")
Back to Journals » Clinical Interventions in Aging » Volume 14
The cGAS/STING pathway: a sensor of senescence-associated DNA damage and trigger of inflammation in early age-related macular degeneration
Received 7 January 2019
Accepted for publication 10 June 2019
Published 11 July 2019 Volume 2019:14 Pages 1277—1283
DOI https://doi.org/10.2147/CIA.S200637
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Zhi-Ying Wu
Yan Wu,1,2,* Qingquan Wei,1,* Jing Yu1,3
1Department of Ophthalmology, Shanghai Tenth People’s Hospital Affiliated with Tongji University, Shanghai, People’s Republic of China; 2Department of Ophthalmology, The First Affiliated Hospital of Soochow University, Suzhou, Jiangsu Province, People’s Republic of China; 3Department of Ophthalmology, Ninghai First Hospital, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Abstract: Age-related macular degeneration (AMD) is the leading cause of irreversible blindness among the elderly. Considering the relatively limited effect of therapy on early AMD, it is important to focus on the pathogenesis of AMD, especially early AMD. Ageing is one of the strongest risk factors for AMD, and analysis of the impact of ageing on AMD development is valuable. Among all the ageing hallmarks, increased DNA damage accumulation is regarded as the beginning of cellular senescence and is related to abnormal expression of inflammatory cytokines, which is called the senescence-associated secretory phenotype (SASP). The exact pathway for DNA damage that triggers senescence-associated hallmarks is poorly understood. Recently, mounting evidence has shown that the cGAS/STING pathway is an important DNA sensor related to proinflammatory factor secretion and is associated with another hallmark of ageing, SASP. Thus, we hypothesized that the cGAS/STING pathway is a vital signalling pathway for early AMD development and that inhibition of STING might be a potential therapeutic strategy for AMD cases.
Keywords: age-related macular degeneration, cGAS/STING pathway, DNA damage, inflammation
Background
Age-related macular degeneration (AMD) is the leading cause of irreversible blindness among the elderly in developed countries.1 As a chronic, degenerative disorder in the macular region of the retina, AMD leads to progressive central vision loss from the early stage (medium-sized drusen and retinal pigment epithelium abnormality) to the late stage (neovascular AMD and geographic atrophy).2 The number of patients with AMD is expected to be approximately 200 million globally by 2020. There are a large number of elderly patients with visual impairment caused by AMD, which is likely to increase with time. The levels of vision described amount to considerable visual compromise and constitute a major public health burden, resulting in increased social isolation, depression, restriction of daily activities, risk of falling and hip fracture.3 And this condition will become a major public health issue with substantial socio-economic losses.
Anti-vascular endothelial growth factor (VEGF) agents (such as ranibizumab, aflibercept, or bevacizumab) are the main treatments for neovascular AMD and provide significant therapeutic effects.4 Ranibizumab 0.5 mg (Lucentis®; Novartis Pharma AG, Basel, Switzerland, and Genentech, South San Francisco, California, USA) was the first anti-VEGF agent to be approved for this indication.5 Aflibercept is a fusion protein containing both VEGFR-1 and VEGFR-2, which binds to VEGF-A thus impeding its activity.6 Bevacizumab is a full-length monoclonal antibody (149 kDa) that was developed for cancer therapy.7 Repetitive intravitreal injections can lead to a severe economic burden and an increased risk of infection, and thus, many clinical trials have been conducted to identify an optimal therapeutic strategy.8,9 There are significant inter-individual differences in response to an anti-VEGF agent. The response to anti-VEGF therapy have been found to be dependent on a variety of factors including patient’s age, lesion characteristics, lesion duration, baseline visual acuity (VA) and the presence of particular genotype risk alleles.10 Some patients undergoing anti-VEGF treatment demonstrated poor or even no response to the drugs, so updated intervention drugs and strategies are still required.11 Geographic atrophy, which is regarded as the end stage of AMD, is estimated to account for one-fifth of cases of legal blindness.
