")
Back to Archived Journals » Journal of Receptor, Ligand and Channel Research » Volume 9
The role of the human Duffy antigen receptor for chemokines in malaria susceptibility: current opinions and future treatment prospects
Authors Ntumngia F, Thomson-Luque R, Pires C, Adams J
Received 15 July 2016
Accepted for publication 22 August 2016
Published 26 September 2016 Volume 2016:9 Pages 1—11
DOI https://doi.org/10.2147/JRLCR.S99725
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Trevor W. Stone
Francis B Ntumngia,* Richard Thomson-Luque,* Camilla V Pires,* John H Adams
Department of Global Health, College of Public Health, University of South Florida, Tampa, FL, USA
*These authors contributed equally to this work.
Abstract: The Duffy antigen receptor for chemokine (DARC) is a nonspecific receptor for several proinflammatory cytokines. It is homologous to the G-protein chemokine receptor superfamily, which is suggested to function as a scavenger in many inflammatory-and proinflammatory-related diseases. G-protein chemokine receptors are also known to play a critical role in infectious diseases; they are commonly used as entry vehicles by infectious agents. A typical example is the chemokine receptor CCR5 or CXCR4 used by HIV for infecting target cells. In malaria, DARC is considered an essential receptor that mediates the entry of the human and zoonotic malaria parasites Plasmodium vivax and Plasmodium knowlesi into human reticulocytes and erythrocytes, respectively. This process is mediated through interaction with the parasite ligand known as the Duffy binding protein (DBP). Most therapeutic strategies have been focused on blocking the interaction between DBP and DARC by targeting the parasite ligand, while strategies targeting the receptor, DARC, have not been intensively investigated. The rapid increase in drug resistance and the lack of new effective drugs or a vaccine for malaria constitute a major threat and a need for novel therapeutics to combat disease. This review explores strategies that can be used to target the receptor. Inhibitors of DARC, which block DBP–DARC interaction, can potentially provide an effective strategy for preventing malaria caused by P. vivax.
Keywords: Plasmodium, vivax, knowlesi, DARC, malaria, treatment
Introduction
The Duffy antigen receptor for chemokines (DARC) was first discovered in the 1950s1 and later defined as a blood group antigen in the late 1960s. It became better known as the cell surface receptor used by the malaria parasites Plasmodium vivax and Plasmodium knowlesi to invade the red blood cells (RBCs).2,3 DARC, a single copy gene located on chromosome 1 (1.q22-1.q23),4 is a glycosylated transmembrane protein of about 35–40 kDa.4–6 It is comprised of an extracellular N-terminal domain (containing the chemokine-binding site) and an intracellular C-terminal domain. Alternatively, it is referred to as cluster of differentiation 234 (CD234) or Fy-glycoprotein (Fy).4 DARC is a minor blood group antigen that has two immunologically distinct and co-dominant alleles referred to as Fya and Fyb, which differ by a single base substitution in codon 42 encoding a glycine in Fya and an aspartic acid in Fyb.7 These two alleles result in four major Duffya, Fyb, Fya+b+, and Fya–b– (also referred to as Fy-null). Four other less reactive phenotypes, Fy3, Fy4, Fy5, and Fy6, have also been described.8,9 The Fy-null phenotype results from a Fyb gene mutation at amino acid position –46 in the erythroid regulatory element of the DARC promoter region. This mutation blocks promoter activity in cells derived from the hemopoietic lineage by disrupting the binding site for the specific erythroid transcription factor GATA1.10 This mutation is responsible for the lack of expression of DARC on the surface of erythroid cells. These DARC polymorphisms form the basis for the Duffy blood group.11,12 The Fya and Fyb antigens are common among Caucasians (Fya 66% and Fyb 83%) and Asians (Fya 99% and Fyb 18.5%) but are far less common in blacks (Fya 10% and Fyb 23%). In fact, the Fya–b– phenotype is present in two-thirds of African-American blacks but is very rare in Caucasians.10,13 Another rare phenotype, Fyb–, has also been described among non-Ashkenazi Jews and Brazilian blacks, but its clinical relevance is still unknown.14
DARC as a receptor for chemokines
DARC, also referred to as the Duffy blood group antigen,15,16 is a promiscuous receptor for several proinflammatory cytokines secreted by immune cells called chemokines, which act as communication signals.17,18 Most chemokine receptors specifically bind to chemokines of a single class: either the CC or the CXC classes. The DARC binds to chemokines of both the CC and CXC classes, the melanoma growth stimulatory activity (MSGA-α/CXCL1), interleukin 8 (CXCL8), regulated upon activation normal T-expressed and secreted (RANTES/CCL5), monocyte chemotactic protein-1 (CCL2), neutrophil activating protein 2 and 3, growth-related gene alpha, epithelial neutrophil activating peptide-78, (CXCL5), and angiogenesis-related platelet factor 1.19–23 DARC is homologous to the chemokine G-protein chemokine receptors, commonly used as entry vehicles by infectious agents.4 However, DARC lacks the DRY motif, which is required for G-protein interaction and signal transduction. It is mainly expressed on the surface of erythroid cells and endothelial cells lining post-capillary venules in the kidneys, spleen, and neuronal cells in both Duffy-positive and Duffy-negative individuals.24,25 DARC is believed to act as a scavenger, reducing excess amounts of toxic chemokines produced in some pathological conditions. It mediates the effects of proinflammatory chemokines on endothelial cells lining post-capillary venules as well as neutrophil emigration to inflammation sites.24,26–28 Nevertheless, its role as a scavenger has been questioned and alternatively suggested to mediate chemokine transcytosis leading to apical retention of intact chemokine.29
DARC and associated diseases
As a receptor for many chemokines, DARC has great clinical significance in many disease conditions. It has been implicated as a major actor in some infectious and inflammatory diseases as well as in cancer. DARC negativity has been shown to influence angiogenesis in animals. There is good evidence in mouse and in vitro models for DARC affecting inflammation, in particular leukocyte trafficking, chemokine levels, and malignancy. This suggests that DARC may play a role in the frequent differences in disease outcome seen in African-Americans.30 It is suggested that the lack of DARC expressivity results in the inability to remove the buildup of angiogenic chemokines, which are believed to contribute to cancer development.31,32 Consequently, the high rate of prostrate cancer in men of African descent has been attributed to the predominance of DARC negativity in this group.32,33 However, other studies have found little or no correlation between DARC expression on erythrocytes and the risk or progression of prostate cancer in men of African descent.32,34 DARC has also been implicated in the clinical outcome of other cancers such as breast cancer and metastasis.35,36 Some other racial differences relying on DARC in endotoxin responses,37 pregnancy,11 renal transplantation,38 and sickle cell39,40 have also been investigated. In HIV infection, DARC -46C/C is associated with a 40% increase in odds of acquiring HIV-1. Interestingly, regulatory variant genotypes of DARC can influence the course of HIV disease. This gives survival advantage to persons of African ancestry with reduced neutrophil counts41,42 as well as reduced plasma levels of HIV-1-suppressive and proinflammatory chemokines such as CCL5/RANTES. It is suggested that DARC influences HIV/AIDS susceptibility by mediating the binding of HIV to RBCs and subsequent viral transfer to HIV target cells.5 On the other hand, some DARC mutations have been found to abrogate receptor expression, leading to a significantly increased susceptibility to HIV-1 infection but, paradoxically, to prolonged survival in HIV-1-infected subjects.43 Other studies have shown that HIV-1 binds to erythrocytes’ DARC, making these RBCs able to transmit HIV to peripheral blood mononuclear cells. Thus, these cells function as a reservoir for HIV-1 or as a receptor for the entry of HIV-1 into CD4 cell subsets, neurons, or endothelial cells. DARC is also clinically relevant in diseases such as asthma and atopy among some populations of African descent,44 Southeast Asian ovalocytosis,45 and other inflammation-related diseases.27,46 Most importantly, DARC plays a critical role in erythrocyte invasion by malaria parasites.
