")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Targeting the D1-N-methyl-D-aspartate receptor complex reduces L-dopa-induced dyskinesia in 6-hydroxydopamine-lesioned Parkinson’s rats
Authors Song L, Zhanzhao Z, Hu R, Cheng J, Li L, Fan Q, Wu N, Gan J, Zhuo M, Liu Z
Received 31 July 2015
Accepted for publication 4 November 2015
Published 3 February 2016 Volume 2016:10 Pages 547—555
DOI https://doi.org/10.2147/DDDT.S93487
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Wei Duan
Lu Song,1,* Zhanzhao Zhang,2,* Rongguo Hu,1 Jie Cheng,1 Lin Li,1 Qinyi Fan,1 Na Wu,1 Jing Gan,1 Mingzhu Zhou,1 Zhenguo Liu1
1Department of Neurology, Xinhua Hospital, 2Department of Plastic and Reconstructive Surgery, Shanghai 9th People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: L-3,4-dihydroxyphenylalanine (L-dopa) remains the most effective therapy for Parkinson’s disease (PD), but its long-term administration is associated with the development of debilitating motor complications known as L-dopa-induced dyskinesia (LID). Enhanced function of dopamine D1 receptor (D1R) and N-methyl-d-aspartate receptor (NMDAR) is believed to participate in the pathogenesis of LID. Given the existence of physical and functional interactions between D1R and NMDAR, we explored the effects of uncoupling D1R and NMDA GluN1 (GluN1) interaction on LID by using the Tat-conjugated interfering peptide (Tat-D1-t2). In this study, we demonstrated in 6-hydroxydopamine (6-OHDA)-lesioned PD rat model that intrastriatal injection of Tat-D1-t2 alleviated dyskinetic behaviors and downregulated the phosphorylation of DARPP-32 at Thr34 induced by levodopa. Moreover, we also showed intrastriatal administration of Tat-D1-t2 elicited alterations in membranous GluN1 and D1R expression. These findings indicate that D1R/GluN1 complexes may be a molecular target with therapeutic potential for the treatment of dyskinesia in Parkinson’s patients.
Keywords: 6-hydroxydopamine, Parkinson’s disease, dyskinesia, L-dopa, D1 receptor, NMDA, protein–protein interaction
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder caused by the loss of nigral dopaminergic neurons and the subsequent massive drop in dopamine (DA) content in the striatum.1 DA replacement therapy with L-3,4-dihydroxyphenylalanine (L-dopa) remains the most effective therapy to alleviate the motor symptoms of PD,1 even though its long-term administration induces development of involuntary movements, known as L-dopa-induced dyskinesia (LID).2 LID represents the most debilitating complication in the vast majority of patients3 and makes the treatment for PD even more difficult, especially during the advanced stages of PD.4
Despite the clinical importance of LID, the underlying molecular mechanisms leading to its development are far from clear. It is believed that the pathological mechanism of LID involves complex alterations of both dopaminergic and non-dopaminergic (eg, glutamatergic) neurotransmitter systems induced by the pulsatile stimulation of short-acting dopaminergic agents.5 Denervation-dependent D1 receptor (D1R) super-sensitivity is widely believed to be an important postsynaptic determinant of LID in animal models, although the exact mechanisms remain to be elucidated.6 There is an increasing evidence supporting the idea that enhancement of glutamate N-methyl-D-aspartate receptor (NMDAR) function may also contribute to the development of LID.7,8
The colocalization of D1R and NMDAR is extensively observed at synaptic, parasynaptic, and nonsynaptic sites in dendritic spines and shafts in striatum,9,10 which provides the structural basis for interaction. In particular, D1R directly couples to the GluN1 subunit of the NMDAR through the carboxyl tails of these receptors.11–13 Association with NMDAR facilitates D1R transportation to the cell surface and inhibits agonist-induced D1R internalization.12–14 The activation of D1R can in turn increase phosphorylation and potentiation of NMDA,15,16 and can also induce rapid distribution of NMDA from intracellular pools to the postsynaptic membrane.17 It is assumed that activation of NMDA promoted the recruitment of D1R to the plasma membrane, which in turn enhances the activity of NMDA and a positive feedback loop is created.18 This loop, if not controlled, might result in overfunction of both D1R and NMDA.19
Together with demonstrating that D1R–GluN1 association and the activation of both receptors are involved in the onset of LID, we aimed to investigate the role of the D1R–GluN1 interaction in LID. We examined the effects of disrupting the direct protein–protein interaction between D1R and GluN1 subunit of NMDAR using the interfering peptide.
