")
Back to Journals » Drug Design, Development and Therapy » Volume 12
The hydroxypropyl–β-cyclodextrin complexation of toltrazuril for enhancing bioavailability
Authors Zhang L, Liu MX, Lu CC, Ren DD, Fan GQ, Liu C, Liu MJ, Shu G, Peng GN, Yuan ZX, Zhong ZJ, Zhang W, Fu HL
Received 20 November 2017
Accepted for publication 14 December 2017
Published 19 March 2018 Volume 2018:12 Pages 583—589
DOI https://doi.org/10.2147/DDDT.S157611
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Li Zhang,1,2,* Mengxi Liu,1,* Chaocheng Lu,1 Dandan Ren,1 Guoqing Fan,1 Chang Liu,1 Mengjiao Liu,1 Gang Shu,1 Guangneng Peng,1 Zhixiang Yuan,1 Zhijun Zhong,1 Wei Zhang,1 Hualin Fu1
1Department of Pharmacy, College of Veterinary Medicine, Sichuan Agricultural University, Chengdu, Sichuan, China; 2Institute of Traditional Chinese Medicine Pharmacology and Toxicology, Sichuan Academy of Chinese Medicine Sciences, Chengdu, Sichuan, China
*These authors contributed equally to this work
Introduction: Toltrazuril (Tol) is used to prevent and combat coccidiosis. However, its low aqueous solubility and poor oral bioavailability limit clinical application.
Methods: To overcome the shortcomings, toltrazuril–hydroxypropyl–β-cyclodextrin inclusion complex (Tol-HP-β-CD) was prepared and characterized. The comparative plasma disposition kinetics of Tol was analyzed after a single orally administered dose of 10 mg/kg Tol or Tol-HP-β-CD in rabbits. Solution-stirring method was selected to prepare the inclusion complex. Complex formation was characterized by thin-layer chromatography, Fourier transform infrared spectrophotometry, and 1H nuclear magnetic resonance spectroscopy. In plasma profile, plasma samples were collected between 1 and 10 days following administration. Plasma Tol concentrations were determined by high-performance liquid chromatography.
Results: In rabbit plasma, the time to peak concentration (Tmax) of Tol-HP-β-CD was shorter than that of Tol (12 h vs 24 h). Cmax (19.92±1.02 µg/mL) and area under the concentration–time curve (AUC0-∞, 1,176.86±70.26 mg/L h) of the Tol-HP-β-CD group significantly increased (p<0.01) than those of the Tol group (Cmax, 8.02±1.04 µg/mL; AUC0-∞, 514.03±66.65 mg/L h).
Conclusion: It can be concluded that the Tol-HP-β-CD increased the aqueous solubility and enhanced the oral bioavailability in rabbits. Complexation with HP-β-CD is a feasible way to prepare a rapidly absorbed and more bioavailable Tol oral product.
Keywords: toltrazuril, coccidian, hydroxypropyl–β-cyclodextrin, inclusion complex, pharmacokinetics
Introduction
Rabbit coccidiosis is a ubiquitous disease caused by one or more of 16 species of the apicomplexan genus Eimeria stiedae.1–4 General clinical symptoms of the disease are characterized by dullness, reduced food consumption, diarrhea or constipation, liver enlargement, ascites, icterus, abdominal distention, and death.3 Coccidiosis in rabbits can be prevented and treated using drugs.1,3,5,6 Toltrazuril (Tol), 1-[3-methyl-4-(4-trifluoromethylsulfanyl-phenoxy)-phenyl]-3-methyl-1,3,5-triazin-2,4,6-trione (Figure 1), is a symmetrical triazinetrione compound that is widely used to prevent and combat coccidiosis.7–10 However, due to poor aqueous solubility, Tol is difficult to be absorbed by the gastrointestinal (GI) tract. The clinical effects of Tol have been discounted because of its solubility in the GI tract.
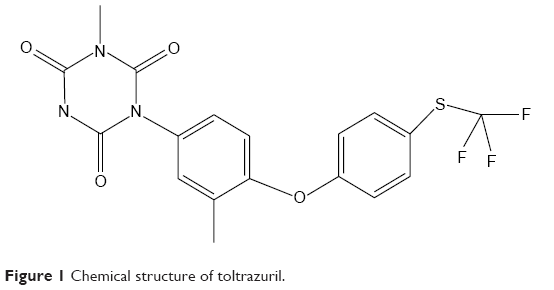 | Figure 1 Chemical structure of toltrazuril. |
The poor aqueous solubility of Tol has been overcome by some techniques, such as solid dispersion, ultrafine power, and nanoemulsion.11–13 As the currently most effective techniques for increasing solubility, Tol solid dispersion only increased the solubility of Tol to 2,000 times,11 which indicates that its solubility still needs to be enhanced significantly through other techniques. In addition, solid dispersion and nanoemulsion are unstable and inconvenient to store, while ultrafine power needs sophisticated equipment to produce.
