological evaluation of the brain tissues in the vehicle-treated animals revealed obvious signs of cognitive deficits, with marked injury and loss of hippocampal neurons, as well as gliocyte proliferation (Figure 4B), compared with the normal rats (Figure 4A). The degree of injury and loss of hippocampal neurons and gliocyte proliferation were significantly alleviated after administering TAC or metformin (Figure 4C–F). Nerve cell arrangements were also orderly after TAC treatment (Figure 4C–F). Moreover, toluidine blue Nissl staining demonstrated the beneficial function of TAC in attenuating the loss of hippocampal neurons, a major pathological characteristic of cognitive deficits (Figure 5). This result showed an important repressor effect of TAC on neuronal damage and loss in type 2 diabetic rats, which contributes to its improvement of cognitive deficits.
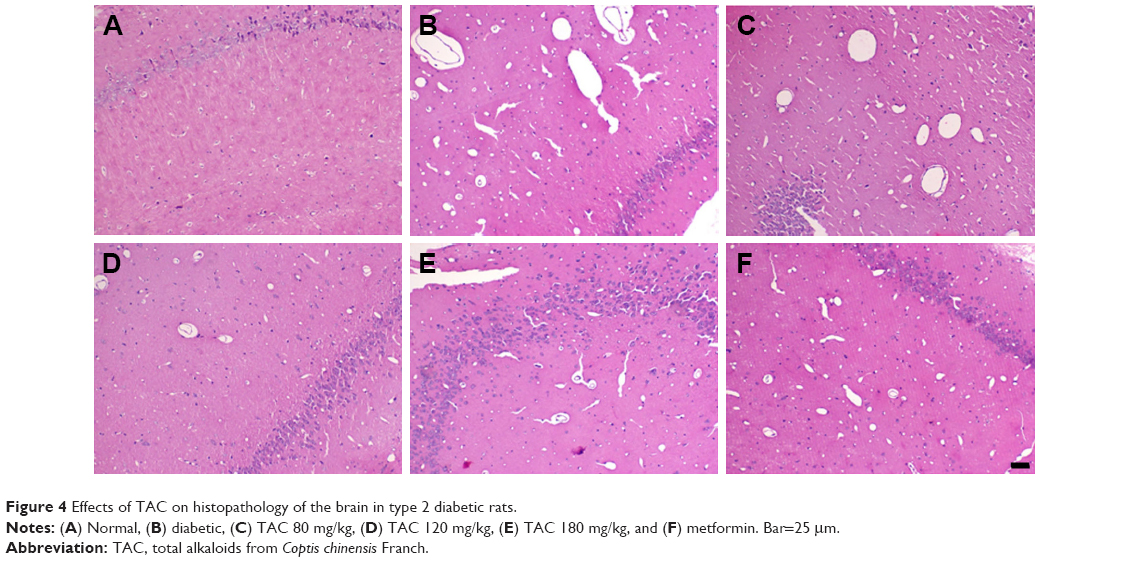 | Figure 4 Effects of TAC on histopathology of the brain in type 2 diabetic rats. |