Materials and methods
Animals
Adult Sprague Dawley female rats (180–220 g) were used in this study. Animals were housed under a 12/12 hours light/dark cycle in a controlled environment at a constant temperature of 23°C and humidity of 50%±10% with food and water available ad libitum. All protocols involving animals were approved by the Institutional Review Board of Xinhua Hospital and were performed according to the guidelines of the National Institutes of Health for the care and use of laboratory animals (NIH publication No 80-23).
Dopaminergic lesions
Rats were anesthetized with 7% chloral hydrate (0.5 mL/100 g, v/w), and then received an injection of a total of 8 μg of 6-OHDA (dissolved in 4 μL of 0.9% physiological saline containing 0.02% ascorbic acid [Sigma–Aldrich, St Louis, MO, USA]) using a stereotaxic apparatus (Narishige, Tokyo, Japan) equipped with a rat adaptor. The coordinates were calculated with reference to bregma for the anterioposterior (AP) and the mediolateral (ML) coordinates using the rat brain atlas20 as follows: 1) AP −3.7 mm, ML −1.7 mm; 2) AP −4.4 mm, ML −1.2 mm. The dorsoventral position of all the injections was −7.8 mm below the dura and the tooth bar was set to −2.4 mm. Three weeks after surgery, rats were tested with a subcutaneous injection of apomorphine at 0.05 mg/kg (Sigma–Aldrich, St Louis, MO, USA). Only those rats displaying a stable apomorphine-induced rotational behavior of at least seven full turns per minute contralateral to the lesioned side were selected for the next experiment. It has been previously demonstrated that rats meeting this criterion have a greater than 90% depletion of striatal DA.21
Drug treatment
Validated PD rats received vehicle or levodopa methylester (Sigma–Aldrich) injection intraperitoneally (ip) at 10 mg/kg in combination with benserazide (12.5 mg/kg) twice daily for 22 days.22 On the final day (day 22), levodopa-induced dyskinetic rats were randomly divided into three groups to receive intrastriatal administration of Tat-fusion interfering peptide (Tat-D1Ri; ChinaPeptides Co., Ltd, Shanghai, People’s Republic of China), Tat-fusion control peptide (Tat-D1Rc; ChinaPeptides), or saline individually. The Tat-D1Ri contains a Tat cell membrane transduction domain (YGRKKRRQRRR) and a core region of GluN1 binding motif (the D1R C-tail from L387 to L416) to prevent the interaction between GluN1 subunit and D1R. A scramble nonsense version of the peptide, Tat-D1Rc, was also generated. Four hours after complete recovery from anesthesia, rats were treated with levodopa and benserazide. For intrastriatal injection, rats were anesthestized with 7% chloral hydrate (0.5 mL/100 g, v/w). A volume of 1 μL of Tat-D1Ri (0.5 nmol), Tat-D1Rc (0.5 nmol),23 or saline was injected at the coordinates: AP =0.5 mm from bregma; ML =−2.5 mm from midline; and DV =−4.2 from the dura surface.
Behavioral assessment
To evaluate the severity of dyskinesia induced by levodopa treatment, each rat was assessed for exhibition of axial, limb, and orolingual movements (ALO AIM) as detailed in Lindenbach et al.24 In brief, three subtypes of AIM were assessed every 20 minutes (a monitoring period of 60 seconds for each) during an entire period of 180 minutes following levodopa treatment. Each of these three subtypes was scored on a four-point severity scale as follows: 0= absent; 1= present during less than half of the observation time; 2= present for more than half of the observation time; 3= present all the times but suppressible by external stimuli; and 4= present all the times and not suppressible by external stimuli. The ALO AIM were tested at 2, 7, 14, and 21 days during the period of levodopa treatment. The rats with low AIM scores were excluded. Only rats with moderate-to-severe AIMs (severity grade ≥2 on each of the three AIM subtypes)25 received intrastriatal injection of Tat-D1Ri, Tat-D1Rc, or saline and underwent behavioral evaluation on the 22nd day. In brief, 4 hours after complete recovery from anesthesia, levodopa was administrated and behavioral assessments were then performed.