β-cyclodextrin (β-CD) is in widespread use because of its unique cavity size, efficiency of drug complexation, and enhancements of drug stability, solubility, and bioavailability.14,15 For its regulatory status, β-CD is listed in numerous pharmacopoeia sources, including the US Pharmacopoeia/National Formulary, European Pharmacopoeia, and Japanese Pharmaceutical Codex.16,17 Hydroxypropyl–β-CD (HP-β-CD) is a hydroxyalkyl β-CD derivative that is studied extensively in drug inclusion complex because of its inclusion ability and high water solubility.18–21 Toxicologic studies have reported on the safety of HP-β-CD in intravenous and oral administrations to the human body,22 and HP-β-CD has been used in clinical formulations to overcome poor solubility issues and enhance bioavailability.23
Not all drugs have properties to be made into complex with HP-β-CD. Tol was found to possess the properties based on a large number of screening research work. To increase the solubility and bioavailability of Tol by inclusion complex formation with HP-β-CD, toltrazuril–hydroxypropyl–β-cyclodextrin inclusion complex (Tol-HP-β-CD) was prepared by solution-stirring method in this study, and thin-layer chromatography (TLC), Fourier transform infrared (FTIR) spectroscopy, and nuclear magnetic resonance (NMR) spectroscopy were employed to characterize the obtained Tol-HP-β-CD. The pharmacokinetic profiles of Tol and Tol-HP-β-CD in rabbits after oral administration were further compared in vivo.
Methods
Materials and reagents
Tol (pharmaceutical grade) was obtained from Hubei Longxiang Pharmaceutical Co., Ltd. (Hubei, China). HP-β-CD (the average relative molecular mass of 1,390.18) was purchased from Xi’an Drake Biological Chemical Industry Co., Ltd. (Xi’an, China). All other reagents were of analytical grade.
New Zealand white rabbits (2.5±0.2 kg) were used for the pharmacokinetic study. All the animals were supplied by the Experimental Animal Center of Sichuan Agricultural University (Chengdu, China). Before the experiment, the animals were acclimatized at 25°C±2°C under natural light/dark conditions for 1 week with free access to food and water. Twelve hours before dosing, the animals were made to fast but were allowed free access to water. All experimental procedures were in accordance with the national standard of the Laboratory Animal-Requirements of Environment and Housing Facilities (GB 14925-2001) and approved by the Sichuan Agricultural University Institutional Animal Care and Use Committee.
Preparation of Tol-HP-β-CD
Solution-stirring method was employed to prepare the Tol-HP-β-CD. In brief, Tol and HP-β-CD (mole ratio of 1:3) were weighed and added to 20% (ω, v/v) aqueous acetonitrile. The pH of previous solution was adjusted to 9 using 2 M NaOH solution, and the mixture was stirred for 3 h at 50°C. The solution was adjusted to 7 by 2 M H3PO4 solution and then filtered through 0.45 μm membrane filters. The filtrate was concentrated using a rotary evaporator (Shanghai Yarong Biochemistry Instrument Factory, Shanghai, China) and further vacuum-dried at 50°C. According to our previous research,24 the average yield is 86.45% and the prepared Tol-HP-β-CD was observed to have a highly enhanced solubility of 9.90 mg/mL compared with Tol.
Characterization
Thin-layer chromatography
Specific amounts of Tol, HP-β-CD, their physical mixture, and inclusion complex were weighed and dispersed in chloroform to prepare the solutions. The same amounts of the physical mixture and inclusion complex were weighed and dissolved in water to prepare an aqueous solution. Appropriate aliquots were spotted in a 1 mm2 spot band located at 2 cm from the bottom of the TLC plate. Development was performed in a chamber tank and saturated with an appropriate eluent for a sufficient period at 25°C. A silica gel plate was used as a stationary phase and ethyl acetate-diethyl ether-petroleum ether (1:3:1, v/v) as a mobile phase. After drying, the TLC plate was observed under a UV lamp at 254 nm.
FTIR spectrophotometry
The Tol-HP-β-CD was characterized by FTIR spectroscopy using Shimadzu FTIR-8400S (Shimadzu Corporation, Kyoto, Japan) for transmittance measurements of KBr pellets in the range of 4,000 cm−1–400 cm−1, with 4 cm−1 resolution and 64 scans.