DARC as a receptor for malaria parasites
The interest on the role of DARC as a scavenger has been diminished slightly, since its establishment as the unique receptor for the human malaria parasite P. vivax2,47,48 and the zoonotic parasite P. knowlesi.3 Evidence for use of DARC for invasion was also reported for the mouse parasite Plasmodium yoelii, although another underlying pathway has been identified.49,50 While P. knowlesi only accounts for zoonotic infections in some regions,51 P. vivax is a major public health problem worldwide.52–54 The World Health Organization reported an estimated 13.8 million cases of clinical illness and 1,400–14,900 deaths due to P. vivax malaria in 2015. Historically, vivax malaria was regarded as causing a benign and often self-limiting infection. There is increasing evidence of clinical severity of disease with a great pathological and economical burden for inhabitants of endemic regions.55–57 Similarly, widespread drug resistance has also been reported.58–60
Erythrocyte invasion by malaria parasites is essential for blood-stage development. This invasion process is mediated by specific ligand–receptor interactions between the parasite (merozoites) and the host erythrocytes.61–63 It is believed that the P. vivax Duffy binding protein (PvDBP) and the P. knowlesi Duffy binding protein alpha on the merozoite surface interact with DARC on the reticulocyte surface precipitating the junction formation step necessary for invasion.3,62,63 Historically, the vital need of the Duffy binding protein (DBP)–DARC interaction was evident from the virtual absence of P. vivax malaria in populations with a high prevalence of DARC negativity2,64,65 and the refractoriness of P. knowlesi merozoites to invade Duffy-negative human erythrocytes.3 This is an indication that the absence of DARC on the erythrocyte surface has a protective advantage against vivax and knowlesi malaria. More additional compelling evidence of the importance of the DBP–DARC interaction was demonstrated by the protective effect against clinical vivax malaria by the Fya allele.66 The vital need of this DBP–DARC interaction during reticulocyte invasion makes DBP a prime target for vaccine-mediated immunity against malaria caused by the parasites.
Variations in the Fy gene have been associated with phenotypic variation in susceptibility to malaria. It has been demonstrated that adherence of the DBP ligand domain (DBPII) to erythrocytes is significantly reduced for erythrocytes in heterozygous individuals carrying one Duffy antigen-negative allele.67 Individuals with the Fya phenotype demonstrated a 30%–80% reduced risk of clinical vivax but not falciparum malaria in a prospective cohort study in the Brazilian Amazon.66 The Fya allele has reached fixation in Southeast Asian populations, areas thought to be the wellspring of P. vivax.68 Conversely, Fyb is present in North and Northern-central European populations and admixed in many populations with strong Northern European influence. This distribution of Fy alleles suggests a selective advantage against P. vivax malaria.66 Importantly, inhibitory antibodies to the DBP ligand domain were much more effective in blocking DBP binding to erythrocytes expressing Fya compared with Fyb. This suggests that the relative frequencies of Fya and Fyb alleles in these populations may affect DBP vaccine efficacy.
Contrary to the established DBP–DARC invasion pathway, there is increasing evidence of a DARC-independent invasion of human reticulocytes by P. vivax.69–73 In Madagascar, with a mixture of Duffy-positive (Fy+) and -negative (Fy–) populations of diverse ethnic backgrounds, there was a significant reduction in the prevalence of clinical vivax malaria in Duffy-negative individuals compared with Duffy-positive individuals.70 Similarly in the Brazilian Amazon, two cases of clinical vivax malaria were observed in samples of Duffy-negative individuals obtained from Rondônia.71 It is not yet clear if these historically anomalous cases represent random isolated events that have always occurred or a new phenomenon related to P. vivax evolving to use an alternate DARC-independent pathway for invasion. Alternatively, DBP might remain the critical invasion ligand using alternate receptors for invasion. A P. vivax DBP homolog erythrocyte binding protein has been identified. This novel ligand is anticipated to be involved in an alternate invasion pathway to DBP.74,75 However, the exact role of this molecule in the invasion of Duffy-negative individuals is still unknown. A recent study suggested that an unusual DNA expansion of DBP in two Duffy-negative P. vivax infections studied suggests that an expansion of DBP may have been selected to allow low-affinity binding to another receptor on Duffy-null erythrocytes.76 Nevertheless, no other receptor other than DARC has been described for P. vivax.
Several in vitro assays have been used to study the interaction between the ligand domains of P. knowlesi and P. vivax DBPs. DBP binds to DARC on host reticulocytes through a conserved cysteine-rich Duffy binding-like (DBL) domain known as region II (DBPII), which is characterized by 12 conserved cysteine residues. The region between cysteines 4 and 7 constitutes the major determinants for receptor recognition.77–80 The binding sites for these two different parasite molecules map to a 35 amino acid region at the N-terminal extracellular domain (ECD1) of DARC.81 A peptide consisting of these 35 amino acid residues as well as a recombinant protein consisting of the N-terminal 60 residues of DARC (nDARC1–60) blocked PvDBP binding to human reticulocytes.81,82 Two tyrosine molecules within the 35 amino acid region (Tyr30 and Tyr41) are post-translationally sulfated, with sulfonation of Tyr41 associated with high-affinity binding of P. vivax DBPII. Specifically, a sulfated recombinant DARC N-terminus construct inhibits the DBPII erythrocyte interaction to a greater extent than an unsulfated construct.82 However, a recent crystal structure of DBPII in contact with nDARC did not show any contact between DBPII and Tyr41 of DARC.83 This suggests that the modification did not directly facilitate DARC–DBPII interaction but probably modified the presentation of DARC or its exposure. Tyrosine sulfation is also critical in the association between the HIV gp120 and the N-terminal domain of CCR5, a receptor for most HIV-1 isolates for invasion.84,85 Similar to DBP, a tyrosine-sulfated peptide based on the amino terminus of CCR5 specifically blocks HIV-1 entry and gp120 association with CCR5.86
The molecular and structural basis of DBP–DARC interaction was recently resolved.83,87 This structure reveals that DARC recognition by DBP is through a receptor-mediated ligand dimerization (Figure 1). This mechanism of interaction suggests that dimerization is critical for, and driven by DARC binding, leading to the formation of a stable high-affinity complex composed of two DBP and two DARC molecules.87 This complex is then believed to precipitate junction formation to initiate entry into the host cell. During this process, DBP dimerization creates a pocket (DARC binding pocket), which fits the N-terminus of DARC. This phenomenon is similar to receptor recognition in DBL domains of other members of the DBL superfamily such as PfEMP1, VAR2CSA, and PfEBA-175.88,89 The specific molecular interactions at the DBP–DARC interface were analyzed, revealing critical contact residues within the N-terminal extracellular domain of DARC. Most importantly is the DARC binding pocket, which has a strong affinity for a sulfotyrosine of DARC.82,83 Key residues for this interaction were located within subdomain 2 of DBPII, with residues F261-T266, L270-K289, and Q356-K367 forming critical contacts with the DARC extracellular domain.83 The residues that play a critical role in this interaction are important targets of protective immunity. Unlike the parasite ligands, ECD1 alone is not sufficient for chemokine binding. The close association of the four extracellular domains of DARC (ECD1–ECD4) through disulfide bonds is required to create an active chemokine-binding pocket.