Over the past decade, there has been growing evidence implicating a role for the complement system in AMD.12 Histopathologic studies have identified various complement components in drusen, in Bruch’s membrane, and in the inner choroid.13 Several treatment strategies that modulate the complement system in AMD are being investigated. These treatments inhibit complement activation by targeting various effectors molecules, such as C3, C5, and factor D.14 Complement inhibition has been reported as a potential therapeutic intervention for geographic atrophy.15 Eculizumab and lampalizumab are inhibitors of the complement cascade, but updated studies and analyses are still required to determine the effect of these drugs. Eculizumab is an inhibitor of terminal complement activation approved by the US Food and Drug Administration. It is approved for the systemic treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Eculizumab successfully controls these diseases by inhibiting C5 an preventing terminal complement activation and formation of membrane attack complex (C5b-9).16 Lampalizumab is an intravitreally administered inhibitor of complement factor D, an important component of the alternative complement pathway.17 Additionally, a new anti-complement therapy has been developed and appears to be the most promising among all the molecules for the treatment of atrophic AMD.18 These results are promising for the detection of a potential method for the prevention and delay of AMD progression. If we can prevent the development of AMD from the early stage to the late stage, the patient would be in the relatively asymptomatic early stage and would show an improved visual outcome and quality of life.
Several clinical trials have focused on the prevention of AMD progression, and the data from Age-Related Eye Disease Study 2 (AREDS2) showed that AREDS formulation in primary analyses did not further reduce the risk of progression to advanced AMD.19 Additionally, several epidemiological studies and meta-analyses demonstrated that omega-3 fatty intake was associated with a lower risk of AMD; however, the conclusion should be validated by more well-designed clinical studies.20,21 Clinical application of nutrition supplementation for the treatment of early AMD is still far from being a reality.
Considering the relatively limited therapeutic effect in early AMD, it is important to focus on the pathogenesis of AMD, especially early AMD. AMD is caused by multiple biological processes, such as inflammation and oxidative stress.22 As a multifactorial chronic disease, AMD incidence is associated with both genetic and environmental factors (genes, ageing, smoking, family history, dietary habits, oxidative stress, and hypertension).23,24 What’s more, several genetic and molecular studies have showed the participation of inflammatory molecules, immune cells, and complement proteins in the development and progression of the disease.25 In addition, different genes (IL-6, IL-8, CFH, CFI, C2, C3, and ARMS2) that play an important role in the inflammatory pathway have been related with AMD risk. Also, the sample cohort has been subjected to a large genotyping analysis of 20 genetic variants which are known to be associated with AMD among European and Asiatic populations. This study revealed that 8 genetic variants (IL-8, CFH, TIMP3, SLC16A8, RAD51B, ARMS2, VEGFA and COL8A1) were significantly related with AMD susceptibility.26–28 Among all the associated risk factors, ageing and smoking are by far the strongest risk factors.29 The results from different observations showed that smoking is significantly associated with the incidence of AMD.6 And smoking cessation is unequivocally cost-effective in terms of its impact on AMD development and progression.30 Both in vivo and in vitro experimental models based on smoke exposure and tobacco extract are available.31–33 Thus, focusing on the impact of ageing on the development of AMD is valuable. More information about the ageing-associated or driven molecular progress of AMD would contribute to the successful prevention or treatment of AMD, and this knowledge might help in other human ageing diseases.
Ageing, which is a time-related, degenerative process beginning in adulthood, occurs in most organisms and eventually ends life.34 Ageing is associated with a series of diseases, including cardiovascular disease, neurodegeneration, carcinoma, and osteoarthritis.35–38 In a previous review, the hallmarks of ageing included genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication.39 Among all the ageing hallmarks, increased DNA damage accumulation was regarded as a beginning of cellular senescence and was related to abnormal expression of inflammatory cytokines, which is called the senescence-associated secretory phenotype (SASP).40 DNA sensors can sense senescence-associated DNA damage and trigger inflammation. cGMP-AMP (cGAMP) synthase (cGAS) and the adaptor stimulator of interferon genes (STING) were shown to be involved in the regulation of senescence.41 In addition, a recent study by Nagaraj Kerur et al indicated that cGAS responded to mitochondrial damage-induced inflammasome activation and thus played an important role in the regulation of geographic atrophy.42 However, the exact role of the cGAS/STING pathway in the development of early AMD as a senescence-associated DNA damage sensor remains unclear.