NMR spectroscopy
1H NMR analysis was performed on a UNITY INOVA400 NMR spectrometer (400 MHz, Varian, USA). Tol was dissolved in CDCl3 (Sigma-Aldrich Inc., St Louis, MO, USA), whereas HP-β-CD and the Tol-HP-β-CD were, respectively, dissolved in D2O (Sigma-Aldrich). They were scanned from 1 to 10 ppm using the following measurement conditions: 9.4 Tesla superconducting magnet and Probe-BBO 400 MHz with Z-gradient and 2H lock.
Pharmacokinetic experiments
Experimental design and sample collection
Twelve healthy rabbits (70 days of age, half males and half females) were allowed to acclimate to the facility for 1 week. The rabbits were then weighed and randomly divided into two groups. One group received 10 mg/kg body weight (bw) Tol, whereas the other group received 10 mg/kg bw Tol-HP-β-CD (containing the equivalent doses of Tol). The preparation of Tol/Tol-HP-β-CD is described as follows: the weighted Tol was added to suitable amount of distilled water, and the solution was shaked strongly to form mixed suspension. The preparation of Tol-HP-β-CD solution was similar to that of Tol mixed suspension. Both groups were orally administered a single dose. Blood sample (1 mL) was collected from the ear vein in heparinized vacutainer tubes prior to treatment immediately before drug administration (baseline) and at 0.25, 0.5, 1, 2, 3, 4, 8, 12, 24, 36, 48, 72, 120, 168, and 240 h after drug administration. Blood samples were centrifuged at 1,788.8× g for 10 min within 1 h after sampling, and plasma was collected and stored at −70°C until analysis.
Sample preparation
Tol was extracted from rabbit plasma using extraction with acetonitrile following Kim et al4 with slight modifications. In brief, the samples were prepared by adding 200 μL of plasma to 300 μL of acetonitrile after unfreezing. The mixture was vortexed for 2 min and centrifuged at 12,000 rpm for 10 min. The supernatant was filtered through a 0.22 μm polytetrafluoroethylene syringe filter. A 20 μL aliquot was used in a high-performance liquid chromatography (HPLC) system for analysis.
Chromatographic conditions
All samples were analyzed on a Shimadzu LC-2010CHT system (Shimadzu Corporation) with a UV detector. HPLC analysis was performed using a 5 μm C18-silica-based stainless-steel Kromasil column (4.6×250 mm; Akzo Nobel, Bohus, Sweden) as a stationary phase. The mobile phase consisted of 0.1% acetic acid aqueous solution and acetonitrile (45:55, v/v), which was ultrasonicated for 10 min and degassed for another 5 min. The flow rate was 1 mL/min at 25°C with the detection wavelength of 240 nm.
Validation
A series of stock solutions of Tol standard were prepared, and each standard (20 μL) was mixed with drug-free plasma (180 μL) to prepare calibration curves ranging from 0.2 to 25.6 μg/mL. The slope of the lines between peak areas and drug concentration was determined using least-squares linear regression. Linearity was established to determine the relationship between Tol concentration and detector response. The detection limit of Tol was established with HPLC analysis of blank plasma fortified with the standard and eventually measured to be 0.05 μg/mL. The regression lines between peak areas and drug concentrations show that the correlation coefficient was 0.9996. The mean extraction recovery from plasma was 90.23%±0.73%. The average relative SDs of intraday and interday precision were 1.51% and 2.12%, respectively.
Data analysis
DAS2.0 (Mathematical Pharmacology Professional Committee of China, Shanghai, China) was used to fit the plasma concentration–time curves obtained after single oral administration in each rabbit, and the pharmacokinetic parameters were calculated using the double-compartment method. The peak concentrations (Cmax) and time to peak occurrence (Tmax) were determined from the plotted concentration–time curve in each rabbit. The area under the concentration–time curve (AUC0-∞) was calculated using the trapezoidal rule.25 Arithmetic mean values and SDs were calculated using Microsoft® Excel 2007. All statistical evaluations were performed with SPSS for Windows version 19.0 (IBM Corporation, Armonk, NY, USA). Some pharmacokinetic parameters were log-transformed before statistical analysis. Statistical significance was set at p<0.05. Pharmacokinetic variables are reported as mean ± SD.
Results and discussion
Characterization of Tol-HP-β-CD
TLC analysis
TLC is a simple and effective method used for the characterization of inclusion complex. Figure 2 shows that the lipophilic analytes of Tol were well separated from the hydrophilic compounds of HP-β-CD. Tol of the chloroform solution (Figure 2A), Tol-HP-β-CD of the aqueous solution (Figure 2B), and physical mixture of the chloroform solution (Figure 2E) showed a spot at the TLC plate with a similar Rf value (Rf =0.5), which were the characteristics of the TLC point of Tol. However, the thin-layer chromatogram of the Tol-HP-β-CD chloroform solution (Figure 2C), HP-β-CD chloroform solution (Figure 2D), and HP-β-CD aqueous solution (Figure 2F) did not show the characteristics of the TLC point of Tol at the same place. This result was attributed to the fact that Tol can be dissolved in chloroform, Tol-HP-β-CD can be dissolved in water, but HP-β-CD cannot be dissolved in chloroform. Similarly, Tol embedded in the HP-β-CD cavity to form the Tol-HP-β-CD could not be dissolved in chloroform. Thus, Figure 2 displays the characteristics of the TLC point of Tol. Simultaneously, HP-β-CD did not interfere with the characteristics of the TLC point of Tol, which proved the formation of the Tol-HP-β-CD.