Coimmunoprecipitation and immunoblot
A group of normal rats also received intrastriatal injection of Tat-D1Ri, Tat-D1Rc, or saline for coimmunoprecipitation experiment. Briefly, animals were deeply anesthetized with 7% chloral hydrate (0.5 mL/100 g, v/w) and decapitated 4 hours after intrastriatal injection of peptide or saline. Rat striatal tissue was homogenized by sonication in an immunoprecipitation lysis buffer (Beyotime, Haimen, People’s Republic of China) plus a protease inhibitor cocktail (Hoffman-La Roche Ltd., Basel, Switzerland), and then centrifuged at 800× g for 10 minutes at 4°C. The supernatant was collected and centrifuged at 11,000× g for 30 minutes at 4°C. The pellet was resuspended in the lysis buffer and used for coimmunoprecipitation. Samples were incubated with a rabbit antibody against D1R (EMD Millipore, Billerica, MA, USA) or a mouse antibody against GluN1 (Millipore) overnight at 4°C. The complex was precipitated with protein G agarose beads or protein A agarose beads by gentle rocking for 3 hours at 4°C. Samples were suspended in a buffer containing 0.5% sodium dodecyl sulfate (SDS) and boiled for 5 minutes. Proteins were resolved by SDS-PAGE (polyacrylamide gel electrophoresis), transferred onto polyvinylidene difluoride membrane (Millipore). Membranes were blocked in 5% nonfat milk for 1 hour at room temperature and incubated with a rabbit primary antibody against D1R (Millipore) or a mouse primary antibody against GluN1 (Millipore) overnight at 4°C. Membranes were then incubated with horseradish peroxidase conjugated secondary antibodies (1:1,000) for 1 hour at room temperature. Immunoblots were developed with the enhanced electrochemiluminescence reagent (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA) and captured by a Bio-Rad molecular imager.
Western blot
Animals were deeply anesthetized with 7% chloral hydrate (0.5 mL/100 g, v/w) and decapitated. Brains were quickly removed and the striatum was dissected. To pellet the cytosol fractions, three to five striatal tissues in every group were homogenized (1:10, w:v) in a homogenization buffer containing 20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1% Triton-100, 1 mM NaF, 100 μM phenylmethylsulfonyl fluoride, and freshly-added protease inhibitor cocktail (Calbiochem, La Jolla, CA, USA). Cytosol fractions used for the detection of DARPP-32 were prepared by centrifugation at 12,000× g for 10 minutes at 4°C. The remaining striatal tissues were homogenized and centrifuged using Membrane Protein Extraction Kit (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s instructions to pellet the membrane fraction. Protein concentrations were determined with a Pierce BCA assay kit (Rockford, IL, USA). Protein samples (20 μg) were separated on a 10% SDS–polyacrylamide gel and electrophoretically transferred to PVDF membranes in Tris–glycine transfer buffer. The membranes were blocked in 5% (w/v) instant nonfat dried milk for 2 hours at room temperature, and incubated with primary antibodies against DARPP-32 (1:1,000; Cell Signaling Technology, Boston, MA, USA), phospho-DARPP-32 at Thr34 (1:500; Cell Signaling Technology), GluN1 (1:500; Millipore), GluN2A (1:500; Millipore), GluN2B (1:500; Millipore), D1R (1:500; Millipore), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1,000; Boster, Wuhan, People’s Republic of China) at 4°C overnight. The membranes were subsequently washed with TBST (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, and 0.05% Tween 20) and incubated with secondary horseradish peroxidase-conjugated IgG (1:1,000) for 1 hour at room temperature. Immunoreactive bands were visualized using chemiluminescence (ECL Kit; Pierce Biotechnology) and captured by a Bio-Rad molecular imager. Protein bands were scanned with Image-Pro plus 6.0 analyses Software. The band densities were calculated with a computerized image analysis system (Image Lab, Bio-Rad Laboratories, Hercules, CA, USA) and normalized with that of GAPDH. For the analysis of GluN1, the optical density (OD) values of blot bands were normalized with PD control.
Statistics
Data are presented as mean ± SEM (standard error of the mean). Statistical analysis was conducted by one-way analysis of variance (ANOVA), followed by least significant difference post hoc comparisons. Analysis for D1R expression was conducted by independent-samples t-test. A P-value of less than 0.05 was considered statistically significant. SPSS17.0 and Graphpad prism 5 were used for statistics and graphics.