Presentation of the hypothesis
Ageing, which is one of the strongest risk factors of AMD, influences both the anatomy and function of the retina.43,44 Retinal pigmented epithelium (RPE) cells, which are a monolayer of cells that provide trophic support to photoreceptors, are regarded as one of the earliest influenced cells in the retina.45 RPE dysregulation is associated with various kinds of retinal diseases, including AMD, diabetic retinopathy and proliferative vitreoretinopathy.46–48 The abnormal expression of growth factors and cytokines secreted by RPE cells is involved in the dysfunction of RPE cells. As reported in previous studies, these growth factors include vascular endothelial cell growth factor (VEGF), pigment epithelium-derived factor (PEDF), and transforming growth factor beta 2 (TGF-β2).49,50 The cytokines include interleukin-6 (IL-6), interleukin-8 (IL-8), interleukin-17A (IL-17A) and interleukin-1β (IL-1β).51–53 SASP is regarded as a key hallmark of ageing, and abnormal expression of various factors, such as IL-1α, IL-1β, IL-6, IL-8, and matrix metalloproteinases (MMP1 and MMP3), is involved.54 There was a wide-ranging overlap between RPE dysfunction-associated factors and SASP-associated factors. Thus, the dysfunction of RPE was, at least partly, caused by cell senescence, and the abnormal expression of growth factors and cytokines were signs of SASP.
Throughout an organism’s lifespan, DNA is exposed to both exogenous and endogenous harmful factors, such as chemicals, radiation and different kinds of metabolic products, leading to different types of DNA damage and genomic instability.55 Many different factors, such as tobacco smoking, which is a strong environmental risk factor, has been reported to be associated to increased DNA damage in retina.56 DNA damage is regarded as both an important hallmark and a key trigger of senescence. A persistent DNA damage response (DDR) in ageing cells leads to senescent DNA damage foci (SDF) and telomere dysfunction-induced foci (TIF).57 DNA damage includes nuclear DNA and mitochondrial DNA (mtDNA) damage. There is a modulated balance between DNA damage and DNA repair in normal cells, and the absence of this balance leads to the accumulation of damaged DNA.58 mtDNA is a closed-loop DNA molecule independent of nuclear DNA in cells. Mitochondrial DNA, which exists without the protection of histone and DNA binding proteins, is vulnerable to oxygen free radical damage. In addition, mtDNA is not easily repaired due to the lack of a repair system. Therefore, mtDNA is more easily affected by influencing factors and accumulates more harmful mutations compared with nuclear DNA.59 A published review has focused on the role of DNA damage in the effect of cellular senescence in AMD. It concluded that oxidative stress can induce DDR and cell senescence, promoting AMD incidence.60 Thus, how the DNA damage signal is detected by the cell could answer questions about the progression of ageing or senescence-associated signs.
Increasing evidence has demonstrated the important role of the cGAS/STING pathway as a cytoplasmic DNA sensor, and its classical function is to promote the production of type I interferons (IFNs) and immune factors, which is important in antiviral and antineoplastic processes.61 Advanced studies have also shown that this pathway is involved in the incidence and progression of autoimmune diseases, carcinoma and ageing.54,61–63 When cellular senescence was considered, the regulatory effect of the cGAS/STING pathway also demonstrated a significant effect. Activation of the cGAS/STING pathway leads to two independent downstream pathways: type I IFNs through interferon regulatory factor 3 (IRF3) and proinflammatory responses through NFκB16.64 Both the IRF3- and NF-κB-dependent pathways lead to the production of inflammatory growth factors and cytokines, leading to SASP.