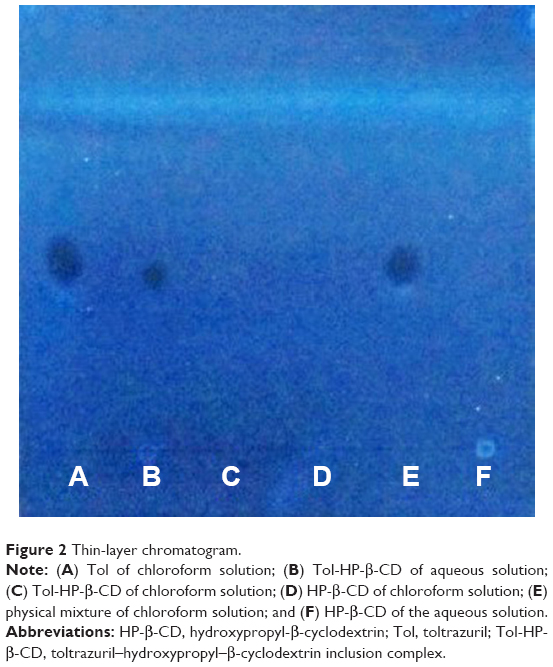 | Figure 2 Thin-layer chromatogram. |
FTIR analysis
The FTIR spectra of Tol, HP-β-CD, the physical mixture, and Tol-HP-β-CD are shown in Figure 3. Tol crystals (Figure 3A) showed a sharp N−H stretch band at 3,340 cm−1, C−F band at 1,395 cm−1, and C=O stretch bands from 1,670 to 1,790 cm−1. The three characteristic peaks of Tol were found in the FTIR spectra of the physical mixture (Figure 3C). The FTIR spectrum of Tol-HP-β-CD (Figure 3D) was almost identical to that of HP-β-CD (Figure 3B) alone. In particular, the C−F (1,395 cm−1) stretch band of the drug completely disappeared in Tol-HP-β-CD spectrum, while it was slightly visible in the physical mixture spectrum. It was predicted that the disappearance of the bands in the Tol-HP-β-CD was dependent on the drug embedded into the HP-β-CD cavity.
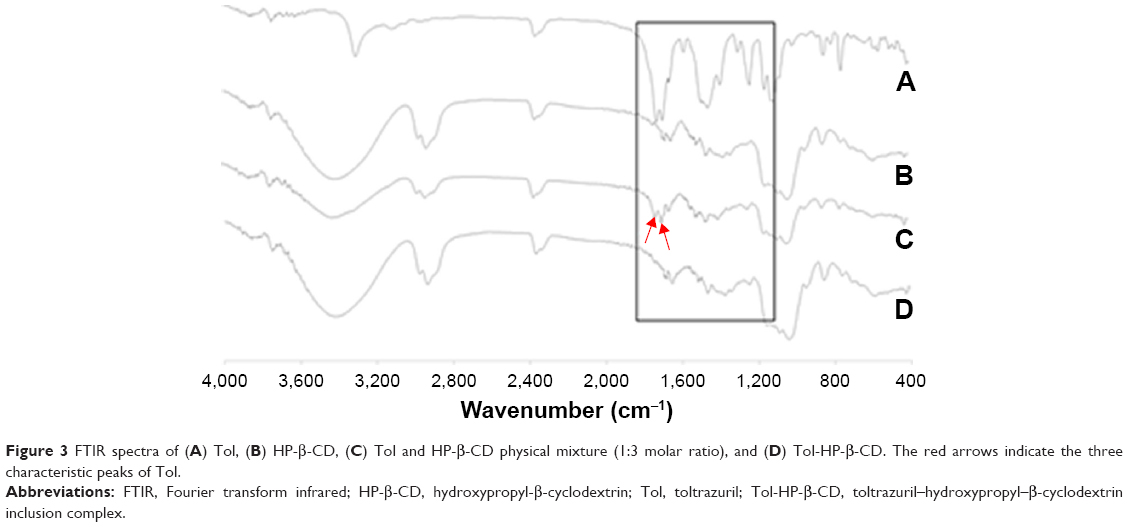 | Figure 3 FTIR spectra of (A) Tol, (B) HP-β-CD, (C) Tol and HP-β-CD physical mixture (1:3 molar ratio), and (D) Tol-HP-β-CD. The red arrows indicate the three characteristic peaks of Tol. |
1H NMR analysis
NMR spectroscopy is a powerful tool to study the formation of inclusion complex between cyclodextrin and various guest molecules because the chemical and electronic environments of protons are affected during complexation and reflected through changes in the δ values.26 In this study, we performed docking studies of Tol in the HP-β-CD cavity (Figure 4). H-3 and H-5 protons of HP-β-CD were positioned inside the cavity, where H-3 was located closer to the wider rim, and H-5 was on the opposite side. After complex formation, δ of H-3 protons shifted to −0.132 ppm, which may be attributed to the formation of some weak bonds between Tol and HP-β-CD molecules. The upfield shifts occurred because of the anisotropic magnetic effect induced by the presence of the aromatic group of the guest molecule.27 The benzene ring in Tol structure was suggested to be included in the HP-β-CD cavity. H-3 protons shifted upfield most significantly, indicating that Tol was inserted into the HP-β-CD cavity from the wider rim. The NMR spectra after inclusion complexation with Tol showed δ values of 7.842 (d, 2H, Ar–H), 7.397 (s, H, Ar–H), 7.289 (m, 3H, Ar–H), 7.187 (d, H, Ar–H), 3.348 (s, 3H, =N−CH3), and 