")
Back to Journals » Drug Design, Development and Therapy » Volume 9
The application of click chemistry in the synthesis of agents with anticancer activity
Authors Ma N, Wang Y, Zhao B, Ye W, Jiang S
Received 29 September 2014
Accepted for publication 12 December 2014
Published 12 March 2015 Volume 2015:9 Pages 1585—1599
DOI https://doi.org/10.2147/DDDT.S56038
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Nan Ma,1–3 Ying Wang,3 Bing-Xin Zhao,3 Wen-Cai Ye,1,3 Sheng Jiang2
1Department of Natural Medicinal Chemistry, China Pharmaceutical University, Nanjing, 2Laboratory of Medicinal Chemistry, Guangzhou Institute of Biomedicine and Health, Chinese Academy of Sciences, 3Institute of Traditional Chinese Medicine and Natural Products, College of Pharmacy, Jinan University, Guangzhou, People’s Republic of China
Abstract: The copper(I)-catalyzed 1,3-dipolar cycloaddition between alkynes and azides (click chemistry) to form 1,2,3-triazoles is the most popular reaction due to its reliability, specificity, and biocompatibility. This reaction has the potential to shorten procedures, and render more efficient lead identification and optimization procedures in medicinal chemistry, which is a powerful modular synthetic approach toward the assembly of new molecular entities and has been applied in anticancer drugs discovery increasingly. The present review focuses mainly on the applications of this reaction in the field of synthesis of agents with anticancer activity, which are divided into four groups: topoisomerase II inhibitors, histone deacetylase inhibitors, protein tyrosine kinase inhibitors, and antimicrotubule agents.
Keywords: topoisomerase II inhibitors, histone deacetylase inhibitors, protein tyrosine kinase inhibitors, antimicrotubule agents
Introduction
Click reaction is a variation of the Huisgen 1,3-dipolar cycloaddition reaction between terminal acetylenes and azides for the construction of triazoles, which is one of the powerful reactions for making carbon–heteroatom–carbon bonds in aqueous environment defined by Kolb et al in 2001.1,2 The Huisgen cycloaddition is the reaction of a dipolarophile with a 1,3-dipolar compound to form the structure of five-membered (hetero)cycles. Dipolarophiles could be multiple-bond compounds such as alkenes, alkynes, carbonyls, and nitriles, which contain carbon, nitrogen, oxygen, or sulfur atoms, while 1,3-dipolar compounds are a class of compounds that can be represented by dipole resonance structures, and they should contain at least one heteroatom.3
Kolb et al described the click reaction as being so wide in scope and easy to perform that chemists could only use readily available reagents to perform the reaction. The click reaction is insensitive to oxygen and water, and the by-product can be easily removed without requiring chromatographic methods and be stereospecific (but not necessarily enantioselective).3 To sum up, click reactions share the following attributes: 1) Many click components are derived from alkenes and alkynes. Carbon–carbon multiple bonds provide both energy and mechanistic pathways to be elaborated into reactive structures for click connections. 2) Most click reactions involve the formation of carbon–heteroatom (mostly N, O, and S) bonds. 3) Click reactions are strongly exothermic, by virtue of either highly energetic reactants or strongly stabilized products. 4) Click reactions are usually fusion processes (leaving no by-products) or condensation ones (producing water as a by-product). 5) Many click reactions are highly tolerant of, and often accelerated by, the existence of water.4
The 1,2,3-triazoles formed by click chemistry are very stable in both metabolic5–7 and chemical degradations. They also show diverse biological activities such as anticancer,8 antibacterial,9 and antiviral.10 All the above characteristics imply the security and stability of 1,2,3-triazoles under physiological conditions.
Since its debut in 1999, click chemistry has stimulated enormous amount of interest in many different research fields and been used increasingly in biomedical research for its good yield, high selectivity, and the tolerance of various kinds of functional groups and reaction conditions. Over the past years, click chemistry has emerged to become one of the most powerful tools in drug discovery, chemical biology, and proteomic applications.4 Many researchers use click chemistry as a synthetic tool for the generation of pharmacologically valuable drugs.11–13
Application of click chemistry in the synthesis of anticancer agents
Cancer, also called malignant neoplasm, is caused by the disorder of cell proliferation mechanism, and has become a major public health burden in developed and developing countries. Although several anticancer agents are in clinical use and have significant curative effects, it is still necessary to screen new molecules with different modes of actions. Enzymes expressed at an estimated 18%–29% within eukaryotic genomes4 and the enzyme inhibitors play a significant role in curing cancer.