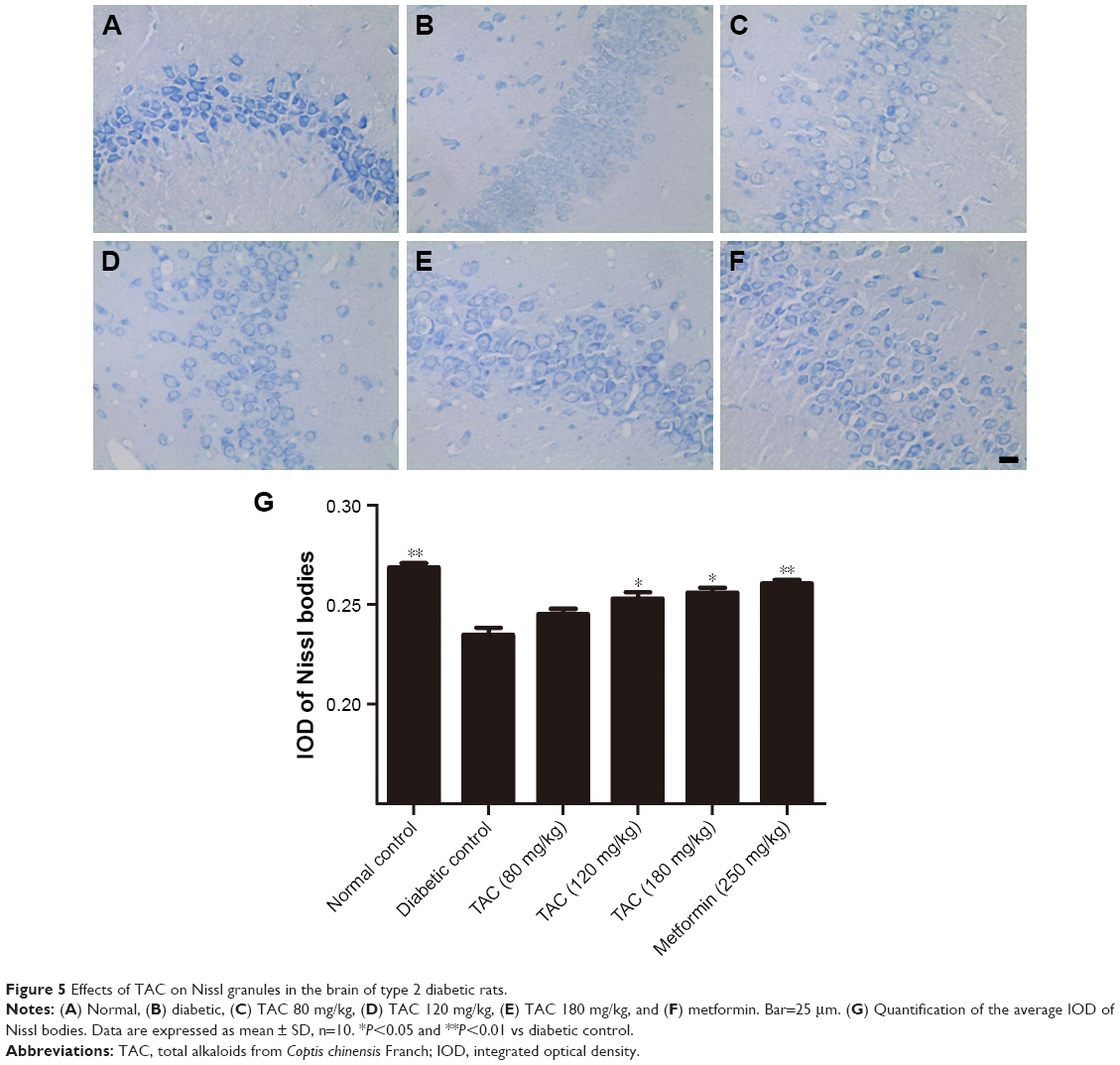 | Figure 5 Effects of TAC on Nissl granules in the brain of type 2 diabetic rats. |
TAC treatment reduces Aβ deposition in type 2 diabetic rats
Type 2 diabetes-induced cognitive deficits are characterized by the presence of senile plaques composed of aggregated extracellular Aβ protein. Thus, in addition to changes in histopathology, we hypothesized that TAC would attenuate the process of Aβ deposition in the brain of type 2 diabetic rats. As expected, obvious Aβ42 deposition was observed in the brain of type 2 diabetic rats (Figure 6B), compared to the normal rats (Figure 6A). However, treatment with TAC (80, 120, and 180 mg/kg) or metformin (250 mg/kg) significantly attenuated the accumulation of Aβ42 in neurons in the hippocampus of type 2 diabetic rats (Figure 6C–F). These results indicated that TAC ameliorates the memory impairment in type 2 diabetic rats due to its inhibitory effect on Aβ deposition (Figure 6A–G).