Results
Effects of intrastriatal administration of Tat-D1Ri on D1R–GluN1 interactions
As the GluN1 binding region in D1R C-termini was identified previously,11,13 we synthesized a Tat-D1Ri to prevent the interaction between GluN1 subunit and D1R. We also synthesized a Tat-D1Rc as control. In order to validate the efficacy and selectivity of Tat-D1Ri in disrupting D1R–GluN1 interactions in striatal neurons of adult rat brains in vivo, Tat-D1Ri, Tat-D1Rc, or saline were locally injected into the rat striatum at a rate of 0.2 μL/min. The intrastriatal injection of Tat-D1Ri caused a reduction in D1R–GluN1 interactions, which was verified by the coimmunoprecipitation experiments (Figure 1). In contrast to Tat-D1Ri, Tat-D1Rc did not alter the D1R–GluN1 interactions (Figure 1).
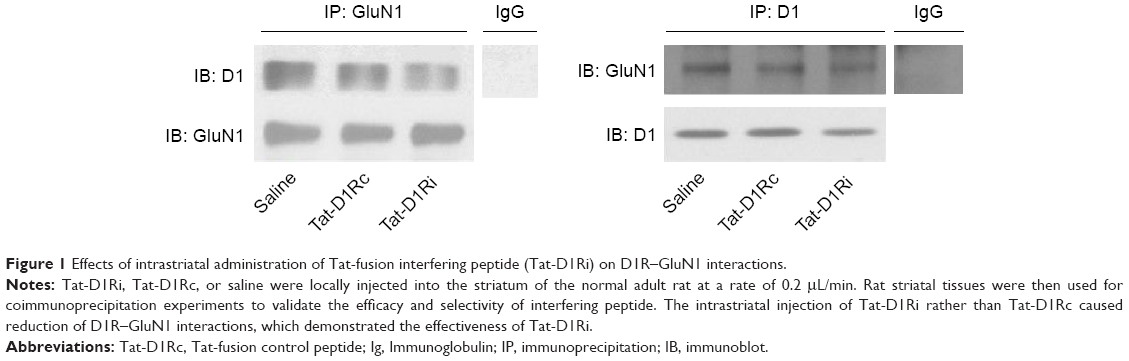 | Figure 1 Effects of intrastriatal administration of Tat-fusion interfering peptide (Tat-D1Ri) on D1R–GluN1 interactions. |
Effects of intrastriatal administration of Tat-D1Ri on established AIMs in 6-OHDA-lesioned rats
After confirmation of the effectiveness of Tat-D1Ri on disrupting the D1R–GluN1 interactions, we tested whether intrastriatal administration of Tat-D1Ri alters levodopa-induced dyskinesia. We subjected 6-OHDA-lesioned rats to twice-daily injection of levodopa (10 mg/kg/each, ip) with benserazide (12.5 mg/kg/each, ip) twice daily for 21 days to produce LID. At day 22, the successful LID rats were randomly divided to receive intrastriatal administration of Tat-D1Ri, Tat-D1Rc, or saline individually. Each rat was then assessed for exhibition of ALO AIM. Interestingly, at day 22, intrastriatal infusion of Tat-D1Ri alleviated the dyskinetic behavior manifested by the reduction of ALO AIM scores (P<0.05, Figure 2). In contrast, intrastriatal infusion of Tat-D1Rc or saline had no effect on dyskinetic behaviors.
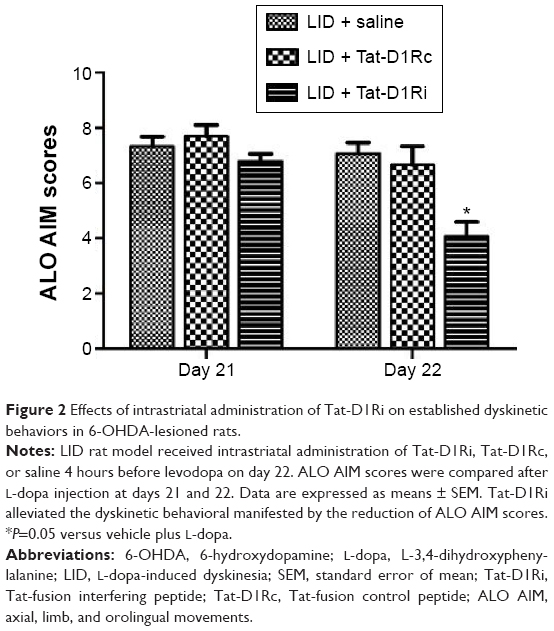 | Figure 2 Effects of intrastriatal administration of Tat-D1Ri on established dyskinetic behaviors in 6-OHDA-lesioned rats. |
Effects of intrastriatal administration of Tat-D1Ri on DARPP-32 phosphorylation level after levodopa treatment in 6-OHDA-lesioned rats
As previous studies have indicated the causal role for striatal hyperphosphorylation of DA- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) at Thr34 in the development of LID in the Parkinson’s model rats,26,27 we examined whether intrastriatal administration of Tat-D1Ri could regulate the phosphorylation level of DARPP-32 at Thr34. We observed no significant changes in the total levels of DARPP-32. The phosphorylation level of DARPP-32 at Thr34 in striatum was upregulated in L-dopa-treated dyskinetic rats with intrastriatal administration of saline, which is in accordance with a previous study.28 Remarkably, the upregulation of phosphorylation of DARPP-32 at Thr34 was apparently prevented in L-dopa-treated dyskinetic rats with intrastriatal administration of Tat-D1Ri, but not with Tat-D1Rc (P<0.05, Figure 3).