Topoisomerase II inhibitors
Topoisomerase enzymes (topoisomerases I and II) are enzymes that control the changes in DNA structure by breaking and rejoining the phosphodiester backbone of DNA strands during the normal cell cycle. In recent years, topoisomerases have become popular targets for cancer chemotherapy drugs.
Podophyllotoxin is an antimicrotubule agent acting at the colchicine-binding site on tubulin.14 Due to its severe toxicities, it cannot be used clinically, but its semisynthetic derivatives, etoposide and teniposide, had been used as effective anticancer drugs for the treatment of lung cancer, Kaposi’s sarcoma, and lymphoma.
Reddy et al reported a series of 4β-[(4-alkyl)-1,2,3-triazol-1-yl] podophyllotoxin derivatives synthesized by employing click chemistry approach in 2011, the majority of which proved to be more potent than etoposide, a clinically therapeutic drug against various cancers. The selected compounds exhibited significant anticancer activity with IC50 values in the range of 0.001–1 μM (Figure 1, 1), which revealed that the presence of methyl, ethyl, and hydroxyl groups in triazole moiety could increase the anticancer activities of those compounds. On the other hand, increasing the length of alkyl chain of those compounds could decrease their anticancer activities. Docking experiments showed a good correlation between calculated interaction energies with the topoisomerase II and the observed IC50 values of all these compounds.15
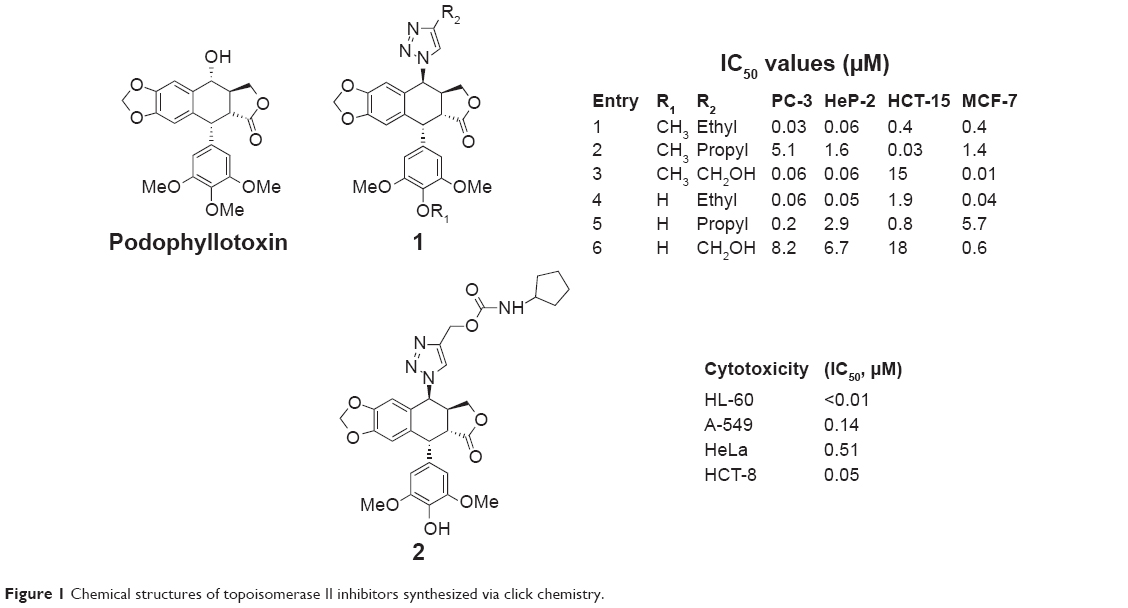 | Figure 1 Chemical structures of topoisomerase II inhibitors synthesized via click chemistry. |
In 2013, Chen et al synthesized the carbamate derivatives of 4β-(1,2,3-triazol-1-yl) podophyllotoxin and then evaluated their cytotoxicities against human cancer cell lines HL-60, A-549, HeLa, and HCT-8. Among the synthetic compounds, 4′-O-demethyl-4β-[(4-hydroxymethyl)-1,2,3-triazol-1-yl]-4-deoxypodophyllotoxin cyclopentylcarbamate (2, Figure 1) showed the most potent cytotoxicities, which induced cell cycle arrest in the G2/M phase accompanied by apoptosis and inhibited the formation of microtubules in A-549 cells. This compound can also cause the inhibition of DNA topoisomerase II.16
Mansonones17,18 are quinone-containing compounds that show good anticancer activities. To investigate the effects of C-9 halogenation and C-3 substitution on mansonone E, Huang et al synthesized two series of novel C-9 chloro- and bromo-substituted mansonone E derivatives with triazole moieties at the C-3 position, which were prepared by using click chemistry. Structure-activity relationship study revealed that the substituent at the C-9 position and the triazole ring exhibited various influences on the cytotoxicity against several human cancer cell lines. Topo II inhibition assay indicated that almost all of the C-9 bromo-substituted derivatives showed better activities than the C-9 chloro-substituted derivatives. The cytotoxic activities of these compounds against A-549, HL-60, K562, and HeLa cells were also evaluated by comparing with etoposide, which was used as a positive control. The result indicated that these compounds (3a–3d, Figure 2) have the potential to be antitumor agents as topoisomerase II inhibitors.19
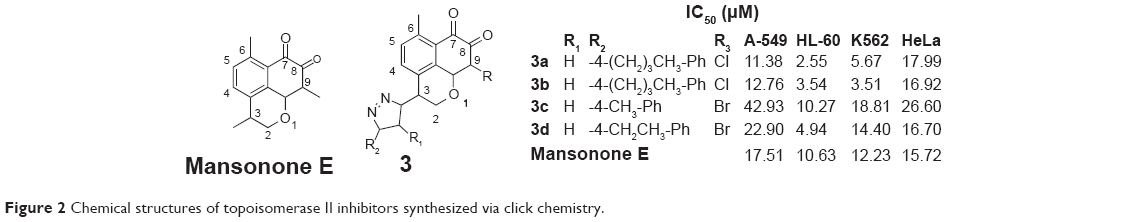 | Figure 2 Chemical structures of topoisomerase II inhibitors synthesized via click chemistry. |
In 2013, Kaushik et al reported the synthesis of a small library of amino acid-linked 1,4-disubstituted 1,2,3-bistriazole conjugates, which were synthesized from the N-protected l-amino acids and propargyl esters through one-pot click reaction. Docking simulation of compounds (4a and 4b, Figure 3) showed inhibition of Escherichia coli topoisomerase II DNA gyrase B through hydrogen-bonding interactions.20
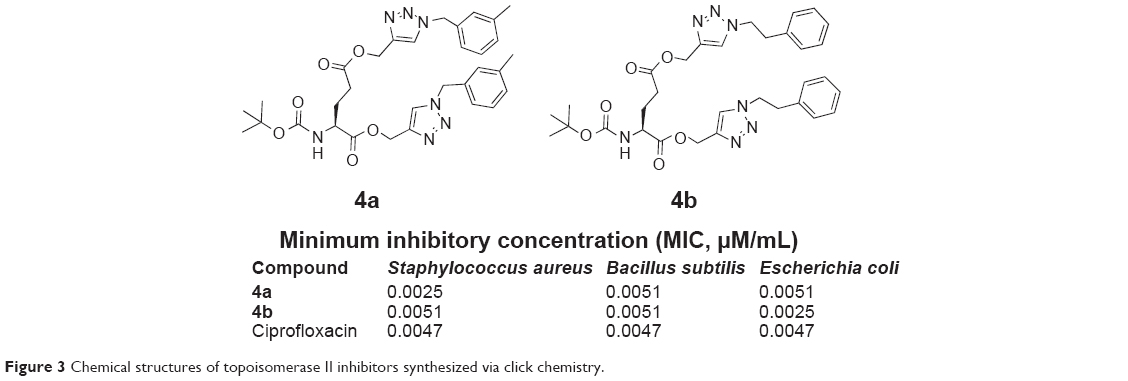 | Figure 3 Chemical structures of topoisomerase II inhibitors synthesized via click chemistry. |
Histone deacetylase inhibitors
Histone deacetylases (HDACs) are a family of enzymes that catalyze the deacetylation of lysine side chain in chromatin. These enzymes are involved in a wide range of biological processes such as cell differentiation, proliferation, angiogenesis, and apoptosis. Histone deacetylase inhibitors (HDACIs) showed the ability to induce cell growth arrest, differentiation, and apoptosis. HDACIs have been clinically validated as a therapeutic strategy for cancer treatment.21–23 The classic pharmacophore for HDACIs consists of three distinct structural motifs: the zinc-binding group, a hydrophobic linker, and a recognition cap group.24
Chen et al modified the cap region of a set of triazolylphenyl-based HDACIs in 2008. They screened the products with a panel of pancreatic cell lines to gain result that the nature of substitution on the phenyl ring plays a main role in their selectivity for HDAC1 versus HDAC6. The triazolylphenyl ligand 5 (Figure 4) had been found to significantly inhibit HDAC6 with an IC50 value of 1.9 nM, which represented a valuable research tool for further chemical modifications.25