Based on the above absorbing viewpoints and studies, we hypothesized that the cGAS/STING pathway is the sensor of senescence-associated DNA damage and trigger of inflammation in early AMD (Figure 1). cGAS/STING functioned as an important bridge to connect ageing-related DNA damage and AMD incidence, and thus, the inhibition of this pathway would help in drug development in the future.
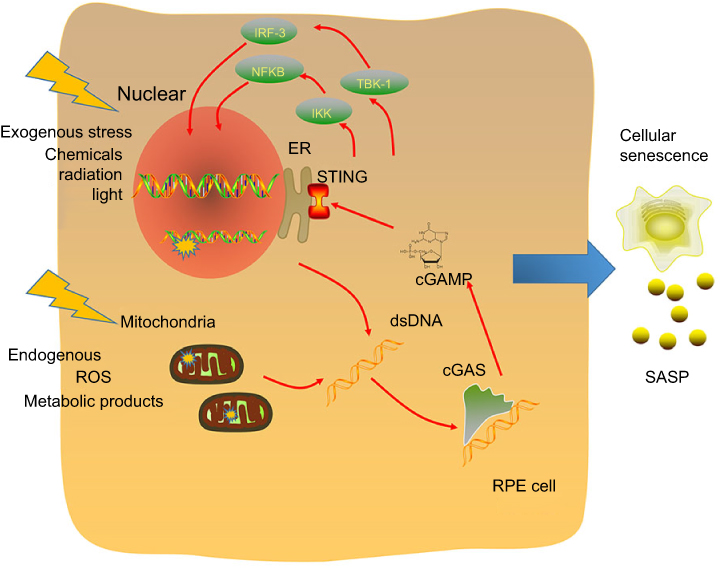 |
Figure 1 Molecular mechanisms of the cGAS/STING pathway as a senescence-associated DNA damage sensor and inflammation trigger. DNA is exposed to both exogenous and endogenous harmful factors, such as chemicals, radiation and different kinds of metabolic products. DNA damage includes nuclear DNA and mitochondrial DNA (mtDNA) damage. The cGAS/STING pathway is a cytoplasmic DNA sensor, and activation of the cGAS/STING pathway leads to two independent downstream pathways: type I IFNs through IRF3 and proinflammatory responses through NFκB16. Both the IRF3- and NF-κB-dependent pathways lead to the production of inflammatory growth factors and cytokines, leading to SASP.Abbreviations: SASP, senescence-associated secretory phenotype; RPE, retinal pigmented epithelium; ER, endoplasmic reticulum; ROS, reactive oxygen species. |
Testing the hypothesis
- To demonstrate the feasibility and stability of this hypothesis, we will first detect the association between RPE cellular senescence and the incidence of ageing and early AMD. Young rats (2 months), ageing rats (24 months) and early AMD rat models (24 months + smoke exposure) will be obtained. Smoke expose will be conducted using a smoking machine for 2 months and following experiments will conducted as a smoke expose group. The average concentration of total suspended particulates was 130 mg/m3 and was monitored twice daily. The retinal structure will be conducted using retina sections and retinal function would be detected with multifocal visual electrophysiology examining system. Next generation sequence will be conducted to detect the general RNA expression in the three groups. The expression of the cell senescence biomarkers SA-β-Gal activity and p16Ink4a will be detected by immunohistochemistry of retinal tissues and Western blot analyses of extracted RPE cells. This aim will be provided evidence for the relationship between RPE cell ageing and the incidence of AMD.
- Second, the DNA damage was linked to the AMD-like phenotype both in-vivo and in-vitro. Both the extracted primary cultured RPE cells from young, old and AMD rat models and in vitro cellular models (normal or tobacco extracted treatment group) will be obtained for detection. Cytoplasmic dsDNA will be detected by staining with a primary antibody against dsDNA. Mitochondrial DNA PCR will be obtained for the detection of cytoplasmic mtDNA. Both cytoplasmic dsDNA and damaged mtDNA will be upregulated in ageing retina and early AMD models.