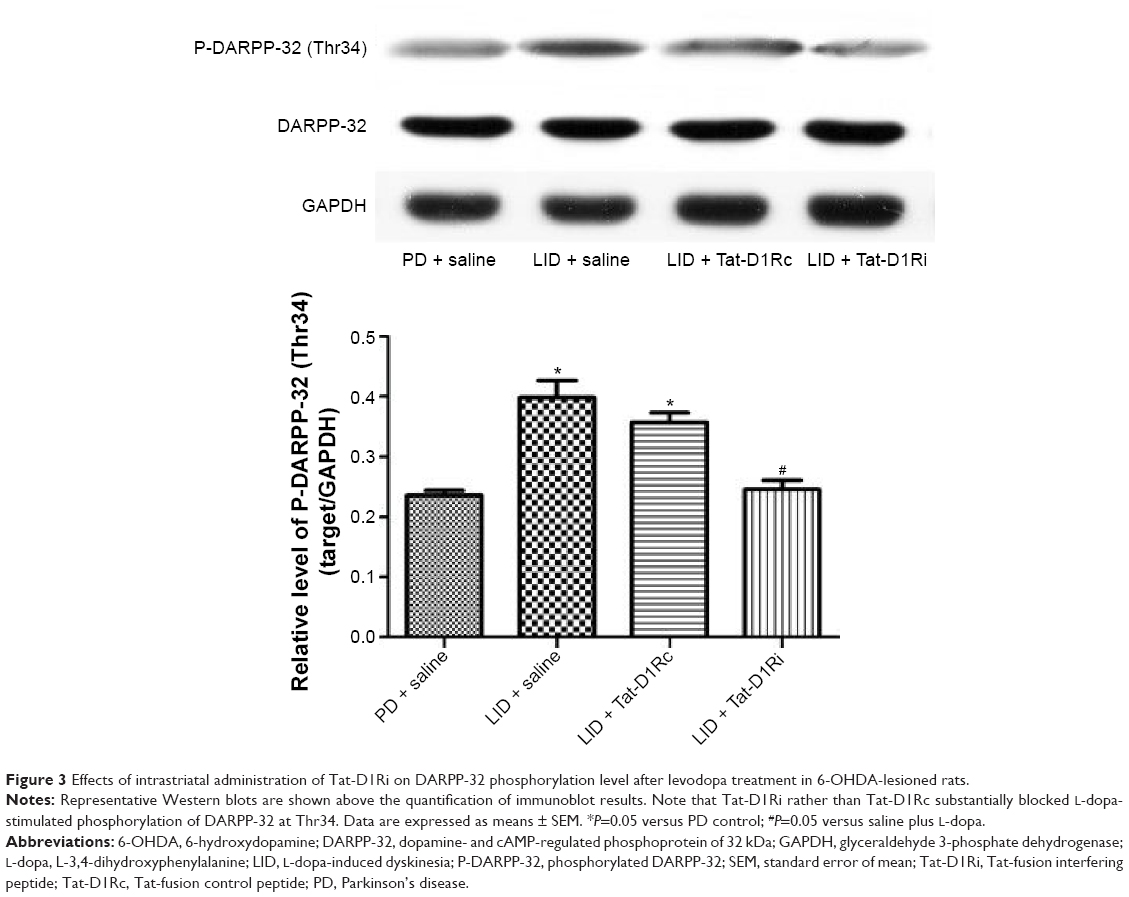 | Figure 3 Effects of intrastriatal administration of Tat-D1Ri on DARPP-32 phosphorylation level after levodopa treatment in 6-OHDA-lesioned rats. |
Effects of intrastriatal administration of Tat-D1Ri on membrane NMDA subunit and D1R expression
We then evaluated the consequences of Tat-D1Ri treatment on NMDA subunit expression. We began by examining GluN1 because it is an obligatory NMDA subunit. As shown in Figure 4, there was no significant alteration of GluN1 expression in the membrane compartments among PD + saline, LID + saline, and LID + Tat-D1Rc groups. Interestingly, intrastriatal administration of Tat-D1Ri induced significant decrease of GluN1 expression in the membrane fraction in L-dopa-treated dyskinetic rats, but no parallel alterations in the expression of NMDA GluN2A (GluN2A) and NMDA GluN2B (GluN2B) were found in all the experimental groups. Considering the interaction of GluN1 with D1R, we then examined whether intrastriatal administration of Tat-D1Ri may influence the D1R expression. As shown in Figure 5, unlike the GluN1 subunit, the amount of D1R located in membrane was increased in L-dopa-treated dyskinetic rats with intrastriatal administration of Tat-D1Ri, when compared to Tat-D1Rc.