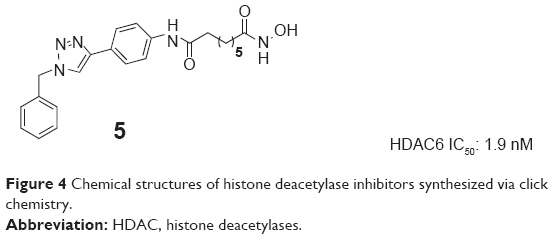 | Figure 4 Chemical structures of histone deacetylase inhibitors synthesized via click chemistry. |
Shen et al reported a new chemical scaffold of HDACIs through click chemistry in 2008. In their design, the precursors corresponding to the “cap” moiety of the HDACI contained an azido group, whereas the zinc-chelating functionality precursors contained an alkyne group. Among the 14 compounds which were synthesized from the combination of two alkyne and seven azido precursors, NSC746457 (6, Figure 5) could inhibit HDAC1 with an IC50 value of 104±30 nM, and it was proved to be quite potent in the cancer cell line screening with GI50 values ranging from 3.92 μM to 10 nM.26 Shen et al also discovered that triazole ring was of suitable size to fit into the narrow active pocket of HDAC protein, and it was correctly positioned to form a π–π interaction with two reserved phenylalanine residues in the docking structure of NSC746457 with HDAC2, indicating that the triazole ring contributed to the binding affinity. In 2011, they reported the further optimization of NSC746457 by using the HDAC2-trichostatin A crystal structure. The optimization was also a click chemistry-based approach, including the replacement of the trans-styryl moiety with a 2-substituted benzo-hetero aromatic ring and the introduction of a substituent onto the central methylene carbon. Among the prepared compounds, isopropyl derivative (compound 7, Figure 5) and t-butyl derivative (8, Figure 5) exhibited excellent potency against HDACs enzyme, with IC50 values of 22 nM and 18 nM, respectively.27 For the in vitro safety tests, NK-HDAC1 (compound 9, Figure 5) was far less toxic to nontransformed cells than tumor cells, while it showed approximately tenfold greater potency than suberoylanilide hydroxamic acid (SAHA) in vitro.28
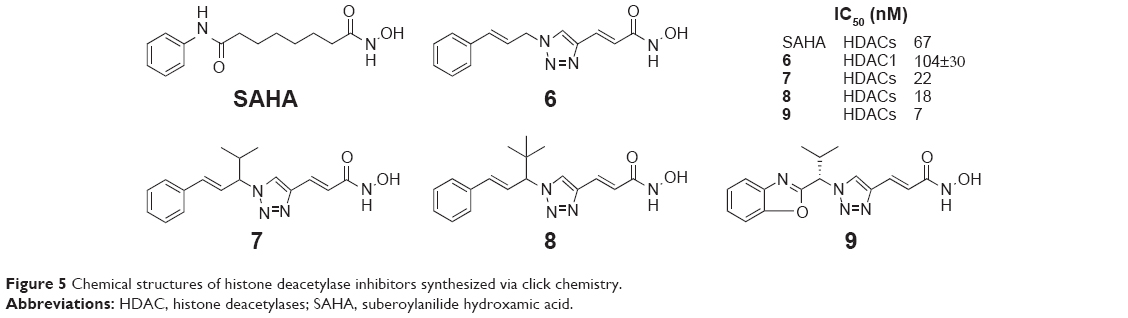 | Figure 5 Chemical structures of histone deacetylase inhibitors synthesized via click chemistry. |
Chen et al established a 1,2,3-triazole ring as a surface recognition cap group-linking moiety in SAHA-like HDACIs. They synthesized several triazole-linked SAHA-like hydroxamates using click chemistry in 2008. In these compounds, the amide bond in SAHA was replaced with a triazole ring. The linker chain length and the aromatic ring of these compounds were both varied. Several compounds (10a–10e, Figure 6) have showed potent inhibition of HDACs.29
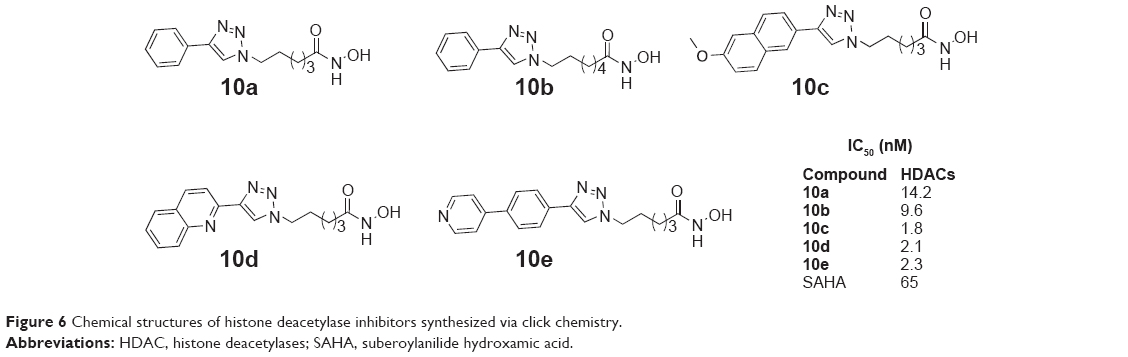 | Figure 6 Chemical structures of histone deacetylase inhibitors synthesized via click chemistry. |
Sun et al envisioned that changing the position of substituents on the triazole ring of 10a (Figure 6) would increase the selectivity for HDAC1. Thus, they synthesized a new series of triazole-based HDAC1 inhibitors using one-pot click chemistry in 2013. These inhibitors showed the features of high potency and selectivity of HDAC1, as well as the ability to inhibit several cancer cells’ growth. The HDAC inhibitory activity data of these compounds confirmed their conjecture. Compound 11 (Figure 7), a representative lead, showed potent inhibition with an IC50 value of 58 nM to HDAC1.30
 | Figure 7 Chemical structures of histone deacetylase inhibitors synthesized via click chemistry. |
In 2009, Horne et al reported the synthesis of HDACIs containing 1,4- or 1,5-disubstituted 1,2,3-triazole analogs to a naturally occurring cyclic tetrapeptide inhibitor of HDACs called apicidin using click chemistry. They aimed to carry out a more controlled study in which apicidin analogs with a fixed cis- or trans-amide isostere could be compared directly in an HDAC inhibition assay to solve the question of which amide configuration of apicidin is present in the dominant bioactive conformation. The 1,4- and 1,5-substituted triazoles acted as surrogates for trans- and cis-amide bonds, respectively. The resultant molecules (12 and 13, Figure 8) showed inhibition of HDAC1 and HDAC3, and proved useful to identify the most bioactive conformation (cis–trans–trans–trans) of the original cyclic tetrapeptide. They established the ability to probe the biologically relevant conformation of a natural peptide ligand by introducing different triazole regioisomers in place of amide bonds in its backbone. The study will help lead to more selective HDAC ligands.31
 | Figure 8 Chemical structures of histone deacetylase inhibitors synthesized via click chemistry. |
Canzoneri et al reported the approach that conjugated a SAHA-like aliphatic-hydroxamate HDAC inhibition group directly to the nuclear localization signal peptide through 1,2,3-triazole moiety to give three compounds (14a–14c, Figure 9). These compounds demonstrated HDAC inhibition similar to SAHA in 2009. Compound 14b showed a threefold improvement in HDAC1/2 inhibition, a threefold increase in HDAC6 selectivity, and a twofold increase in HDAC8 selectivity when compared to SAHA.32