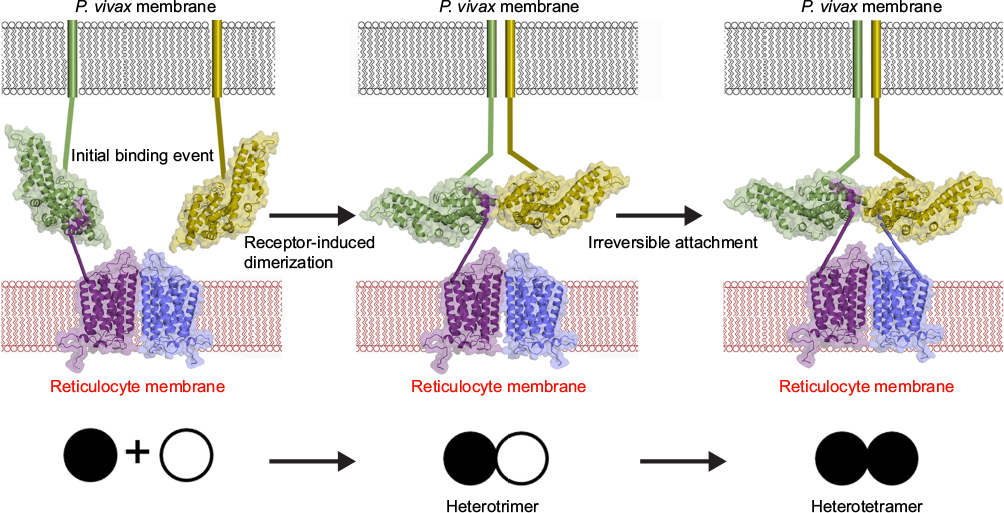 | Figure 1 A model for DBP–DARC interaction during invasion. Notes: DBP binds to DARC via a model of receptor-mediated ligand dimerization. Dimerization of DBP is induced upon receptor binding and drives recognition of DARC. An initial binding event is followed by receptor-induced dimerization (DBPII–DARC heterotrimer). This brings a second DBPII molecule in close proximity to a second DARC ectodomain in the DARC homodimer. A second binding event creates the DBPII–DARC heterotetramer. DBPII molecules: green and yellow. DARC19–30 molecules: purple and blue. A schematic for the stepwise assembly is shown at the bottom. Closed circle: bound DBPII and open circle: unbound DBPII. Adapted from Batchelor JD, Malpede BM, Omattage NS, DeKoster GT, Henzler-Wildman KA, Tolia NH. Red blood cell invasion by Plasmodium vivax: structural basis for DBP engagement of DARC. PLoS Pathog. 2014;10(1):e1003869.83 Abbreviations: DBP, Duffy binding protein; DARC, Duffy antigen receptor for chemokine; P. vivax, Plasmodium vivax. |
Current opinion and future therapeutic prospects
Chemokines and their receptors play a major role in facilitating the entry and transmission of intracellular pathogens, typical examples being in HIV and P. vivax infections. As such, they represent attractive targets for novel therapeutics.90–92 In order to exploit the potential of these chemokine receptors as drug targets, there is a need to identify the specific ligands and receptors that are rate limiting in a given disease setting and develop reagents to block their interaction. Methods that can prevent receptor–ligand interaction such as interfering with signaling pathways that are induced upon receptor activation and modification of receptor trafficking pathways can be explored to develop therapeutics.
In P. vivax and P. knowlesi, until other parasite proteins and their subsequent unknown receptors are identified, the sole dependence on DARC makes this outstanding nonspecific multireceptor for chemokines an invaluable target to explore procedures to inhibit blood-stage propagation of P. vivax. Extensive effort has been put into designing therapeutic strategies focused on the interaction between DBPII and DARC. This is mainly based on the evidence that some individuals are able to develop long-lasting and strain-transcending inhibitory antibodies against DBPII. Notwithstanding, strategies targeting the other counterpart of this host–parasite interaction in the blood stream, that is DARC, have not been intensively explored. To date, it is mostly established that P. vivax and P. knowlesi invasion depends on the parasite Duffy binding protein DBL domain (PvDBPII or P. knowlesi Duffy binding protein alpha) engaging DARC on RBCs. Inhibition of this key interaction provides an excellent opportunity for parasite inhibition. Individuals in endemic regions produce anti-DBP antibodies capable of blocking DBP–DARC interaction and parasite invasion of reticulocytes.93–96 Epitopes that are targets of these neutralizing antibodies mapped to the dimer interface, DARC binding pocket and surround the DARC binding pocket,87,97 suggesting that these protective antibodies target DBP functional regions, and interfere with dimerization or prevent receptor binding. This shows the importance of the DARC binding pocket and dimer formation for parasite survival. The structural determinants for DBP–DARC interaction have been identified.83,87 These regions, especially the DARC epitopes that make contact with DBP, represent critical regions that can be exploited for rational design of potent neutralizing therapeutics aimed at disrupting erythrocyte binding. Specific examples include designing vaccines and small molecule inhibitors that can compete with DBP for binding to DARC. These molecules if targeted to the dimer interface and receptor-binding pocket, they could prevent dimer formation and consequently merozoite invasion. Targeting DBP–DARC is effective against P. vivax, as natural selection of a Duffy-null phenotype has largely eliminated P. vivax in West Africa.2 As receptor-mediated ligand dimerization is a general mechanism by which EBL proteins engage receptors, disrupting dimerization could be a viable strategy for therapeutic intervention against other Plasmodium species.
Vaccines as a therapeutic tool
Vaccines are considered the best means of control of infectious diseases. Over 70 different vaccines against P. falciparum are in development, and ~23 are currently undergoing clinical trials. The most promising vaccine against P. falciparum, RTS,S/AS01, has completed Phase 3 clinical trials. Unfortunately, the same advances have not been seen for vaccines against P. vivax. A series of P. vivax merozoite antigens that offer great potential as vaccine candidates has been identified and immunologically characterized. These include the apical membrane antigen (AMA-1),98 DBP,99,100 reticulocyte binding proteins,101,102 and merozoite surface proteins.103–105 However, the development of a P. vivax vaccine has been hampered by technical difficulties. P. vivax preferentially invades reticulocytes, which account for only 1%–2% of total peripheral blood circulation. Because of the difficulty in obtaining enough reticulocytes, long-term culture for P. vivax has been a major challenge and a drawback to adequately study the biology of the parasite. Despite these challenges, a number of potential strategies are being explored for the development of a P. vivax malaria vaccine.106–109
DBP is a leading vaccine candidate for blood-stage P. vivax malaria. Individuals in endemic regions produce anti-DBP antibodies, which block DARC binding and reticulocyte invasion.93,96 However, this antibody response is generally weak and bias toward the development of strain-specific immunity.95,97,100 Despite this, a few elite responders are able to produce strain-transcending inhibitory antibody responses,96,110 suggesting the existence of conserved neutralizing epitopes on DBP. Some studies have reported strategies to overcoming strain-specific immunity in P. vivax, including a combination vaccine involving multiple-variant DBP alleles109 or a synthetic DBP antigen devoid of the dominant polymorphic B-cell epitopes.108,111 These studies, which are aimed at focusing on immune response to functional conserved neutralizing epitopes on DBP, could be optimized to target especially those residues that make contact with the DARC binding pocket. Notwithstanding, the efficacy of a DBP-based vaccine may differ among populations with varying Fy phenotypes.