 | Figure 4 Effects of intrastriatal administration of Tat-D1Ri on membrane NMDA subunit expression. |
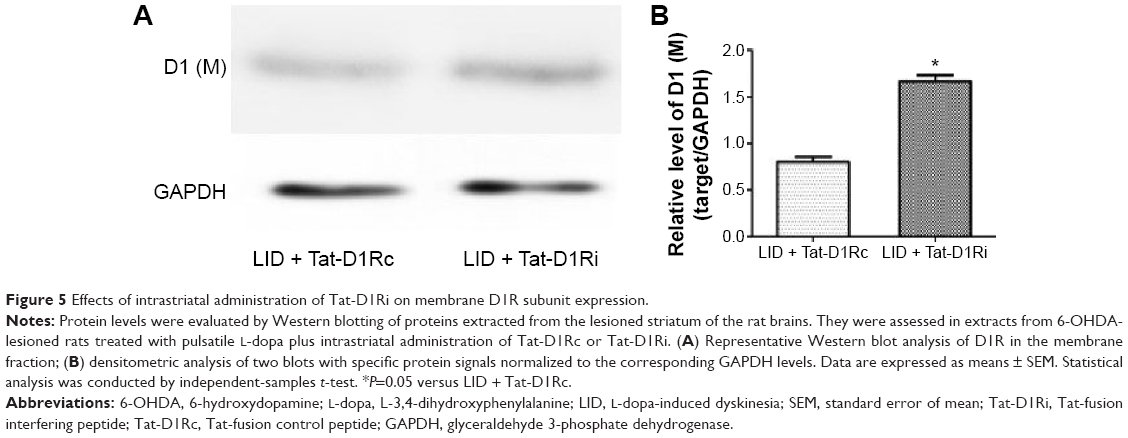 | Figure 5 Effects of intrastriatal administration of Tat-D1Ri on membrane D1R subunit expression. |
Discussion
This study investigated the effect of intrastriatal administration of Tat-D1Ri on behavioral and neurochemical changes in LID rat. We first verified the effectiveness of Tat-D1Ri by showing that the intrastriatal administration of Tat-D1Ri can decrease the interaction of D1R with NMDA (Figure 1). Then, we demonstrated that intrastriatal administration of Tat-D1Ri can reduce established dyskinetic severity and phosphorylation level of DARPP-32 at Thr34 in L-dopa-treated dyskinetic rats. Our next aim was to investigate whether the Tat-D1Ri administration could influence the expression of NMDAR and D1R. We found intrastriatal administration of Tat-D1Ri did not alter GluN2A and GluN2B expression, but downregulated the expression of GluN1 subunit and slightly increased membrane D1R expression in L-dopa-treated dyskinetic rats.