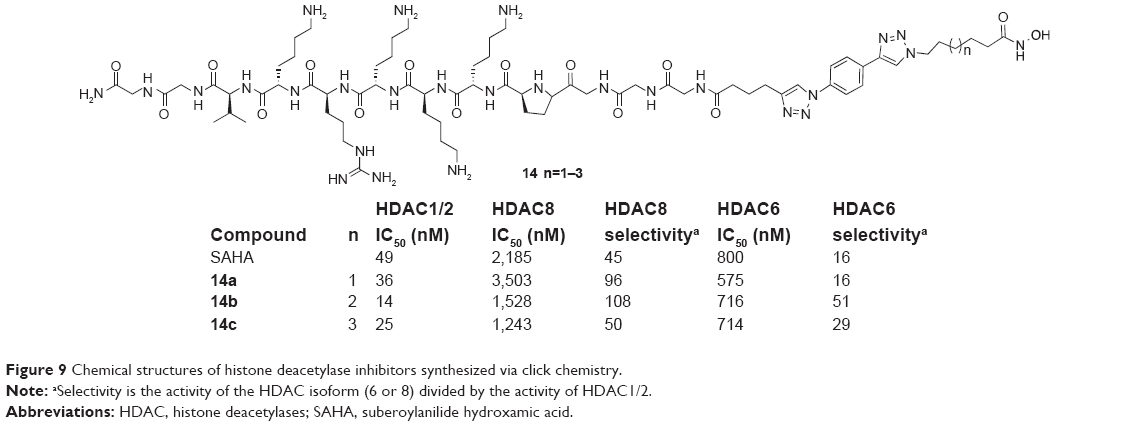 | Figure 9 Chemical structures of histone deacetylase inhibitors synthesized via click chemistry. |
Suzuki et al established a Cu(I) complex of HDAC8 accelerating a selective reaction between an azide and an alkyne to form potent HDACIs (15 and 16, Figure 10) with IC50 values of 0.51 μM and 4.0 μM at HDAC8, respectively, via in situ click chemistry in 2010.33
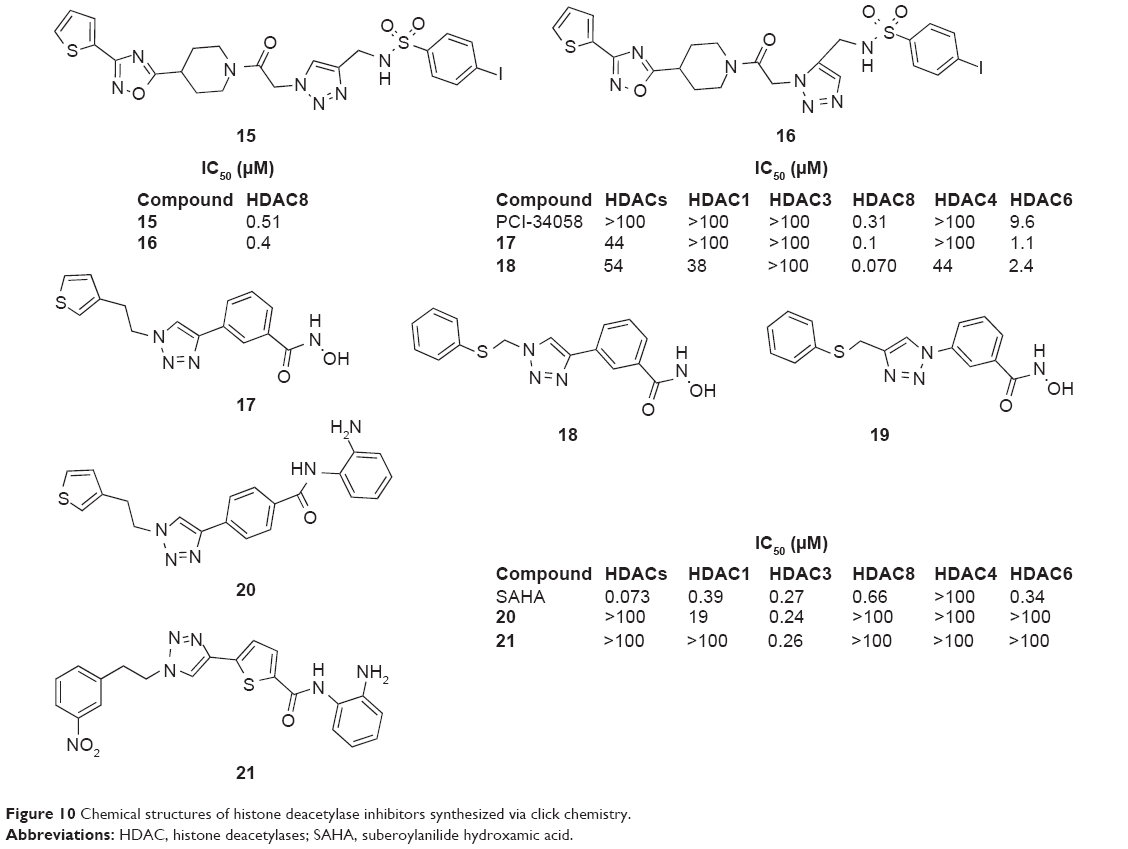 | Figure 10 Chemical structures of histone deacetylase inhibitors synthesized via click chemistry. |
Suzuki et al then reported two potent HDAC8-selective inhibitors, C142 and C149 (17 and 18, Figure 10), synthesized by click chemistry in 2012. They linked the zinc-binding group that coordinates with the active-site zinc ion to the capping structure, which interacts with residues on the rim of the active site via a triazole moiety in the presence of Cu(I) catalyst. The compound C149 showed more potent activity than the known HDAC8 inhibitor PCI-34058 (IC50 =0.31 μM) with an IC50 value of 0.070 μM. They also found that the inhibitors caused selective acetylation of cohesin in cells and exerted growth inhibitory effects on T-cell lymphoma and neuroblastoma cells (GI50 =3–80 μM), which suggests that HDAC8-selective inhibitors have the potential as anticancer agents.34 Based on these results, in 2014, Suzuki et al reported a series of C149 derivatives that were designed and synthesized as HDAC8-selective inhibitors by converting the triazole ring of C149 into various aromatic rings: benzene ring, thiazole ring, oxadiazole ring, triazole ring, and thiophene ring. Among the derivatives, oxadiazole derivative increased cohesin (HDAC8 substrate) acetylation and showed greater growth inhibitory activity than C149 against T-cell lymphoma cells. The triazole derivative (19, Figure 10) showed the most potent HDAC8 inhibitory activity with an IC50 value of 0.053 μM. These findings are useful for the further development of HDAC8-selective inhibitors and should pave the way for the development of new anticancer drugs.35
To discover HDAC3-selective inhibitors, they performed a further click chemistry approach to find a series of HDAC3-selective inhibitors in 2013. Two compounds, T247 and T326 (20 and 21, Figure 10), were screened out with good selectivity for HDAC3 and did not strongly inhibit other isozymes. They also induced a dose-dependent selective increase of NF-κB acetylation in human colon cancer HCT-116 cells, indicating selective inhibition of HDAC3 in the cells. The result suggested the usefulness of the click chemistry approach to find isozyme-selective HDACIs.36
Largazole 3 was a natural macrocyclic depsipeptide reported by Taori et al in 2008, which showed promising HDAC1 inhibitory activity and selectivity.37 Li et al replaced the 4-methylthiazoline moiety of largazole with nitrogen functionality via click chemistry and obtained a series of new largazole analogs (22, Figure 11), which showed good selectivity for HDAC1 (IC50 =0.1 μM) over HDAC9 (IC50 =34.6 μM) compared to largazole in 2012. The experimental results also indicated that the introduction of appropriate aromatic groups into the largazole skeleton was a useful optimizing tool for this unique class of anticancer agents.38
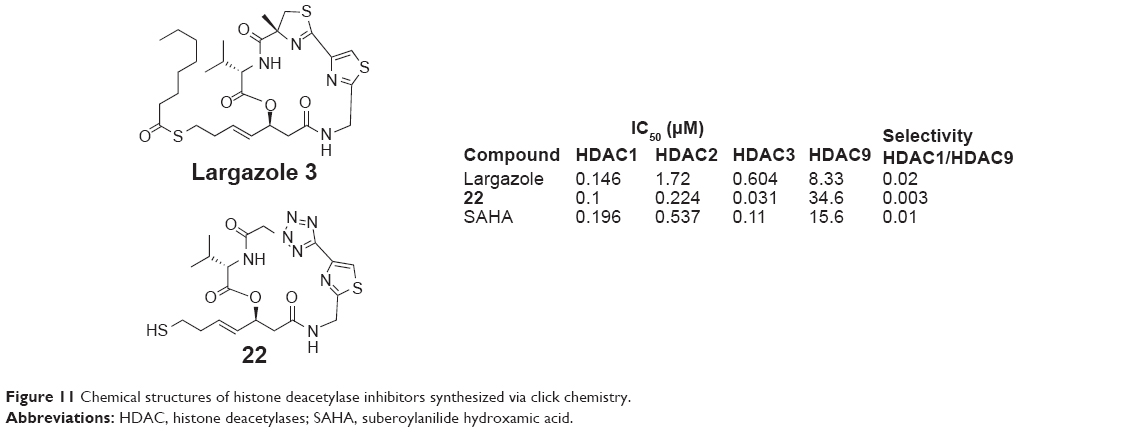 | Figure 11 Chemical structures of histone deacetylase inhibitors synthesized via click chemistry. |
In 2012, Spencer et al reported a small library of ferrocene-based 1,2,3-triazole-containing hydroxamic acids synthesized by employing click chemistry. Compound 23 (Figure 12), 7-(4-ferrocenyl-1H-1,2,3-triazol-1-yl)-N-hydroxyheptanamide, showed good HDAC inhibition with low IC50 values. The potency was slightly lower than that of SAHA. It was also shown to have appreciable growth inhibition in breast cancer cells (MCF-7).39
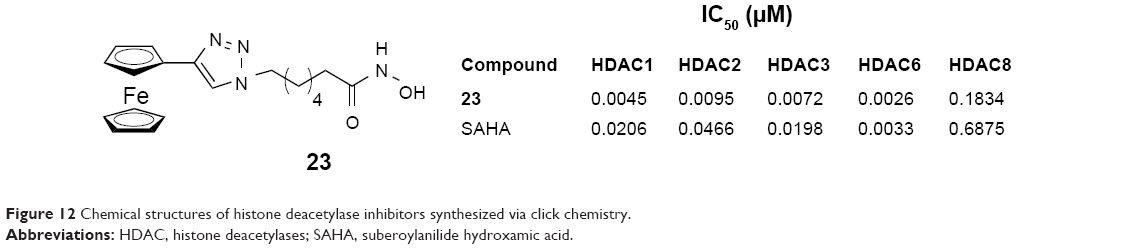 | Figure 12 Chemical structures of histone deacetylase inhibitors synthesized via click chemistry. |
Meng et al reported a 64Cu-labeled HDAC imaging probe in 2013. It was obtained by the introduction of a metal chelator through click reaction of HDACI CUDC-101 (Figure 13) and then radiolabeled with 64Cu (24, Figure 13). It was identified as a positron emission tomography (PET) imaging probe to noninvasively visualize HDAC expression in vivo. The IC50 of this compound to HDACs was shown to be in the nanomolar range by enzymatic assay, which suggested that 24 has the potential to be used in clinical applications.40
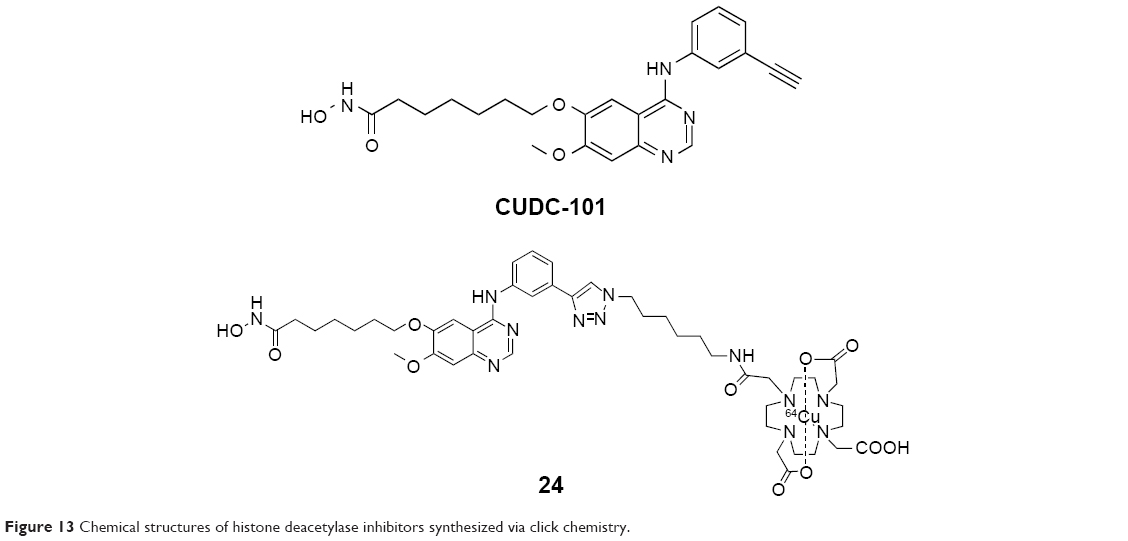 | Figure 13 Chemical structures of histone deacetylase inhibitors synthesized via click chemistry. |
Protein tyrosine kinase inhibitors
Protein tyrosine kinases (PTKs) catalyze the phosphorylation of phenolic group of tyrosine residue in many substrate proteins by the transfer of c-phosphate moiety of ATP. PTKs play a crucial role in the signal transduction pathways. Aberrant regulation of kinase activity has been implicated in many diseases including cancer.