Gene knockouts and targeted gene mutagenesis
One of the greatest ways to validate a target is to study the effect of the deletion of the gene of interest in vivo (receptor “knock out”). Several chemokine receptors have been validated as successful targets of anti-inflammatory therapies and anti-HIV infectivity strategy. CCR5 is the best example as a target, with a natural gene “knock out”. A natural deletion of a 32 base pair region on an allele of CCR5 (Δ32-CCR5) results in a lack of surface expression of CCR5. While individuals homozygous for the Δ32-CCR5 are resistant to HIV infection, those heterozygous for the deletion develop a delayed progression to disease.112 Interestingly, individuals who are homozygote for the Δ32-CCR5 mutation do not appear to have any adverse health issues. A long-standing goal of biomedical research is to develop efficient and reliable ways to make precise, targeted changes to the genome of living cells similar to the natural Δ32-CCR5 mutation. A new biotechnology tool for genome editing, CRISPR, has gained wide interest in the scientific world and researchers believe that it could transform the field of biology.113 CRISPR allows the ability to edit genomes with unprecedented precision, efficiency, and flexibility. An example is engineered monkeys with targeted mutations to prevent HIV infections in human cells.114 However, this application is limited because it provides only temporary inhibition of gene function and unpredictable off-target effects.115 Considering the fact that natural Duffy negativity does not create any adverse health problems to humans, this technology can be exploited to delete the Duffy positivity footprint in peripheral blood erythrocytes. Developing a strategy to target DARC expression in stem cells could be an interesting way to limit DARC in peripheral blood erythrocytes, without necessarily affecting DARC expression in endothelial cells.
An alternate DARC-related approach to vaccines is the use of novel agents specific for individual malaria species targeting their pathways of invasion. A typical example includes CCR5 blockers, already in use for HIV treatment, which could potentially block DARC, therefore inhibiting invasion of the target cell.116,117 Likewise, many studies have suggested artificial aberration of the host pathway by target mutagenesis of either RBC receptors to abolish or reduce susceptibility of the host to malaria. Zinc finger array precursors of zinc finger nucleases, which are artificial hybrid restriction enzymes, are becoming powerful tools for primary editing of host genomes as a strategy to halt pathogen infectivity. This strategy has been established in HIV-1 resistance in CD4+ T cells by disrupting the coding region of CCR5 upstream of the Δ32 mutation.118,119 Similarly, with appropriate optimization in vitro to enhance specificity to DARC, this strategy could potentially be applied to the development of an experimental gene-based Malaria vaccine.120 Alternatively, meganucleases and transcription activator-like effector nucleases121 that recognize longer stretches of DARC and DNA, especially the N-terminal region, may serve the specific purpose of abrogating invasion of RBCs by P. vivax. The feasibility of this approach is supported by existing evidence pointing to resistance of RBCs of naturally selected Duffy-negative blacks to P. vivax infection10 and the fact that DARC negativity will have no adverse effect on human health.
Small molecule agonists and antagonists as inhibitors of receptor-ligand interaction
The interaction between chemokine receptors and their ligands involves initial interaction with the N-terminal extracellular domains of the receptor, generally mediated by electrostatic forces. Modifications of these regions, using chemokine analogues, either truncations or extension of the amino terminus,122–124 have been found to retain affinity for the receptors, while impairing signaling. Chemokine receptor antagonists are still in early stages of development.125 Clinical trials using chemokine receptor antagonist have been reported for many diseases. Yet, the most advanced studies have been performed with CCR5 inhibitors used as retrovirals against HIV. Through screening of small molecules, Pfizer was able to identify a small molecule inhibitor that could block the gp120 binding to the chemokine receptor CCR5.126 The sites bound by most of these analogues are not yet identified. However, TAK-779, an antagonist of CCR5 with potent anti-HIV activity, binds to a cavity formed between helices located near the extracellular surface of the receptor, which is different from the extracellular loop known as the ligand binding site.116,117 Many other studies have reported high-affinity antagonists for a series of chemokine receptors, including CCR1, 2, and 5 and CXCR 2 and 4.117,127,128
The biology of DARC is very closely related to that of the HIV co-receptors CXCR4 and CCR5. Dimerization following ligand activation has been reported for DARC as well as CCR2, CCR5, and CXCR4.87,129,130 Considering the similarities in the interactions between antagonists and receptors within the G-protein-coupled receptor superfamily, antagonists/inhibitors, that have been developed for other members will help the rational design of agonists and antagonists of chemokine receptors such as DARC to prevent P. vivax malaria. A similar strategy aimed at identifying small molecule inhibitors of DARC, especially the extracellular N-terminal region bound by DBP, will be greatly useful in preventing P. vivax infection of human reticulocytes. In the absence of a continuous culture system for P. vivax, short-term in vitro vivax cultures131 and standard in vitro assays such as COS 780 and flow cytometry-based binding assays132 as well as the nDARC assay82 could serve as useful surrogates to screen small molecule libraries to identify potential inhibitory molecules to DBP–DARC interaction. It has been shown that chemokines such as CXCL1 and CXCL8, DBP, and anti-Fy6, a DARC antibody, all bind to similar molecular determinants on DARC and are able to block DBP–DARC interaction by P. knowlesi.15,81,133,134 This suggests that molecules, which compete with DARC for binding, could be used as therapeutics for vivax malaria. Novel small molecule inhibitors of DARC such as monoclonal antibodies, soluble receptors, or variant versions of the protein, specifically targeting the DARC binding pocket, can be exploited. This could serve as potential effective strategy for antimalarial therapy either alone or in combination with existing antimalarial drugs to develop new drugs to block DBP–DARC interaction, thereby preventing reticulocyte invasion and consequently P. vivax malaria.135 It is generally suggested that for G-protein-coupled receptors, small molecule agonists or antagonists may exert their effects by stabilizing either an active or a non-active form of the receptor, rather than simply blocking the physical interaction between the receptor and the ligand.136
Conclusion
Emerging resistance of P. vivax to current antimalarial drugs demonstrates an urgent need to develop new and alternative approaches to prevent this widespread cause of malaria. Inhibitors of DARC can serve as potential effective strategy for preventing malaria caused by P. vivax. Blocking DARC with small molecule inhibitors is a viable, attractive, and increasingly potential new therapeutic approach to prevent vivax-induced malaria.137 Historically, it has been demonstrated that populations in West Africa do not express DARC on their erythrocytes and as such are resistant to vivax malaria.2 In HIV infection, a 32 pb deletion (Δ32) in the coding sequence of CCR5, the co-receptor for HIV, inhibits expression of this receptor on HIV target cells, thereby preventing HIV infection. Despite the presence of DARC-negative as well as CCR5-negative phenotypes in such individuals, these genetic deficiencies do not seem to play any adverse physiological effects on these individuals. This suggests that blocking DARC will not lead to any deleterious consequences in humans. The parallels of the Δ32-CCR5 mutation as a protective factor in HIV infection and DARC negativity as a protective factor in P. vivax infection indicate that any inhibitors of DARC will be effective in preventing P. vivax malaria.