2.315 ppm (s, 3H, Ar–CH3) for Tol. These results indicated that Tol should be included in the HP-β-CD cavity without any chemical modification.28 Particularly, H protons in group =N−CH3 shifted mildly while some of the H protons in the benzene ring attached to the sulfur atom shifted to 0.288 ppm significantly. Based on the aforementioned analysis, inclusion complex geometry was proposed for Tol-HP-β-CD (Figure 5).
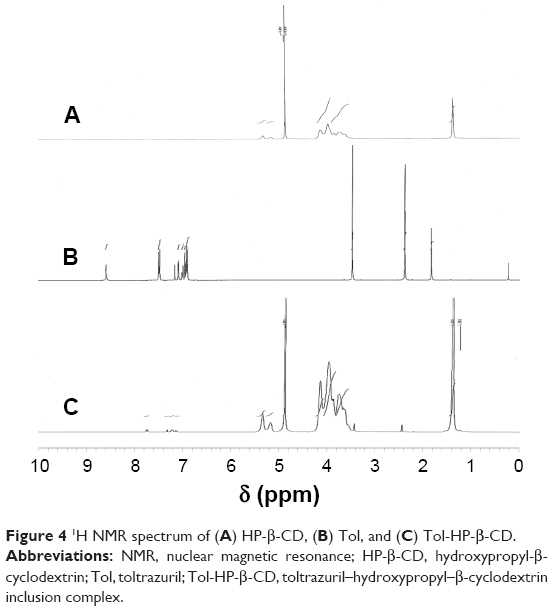 | Figure 4 1H NMR spectrum of (A) HP-β-CD, (B) Tol, and (C) Tol-HP-β-CD. |
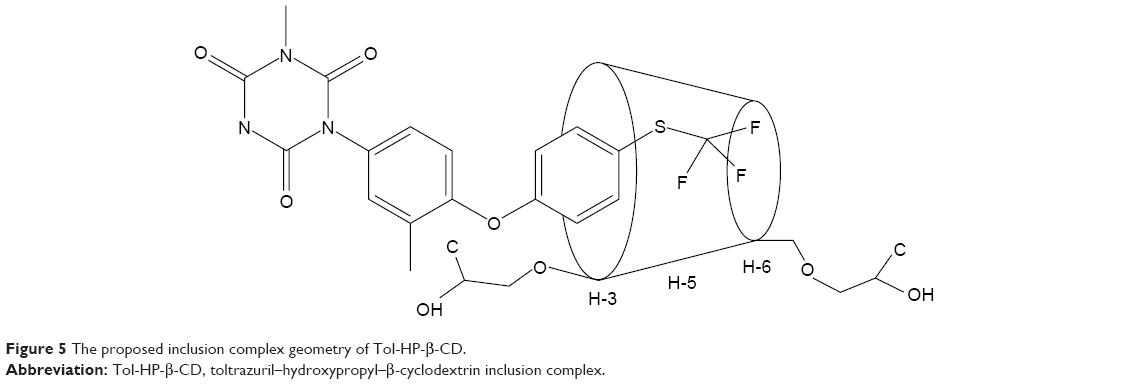 | Figure 5 The proposed inclusion complex geometry of Tol-HP-β-CD. |
Pharmacokinetic applications
The pharmacokinetic parameters of Tol were assessed by fitting a double-compartment model to the individual concentration–time data for plasma. The semilogarithmic plots of plasma concentration–time profiles obtained in rabbits following single oral administration of Tol and Tol-HP-β-CD at a dose of 10 mg/kg bw over a 10-day period are shown in Figure 6. The pharmacokinetic parameters (mean ± SD) are presented in Table 1. After oral administration, Cmax of Tol and Tol-HP-β-CD in plasma of rabbits calculated in this study were 8.02 (at 24.0 h) and 19.92 μg/mL (at 12.0 h), respectively. After reaching the peak values, a more rapid decline in the concentration–time curve in the Tol-HP-β-CD group was observed than that in the Tol group. The AUC0-∞ of Tol-HP-β-CD (1,181.4 mg/L h) was 2.48-fold higher (p<0.01) than that of Tol (474.94 mg/L h). In the Tol-HP-β-CD group, Cmax (19.92±1.02 μg/mL) was extremely higher (p<0.01) and the absorption half-life (t1/2ab; 20.82±2.62 h) was shorter (p<0.01) than those in the Tol group (Cmax =8.02±1.04 μg/mL, t1/2ab =33.30±4.26 h). However, the mean residence time (MRT) was similar in both groups (51.26±4.83 h vs 52.18±7.42 h; p>0.05). The differences in Cmax, Tmax, AUC0-∞, and t1/2ab were generally significant, and no statistical difference was observed for MRT. Generally, inclusion complex has a sustained release effect on the drug release. Especially in the later stages, drug release is slower than it was initially as the amount of drug is reduced and HP-β-CD molecules are relatively abundant. Therefore, after Tol-HP-β-CD was orally administered, the pharmacokinetic