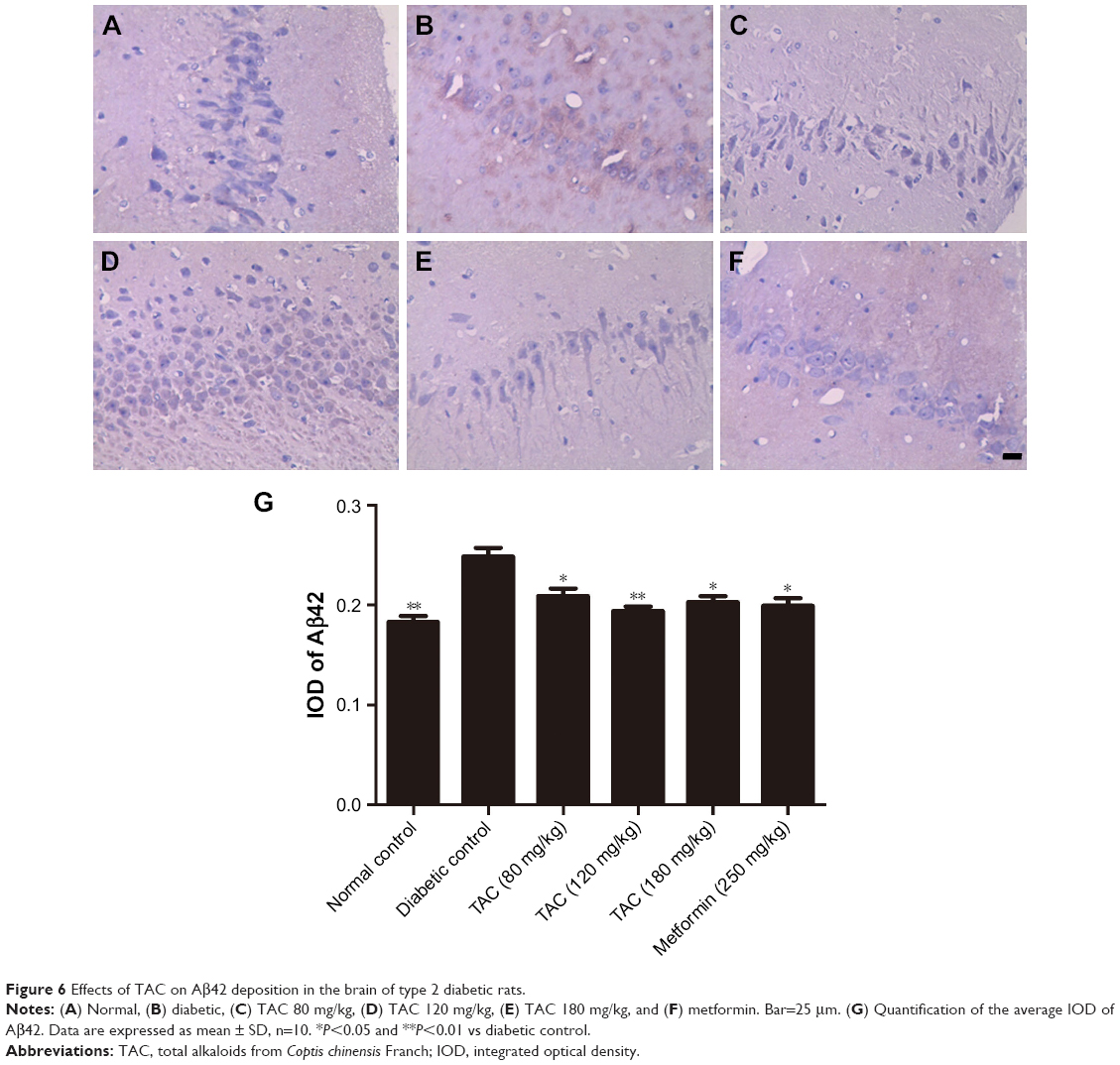 | Figure 6 Effects of TAC on Aβ42 deposition in the brain of type 2 diabetic rats. |
TAC treatment regulates insulin signaling in the brain of type 2 diabetic rats
As impaired IRS/PI3K/Akt insulin signaling and overactivation of GSK3β are involved in type 2 diabetes-induced cognitive deficits, we investigated whether TAC altered the IRS/PI3K/Akt/GSK3β signaling pathway in the brain of type 2 diabetic rats. As shown in Figure 7, reduced phosphorylation levels of IRS1, PI3K, Akt, and GSK3β were observed in the brain of type 2 diabetic rats, compared to the normal rats. TAC (80 and 180 mg/kg) treatment significantly reversed the reduced phosphorylation levels of IRS1, PI3K, Akt, and GSK3β. The resulting efficacy of TAC at 180 mg/kg on IRS/PI3K/Akt/GSK3β signaling was similar to that of metformin (Figure 7). This result indicated that TAC improved the type 2 diabetes-induced cognitive deficits, in part, by enhancing the IRS/PI3K/Akt signaling and inhibiting the overactivation of GSK3β.