Src family kinases, one genre of PTKs, play an important role in the regulation of normal cellular signal transduction pathways, such as cell division, growth factor signaling, differentiation, survival, adhesion, migration, and invasion.41 Src mutations and/or overexpression had been correlated with tumor growth, metastasis, and angiogenesis.42,43 Src kinase has been implicated in the development of a variety of cancers.44,45
For studying the evaluation of 1,4-disubstituted 1,2,3-triazoles as a novel template for Src kinase inhibition, Kumar et al hypothesized that substitution at N1 and position 4 of 1,2,3-triazoles with hydrophobic residues may occupy and interact with the hydrophobic-binding pocket of Src ATP-binding site similar to that of 3-phenylpyrazolo-pyrimidines. They synthesized two classes of 1,4-disubstituted 1,2,3-triazoles using one-pot reaction of α-tosyloxy ketones/α-halo ketones in the presence of aqueous polyethylene glycol (1:1, v/v) via the click chemistry approach in 2010. Compounds 25a–25c and 26a and 26b (Figure 14) exhibited modest Src kinase inhibitory activity among the synthesized 1,2,3-triazoles with IC50 values in the range of 32–43 μM. The insertion of C6H5– and 4-CH3C6H4– at the triazole functional group and less bulkier group at the ketone position shows better inhibitory activity for Src kinase.46
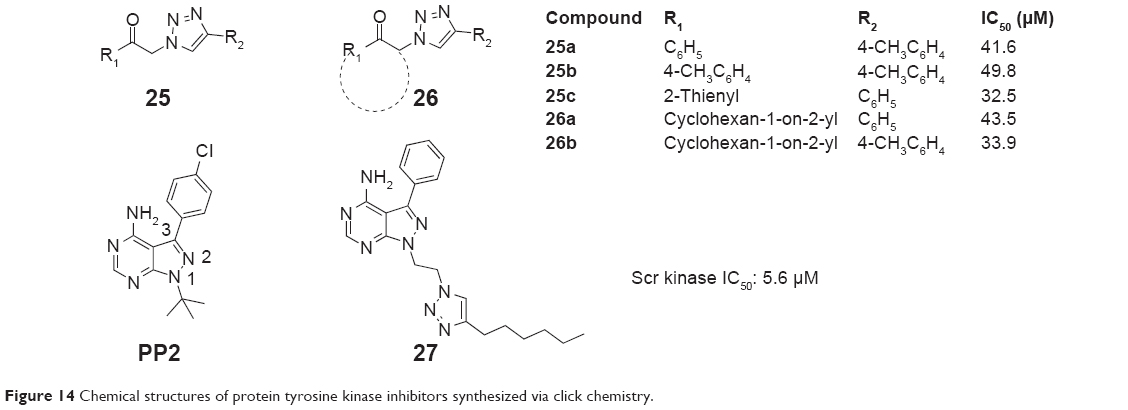 | Figure 14 Chemical structures of protein tyrosine kinase inhibitors synthesized via click chemistry. |
In 2011, Kumar et al synthesized a series of two classes of 3-phenylpyrazolopyrimidine-1,2,3-triazole conjugates using the click chemistry approach to investigate whether variation of N1 substitution in 3-phenylpyrazolopyrimidines with different 1,2,3-triazoles-containing hydrophobic residues can occupy and/or interact with amino acids of the cavity formed from side chains of helix αC and helix αD and contribute to the enhancement of Src kinase inhibitory potency. All the compounds that they synthesized were evaluated for inhibition of Src kinase and human ovarian adenocarcinoma (SK-Ov-3), breast carcinoma (MDA-MB-361), and colon adenocarcinoma (HT-29). Hexyl triazolyl-substituted 3-phenylpyrazolopyrimidine (27, Figure 14) exhibited inhibition of Src kinase with an IC50 value of 5.6 μM.47
The cellular form of Abelson (Abl) tyrosine kinase is a close relative of the Src family of tyrosine kinases. Abl tyrosine kinase is directly implicated in chronic myelogenous leukemia. Kalesh et al reported the rapid synthesis of Abl tyrosine kinase inhibitors, which contained the head portion of Imatinib, short-chain azides, and an alkyne group, using the highly modular and efficient click chemistry in 2009. Among these compounds, 28 and 29 (Figure 15) showed better inhibition against Src kinase with IC50 values of 0.7 μM and 1.12 μM by enzymatic screening, respectively, which had comparable potency to Imatinib (a Food and Drug Administration-approved drug for the treatment of chronic myeloid leukemia).48
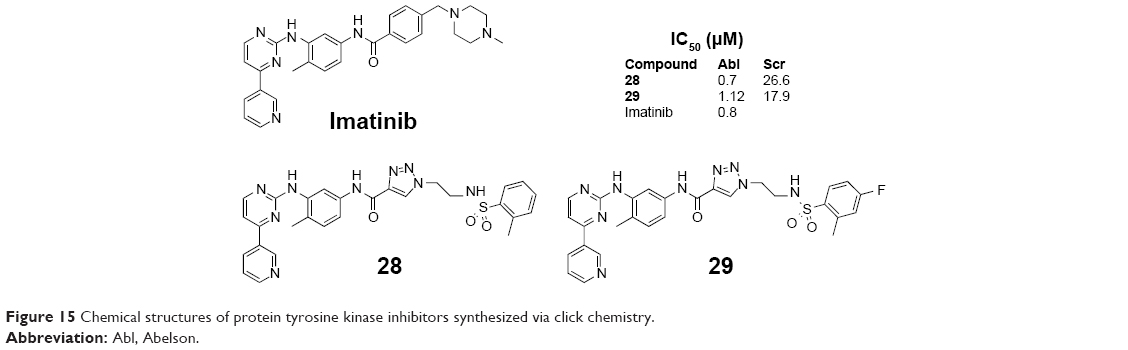 | Figure 15 Chemical structures of protein tyrosine kinase inhibitors synthesized via click chemistry. |
Li et al designed a series of 3-(1H-1,2,3-triazol-1-yl)benzamide derivatives as new Bcr-Abl inhibitors by using combinational strategies of bio-isosteric replacement, scaffold hopping, and conformational constraint with the applications of click chemistry in 2012. The 1,2,3-triazole was utilized as a new linker between the heterocyclic moiety (the so-called head region) and the methylphenyl group (middle part) of the inhibitors. The new Bcr-Abl inhibitors showed significant inhibition against a broad spectrum of Bcr-Abl mutants including the most refractory gatekeeper T315I mutation. The most potent compound 30 (Figure 16) strongly inhibited the kinase activities of Bcr-AblWT and Bcr-AblT315I with mean IC50 values of 0.60 nM and 1.12 nM, respectively.49
 | Figure 16 Chemical structures of protein tyrosine kinase inhibitors synthesized via click chemistry. |
Peruzzotti et al employed in situ click chemistry between azides and alkyne fragments for probing the ligand-binding site of Abl in 2012. This allowed the possible use of this strategy for the discovery of new scaffolds useful for the inhibition of the large family of tyrosine kinases.50
Epidermal growth factor receptor (EGFR) kinase is believed to undergo homo- or heterodimerization followed by activation of the intrinsic PTK. EGFR can overexpress in breast, ovarian, and other human cancers51 and is a well-validated target used in anticancer drug discovery now.52–55 Its family has four members: human epidermal growth factor receptor-1 (HER1), HER2, HER3, and HER4. HER2 is a transmembranous glycoprotein (p185neu) with intrinsic tyrosine kinase activity. It is encoded by the HER2 protooncogene located on the long arm of chromosome 17 (17q21).56 The overexpression of HER2 is identified on many tumor cells. Statistically, overexpression of HER2 occurs in a number of human cancers, including 25%–30% of breast cancer,57 28% of pulmonary adenocarcinoma, and 17% of colorectal adenocarcinoma.58
The 1,2,3-triazole was discovered as an inhibitor of HER2 tyrosine kinase through computer-aided drug design approach and searched from molecular libraries by Cheng et al. In 2007, Cheng et al synthesized a series of 4-aryl-5-cyano-2H-1,2,3-triazole derivatives bearing a variety of groups at the 4-position of phenyl by click chemistry. The derivatives showed bioactivity at HER2 tyrosine kinase. Compound 31 (Figure 17) has the lowest IC50 value of 6.6 μM for inhibiting HER2 tyrosine kinase phosphorylation in breast cancer cells and the IC50 value of 30.9 μM for breast cancer MDA-MB-453 cell growth inhibition.59
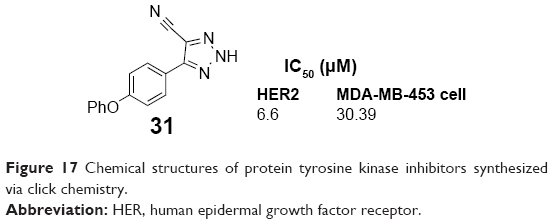 | Figure 17 Chemical structures of protein tyrosine kinase inhibitors synthesized via click chemistry. |
Kobus et al reported an 18F-labeled tyrosine kinase inhibitor, 6-(4-N, N-dimethylaminocrotonyl)amido-4-(3-chloro-4-fluoro)phenylamino-7-{3-[4-(2-[18F]fluoroethyl)-2,3,4-triazol-1-yl]propoxy} quinazoline (32, Figure 18), synthesized via Huisgen 1,3-dipolar cycloaddition for assessing the EGFR overexpression in tumors in 2009. They applied a two-step click approach that can be adapted to a fully automated synthesis module. PET images of PC9 tumor xenograft using the novel biomarker showed promising results to visualize EGFR overexpression.60
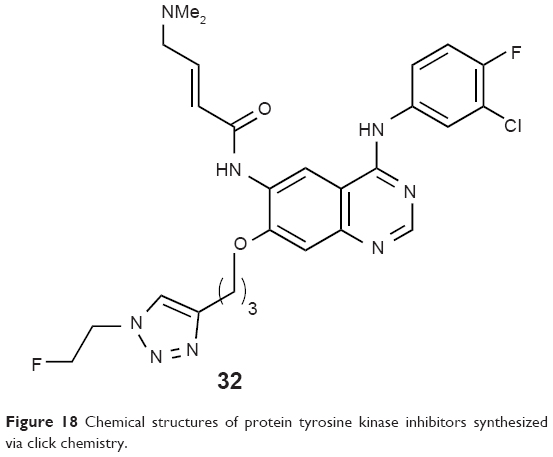 | Figure 18 Chemical structures of protein tyrosine kinase inhibitors synthesized via click chemistry. |