- Third, cGAS/STING and its downstream IRF3- and NF-κB-dependent pathway-related key proteins will be detected in an in vitro model. Tobacco extract treatment will be used in the in vitro model construction, and the dose-response effect of tobacco extract will be also detected. For the SASP-associated factors, the cellular and secreted growth factors, including IL-1α, IL-1β, IL-6, IL-8, and matrix metalloproteinases (MMP1 and MMP3), will be detected by Western blots and ELISAs.
- In the end, we assessed inhibition of the cGAS/STING pathway as a treatment of early AMD. As reported in a recent study, covalent small molecules can inhibit STING,65 and the specific STING inhibitors will be used both in-vivo and in-vitro to test their potential therapeutic effects. If an inhibitor could reverse retinal ageing signs and release SASP markers, these molecules will be a potential drug for early AMD.
Conclusion
Ageing is one of the strongest risk factors for AMD incidence, but the exact role of ageing remains unclear. DNA damage, including nuclear and mtDNA damage, is important in the progression of senescence. The extract pathway for DNA damage in the trigger of senescence-associated hallmarks is poorly understood. Recently, mounting evidence has shown that the cGAS/STING pathway is an important DNA sensor related to proinflammatory factor secretion and is associated with another hallmark of ageing, SASP. Thus, we hypothesized that the cGAS/STING pathway is a vital signalling pathway for early AMD development and that inhibition of STING might be a potential therapeutic strategy for AMD cases.
Acknowledgments
This work was supported in whole or in part by the Project supported by the National Science Foundation for Young Scientists of China (Grant No. 81700804) and the Foundation for Young Medical Talents of Jiangsu Province, 2016 (Grant No. QNRC2016211).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mitchell P, Liew G, Gopinath B, Wong TY. Age-related macular degeneration. Lancet. 2018;392(10153):1147–1159. doi:10.1016/S0140-6736(18)31550-2
2. Kaur G, Tan LX, Rathnasamy G, et al. Aberrant early endosome biogenesis mediates complement activation in the retinal pigment epithelium in models of macular degeneration. Proc Natl Acad Sci U S A. 2018;115(36):9014–9019. doi:10.1073/pnas.1805039115
3. Owen CG, Fletcher AE, Donoghue M, Rudnicka AR. How big is the burden of visual loss caused by age related macular degeneration in the United Kingdom? Br J Ophthalmol. 2003;87(3):312–317. doi:10.1136/bjo.87.3.312
4. Wang C, Seo SJ, Kim JS, et al. Intravitreal implantable magnetic micropump for on-demand VEGFR-targeted drug delivery. J Control Release. 2018;283:105–112. doi:10.1016/j.jconrel.2018.05.030
5. Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1432–1444. doi:10.1056/NEJMoa062655
6. Saunier V, Merle BMJ, Delyfer MN, et al. Incidence of and risk factors associated with age-related macular degeneration: four-year follow-up from the ALIENOR study. JAMA Ophthalmol. 2018;136(5):473–481. doi:10.1001/jamaophthalmol.2018.0504
7. Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3(5):391–400. doi:10.1038/nrd1381
8. Buitendijk GHS, Schauwvlieghe AME, Vingerling JR, Schlingemann RO, Klaver CCW, Comparing Bevacizumab to Ranibizumab in Age-related macular degeneration Trial Research G. Antiplatelet and anticoagulant drugs do not affect visual outcome in neovascular age-related macular degeneration in the BRAMD trial. Am J Ophthalmol. 2018;187:130–137. doi:10.1016/j.ajo.2018.01.003
9. Mehta H, Tufail A, Daien V, et al. Real-world outcomes in patients with neovascular age-related macular degeneration treated with intravitreal vascular endothelial growth factor inhibitors. Prog Retin Eye Res. 2018;65:127–146. doi:10.1016/j.preteyeres.2017.12.002
10. Amoaku WM, Chakravarthy U, Gale R, et al. Defining response to anti-VEGF therapies in neovascular AMD. Eye (Lond). 2015;29(6):721–731. doi:10.1038/eye.2015.48
11. Delplace V, Payne S, Shoichet M. Delivery strategies for treatment of age-related ocular diseases: from a biological understanding to biomaterial solutions. J Control Release. 2015;219:652–668. doi:10.1016/j.jconrel.2015.09.065
12. Khandhadia S, Cipriani V, Yates JR, Lotery AJ. Age-related macular degeneration and the complement system. Immunobiology. 2012;217(2):127–146. doi:10.1016/j.imbio.2011.07.019
13. Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134(3):411–431.