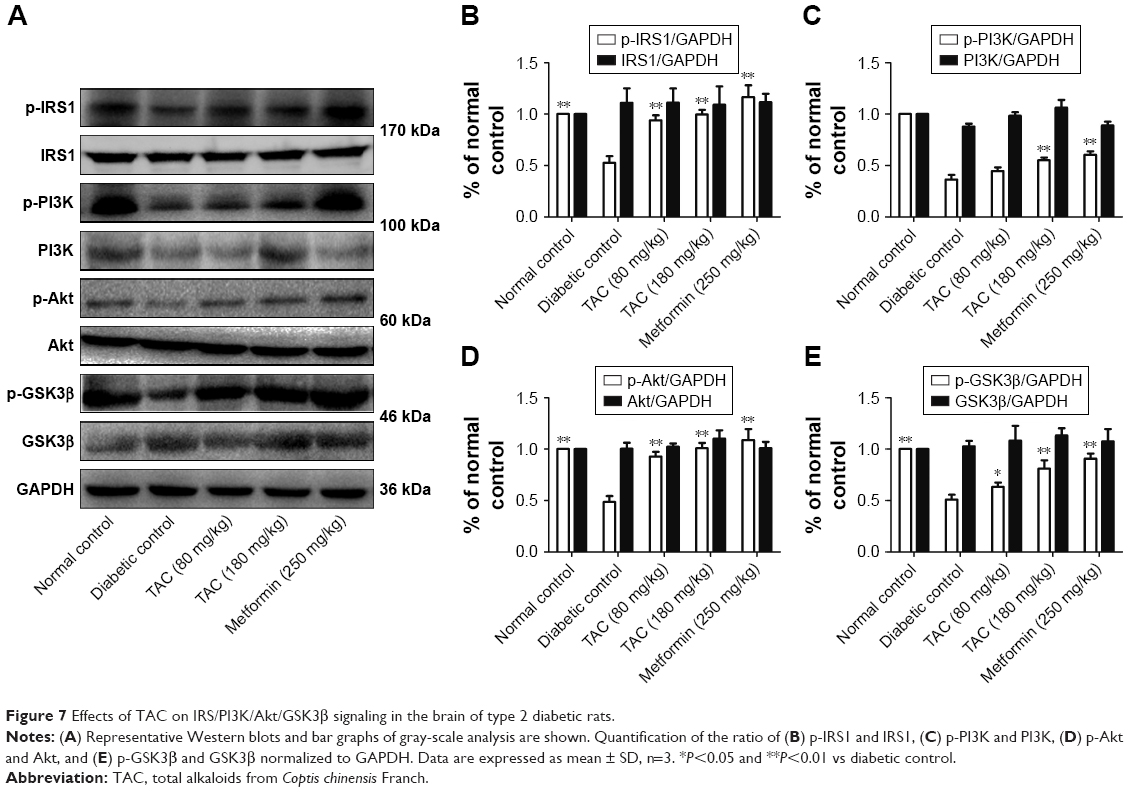 | Figure 7 Effects of TAC on IRS/PI3K/Akt/GSK3β signaling in the brain of type 2 diabetic rats. |
Discussion
Although the pathogenesis of diabetic cognitive deficits is still not well understood, pharmacological treatment for this disease warrants exploration. Natural products are historically invaluable as a source of therapeutic reagents. Recently, growing concerns have been paid to the development of new therapeutic reagents from natural products or traditional medicinal plants due to their effectiveness and wider safety range. C. chinensis, a well-known traditional Chinese herbal medicine, has been found to have antidiabetic effect. In the present study, we found that the total alkaloids, the main active ingredients of C. chinensis, potentially improved the cognitive deficits in STZ- and high-glucose/high-fat diet-induced type 2 diabetic rats.