Acknowledgments
This work was supported in part by the National Institutes of Health, USA, grants R01AI064478 (JHA) and R21AI107455 (FBN). The authors thank Samantha Barnes for critical reading of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Cutbush M, Mollison PL. The Duffy blood group system. Heredity (Edinb). 1950;4(3):383–389. | ||
Miller LH, Mason SJ, Clyde DF, McGinniss MH. The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N Engl J Med. 1976;295(6):302–304. | ||
Miller LH, Mason SJ, Dvorak JA, McGinniss MH, Rothman IK. Erythrocyte receptors for (Plasmodium knowlesi) malaria: Duffy blood group determinants. Science. 1975;189(4202):561–563. | ||
Chaudhuri A, Polyakova J, Zbrzezna V, Williams K, Gulati S, Pogo AO. Cloning of glycoprotein D cDNA, which encodes the major subunit of the Duffy blood group system and the receptor for the Plasmodium vivax malaria parasite. Proc Natl Acad Sci U S A. 1993;90(22):10793–10797. | ||
He W, Neil S, Kulkarni H, et al. Duffy antigen receptor for chemokines mediates trans-infection of HIV-1 from red blood cells to target cells and affects HIV-AIDS susceptibility. Cell Host Microbe. 2008;4(1):52–62. | ||
de Brevern AG, Wong H, Tournamille C, Colin Y, Le Van Kim C, Etchebest C. A structural model of a seven-transmembrane helix receptor: the Duffy antigen/receptor for chemokine (DARC). Biochim Biophys Acta. 2005;1724(3):288–306. | ||
Iwamoto S, Omi T, Kajii E, Ikemoto S. Genomic organization of the glycoprotein D gene: Duffy blood group Fya/Fyb alloantigen system is associated with a polymorphism at the 44-amino acid residue. Blood. 1995;85(3):622–626. | ||
Behzad O, Lee CL, Gavin J, Marsh WL. A new anti-erythrocyte antibody in the Duffy system: anti-Fy4. Vox Sang. 1973;24(4):337–342. | ||
Colledge KI, Pezzulich M, Marsh WL. Anti-Fy5, and antibody disclosing a probable association between the Rhesus and Duffy blood group genes. Vox Sang. 1973;24(3):193–199. | ||
Tournamille C, Colin Y, Cartron JP, Le Van Kim C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat Genet. 1995;10(2):224–228. | ||
Marsh WL, Ehrich CC. The Duffy blood group system: a review of recent developments. Infusionsther Klin Ernahr. 1975;2(4):280–289. | ||
Meny GM. The Duffy blood group system: a review. Immunohematology. 2010;26(2):51–56. | ||
Sanger R, Race RR, Jack J. The Duffy blood groups of New York negroes: the phenotype Fy (a-b-). Br J Haematol. 1955;1(4):370–374. | ||
Parasol N, Reid M, Rios M, Castilho L, Harari I, Kosower NS. A novel mutation in the coding sequence of the FY*B allele of the Duffy chemokine receptor gene is associated with an altered erythrocyte phenotype. Blood. 1998;92(7):2237–2243. | ||
Horuk R, Chitnis CE, Darbonne WC, et al. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science. 1993;261(5125):1182–1184. | ||
Neote K, Mak JY, Kolakowski LF Jr, Schall TJ. Functional and biochemical analysis of the cloned Duffy antigen: identity with the red blood cell chemokine receptor. Blood. 1994;84(1):44–52. | ||
Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. | ||
Demogines A, Truong KA, Sawyer SL. Species-specific features of DARC, the primate receptor for Plasmodium vivax and Plasmodium knowlesi. Mol Biol Evol. 2012;29(2):445–449. | ||
Horuk R, Colby TJ, Darbonne WC, Schall TJ, Neote K. The human erythrocyte inflammatory peptide (chemokine) receptor. Biochemical characterization, solubilization, and development of a binding assay for the soluble receptor. Biochemistry. 1993;32(22):5733–5738. | ||
Darbonne WC, Rice GC, Mohler MA, et al. Red blood cells are a sink for interleukin 8, a leukocyte chemotaxin. J Clin Invest. 1991;88(4):1362–1369. | ||
Waśniowska K, Czerwinski M, Jachymek W, Lisowska E. Expression and binding properties of a soluble chimeric protein containing the N-terminal domain of the Duffy antigen. Biochem Biophys Res Commun. 2000;273(2):705–711. | ||
McMorran BJ, Wieczorski L, Drysdale KE, et al. Platelet factor 4 and Duffy antigen required for platelet killing of Plasmodium falciparum. Science. 2012;338(6112):1348–1351. | ||
Tournamille C, Filipe A, Wasniowska K, et al. Structure-function analysis of the extracellular domains of the Duffy antigen/receptor for chemokines: characterization of antibody and chemokine binding sites. Br J Haematol. 2003;122(6):1014–1023. | ||
Peiper SC, Wang ZX, Neote K, et al. The Duffy antigen/receptor for chemokines (DARC) is expressed in endothelial cells of Duffy negative individuals who lack the erythrocyte receptor. J Exp Med. 1995;181(4):1311–1317. | ||
Horuk R, Martin A, Hesselgesser J, et al. The Duffy antigen receptor for chemokines: structural analysis and expression in the brain. J Leukoc Biol. 1996;59(1):29–38. | ||
Hadley TJ, Lu ZH, Wasniowska K, et al. Postcapillary venule endothelial cells in kidney express a multispecific chemokine receptor that is structurally and functionally identical to the erythroid isoform, which is the Duffy blood group antigen. J Clin Invest. 1994;94(3):985–991. | ||
Lee JS, Frevert CW, Wurfel MM, et al. Duffy antigen facilitates movement of chemokine across the endothelium in vitro and promotes neutrophil transmigration in vitro and in vivo. J Immunol. 2003;170(10):5244–5251. | ||
Williams MR, Azcutia V, Newton G, Alcaide P, Luscinskas FW. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol. 2011;32(10):461–469. | ||
Pruenster M, Mudde L, Bombosi P, et al. The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat Immunol. 2009;10(1):101–108. | ||
Horne K, Woolley IJ. Shedding light on DARC: the role of the Duffy antigen/receptor for chemokines in inflammation, infection and malignancy. Inflamm Res. 2009;58(8):431–435. | ||
Moore BB, Arenberg DA, Stoy K, et al. Distinct CXC chemokines mediate tumorigenicity of prostate cancer cells. Am J Pathol. 1999;154(5):1503–1512. | ||
Shen H, Schuster R, Stringer KF, Waltz SE, Lentsch AB. The Duffy antigen/receptor for chemokines (DARC) regulates prostate tumor growth. FASEB J. 2006;20(1):59–64. | ||
Lentsch AB. The Duffy antigen/receptor for chemokines (DARC) and prostate cancer. A role as clear as black and white? FASEB J. 2002;16(9):1093–1095. | ||