The interfering peptide (Tat-D1Ri) used in this study was synthesized with a Tat cell membrane transduction domain (YGRKKRRQRRR) and a core region of D1R–GluN1 binding motif. The arginine-enriched Tat domain renders cell permeability29 and the binding motif may compete with endogenous D1Rs for GluN1 binding. The Tat-D1Ri peptide was shown to inhibit the interaction between D1R and GluN1 subunit of NMDA in the recording pipettes in a previous study,11 and was used in other studies to prevent the physical interaction of D1R with GluN1 subunit of NMDA.30,31
LID has been viewed as an aberrant form of motor learning resulting from DA and glutamate-dependent molecular alterations at corticostriatal synapses.32,33 Given the interaction of D1R and NMDA, we used the interfering peptide to observe its impact on LID. ALO score has been used to evaluate the severity of dyskinesia and potential antidyskinetic drugs for PD.34 The reduction of ALO score induced by the intrastriatal administration of Tat-D1Ri, but not Tat-D1Rc, indicated the ability of Tat-D1Ri to reduce dyskinetic behavioral responses to levodopa. The pathogenesis of cocaine addiction is also believed to involve the abnormal synaptic plasticity controlled by DA and glutamate systems in MSN.35 It has been recently reported that the dissociation of D1R/GluN1 interactions remarkably prevents the development of psychomotor sensitization to cocaine.36
DARPP-32 is a key component of the canonical cAMP/cAMP-dependent protein kinase (PKA) signaling cascade activated by D1R.37 It also has been shown that LID is decreased by selective inactivation of DARPP-32 in striatonigral MSNs.38 The phosphorylation of DARPP-32 at Thr35 has been causally linked to the emergence of LID.26 As shown in this study, the effect of the intrastriatal administration of Tat-D1Ri on downregulation of phosphorylation of DARPP-32 at Thr35 was in accordance with the behavioral result, which indicated disruption of D1R–GluN1 interaction may alleviate LID.
As shown in this study, the membrane expression of GluN1, GluN2A, and GluN2B was similar between PD + saline and LID + saline groups, which is in agreement with a previous work.39 The intrastriatal administration of Tat-D1Ri induced significant decrease of GluN1 expression in the membrane fraction in L-dopa-treated dyskinetic rats, while leaving the expression of GluN2A and GluN2B subunits unchanged. The decrease of GluN1 expression may result in the redution of NMDA function. The elevated NMDA tone has been verified in LID animal model and PD patients expressing LID.40 Amantadine is licensed for the treatment of dyskinesia in PD, and its effectiveness supports the involvement of enhanced NMDA signaling in LID.41 DARPP-32 can also be activated by glutamate via different signaling cascades, partly by the NMDA-dependent pathway.42 It has been reported that uncoupling of the D1R–NMDA complex significantly inhibits NMDA-dependent long-term potentiation and induces working memory deficits.30 The effect of intrastriatal administration of Tat-D1Ri on LID may be related to tuning the function of NMDA.
Our results showed uncoupling of the D1R–NMDA complex by intrastriatal administration of Tat-D1Ri promoted the membrane expression of DA D1Rs in LID rat. It seems to be in contradiction with the notion that the NMDA activation can increase membrane expression of DA D1R via a direct protein–protein interaction between D1-t2 and GluN1-C1.13 However, due to the lack of obvious functional deficits in the brain DA system observed in NR1-KD mice, which shows a 90% reduction in GluN1 expression,43 it has been proposed that the observed increase in D1R surface expression, induced by NMDA activation, may be transient or restricted to certain neuronal populations in vivo.44 In addition, postsynaptic density-95 (PSD-95) and GluN1 bind an overlapping region on the carboxyl tails of D1R.45 PSD-95 functionally uncouples D1R trafficking and signaling from modulation by NMDA, which may be achieved through a direct, physical obstruction of D1R–GluN1 binding.45 Morever, PSD-95 is reported to play a direct role in governing the surface stabilization of D1R in the extrasynaptic compartment, as well as in the vicinity of corticostriatal synapses.23 Hence, the impact of intrastriatal administration of Tat-D1Ri on D1R surface expression may result from a competition between GluN1 and PSD-95 for binding to D1R, but this assumption requires further investigation.
Conclusion
In conclusion, our experiments provide evidence that disruption of D1R/GluN1 interaction by the interfering peptide may alleviate LID by downregulation of NMDA function. As simply blocking of particular receptors is often related to serious side effects, identifying drugs and peptides selectively targeting receptor–receptor interactions may represent a more efficient approach to coordinate and fine-tune pathological imbalances in neurotransmitter signaling.46 Given the oligomeric nature of D1R and NMDA, development of pharmacological agents to modulate the crosstalk between DA and glutamate should thus be considered for treating LID, which involves malfunctioning of D1R and NMDAR system.