Increasing evidence suggested that hyperglycemia can be considered as a determinant of cognitive decline in patients with diabetes,16 implicating that intensive blood glucose control is imperative for ameliorating diabetes-induced cognitive deficits. A previous report has shown that hypoglycemic agents and insulin sensitizers reduce the risk of cognitive dysfunction under diabetic conditions.17 In this study, TAC treatment markedly reduced the excessive elevated level of blood glucose and nonenzymatic glycosylation products including GHb and GSP, which suggested that TAC could be used as a good hypoglycemic reagent or an insulin sensitizer. The hypoglycemic action of TAC was mainly attributed to the total alkaloids, especially berberine.18 Based on these data, the effective blood glucose control of TAC facilitated its alleviation of cognitive impairment induced by diabetes, which was further confirmed by behavioral experiments including Morris water maze test and Y-maze test. Furthermore, an epidemiological study has reported an association between Alzheimer’s disease and some vascular risk factors, including hyperlipidemia.19 Therefore, hyperlipidemia-induced intracranial vascular dysfunction may be a contributing factor to cognitive deficits.20 Our results indicated that TAC also attenuated the increased levels of NEFA, TC, TG, and ApoB in type 2 diabetic rats. The hypolipidemic effect of TAC may contribute to its anti-dementia effect through improving the intracranial vascular dysfunction.
Some clinical studies showed that Aβ accumulation was more obvious in the brain of patients with cognitive decline.21 Aβ protein can trigger apoptotic cascades in neurons and then lead to neurodegeneration, DNA fragmentation, and apoptosis proteins expression, which are the leading causes of cognitive deficits.22 Moreover, chronic hyperglycemia can promote Aβ accumulation23 and enhance the susceptibility of nerve cells and brain microvascular endothelial cells to Aβ toxicity.24 In our study, we found a significant Aβ deposition in the brains of type 2 diabetic rats. As expected, neuronal damage and loss were also observed in the brains of type 2 diabetic rats by H&E staining and toluidine blue Nissl staining. These changes were consistent with the results obtained from the Morris water maze test and Y-maze test. TAC treatment suppressed the Aβ deposition and neuronal loss, which was beneficial in restoring the learning and memory in diabetic rats. Additionally, ApoA1 has been shown in vivo to participate in the amylogenesis process by binding to Aβ protein and to involve in the regeneration process of neuronal cells after injury.25 TAC treatment elevated the serum level of ApoA1 and decreased the content of ApoB in type 2 diabetic rats. This result may help explain the underlying mechanism and effect of TAC on attenuating Aβ deposition and neuronal loss as well as cognitive deficits.
Recent epidemiological evidence also suggests that insulin resistance in central nervous system is a risk factor for cognitive decline.26 Because elevations in insulin lead to accumulation of Aβ, which then further impairs insulin signaling, impairment in brain insulin signaling can initiate a vicious spiral of decline in brain metabolism, synaptic dysfunction, and cognitive impairment.27 Downstream targets of insulin signaling pathway, for example, p-Ser302 IRS1, are downregulated and involved in the process of insulin resistance.28 Akt, a key kinase of insulin signaling, mediates the effect of insulin via important intracellular signaling cascades including the PI3K/Akt pathway.29 This study identified reductions of p-Ser302 IRS1 and p-Thr308 Akt in the hippocampus of type 2 diabetic rats, suggesting an impaired insulin signaling in the brain. TAC treatment corrected the insulin resistance in the brain by regulating IRS1/PI3K/Akt signaling, which was beneficial for the improvement of memory impairment.