To facilitate clinical evaluation of primary tumors and/or metastases using the EGFR-specific imaging agent, Pisaneschi et al synthesized a small array of fluorine-containing compounds based on a 3-cyanoquinoline core in 2010. Compound 33 (Figure 19), incorporating 20-fluoroethyl-1,2,3-triazole, was selected for evaluation as a radioligand based on its high affinity for EGFR kinase (IC50 =1.81±0.18 nM), good cellular potency (IC50 =21.97±9.06 nM), low lipophilicity, and good metabolic stability. Experimental data of the compound showed good stability in vivo and a fourfold higher uptake in high EGFR-expressing A431 tumor xenografts compared to low EGFR-expressing HCT-116 tumor xenografts.61
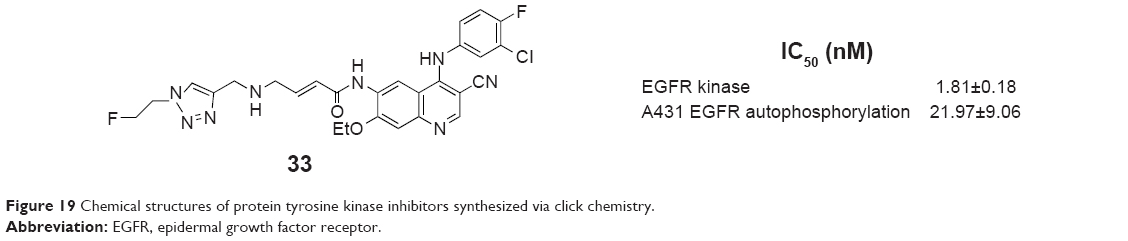 | Figure 19 Chemical structures of protein tyrosine kinase inhibitors synthesized via click chemistry. |
The family of fibroblast growth factor receptor (FGFR) is one of the 20 subclasses of receptor tyrosine kinases, consisting of four homologous receptors numbered FGFR1–4.62 FGFR3 can act as an oncogene in various cancers.63–65 Le Corre et al reported a library of pyrido[2,3-d]pyrimidines as inhibitors of FGFR3 tyrosine kinase allowing possible interactions with an unexploited region of the ATP-binding site in 2010. This library was built up with an efficient step of click chemistry giving easy access to triazole-based compounds bearing a large panel of substituents. Among the 27 analogs they synthesized, more than half exhibited 55%–89% inhibition of in vitro FGFR3 kinase activity at 2 μM, and one (34, Figure 20) was able to inhibit auto-phosphorylation of mutant FGFR3-K650M in transfected HEK cells. Investigation also showed that the targeted region of the ATP-binding site accepts 1,2,3-triazole moiety, with a preference for positively charged substituents.66
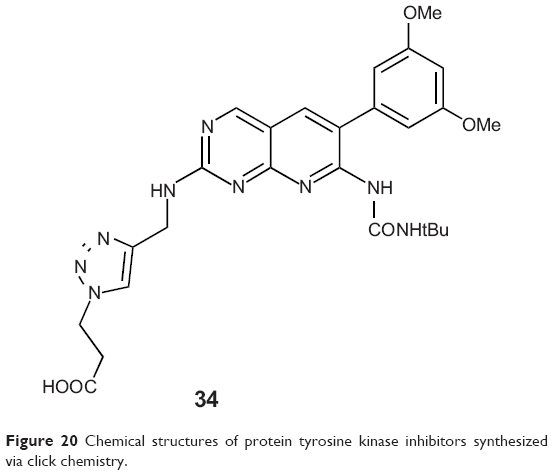 | Figure 20 Chemical structures of protein tyrosine kinase inhibitors synthesized via click chemistry. |
Antimicrotubule agents
The tubulin system plays a key role during mitosis, and disturbing its dynamic equilibrium can prevent cell division and induce apoptosis. Microtubules, dynamic protein polymers composed of α-tubulin and β-tubulin heterodimers, are major components of the cytoskeleton with an important role in a variety of cellular functions. They have become a well-established cellular target for anticancer drugs in recent years.67,68 Searching for novel microtubule-binding agents that have enhanced tumor specificity, reduced neurotoxicity, and insensitivity to chemoresistance mechanisms, is the main objective.69
Stefely et al reported a series of N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)arylamides as a new scaffold that provides rapid access to antimicrotubule agents by copper-catalyzed azide–alkyne cycloaddition in 2010. These compounds all afforded inhibitors of cancer cell growth. Among the inhibitors, compound 35b (Figure 21), led by the initial hit compound 35a, had an IC50 of 46 nM against MCF-7 human breast tumor cells.70
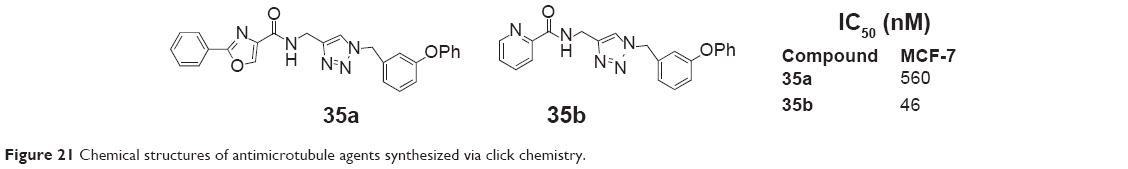 | Figure 21 Chemical structures of antimicrotubule agents synthesized via click chemistry. |
Steganacin and podophyllotoxin are two naturally occurring ligands that share the capability to disrupt tubulin assembly. Imperio et al had used the ruthenium-catalyzed [3+2] azide–alkyne cycloaddition to replace the lactone ring of both the structures with a 1,5-disubstituted triazole in few synthetic steps in 2007 (36a and 36b, Figure 22). These compounds were found to be less cytotoxic while retaining antitubulin activity compared to podophyllotoxin.71
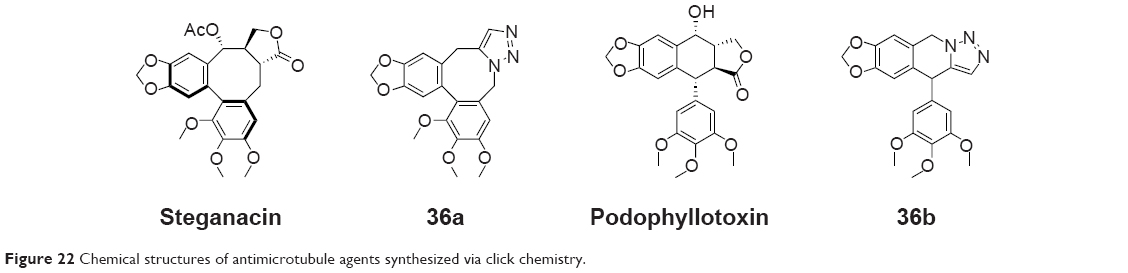 | Figure 22 Chemical structures of antimicrotubule agents synthesized via click chemistry. |
In 2011, Manach et al synthesized possible taxol (microtubule-stabilizing antitubulin agents) substitutes (37a and 37b, Figure 23) by a stereoselective β-glycosylation of l-glucurono-γ-lactone followed by a click cycloaddition of aromatic structures. This synthetic route is very simple and efficient and can provide fast access to a small library of compounds.72
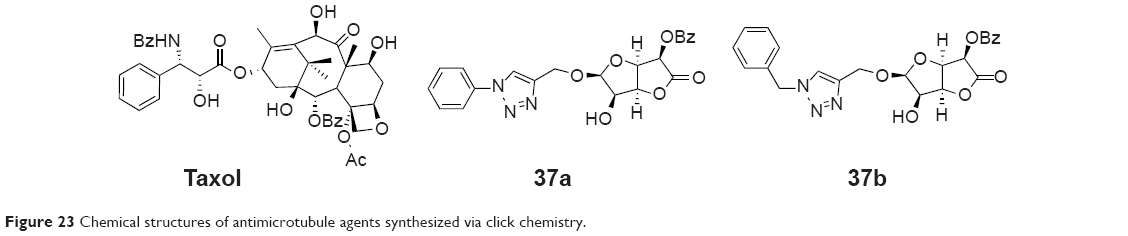 | Figure 23 Chemical structures of antimicrotubule agents synthesized via click chemistry. |
Odlo et al reported a series of cis-restricted 1,5-disubstituted 1,2,3-triazole analogs of combretastatin A (an attractive lead compound in the development of new anticancer agents) using click chemistry in 2008. Compound 38 (Figure 24), 2-methoxy-5-(1-(3,4,5-trimethoxyphenyl)-1H-1,2,3-triazol-5-yl)aniline, inhibited tubulin polymerization with an IC50 value of 4.8 μM. Molecular modeling experiments revealed that the triazole moiety interacts with β-tubulin by means of hydrogen bonding with several amino acids.73
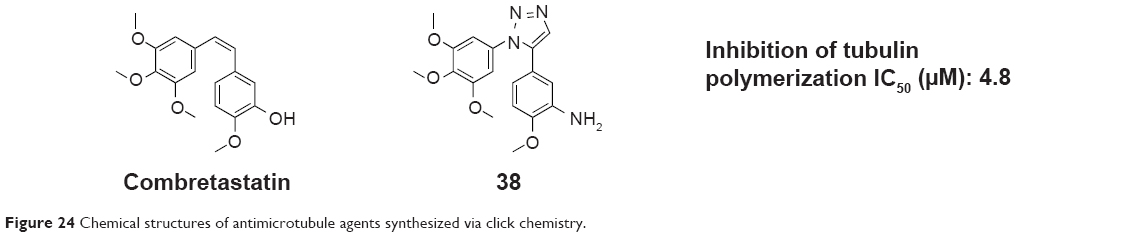 | Figure 24 Chemical structures of antimicrotubule agents synthesized via click chemistry. |
Chalcones (chalcone 1, Figure 25) are open-chained molecules consisting of two aromatic rings linked by a three-carbon enone fragment, and some of them have the ability to inhibit tubulin polymerization. Mesenzani et al reported a small class of antitubulin agents using click chemistry in 2010. They replaced the olefinic portion of chalcones with metabolically stable and chemically inert heterocyclic rings. Compound 39 (Figure 25) retained antitubulin activity. The biologic data showed that click chalcones were inactive, and suggested that the olefinic ring might not be merely a structural linker.74
 | Figure 25 Chemical structures of antimicrotubule agents synthesized via click chemistry. |
To understand the precise binding mode of the diketo-piperazine-based antimicrotubule agent plinabulin on tubulin in 2010, Yamazaki et al reported a new bioactive photoaffinity probe KPU-252-B1 (40, Figure 26), possessing a biotin tag on the oxazole ring of a potent plinabulin derivative KPU-244, which was synthesized via the Cu(I)-catalyzed Huisgen’s cycloaddition reaction. The results revealed that the probe KPU-252-B1 was recognized to be located around the intradimer space between the α- and β-tubulin units, which was near the colchicine-binding site. The probe showed significant binding affinity toward tubulin and cytotoxicity against HT-29 cells.75
 | Figure 26 Chemical structures of antimicrotubule agents synthesized via click chemistry. |
Conclusion