parameters such as Cmax, Tmax, and AUC0-∞ were all significantly different from those of Tol. However, MRT of Tol-HP-β-CD did not show a significant decrease relative to Tol. The pharmacokinetics of Tol alone has been previously studied in most animal species29–33 including rabbits. In this study, the pharmacokinetics of Tol after oral administration of Tol or Tol-HP-β-CD in both groups was best described by a double-compartment model, which was similar to previous findings.4,34 The absorption of chemicals from the GI tract depends on the physiochemical properties of compounds, such as lipid solubility and dissociation rate. Although an increase in lipid solubility generally increases the absorption of chemicals, extremely lipid-soluble chemicals may have poor oral bioavailability because highly lipophilic molecules can become stuck in the lipid portion of the plasma membrane,35 whereas highly lipophilic compounds are more difficult to dissolve in GI fluids.36 Different pharmacokinetic values between Tol-free drug and Tol-HP-β-CD might be explained by prior processes before absorption, mainly, binding to plasma membrane and poor solubility in GI fluids. Therefore, solubility was the key factor affecting Tol absorption and bioavailability. After the inclusion of Tol into HP-β-CD, the solubility of Tol was significantly increased by its improved hydrophilicity. In our previous research, solubility of Tol was enhanced by more than 24,000 times, from 4.12×10−4 to 9.90 mg/mL, after forming Tol-HP-β-CD, significantly higher than Tol solid dispersions of 2,000 times.11 In addition, HP-β-CD can also reduce the viscosity of intestinal mucus layer and include mucosal tissue components such as cholesterol and phospholipids, thereby changing the lipid barrier of the absorption site to promote drug absorption.37 So, once the Tol-HP-β-CD passed by the GI tract, high Tol concentration might contribute to faster absorption of Tol. Significant solubility enhancement might be responsible for the high Cmax and AUC0-∞ value of Tol-HP-β-CD than those of the free Tol, indicating a better absorption of Tol entrapped in HP-β-CD in the GI tract. Thus, the pharmaceutical properties of absorption and bioavailability of Tol could be partially improved by complexation with HP-β-CD.