Nemesure B, Wu SY, Hennis A, Leske MC. Distribution of Duffy antigen receptor for chemokines (DARC) and risk of prostate cancer in Barbados, West Indies. J Immigr Minor Health. 2015;17(3):679–683. | ||
Zijlstra A, Quigley JP. The DARC side of metastasis: shining a light on KAI1-mediated metastasis suppression in the vascular tunnel. Cancer Cell. 2006;10(3):177–178. | ||
Wang J, Ou ZL, Hou YF, et al. Enhanced expression of Duffy antigen receptor for chemokines by breast cancer cells attenuates growth and metastasis potential. Oncogene. 2006;25(54):7201–7211. | ||
Mayr FB, Spiel AO, Leitner JM, et al. Racial differences in endotoxin-induced tissue factor-triggered coagulation. J Thromb Haemost. 2009;7(4):634–640. | ||
Watorek E, Boratynska M, Halon A, Klinger M. Anti-Fya antibodies as the cause of an unfortunate post-transplant course in renal transplant recipient. Ann Transplant. 2008;13(1):48–52. | ||
Afenyi-Annan A, Kail M, Combs MR, Orringer EP, Ashley-Koch A, Telen MJ. Lack of Duffy antigen expression is associated with organ damage in patients with sickle cell disease. Transfusion. 2008;48(5):917–924. | ||
Nebor D, Durpes MC, Mougenel D, et al. Association between Duffy antigen receptor for chemokines expression and levels of inflammation markers in sickle cell anemia patients. Clin Immunol. 2010;136(1):116–122. | ||
Kulkarni H, Marconi VC, He W, et al. The Duffy-null state is associated with a survival advantage in leukopenic HIV-infected persons of African ancestry. Blood. 2009;114(13):2783–2792. | ||
Reich D, Nalls MA, Kao WH, et al. Reduced neutrophil count in people of African descent is due to a regulatory variant in the Duffy antigen receptor for chemokines gene. PLoS Genet. 2009;5(1):e1000360. | ||
Walton RT, Rowland–Jones SL. HIV and chemokine binding to red blood cells–DARC matters. Cell Host Microbe. 2008;4(1):3–5. | ||
Vergara C, Tsai YJ, Grant AV, et al. Gene encoding Duffy antigen/receptor for chemokines is associated with asthma and IgE in three populations. Am J Respir Crit Care Med. 2008;178(10):1017–1022. | ||
Woolley IJ, Hutchinson P, Reeder JC, Kazura JW, Cortes A. Southeast Asian ovalocytosis is associated with increased expression of Duffy antigen receptor for chemokines (DARC). Immunohematology. 2009;25(2):63–66. | ||
Geleff S, Draganovici D, Jaksch P, Segerer S. The role of chemokine receptors in acute lung allograft rejection. Eur Respir J. 2010;35(1):167–175. | ||
Barnwell JW, Nichols ME, Rubinstein P. In vitro evaluation of the role of the Duffy blood group in erythrocyte invasion by Plasmodium vivax. J Exp Med. 1989;169(5):1795–1802. | ||
Wertheimer SP, Barnwell JW. Plasmodium vivax interaction with the human Duffy blood group glycoprotein: identification of a parasite receptor-like protein. Exp Parasitol. 1989;69(4):340–350. | ||
Xu L, Niu J, Chaudhuri A. The domain on the mouse Duffy protein for Plasmodium yoelii binding and invasion to mouse erythrocytes. Mol Biochem Parasitol. 2006;146(2):142–150. | ||
Swardson-Olver CJ, Dawson TC, Burnett RC, Peiper SC, Maeda N, Avery AC. Plasmodium yoelii uses the murine Duffy antigen receptor for chemokines as a receptor for normocyte invasion and an alternative receptor for reticulocyte invasion. Blood. 2002;99(8):2677–2684. | ||
Cox-Singh J, Singh B. Knowlesi malaria: newly emergent and of public health importance? Trends Parasitol. 2008;24(9):406–410. | ||
Price RN, Tjitra E, Guerra CA, Yeung S, White NJ, Anstey NM. Vivax malaria: neglected and not benign. Am J Trop Med Hyg. 2007;77(Suppl 6):79–87. | ||
Mendis K, Sina BJ, Marchesini P, Carter R. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg. 2001;64(Suppl 1–2):97–106. | ||
Galinski MR, Barnwell JW. Plasmodium vivax: who cares? Malar J. 2008;7(Suppl 1):S9. | ||
Kochar DK, Das A, Kochar SK, et al. Severe Plasmodium vivax malaria: a report on serial cases from Bikaner in northwestern India. Am J Trop Med Hyg. 2009;80(2):194–198. | ||
Sharma A, Khanduri U. How benign is benign tertian malaria? J Vector Borne Dis. 2009;46(2):141–144. | ||
Alexandre MA, Ferreira CO, Siqueira AM, et al. Severe Plasmodium vivax malaria, Brazilian Amazon. Emerg Infect Dis. 2010;16(10):1611–1614. | ||
Rijken MJ, Boel ME, Russell B, et al. Chloroquine resistant vivax malaria in a pregnant woman on the western border of Thailand. Malar J. 2011;10:113. | ||
Ketema T, Bacha K, Birhanu T, Petros B. Chloroquine-resistant Plasmodium vivax malaria in Serbo town, Jimma zone, south-west Ethiopia. Malar J. 2009;8:177. | ||
Mohan K, Maithani MM. Congenital malaria due to chloroquine-resistant Plasmodium vivax: a case report. J Trop Pediatr. 2010;56(6):454–455. | ||
Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell. 2006;124(4):755–766. | ||
Adams JH, Hudson DE, Torii M, et al. The Duffy receptor family of Plasmodium knowlesi is located within the micronemes of invasive malaria merozoites. Cell. 1990;63(1):141–153. | ||
Adams JH, Sim BK, Dolan SA, Fang X, Kaslow DC, Miller LH. A family of erythrocyte binding proteins of malaria parasites. Proc Natl Acad Sci U S A. 1992;89(15):7085–7089. | ||
Guerra CA, Howes RE, Patil AP, et al. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010;4(8):e774. | ||
Miller LH, McGinniss MH, Holland PV, Sigmon P. The Duffy blood group phenotype in American blacks infected with Plasmodium vivax in Vietnam. Am J Trop Med Hyg. 1978;27(6):1069–1072. | ||
King CL, Adams JH, Xianli J, et al. Fy(a)/Fy(b) antigen polymorphism in human erythrocyte Duffy antigen affects susceptibility to Plasmodium vivax malaria. Proc Natl Acad Sci U S A. 2011;108(50):20113–20118. | ||
Michon P, Woolley I, Wood EM, Kastens W, Zimmerman PA, Adams JH. Duffy-null promoter heterozygosity reduces DARC expression and abrogates adhesion of the P. vivax ligand required for blood-stage infection. FEBS Lett. 2001;495(1–2):111–114. | ||
Howes RE, Patil AP, Piel FB, et al. The global distribution of the Duffy blood group. Nat Commun. 2011;2:266. | ||
Ryan JR, Stoute JA, Amon J, et al. Evidence for transmission of Plasmodium vivax among a Duffy antigen negative population in Western Kenya. Am J Trop Med Hyg. 2006;75(4):575–581. | ||
Menard D, Barnadas C, Bouchier C, et al. Plasmodium vivax clinical malaria is commonly observed in Duffy-negative Malagasy people. Proc Natl Acad Sci U S A. 2010;107(13):5967–5971. | ||