Click chemistry is one of the powerful tools for the construction of heteroatom links (C–X–C) for chemistry and biology in anticancer agents development and investigation due to the reliability, specificity, and biocompatibility of 1,2,3-triazole. Click chemistry has the potential to shorten procedures and render more efficient lead identification and optimization procedures in medicinal chemistry. Research and development in this field are still increasing exponentially. The review discusses the use of click chemistry in the area of synthesis of agents with anticancer activity and intends to demonstrate the diversity of applications of this chemical strategy in the area of drug discovery. However, we are still in the early developmental stages of this concept-driven research. Though this reaction can be used for integration or linkage of functional molecules by 1,2,3-triazole, which may form the basis of small-molecule pharmaceutical leads and improve their pharmacological activities, 1,2,3-triazole mainly acts as a linker more than a bioisostere in the current studies, indicating that 1,2,3-triazole may not be a good bioisostere. And the fact that there have been few drugs that bear 1,2,3-triazole ring currently being sold on the market indicates the limitations in the use of 1,2,3-triazole as bioisostere and linker in the drug discovery. Not only that, the copper safety in the body and the biocompatibility of 1,2,3-triazoles also make this reaction far from perfect in terms of its wide application in medicinal chemistry at this stage. What is more, the reaction’s narrow application that mostly focuses on the reactions between terminal acetylenes and azides, the single type of catalyst used in the reaction, and the easily oxidized Cu(I) also limit the overall development of click chemistry. Therefore, further studies in this field should be carried out, and other cores similar to 1,2,3-trizole that exhibit more biocompatibility should also be designed based on the concept of click chemistry for the better applications of this chemical strategy.
Disclosure
The authors report no conflicts of interest in this work.
References
Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed. 2001;40(11):2004–2021. | ||
Wu P, Feldman AK, Nugent AK, et al. Efficiency and fidelity in a click-chemistry route to triazoledendrimers by the copper (I)-catalyzed ligation of azides and alkynes. Angew Chem Int Ed. 2004;43(30):3928–3932. | ||
Huisgen R. 1,3-dipolar cycloadditions. Past and future. Angew Chem Int Ed Engl. 1963;2(10):565–598. | ||
Thirumurugan P, Matosiuk D, Jozwiak K. Click chemistry for drug development and diverse chemical – biology applications. Chem Rev. 2013;113(7):4905–4979. | ||
Chemama M, Fonvielle M, Arthur M, Valéry JM, Quelquejeu ME. Synthesis of stable aminoacyl-tRNA analogues containing triazole as a bioisoster of esters. Chemistry. 2009;15(8):1929–1938. | ||
Fray MJ, Bull DJ, Carr CL, Gautier ECL, Mowbray CE, Stobie A. Structure-activity relationships of 1,4-dihydro-(1H, 4H)-quinoxaline-2,3-diones as N-methyl-D-aspartate (glycine site) receptor antagonists. 1. Heterocyclic substituted 5-alkyl derivatives. J Med Chem. 2001;44(12):1951–1962. | ||
Taddei M, Ferrini S, Giannotti L, et al. Synthesis and evaluation of new Hsp90 inhibitors based on a 1,4,5-trisubstituted 1,2,3-triazole scaffold. J Med Chem. 2014;57(6):2258–2274. | ||
Altimari JM, Niranjan B, Risbridger GP, et al. Synthesis and preliminary investigations into novel 1,2,3-triazole-derived androgen receptor antagonists inspired by bicalutamide. Bioorg Med Chem Lett. 2014;24(21):4948–4953. | ||
Vatmurge NS, Hazra BG, Pore VS, Shirazi F, Chavan PS, Deshpande MV. Synthesis and antimicrobial activity of beta-lactam-bile acid conjugates linked via triazole. Bioorg Med Chem Lett. 2008;18:2043–2047. | ||
Whiting M, Tripp JC, Lin YC, et al. Rapid discovery and structure-activity profiling of novel inhibitors of human immunodeficiency virus type 1 protease enabled by the copper (I)-catalyzed synthesis of 1,2,3-triazoles and their further functionalization. J Med Chem. 2006;49(26):7697–7710. | ||
Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discov Today. 2003;8(24):1128–1137. | ||
Moses JE, Moorhouse AD. The growing applications of click chemistry. Chem Rev. 2007;36(8):1249–1262. | ||
Finn MG, Fokin VV. Click chemistry: function follows form. Chem Soc Rev. 2010;39(4):1231–1232. | ||
Jordan A, Hadfield JA, Lawrence NJ, McGown AT. Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med Res Rev. 1998;18(4):259–296. | ||
Reddy DM, Srinivas J, Chashoo G, Saxena AK, Kumar HMS. 4β-[(4-Alkyl)-1,2,3-triazol-1-yl] podophyllotoxins as anticancer compounds: design, synthesis and biological evaluation. Eur J Med Chem. 2011;46:1983–1991. | ||
Liu JF, Sang CY, Xu XH, et al. Synthesis and cytotoxic activity on human cancer cells of carbamate derivatives of 4β-(1,2,3-triazol-1-yl)podophyllotoxin. Eur J Med Chem. 2013;64:621–628. | ||
Wu WB, Ou JB, Huang ZH, et al. Synthesis and evaluation of mansonone F derivatives as topoisomerase inhibitors. Eur J Med Chem. 2011;46(8):3339–3347. | ||
Liu Z, Huang SL, Li MM, Huang ZS, Lee KS. Inhibition of thioredoxinreductase by mansonone F analogues: implications for anticancer activity. Chem Biol Interact. 2009;177(1):48–57. | ||
Huang ZH, Zhuo ST, Li CY, Xie HT, Li D, Tan JH. Design, synthesis and biological evaluation of novel mansonone E derivatives prepared via CuAAC click chemistry as topoisomerase II inhibitors. Eur J Med Chem. 2013;68:58–71. | ||
Kaushik CP, Lal K, Kumar A, Kumar S. Synthesis and biological evaluation of amino acid-linked 1,2,3-bistriazole conjugates as potential antimicrobial agents. Med Res Rev. 2013;23:1–10. | ||
Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. | ||
Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–784. | ||
Emanuele S, Lauricella M, Tesoriere G. Histone deacetylase inhibitors: apoptotic effects and clinical implications. (Review). Int J Oncol. 2008;33(4):637-646. | ||
Miller TA, Witter DJ, Belvedere S. Histone deacetylase inhibitors. J Med Chem. 2003;46(24):5097–5116. | ||
Chen Y, Lopez-Sanchez M, Savoy DN, Billadeau DD, Dow GS, Kozikowski AP. A series of potent and selective, triazolyphenyl-based histone deacetylasea inhibitors with activity against pancreatic cancer cells and plasmodium falciparum. J Med Chem. 2008;51(12):3437–3448. | ||
Shen J, Woodward R, Kedenburg JP, et al. Histone deacetylase inhibitors through click chemistry. J Med Chem. 2008;51(23):7417–7427. | ||
Hou J, Feng C, Li Z, et al. Structure-based optimization of click-based histone deacetylase inhibitors. Eur J Med Chem. 2011;46(8):3190–3200. | ||
Hou J, Li Z, Fang Q, et al. Discovery and extensive in vitro evaluations of NK-HDAC-1: a chiral histone deacetylase inhibitor as a promising lead. J Med Chem. 2012;55(7):3066–3075. | ||
Chen PC, Patil V, Guerrant W, Green P, Oyelere AK. Synthesis and structure-activity relationship of histone deacetylase (HDAC) inhibitors with triazole-linked cap group. Bioorg Med Chem. 2008;16:4839–4853. | ||
Sun Q, Yao Y, Liu C, et al. Design, synthesis, and biological evaluation of novel histone deacetylase 1 inhibitors through click chemistry. Bioorg Med Chem Lett. 2013;23(11):3295–3299. | ||
Horne WS, Olsen CA, Beierle JM, Montero A, Ghadiri MR. Probing the bioactive conformation of an archetypal natural product HDAC inhibitor with conformationally homogeneous triazole-modified cyclic tetrapeptides. Angew Chem Int Ed. 2009;48(26):4718–4724. | ||