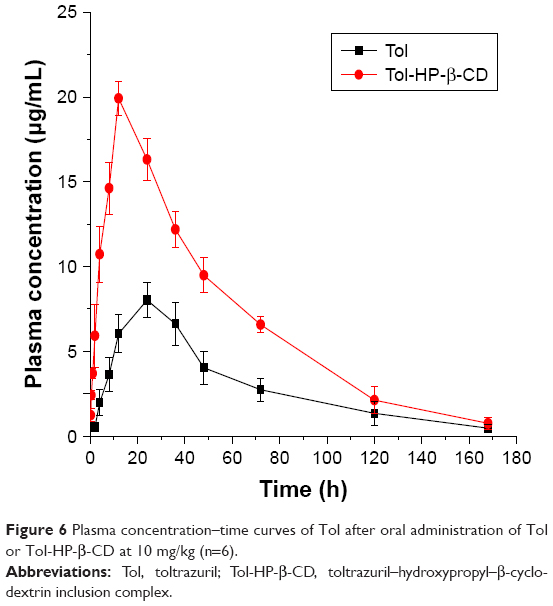 | Figure 6 Plasma concentration–time curves of Tol after oral administration of Tol or Tol-HP-β-CD at 10 mg/kg (n=6). |
 | Table 1 Pharmacokinetic parameters (mean ± SD) of Tol after a single oral administration of Tol or Tol-HP-β-CD inclusion complex at a dose of 10 mg/kg bw in rabbits (n=6) |
Conclusion
In this study, the Tol-HP-β-CD was successfully prepared for the enhancement of aqueous solubility of Tol. TLC, FTIR spectra, and 1H NMR spectroscopy were applied to characterize the formation of Tol-HP-β-CD. After single oral administration of Tol and Tol-HP-β-CD in rabbits, the pharmacokinetic profile illustrated that Tol-HP-β-CD exhibited a better oral bioavailability than free Tol, indicating that HP-β-CD could enhance the solubility, absorption, and bioavailability of Tol. Taken the shortage of Tol for clinical application, as well as the easy and environment-friendly preparation of Tol-HP-β-CD into consideration, the prepared inclusion complex should be a promising and available formulation design for Tol.
Disclosure
The authors report no conflicts of interest in this work.
References
Pakandl M. Coccidia of rabbit: a review. Folia Parasitol (Praha). 2009;56(3):153–166. | ||
Polozowski A. [Coccidiosis of rabbits and its control]. Wiad Parazytol. 1993;39(1):13–28. Polish. | ||
Cam Y, Atasever A, Eraslan G, et al. Eimeria stiedae: experimental infection in rabbits and the effect of treatment with toltrazuril and ivermectin. Exp Parasitol. 2008;119(1):164–172. | ||
Kim MS, Lim JH, Hwang YH, Park BK, Song IB, Yun HI. Plasma disposition of toltrazuril and its metabolites, toltrazuril sulfoxide and toltrazuril sulfone, in rabbits after oral administration. Vet Parasitol. 2010;169(1–2):51–56. | ||
Pan BL, Zhang YF, Suo X, Xue Y. Effect of subcutaneously administered diclazuril on the output of Eimeria species oocysts by experimentally infected rabbits. Vet Rec. 2008;162(5):153–155. | ||
Fitzgerald PR. Efficacy of monensin or amprolium in the prevention of hepatic coccidiosis in rabbits. J Protozool. 1972;19(2):332–334. | ||
Le Sueur C, Mage C, Mundt HC. Efficacy of toltrazuril (Baycox 5% suspension) in natural infections with pathogenic Eimeria spp. in housed lambs. Parasitol Res. 2009;104(5):1157–1162. | ||
Gottstein B, Eperon S, Dai WJ, Cannas A, Hemphill A, Greif G. Efficacy of toltrazuril and ponazuril against experimental Neospora caninum infection in mice. Parasitol Res. 2001;87(1):43–48. | ||
Harder A, Haberkorn A. Possible mode of action of toltrazuril: studies on two Eimeria species and mammalian and Ascaris suum enzymes. Parasitol Res. 1989;76(1):8–12. | ||
Mehlhorn H, Schmahl G, Haberkorn A. Toltrazuril effective against a broad spectrum of protozoan parasites. Parasitol Res. 1988;75(1):64–66. | ||
Qi WW, Li JJ, Liu YQ, et al. Preparation of toltrazuril solid dispersion. Chin J Vet Drug. 2009;43(4):34–37. | ||
Li B. Preparation and Research on the Anticoccidial Effect of Toltrazuril Ultrafine Powder. Nanjing: Nanjing Agricultural University; 2010. | ||
Zhang ZM, Deng XM, Shen ZQ, et al. Preparation of toltrazuril nanoemulsion and quality evaluation. J Domestic Animal Ecol. 2013;34(10):55–57. | ||
Challa R, Ahuja A, Ali J, Khar RK. Cyclodextrins in drug delivery: an updated review. AAPS Pharm Sci Tech. 2005;6(2):E329–E357. | ||
Wempe MF, Wacher VJ, Ruble KM, et al. Pharmacokinetics of raloxifene in male Wistar-Hannover rats: influence of complexation with hydroxybutenyl-beta-cyclodextrin. Int J Pharm. 2008;346(1–2):25–37. | ||
Hwang YY, Shin DC, Nam YS, et al. Characterization, stability, and pharmacokinetics of sibutramine/β-cyclodextrin inclusion complex. J Ind Eng Chem. 2012;18(1):1412–1417. | ||
Brewster ME, Loftsson T. Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev. 2007;59(7):645–666. | ||