14. Zarbin MA, Rosenfeld PJ. Pathway-based therapies for age-related macular degeneration: an integrated survey of emerging treatment alternatives. Retina. 2010;30(9):1350–1367. doi:10.1097/IAE.0b013e3181f57e30
15. Yehoshua Z, de Amorim Garcia Filho CA, Nunes RP, et al. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology. 2014;121(3):693–701. doi:10.1016/j.ophtha.2013.09.044
16. Thomas TC, Rollins SA, Rother RP, et al. Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol Immunol. 1996;33(17–18):1389–1401.
17. Rhoades W, Dickson D, Do DV. Potential role of lampalizumab for treatment of geographic atrophy. Clin Ophthalmol. 2015;9:1049–1056. doi:10.2147/OPTH.S59725
18. Narayanan R, Kuppermann BD. Hot topics in dry AMD. Curr Pharm Des. 2017;23(4):542–546. doi:10.2174/1381612822666161221154424
19. Age-Related Eye Disease Study 2 Research G. Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the age-related eye disease study 2 (AREDS2) randomized clinical trial. JAMA. 2013;309(19):2005–2015. doi:10.1001/jama.2013.4997
20. Wu J, Cho E, Giovannucci EL, et al. Dietary intake of alpha-linolenic acid and risk of age-related macular degeneration. Am J Clin Nutr. 2017;105(6):1483–1492. doi:10.3945/ajcn.116.143453
21. Wu J, Cho E, Giovannucci EL, et al. Dietary intakes of eicosapentaenoic acid and docosahexaenoic acid and risk of age-related macular degeneration. Ophthalmology. 2017;124(5):634–643. doi:10.1016/j.ophtha.2016.12.033
22. Datta S, Cano M, Ebrahimi K, Wang L, Handa JT. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog Retin Eye Res. 2017;60:201–218. doi:10.1016/j.preteyeres.2017.03.002
23. Corso-Diaz X, Jaeger C, Chaitankar V, Swaroop A. Epigenetic control of gene regulation during development and disease: A view from the retina. Prog Retin Eye Res. 2018;65:1–27. doi:10.1016/j.preteyeres.2018.03.002
24. Toomey CB, Johnson LV, Bowes Rickman C. Complement factor H in AMD: bridging genetic associations and pathobiology. Prog Retin Eye Res. 2018;62:38–57. doi:10.1016/j.preteyeres.2017.09.001
25. Cascella R, Ragazzo M, Strafella C, et al. Age-related macular degeneration: insights into inflammatory genes. J Ophthalmol. 2014;2014:582842. doi:10.1155/2014/582842
26. Corominas J, Colijn JM, Geerlings MJ, et al. Whole-exome sequencing in age-related macular degeneration identifies rare variants in COL8A1, a component of Bruch’s membrane. Ophthalmology. 2018;125(9):1433–1443. doi:10.1016/j.ophtha.2018.03.040
27. Yan Q, Ding Y, Liu Y, et al. Genome-wide analysis of disease progression in age-related macular degeneration. Hum Mol Genet. 2018;27(5):929–940. doi:10.1093/hmg/ddy002
28. Cascella R, Strafella C, Longo G, et al. Uncovering genetic and non-genetic biomarkers specific for exudative age-related macular degeneration: significant association of twelve variants. Oncotarget. 2018;9(8):7812–7821. doi:10.18632/oncotarget.23241
29. DeAngelis MM, Owen LA, Morrison MA, et al. Genetics of age-related macular degeneration (AMD). Hum Mol Genet. 2017;26(R1):R45–R50. doi:10.1093/hmg/ddx228
30. Hurley SF, Matthews JP, Guymer RH. Cost-effectiveness of smoking cessation to prevent age-related macular degeneration. Cost Eff Resour Alloc. 2008;6:18. doi:10.1186/1478-7547-6-18