Canzoneri JC, Chen PC, Oyelere AK. Design and synthesis of novel histone deacetylase inhibitor derived from nuclear localization signal peptide. Bioorg Med Chem Lett. 2009;19(23):6588–6590. | ||
Suzuki T, Ota Y, Kasuya Y, et al. An unexpected example of protein-templated click chemistry. Angew Chem Int Ed. 2010;49(38):6817–6820. | ||
Suzuki T, Ota Y, Ri M, et al. Rapid discovery of highly potent and selective inhibitors of histone deacetylase 8 using click chemistry to generate candidate libraries. J Med Chem. 2012;55(22):9562–9575. | ||
Suzuki T, Muto N, Bando M, et al. Design, synthesis, and biological activity of NCC149 derivatives as histone deacetylase 8-selective inhibitors. Chem Med Chem. 2014;9(3):657–664. | ||
Suzuki T, Kasuya Y, Itoh Y, et al. Identification of highly selective and potent histone deacetylase 3 inhibitors using click chemistry-based combinatorial fragment assembly. PLoS One. 2013;8(7):e68669. | ||
Taori K, Paul VJ, Luesch H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J Am Chem Soc. 2008;130(6):1806–1807. | ||
Li X, Tu Z, Li H, et al. Biological Evaluation of new largazole analogues: alteration of macrocyclic scaffold with click chemistry. ACS Med Chem Lett. 2012;4(1):132–136. | ||
Spencer J, Amin J, Wang M, et al. Click JAHAs: conformationally restricted ferrocene-based histone deacetylase inhibitors. Med Chem Comm. 2012;3(1):61–64. | ||
Meng Q, Li F, Jiang S, Li Z. Novel 64Cu-Labeled CUDC-101 for in vivo PET Imaging of histone deacetylases. ACS Med Chem Lett. 2013;4(9):858–862. | ||
Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22(4):337–358. | ||
Martin GS. The hunting of the Src. Nat Rev Mol Cell Biol. 2001;2:467–475. | ||
Schlessinger J. New roles for Src kinases in control of cell survival and angiogenesis. Cell. 2000;100(3):293–296. | ||
Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002;1602(2):114–130. | ||
Fizazi K. The role of Src in prostate cancer. Ann Oncol. 2007;18(11):1765–1773. | ||
Kumar D, Reddy VB, Kumar A, Mandal D, Tiwari R, Parang K. Click chemistry inspired one-pot synthesis of 1,4-disubstituted 1,2,3-triazoles and their Src kinase inhibitory activity. Bioorg Med Chem Lett. 2011;21(1):449–452. | ||
Kumar A, Ahmad I, Chhikara BS, Tiwari R, Mandal D, Parang K. Synthesis of 3-phenylpyrazolopyrimidine-1,2,3-triazole conjugates and evaluation of their Src kinase inhibitory and anticancer activities. Bioorg Med Chem Lett. 2011;21(5):1342–1346. | ||
Kalesh KA, Liu K, Yao SQ. Rapid synthesis of Abelson tyrosine kinase inhibitors using click chemistry. Org Biomol Chem. 2009;7(24):5129–5136. | ||
Li Y, Shen M, Zhang Z, et al. Design, synthesis, and biological evaluation of 3-(1 H-1,2,3-triazol-1-yl) benzamide derivatives as potent pan Bcr-Abl inhibitors including the threonine315→ isoleucine315 mutant. J Med Chem. 2012;55(22):10033–10046. | ||
Peruzzotti C, Borrelli S, Ventura M, et al. Probing the binding site of Abl tyrosine kinase using in situ click chemistry. ACS Med Chem Lett. 2013;4(2):274–277. | ||
Bazley LA, Gullick WJ. The epidermal growth factor receptor family. Endocr Relat Cancer. 2005;12(suppl 1):S17–S27. | ||
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. | ||
Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–354. | ||
Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–225. | ||
Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355–365. | ||
Maguire HC Jr, Greene MI. The neu (c-erbB-2) oncogene. Semin Oncol. 1989;16:148. | ||
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. | ||
Koeppen HK, Wright BD, Burt AD, et al. Overexpression of HER2/neu in solid tumours: an immunohistochemical survey. Histopathology. 2001;38(2):96–104. | ||
Cheng ZY, Li WJ, He F, Zhou JM, Zhu XF. Synthesis and biological evaluation of 4-aryl-5-cyano-2-H-1,2,3-triazoles as inhibitor of HER2 tyrosine kinase. Bioorg Med Chem. 2007;15(3):1533–1538. | ||
Kobus D, Giesen Y, Ullrich R, Backes H, Neumaier B. A fully automated two-step synthesis of an 18F-labelled tyrosine kinase inhibitor for EGFR kinase activity imaging in tumors. Appl Radiat Isot. 2009;67:1977–1984. | ||
Pisaneschi F, Nguyen QD, Shamsaei E, et al. Development of a new epidermal growth factor receptor positron emission tomography imaging agent based on the 3-cyanoquinoline core: synthesis and biological evaluation. Bioorg Med Chem Lett. 2010;18(18):6634–6645. | ||
Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235. | ||
van Rhijn BWG, Lurkin I, Radvanyi F, Kirkels WJ, van der Kwast TH, Zwarthoff EC. The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Res. 2001;61(4):1265–1268. | ||
Chesi M, Brents LA, Ely SA, et al. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001;7(3):729–736. | ||
Rosty C, Aubriot MH, Cappellen D, et al. Clinical and biological characteristics of cervical neoplasias with FGFR3 mutation. Mol Cancer. 2005;4(1):15. | ||
Le Corre L, Girard AL, Aubertin J, et al. Synthesis and biological evaluation of a triazole-based library of pyrido [2,3-d] pyrimidines as FGFR3 tyrosine kinase inhibitors. Org Biomol Chem. 2010;8(9):2164–2173. | ||
Perez EA. Microtubule inhibitors: differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol Cancer Ther. 2009;8(8):2086–2095. | ||
Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–265. | ||
Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9(10):790–803. | ||
Stefely JA, Palchaudhuri R, Miller PA, et al. N-((1-Benzyl-1 H-1,2,3-triazol-4-yl)methyl)arylamide as a new scaffold that provides rapid access to antimicrotubule agents: synthesis and evaluation of antiproliferative activity against select cancer cell lines. J Med Chem. 2010;53(8):3389–3395. | ||
Imperio D, Pirali T, Galli U, et al. Replacement of the lactone moiety on podophyllotoxin and steganacin analogues with a 1,5-disubstituted 1,2,3-triazole via ruthenium-catalyzed click chemistry. Bioorg Med Chem Lett. 2007;15(21):6748–6757. | ||
Manach CL, Baron A, Guillot R, Vauzeilles B, Beau JM. Design and synthesis by click triazole formation of paclitaxel mimics with simplified core and side-chain structures. Tetrahedron Lett. 2011;52(13):1462–1465. | ||
Odlo K, Hentzen J, dit Chabert JF, et al. 1,5-disubstituted 1,2,3-triazoles as cis-restricted analogues of combretastatin A-4: synthesis, molecular modeling and evaluation as cytotoxic agents and inhibitors of tubulin. Bioorg Med Chem Lett. 2008;16(9):4829–4838. | ||
Mesenzani O, Massarotti A, Giustiniano M, et al. Replacement of the double bond of antitubulin chalcones with triazoles and tetrazoles: synthesis and biological evaluation. Bioorg Med Chem Lett. 2011;21(2): 764–768. | ||
Yamazaki Y, Kido Y, Hidaka K, et al. Tubulin photoaffinity labeling study with a plinabulin chemical probe possessing a biotin tag at the oxazole. Bioorg Med Chem Lett. 2011;19(1):595–602. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.