Srivalli KM, Mishra B. Improved aqueous solubility and antihypercholesterolemic activity of ezetimibe on formulating with hydroxypropyl-β-cyclodextrin and hydrophilic auxiliary substances. AAPS Pharm Sci Tech. 2016;17(2):272–283. | ||
Gould S, Scott RC. 2-Hydroxypropyl-beta-cyclodextrin (HP-beta-CD): a toxicology review. Food Chem Toxicol. 2005;43(10):1451–1459. | ||
Misiuk W, Zalewska M. Investigation of inclusion complex of trazodone hydrochloride with hydroxypropyl-β-cyclodextrin. Carbohyd Polym. 2009;77(3):482–488. | ||
Folch Cano C, Yazdani-Pedram M, Olea-Azar C. Inclusion and functionalization of polymers with cyclodextrins: current applications and future prospects. Molecules. 2014;19(9):14066–14079. | ||
Frömming K, Szejtli J. Cyclodextrins in Pharmacy. Dordrecht: Springer; 1994. | ||
Barone JA, Moskovitz BL, Guarnieri J, et al. Enhanced bioavailability of itraconazole in hydroxypropyl-beta-cyclodextrin solution versus capsules in healthy volunteers. Antimicrob Agents Chemother. 1998;42(7):1862–1865. | ||
Lu CC, Yang J, Zhang W, et al. Preparation and characterization of toltrazuril–hydroxypropyl-β-cyclodextrin inclusion complex. J Process Eng. 2012;12(6):982–988. | ||
Perrier D, Gibaldi M. General derivation of the equation for time to reach a certain fraction of steady state. J Pharm Sci. 1982;71(4):474–475. | ||
Garnero C, Zoppi A, Genovese D, Longhi M. Studies on trimethoprim:hydroxypropyl-beta-cyclodextrin: aggregate and complex formation. Carbohydr Res. 2010;345(17):2550–2556. | ||
Veiga FJ, Fernandes CM, Carvalho RA, Geraldes CF. Molecular modelling and 1H-NMR: ultimate tools for the investigation of tolbutamide: beta-cyclodextrin and tolbutamide: hydroxypropyl-beta-cyclodextrin complexes. Chem Pharm Bull (Tokyo). 2001;49(10):1251–1256. | ||
Mahajan HS, Pingale MH, Agrawal KM. Solubility and dissolution enhancement of saquinavir mesylate by inclusion complexation technique. J Incl Phenom Macro. 2013;76(2):1–6. | ||
Prado ME, Ryman JT, Boileau MJ, Martin-Jimenez T, Meibohm B. Pharmacokinetics of ponazuril after oral administration to healthy llamas (Lama glama). Am J Vet Res. 2011;72(10):1386–1389. | ||
Lim J, Park B, Kim M, et al. Pharmacokinetics of toltrazuril after oral administrations in broilers. J Vet Clin. 2001;24(2):308–311. | ||
Lim JH, Kim MS, Hwang YH, Song IB, Park BK, Yun HI. Pharmacokinetics of toltrazuril and its metabolites, toltrazuril sulfoxide and toltrazuril sulfone, after a single oral administration to pigs. J Vet Med Sci. 2010;72(8):1085–1087. | ||
Tobin T, Dirikolu L, Harkins JD, et al. Preliminary pharmacokinetics of diclazuril and toltrazuril in the horse. In: Norwood G, editor. Proceedings of the 43rd Annual Convention of the American Association of Equine Practitioners: Phoenix, Arizona, December 7–10, 1997. Lexington: American Association of Equine Practitioners; 1997:15–16. | ||
Dirikolu L, Karpiesiuk W, Lehner AF, Hughes C, Granstrom DE, Tobin T. Synthesis and detection of toltrazuril sulfone and its pharmacokinetics in horses following administration in dimethylsulfoxide. J Vet Pharmacol Ther. 2009;32(4):368–378. | ||
Hu L, Liu C, Shang C, Yang X, Yang J. Pharmacokinetics and improved bioavailability of toltrazuril after oral administration to rabbits. J Vet Pharmacol Ther. 2010;33(5):503–506. | ||
Martinez MN, Amidon GL. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J Clin Pharmacol. 2002;42(6):620–643. | ||
Houston JB, Upshall DG, Bridges JW. A re-evaluation of the importance of partition coefficients in the gastrointestinal absorption of anutrients. J Pharmacol Exp Ther. 1974;189(1):244–254. | ||
Yi J, Wu QP, Zhang FL, et al. Promotion of enhancers on duodenal absorption of paeoniflorin in rats. Drug Eval Res. 2014;37(2):141–144. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.