31. Espinosa-Heidmann DG, Suner IJ, Catanuto P, Hernandez EP, Marin-Castano ME, Cousins SW. Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Invest Ophthalmol Vis Sci. 2006;47(2):729–737. doi:10.1167/iovs.05-0719
32. Woodell A, Jones BW, Williamson T, et al. A targeted inhibitor of the alternative complement pathway accelerates recovery from smoke-induced ocular injury. Invest Ophthalmol Vis Sci. 2016;57(4):1728–1737. doi:10.1167/iovs.15-18471
33. Vu KT, Hulleman JD. An inducible form of Nrf2 confers enhanced protection against acute oxidative stresses in RPE cells. Exp Eye Res. 2017;164:31–36. doi:10.1016/j.exer.2017.08.001
34. Zhang L, Vijg J. Somatic mutagenesis in mammals and its implications for human disease and aging. Annu Rev Genet. 2018;52:397–419. doi:10.1146/annurev-genet-120417-031501
35. Wyss-Coray T. Ageing, neurodegeneration and brain rejuvenation. Nature. 2016;539(7628):180–186. doi:10.1038/nature20411
36. Hamczyk MR, Del Campo L, Andres V. Aging in the cardiovascular system: lessons from hutchinson-gilford progeria syndrome. Annu Rev Physiol. 2018;80:27–48. doi:10.1146/annurev-physiol-021317-121454
37. Mertens J, Reid D, Lau S, Kim Y, Gage FH. Aging in a dish: iPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu Rev Genet. 2018;52:271–293. doi:10.1146/annurev-genet-120417-031534
38. Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12(7):412–420. doi:10.1038/nrrheum.2016.65
39. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi:10.1016/j.cell.2013.05.039
40. Wang AS, Dreesen O. Biomarkers of cellular senescence and skin aging. Front Genet. 2018;9:247. doi:10.3389/fgene.2018.00173
41. Gluck S, Ablasser A. Innate immunosensing of DNA in cellular senescence. Curr Opin Immunol. 2018;56:31–36. doi:10.1016/j.coi.2018.09.013
42. Kerur N, Fukuda S, Banerjee D, et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med. 2018;24(1):50–61. doi:10.1038/nm.4450
43. Nadal-Nicolas FM, Vidal-Sanz M, Agudo-Barriuso M. The aging rat retina: from function to anatomy. Neurobiol Aging. 2018;61:146–168. doi:10.1016/j.neurobiolaging.2017.09.021
44. El-Sayyad HI, Khalifa SA, El-Sayyad FI, Mousa SA, Mohammed EA. Analysis of fine structure and biochemical changes of retina during aging of Wistar albino rats. Clin Experiment Ophthalmol. 2014;42(2):169–181. doi:10.1111/ceo.12123
45. Fisher CR, Ferrington DA. Perspective on AMD pathobiology: a bioenergetic crisis in the RPE. Invest Ophthalmol Vis Sci. 2018;59(4):AMD41–AMD47. doi:10.1167/iovs.18-24289
46. Tamiya S, Kaplan HJ. Role of epithelial-mesenchymal transition in proliferative vitreoretinopathy. Exp Eye Res. 2016;142:26–31. doi:10.1016/j.exer.2015.02.008
47. Xia T, Rizzolo LJ. Effects of diabetic retinopathy on the barrier functions of the retinal pigment epithelium. Vision Res. 2017;139:72–81. doi:10.1016/j.visres.2017.02.006
48. Zhu W, Meng YF, Xing Q, Tao JJ, Lu J, Wu Y. Identification of lncRNAs involved in biological regulation in early age-related macular degeneration. Int J Nanomedicine. 2017;12:7589–7602. doi:10.2147/IJN.S140275
49. Nagineni CN, Samuel W, Nagineni S, et al. Transforming growth factor-beta induces expression of vascular endothelial growth factor in human retinal pigment epithelial cells: involvement of mitogen-activated protein kinases. J Cell Physiol. 2003;197(3):453–462. doi:10.1002/jcp.10378