Cavasini CE, Mattos LC, Couto AA, et al. Plasmodium vivax infection among Duffy antigen-negative individuals from the Brazilian Amazon region: an exception? Trans R Soc Trop Med Hyg. 2007;101(10):1042–1044. | ||
Wurtz N, Mint Lekweiry K, Bogreau H, et al. Vivax malaria in Mauritania includes infection of a Duffy-negative individual. Malar J. 2011;10:336. | ||
Fru-Cho J, Bumah VV, Safeukui I, Nkuo-Akenji T, Titanji VP, Haldar K. Molecular typing reveals substantial Plasmodium vivax infection in asymptomatic adults in a rural area of Cameroon. Malar J. 2014;13:170. | ||
Hester J, Chan ER, Menard D, et al. De novo assembly of a field isolate genome reveals novel Plasmodium vivax erythrocyte invasion genes. PLoS Negl Trop Dis. 2013;7(12):e2569. | ||
Menard D, Chan ER, Benedet C, et al. Whole genome sequencing of field isolates reveals a common duplication of the Duffy binding protein gene in Malagasy Plasmodium vivax strains. PLoS Negl Trop Dis. 2013;7(11):e2489. | ||
Gunalan K, Lo E, Hostetler JB, et al. Role of Plasmodium vivax Duffy-binding protein 1 in invasion of Duffy-null Africans. Proc Natl Acad Sci U S A. 2016;113(22):6271–6276. | ||
VanBuskirk KM, Sevova E, Adams JH. Conserved residues in the Plasmodium vivax Duffy-binding protein ligand domain are critical for erythrocyte receptor recognition. Proc Natl Acad Sci U S A. 2004;101(44):15754–15759. | ||
Ranjan A, Chitnis CE. Mapping regions containing binding residues within functional domains of Plasmodium vivax and Plasmodium knowlesi erythrocyte-binding proteins. Proc Natl Acad Sci U S A. 1999;96(24):14067–14072. | ||
Chitnis CE, Miller LH. Identification of the erythrocyte binding domains of Plasmodium vivax and Plasmodium knowlesi proteins involved in erythrocyte invasion. J Exp Med. 1994;180(2):497–506. | ||
Hans D, Pattnaik P, Bhattacharyya A, et al. Mapping binding residues in the Plasmodium vivax domain that binds Duffy antigen during red cell invasion. Mol Microbiol. 2005;55(5):1423–1434. | ||
Chitnis CE, Chaudhuri A, Horuk R, Pogo AO, Miller LH. The domain on the Duffy blood group antigen for binding Plasmodium vivax and P. knowlesi malarial parasites to erythrocytes. J Exp Med. 1996;184(4):1531–1536. | ||
Choe H, Moore MJ, Owens CM, et al. Sulphated tyrosines mediate association of chemokines and Plasmodium vivax Duffy binding protein with the Duffy antigen/receptor for chemokines (DARC). Mol Microbiol. 2005;55(5):1413–1422. | ||
Batchelor JD, Malpede BM, Omattage NS, DeKoster GT, Henzler-Wildman KA, Tolia NH. Red blood cell invasion by Plasmodium vivax: structural basis for DBP engagement of DARC. PLoS Pathog. 2014;10(1):e1003869. | ||
Farzan M, Mirzabekov T, Kolchinsky P, et al. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell. 1999;96(5):667–676. | ||
Cimbro R, Gallant TR, Dolan MA, et al. Tyrosine sulfation in the second variable loop (V2) of HIV-1 gp120 stabilizes V2-V3 interaction and modulates neutralization sensitivity. Proc Natl Acad Sci U S A. 2014;111(8):3152–3157. | ||
Farzan M, Vasilieva N, Schnitzler CE, et al. A tyrosine-sulfated peptide based on the N terminus of CCR5 interacts with a CD4-enhanced epitope of the HIV-1 gp120 envelope glycoprotein and inhibits HIV-1 entry. J Biol Chem. 2000;275(43):33516–33521. | ||
Batchelor JD, Zahm JA, Tolia NH. Dimerization of Plasmodium vivax DBP is induced upon receptor binding and drives recognition of DARC. Nat Struct Mol Biol. 2011;18(8):908–914. | ||
Tolia NH, Enemark EJ, Sim BK, Joshua-Tor L. Structural basis for the EBA-175 erythrocyte invasion pathway of the malaria parasite Plasmodium falciparum. Cell. 2005;122(2):183–193. | ||
Srivastava A, Gangnard S, Round A, et al. Full-length extracellular region of the var2CSA variant of PfEMP1 is required for specific, high-affinity binding to CSA. Proc Natl Acad Sci U S A. 2010;107(11):4884–4889. | ||
Rucker J, Edinger AL, Sharron M, et al. Utilization of chemokine receptors, orphan receptors, and herpesvirus-encoded receptors by diverse human and simian immunodeficiency viruses. J Virol. 1997;71(12):8999–9007. | ||
Horuk R. The interleukin-8-receptor family: from chemokines to malaria. Immunol Today. 1994;15(4):169–174. | ||
Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. | ||
Michon P, Fraser T, Adams JH. Naturally acquired and vaccine-elicited antibodies block erythrocyte cytoadherence of the Plasmodium vivax Duffy binding protein. Infect Immun. 2000;68(6):3164–3171. | ||
Grimberg BT, Udomsangpetch R, Xainli J, et al. Plasmodium vivax invasion of human erythrocytes inhibited by antibodies directed against the Duffy binding protein. PLoS Med. 2007;4(12):e337. | ||
Ceravolo IP, Sanchez BA, Sousa TN, et al. Naturally acquired inhibitory antibodies to Plasmodium vivax Duffy binding protein are short-lived and allele-specific following a single malaria infection. Clin Exp Immunol. 2009;156(3):502–510. | ||
King CL, Michon P, Shakri AR, et al. Naturally acquired Duffy-binding protein-specific binding inhibitory antibodies confer protection from blood-stage Plasmodium vivax infection. Proc Natl Acad Sci U S A. 2008;105(24):8363–8368. | ||
Chootong P, Ntumngia FB, VanBuskirk KM, et al. Mapping epitopes of the Plasmodium vivax Duffy binding protein with naturally acquired inhibitory antibodies. Infect Immun. 2010;78(3):1089–1095. | ||
Kocken CH, Dubbeld MA, Van Der Wel A, et al. High-level expression of Plasmodium vivax apical membrane antigen 1 (AMA-1) in Pichia pastoris: strong immunogenicity in Macaca mulatta immunized with P. vivax AMA-1 and adjuvant SBAS2. Infect Immun. 1999;67(1):43–49. | ||
Singh S, Pandey K, Chattopadhayay R, et al. Biochemical, biophysical, and functional characterization of bacterially expressed and refolded receptor binding domain of Plasmodium vivax Duffy-binding protein. J Biol Chem. 2001;276(20):17111–17116. | ||
Xainli J, Cole-Tobian JL, Baisor M, et al. Epitope-specific humoral immunity to Plasmodium vivax Duffy binding protein. Infect Immun. 2003;71(5):2508–2515. | ||
Galinski MR, Medina CC, Ingravallo P, Barnwell JW. A reticulocyte-binding protein complex of Plasmodium vivax merozoites. Cell. 1992;69(7):1213–1226. | ||