Furthermore, we revealed that TAC obviously enhanced the phosphorylation of GSK3β at Ser9. A major target of Akt, GSK-3β is inhibited when it is phosphorylated at Ser9, resulting in glycogen synthesis and dephosphorylation of tau protein.30 On the other hand, activated GSK-3β is implicated in the process of Aβ deposition31 and disturbs the growth and survival of neurons.32 Therefore, GSK-3β is the bridge connecting insulin signaling and maintenance of neuronal functions. In this study, we found that GSK3β activity was inhibited after TAC treatment, indicating that TAC exerted an inhibitory effect on Aβ deposition, neuronal loss, and cognitive deficits, in part, by the regulation of GSK3β activity. This result is consistent with a previous report, which showed that GSK-3β inhibition ameliorated diabetes-induced cognitive deficits in rats.33
Meanwhile, we have also identified several compounds in the TAC, such as berberine. Previous studies have demonstrated the regulation of berberine on several kinases and biomarkers that play crucial roles in the development of diabetic cognitive deficits, such as Akt and GSK3β, as well as Aβ.34,35 Therefore, the improvement of cognitive deficits by TAC is largely due to berberine. Moreover, many natural products exhibit their activities through regulating multiple targets. For example, several proteins, such as AchE, AMPK, and PPAR, are identified as the targets of berberine.36,37 Taking into account the general agreement that molecules interfering simultaneously with multiple targets might be more effective than single-target agents,38 other potential mechanisms should be explored in further study. Taken together, the findings of the present study provide pharmacodynamic and molecular biological evidence for the preventive effects of TAC on diabetes-induced cognitive deficits.
Conclusion
Overall, our findings demonstrated the protective effect of TAC on cognitive deficits in STZ- and high-glucose and high-fat diet-induced type 2 diabetic rats. This action may be attributed to its ability to reverse pathological changes of the brain including loss of neurons and Aβ deposition, as well as regulate the IRS1/PI3K/Akt insulin signaling and GSK3β activity. The data thus suggest that TAC may be beneficial for the treatment of diabetic cognitive deficits.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81302912), China Postdoctoral Science Foundation (2015M582560), and Fundamental Research Funds for the Central Universities (2017NZYQN35).
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: a lifespan perspective. Lancet Neurol. 2008;7(2):184–190. | ||
Mijnhout GS, Scheltens P, Diamant M, et al. Diabetic encephalopathy: A concept in need of a definition. Diabetologia. 2006;49(6):1447–1448. | ||
Luchsinger JA, Adiposity LJA. Adiposity, hyperinsulinemia, diabetes and Alzheimer’s disease: an epidemiological perspective. Eur J Pharmacol. 2008;585(1):119–129. | ||
Sims-Robinson C, Kim B, Rosko A, Feldman EL. How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol. 2010;6(10):551–559. | ||
Kodl CT, Seaquist ER. Cognitive dysfunction and diabetes mellitus. Endocr Rev. 2008;29(4):494–511. | ||
Cameron NE, Eaton SE, Cotter MA, Tesfaye S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia. 2001;44(11):1973–1988. | ||
Biessels GJ, van der Heide LP, Kamal A, Bleys RL, Gispen WH. Ageing and diabetes: implications for brain function. Eur J Pharmacol. 2002;441(1–2):1–14. | ||
Biessels GJ, Reagan LP. Hippocampal insulin resistance and cognitive dysfunction. Nat Rev Neurosci. 2015;16(11):660–671. | ||
di Raimondo D, Tuttolomondo A, Buttà C, Miceli S, Licata G, Pinto A. Effects of ACE-inhibitors and angiotensin receptor blockers on inflammation. Curr Pharm Des. 2012;18(28):4385–4413. | ||
Tuttolomondo A, Pecoraro R, Simonetta I, et al. Neurological complications of Anderson-Fabry disease. Curr Pharm Des. 2013;19(33):6014–6030. | ||
Lin Z, Gu J, Xiu J, Mi T, Dong J, Tiwari JK. Traditional Chinese Medicine for Senile Dementia. Evid Based Complement Alternat Med. 2012;2012:692621. | ||
Limón ID, Mendieta L, Díaz A, et al. Neuroprotective effect of alpha-asarone on spatial memory and nitric oxide levels in rats injected with amyloid-beta((25–35)). Neurosci Lett. 2009;453(2):98–103. | ||