50. Liegl R, Koenig S, Siedlecki J, Haritoglou C, Kampik A, Kernt M. Temsirolimus inhibits proliferation and migration in retinal pigment epithelial and endothelial cells via mTOR inhibition and decreases VEGF and PDGF expression. PLoS One. 2014;9(2):e88203. doi:10.1371/journal.pone.0088203
51. Wang L, Li P, Tian Y, et al. Human umbilical cord mesenchymal stem cells: subpopulations and their difference in cell biology and effects on retinal degeneration in RCS rats. Curr Mol Med. 2017;17(6):421–435. doi:10.2174/1566524018666171205140806
52. Qin T, Gao S. Inhibition of proteasome activity upregulates IL-6 expression in RPE cells through the activation of P38 MAPKs. J Ophthalmol. 2018;2018:5392432. doi:10.1155/2018/5392432
53. Zhang S, Yu N, Zhang R, Zhang S, Wu J. Interleukin-17A induces IL-1beta secretion from RPE cells via the NLRP3 inflammasome. Invest Ophthalmol Vis Sci. 2016;57(2):312–319. doi:10.1167/iovs.15-17578
54. Ghosh K, Capell BC. The senescence-associated secretory phenotype: critical effector in skin cancer and aging. J Invest Dermatol. 2016;136(11):2133–2139. doi:10.1016/j.jid.2016.06.621
55. Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of vascular aging. Circ Res. 2018;123(7):849–867. doi:10.1161/CIRCRESAHA.118.311378
56. Marazita MC, Dugour A, Marquioni-Ramella MD, Figueroa JM, Suburo AM. Oxidative stress-induced premature senescence dysregulates VEGF and CFH expression in retinal pigment epithelial cells: implications for age-related macular degeneration. Redox Biol. 2016;7:78–87. doi:10.1016/j.redox.2015.11.011
57. Martinez-Zamudio RI, Robinson L, Roux PF, Bischof O. SnapShot: cellular senescence pathways. Cell. 2017;170(4):816–816 e811. doi:10.1016/j.cell.2017.07.049
58. Chroma K, Mistrik M, Moudry P, et al. Tumors overexpressing RNF168 show altered DNA repair and responses to genotoxic treatments, genomic instability and resistance to proteotoxic stress. Oncogene. 2017;36(17):2405–2422. doi:10.1038/onc.2016.392
59. Kujoth GC, Hiona A, Pugh TD, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309(5733):481–484. doi:10.1126/science.1112125
60. Blasiak J, Piechota M, Pawlowska E, Szatkowska M, Sikora E, Kaarniranta K. Cellular senescence in age-related macular degeneration: can autophagy and dna damage response play a role? Oxid Med Cell Longev. 2017;2017:5293258. doi:10.1155/2017/5293258
61. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142–1149. doi:10.1038/ni.3558
62. Ahlers LR, Goodman AG. Nucleic acid sensing and innate immunity: signaling pathways controlling viral pathogenesis and autoimmunity. Curr Clin Microbiol Rep. 2016;3(3):132–141. doi:10.1007/s40588-016-0043-5
63. Mathur V, Burai R, Vest RT, et al. Activation of the STING-dependent type i interferon response reduces microglial reactivity and neuroinflammation. Neuron. 2017;96(6):1290–1302 e1296. doi:10.1016/j.neuron.2017.11.032
64. Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114(23):E4612–E4620. doi:10.1073/pnas.1705499114
65. Haag SM, Gulen MF, Reymond L, et al. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559(7713):269–273. doi:10.1038/s41586-018-0287-8
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.