Galinski MR, Xu M, Barnwell JW. Plasmodium vivax reticulocyte binding protein-2 (PvRBP-2) shares structural features with PvRBP-1 and the Plasmodium yoelii 235 kDa rhoptry protein family. Mol Biochem Parasitol. 2000;108(2):257–262. | ||
Galinski MR, Ingravallo P, Corredor-Medina C, Al-Khedery B, Povoa M, Barnwell JW. Plasmodium vivax merozoite surface proteins-3beta and-3gamma share structural similarities with P. vivax merozoite surface protein-3alpha and define a new gene family. Mol Biochem Parasitol. 2001;115(1):41–53. | ||
Lima-Junior JC, Tran TM, Meyer EV, et al. Naturally acquired humoral and cellular immune responses to Plasmodium vivax merozoite surface protein 9 in Northwestern Amazon individuals. Vaccine. 2008;26(51):6645–6654. | ||
Dutta S, Ware LA, Barbosa A, Ockenhouse CF, Lanar DE. Purification, characterization, and immunogenicity of a disulfide cross-linked Plasmodium vivax vaccine candidate antigen, merozoite surface protein 1, expressed in Escherichia coli. Infect Immun. 2001;69(9):5464–5470. | ||
Arevalo-Herrera M, Chitnis C, Herrera S. Current status of Plasmodium vivax vaccine. Hum Vaccin. 2010;6(1):124–132. | ||
Valencia SH, Rodriguez DC, Acero DL, Ocampo V, Arevalo-Herrera M. Platform for Plasmodium vivax vaccine discovery and development. Mem Inst Oswaldo Cruz. 2011;106(Suppl 1):179–192. | ||
Ntumngia FB, Adams JH. Design and immunogenicity of a novel synthetic antigen based on the ligand domain of the Plasmodium vivax Duffy binding protein. Clin Vaccine Immunol. 2012;19(1):30–36. | ||
Ntumngia FB, Schloegel J, McHenry AM, et al. Immunogenicity of single versus mixed allele vaccines of Plasmodium vivax Duffy binding protein region II. Vaccine. 2013;31(40):4382–4388. | ||
Cole-Tobian JL, Michon P, Biasor M, et al. Strain-specific Duffy binding protein antibodies correlate with protection against infection with homologous compared to heterologous Plasmodium vivax strains in Papua New Guinean children. Infect Immun. 2009;77(9):4009–4017. | ||
Chen E, Salinas ND, Ntumngia FB, Adams JH, Tolia NH. Structural analysis of the synthetic Duffy binding protein (DBP) antigen DEKnull relevant for Plasmodium vivax malaria vaccine design. PLoS Negl Trop Dis. 2015;9(3):e0003644. | ||
Dean M, Carrington M, Winkler C, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996;273(5283):1856–1862. | ||
Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. | ||
Hu W, Kaminski R, Yang F, et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A. 2014;111(31):11461–11466. | ||
Alic N, Hoddinott MP, Foley A, Slack C, Piper MD, Partridge L. Detrimental effects of RNAi: a cautionary note on its use in Drosophila ageing studies. PLoS One. 2012;7(9):e45367. | ||
Dragic T, Trkola A, Thompson DA, et al. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc Natl Acad Sci U S A. 2000;97(10):5639–5644. | ||
Baba M, Nishimura O, Kanzaki N, et al. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci U S A. 1999;96(10):5698–5703. | ||
Perez EE, Wang J, Miller JC, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26(7):808–816. | ||
Wilen CB, Wang J, Tilton JC, et al. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 2011;7(4):e1002020. | ||
Kajumbula H, Byarugaba W, Wayengera M. Targeting wild-type erythrocyte receptors for Plasmodium falciparum and vivax merozoites by zinc finger nucleases in- silico: towards a genetic vaccine against malaria. Genet Vaccines Ther. 2012;10(1):8. | ||
Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39(21):9283–9293. | ||
Clark-Lewis I, Schumacher C, Baggiolini M, Moser B. Structure-activity relationships of interleukin-8 determined using chemically synthesized analogs. Critical role of NH2-terminal residues and evidence for uncoupling of neutrophil chemotaxis, exocytosis, and receptor binding activities. J Biol Chem. 1991;266(34):23128–23134. | ||
Gong JH, Clark-Lewis I. Antagonists of monocyte chemoattractant protein 1 identified by modification of functionally critical NH2-terminal residues. J Exp Med. 1995;181(2):631–640. | ||
Proudfoot AE, Power CA, Hoogewerf AJ, et al. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J Biol Chem. 1996;271(5):2599–2603. | ||
Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov. 2009;8(1):23–33. | ||
Dorr P, Westby M, Dobbs S, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49(11):4721–4732. | ||
Ng HP, May K, Bauman JG, et al. Discovery of novel non-peptide CCR1 receptor antagonists. J Med Chem. 1999;42(22):4680–4694. | ||
Liang M, Mallari C, Rosser M, et al. Identification and characterization of a potent, selective, and orally active antagonist of the CC chemokine receptor-1. J Biol Chem. 2000;275(25):19000–19008. | ||
Rodriguez-Frade JM, Vila-Coro AJ, de Ana AM, Albar JP, Martinez-A C, Mellado M. The chemokine monocyte chemoattractant protein-1 induces functional responses through dimerization of its receptor CCR2. Proc Natl Acad Sci U S A. 1999;96(7):3628–3633. | ||
Rodriguez-Frade JM, Vila-Coro AJ, Martin A, et al. Similarities and differences in RANTES- and (AOP)-RANTES-triggered signals: implications for chemotaxis. J Cell Biol. 1999;144(4):755–765. | ||
Russell B, Suwanarusk R, Borlon C, et al. A reliable ex vivo invasion assay of human reticulocytes by Plasmodium vivax. Blood. 2011;118(13):e74–e81. | ||
Tran TM, Moreno A, Yazdani SS, Chitnis CE, Barnwell JW, Galinski MR. Detection of a Plasmodium vivax erythrocyte binding protein by flow cytometry. Cytometry A. 2005;63(1):59–66. | ||
Hesselgesser J, Chitnis CE, Miller LH, et al. A mutant of melanoma growth stimulating activity does not activate neutrophils but blocks erythrocyte invasion by malaria. J Biol Chem. 1995;270(19):11472–11476. | ||
Chitnis CE. Molecular insights into receptors used by malaria parasites for erythrocyte invasion. Curr Opin Hematol. 2001;8(2):85–91. | ||
Chitnis CE, Sharma A. Targeting the Plasmodium vivax Duffy-binding protein. Trends Parasitol. 2008;24(1):29–34. | ||
Schwartz TW, Rosenkilde MM. Is there a ‘lock’ for all agonist ‘keys’ in 7TM receptors? Trends Pharmacol Sci. 1996;17(6):213–216. | ||
Handel TM, Horuk R. Duffy antigen inhibitors: useful therapeutics for malaria? Trends Parasitol. 2010;26(7):329–333. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.