Geng Y, Li C, Liu J, et al. Beta-asarone improves cognitive function by suppressing neuronal apoptosis in the beta-amyloid hippocampus injection rats. Biol Pharm Bull. 2010;33(5):836–843. | ||
Li JC, Shen XF, Meng XL. A traditional Chinese medicine JiuHuangLian (Rhizoma coptidis steamed with rice wine) reduces oxidative stress injury in type 2 diabetic rats. Food Chem Toxicol. 2013;59:222–229. | ||
Howes MJ, Houghton PJ. Plants used in Chinese and Indian traditional medicine for improvement of memory and cognitive function. Pharmacol Biochem Behav. 2003;75(3):513–527. | ||
Wessels AM, Scheltens P, Barkhof F, Heine RJ. Hyperglycaemia as a determinant of cognitive decline in patients with type 1 diabetes. Eur J Pharmacol. 2008;585(1):88–96. | ||
Ryan CM, Freed MI, Rood JA, Cobitz AR, Waterhouse BR, Strachan MW. Improving metabolic control leads to better working memory in adults with type 2 diabetes. Diabetes Care. 2006;29(2):345–351. | ||
Yin J, Xing H, Ye J. Efficacy of berberine in patients with type 2 diabetes mellitus. Metabolism. 2008;57(5):712–717. | ||
Viswanathan A, Rocca WA, Tzourio C. Vascular risk factors and dementia: how to move forward? Neurology. 2009;72(4):368–374. | ||
Roher AE, Tyas SL, Maarouf CL, et al. Intracranial atherosclerosis as a contributing factor to Alzheimer’s disease dementia. Alzheimers Dement. 2011;7(4):436–444. | ||
Blennow K, Mattsson N, Schöll M, Hansson O, Zetterberg H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci. 2015;36(5):297–309. | ||
Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003;34(4–5):385–397. | ||
Huang HJ, Liang KC, Chen CP, Chen CM, Hsieh-Li HM. Intrahippocampal administration of A beta(1–40) impairs spatial learning and memory in hyperglycemic mice. Neurobiol Learn Mem. 2007;87(4):483–494. | ||
Carvalho C, Katz PS, Dutta S, Katakam PV, Moreira PI, Busija DW. Increased susceptibility to amyloid-β toxicity in rat brain microvascular endothelial cells under hyperglycemic conditions. J Alzheimers Dis. 2014;38(1):75–83. | ||
Merched A, Xia Y, Visvikis S, Serot JM, Siest G. Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer’s disease. Neurobiol Aging. 2000;21(1):27–30. | ||
Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J Cell Mol Med. 2011;15(9):1807–1821. | ||
Zhao WQ, Townsend M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim Biophys Acta. 2009;1792(5):482–496. | ||
Giraud J, Leshan R, Lee YH, White MF. Nutrient-dependent and insulin-stimulated phosphorylation of insulin receptor substrate-1 on serine 302 correlates with increased insulin signaling. J Biol Chem. 2004;279(5):3447–3454. | ||
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. | ||
Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol. 2011;225(1):54–62. | ||
Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J Neurochem. 2008;104(6):1433–1439. | ||
Takeda S, Sato N, Rakugi H, Morishita R. Molecular mechanisms linking diabetes mellitus and Alzheimer disease: beta-amyloid peptide, insulin signaling, and neuronal function. Mol Biosyst. 2011;7(6):1822–1827. | ||
Datusalia AK, Sharma SS. Amelioration of diabetes-induced cognitive deficits by GSK-3β inhibition is attributed to modulation of neurotransmitters and neuroinflammation. Mol Neurobiol. 2014;50(2):390–405. | ||
Yu G, Li Y, Tian Q, et al. Berberine attenuates calyculin A-induced cytotoxicity and Tau hyperphosphorylation in HEK293 cells. J Alzheimers Dis. 2011;24(3):525–535. | ||
Durairajan SS, Liu LF, Lu JH, et al. Berberine ameliorates β-amyloid pathology, gliosis, and cognitive impairment in an Alzheimer’s disease transgenic mouse model. Neurobiol Aging. 2012;33(12):2903–2919. | ||
Ji HF, Shen L. Berberine: a potential multipotent natural product to combat Alzheimer’s disease. Molecules. 2011;16(8):6732–6740. | ||
Pang B, Zhao LH, Zhou Q, et al. Application of berberine on treating type 2 diabetes mellitus. Int J Endocrinol. 2015;2015:905749. | ||
Petrelli A, Giordano S. From single- to multi-target drugs in cancer therapy: when aspecificity becomes an advantage. Curr Med Chem. 2008;15(5):422–432. |
Supplementary material
 | Figure S1 The fingerprint chromatogram of TAC. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.