Acknowledgment
The study was supported by the Projects of National Science Foundation of China (Nos 81171203, 81171204, 81200871, 81400925, and 81471148).
Disclosure
The authors report no conflicts of interest in this work.
References
Obeso JA, Olanou CW, Nutt JG. Levodopa motor complications in Parkinson’s disease. Trends Neurosci. 2000;23(Suppl 1):S2–S7. | ||
Bezard E, Brotchie JM, Gross CE. Pathophysiology of levodopa-induced dyskinesia: potential for new therapies. Nat Rev Neurosci. 2001;2(8):577–588. | ||
Bhidayasiri R, Truong DD. Motor complications in Parkinson disease: clinical manifestations and management. J Neurol Sci. 2008;266(1–2):204–215. | ||
Huot P, Johnston TH, Koprich J B, Fox SH, Brotchie JM. The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol Rev. 2013;65(1):171–222. | ||
Olanow CW, Obeso JA, Stocchi F. Continuous dopamine-receptor treatment of Parkinson’s disease: scientific rationale and clinical implications. Lancet Neurol. 2006;5(8):677–687. | ||
Cenci M, Ohlin K, Rylander D. Plastic effects of L-DOPA treatment in the basal ganglia and the relevance to the development of dyskinesia.Parkinsonism Relat Disord. 2009;15(Suppl 3):S59–S63. | ||
Robelet S, Melon C, Guillet B, Salin P, Kerkerian-Le Goff L. Chronic L-DOPA treatment increases extracellular glutamate levels and GLT1 expression in the basal ganglia in a rat model of Parkinson’s disease. Eur J Neurosci. 2004;20(5):1255–1266. | ||
Brotchie JM. Nondopaminergic mechanisms in levodopa-induced dyskinesia. Mov Dis. 2005;20(8):919–931. | ||
Hara Y, Pickel VM. Overlapping intracellular and differential synaptic distributions of dopamine D1 and glutamate N-methyl-D-aspartate receptors in rat nucleus accumbens. J Comp Neurol. 2005;492(4):442–455. | ||
Pickel VM, Colago EE, Mania I, Molosh AI, Rainnie DG. Dopamine D1 receptors co-distribute with N-methyl-D-aspartic acid type-1 subunits and modulate synaptically-evoked N-methyl-D-aspartic acid currents in rat basolateral amygdala. Neuroscience. 2006;142(3):671–690. | ||
Lee FJ, Xue S, Pei L, et al. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell. 2002;111(2):219–230. | ||
Fiorentini C, Gardoni F, Spano P, Di Luca M, Missale C. Regulation of dopamine D1 receptor trafficking and desensitization by oligomerization with glutamate N-methyl-D-aspartate receptors. J Biol Chem. 2003;278(22):20196–20202. | ||
Pei L, Lee FJ, Moszczynska A, Vukusic B, Liu F. Regulation of dopamine D1 receptor function by physical interaction with the NMDA receptors. J Neurosci. 2004;24(5):1149–1158. | ||
Scott L, Kruse MS, Forssberg H, Brismar H, Greengard P, Aperia A. Selective up-regulation of dopamine D1 receptors in dendritic spines by NMDA receptor activation. Proc Natl Acad Sci U S A. 2002;99(3):1661–1664. | ||
Lachowicz JE, Sibley DR. Molecular characteristics of mammalian dopamine receptors. Pharmacol Toxicol. 1997;81(3):105–113. | ||
Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78(1):189–225. | ||
Dunah AW, Standaert DG. Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J Neurosci. 2001;21(15):5546–5558. | ||
Cepeda C, Levine MS. Where do you think you are going? The NMDA-D1 receptor trap. Sci STKE. 2006;2006(333):pe20. | ||
Bozzi Y, Borrelli E. Dopamine in neurotoxicity and neuroprotection: what do D2 receptors have to do with it? Trends Neurosci. 2006;29(3):167–174. | ||
Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego: Academic Press; 2007. | ||
Papa SM, Engber TM, Kask AM, Chase TN. Motor fluctuations in levodopa treated parkinsonian rats: relation to lesion extent and treatment duration. Brain Res. 1994;662(1–2):69–74. | ||
Putterman DB, Munhall AC, Kozell LB, Belknap JK, Johnson SW. Evaluation of levodopa dose and magnitude of dopamine depletion as risk factors for levodopa-induced dyskinesia in a rat model of Parkinson’s disease. J Pharmacol Exp Ther. 2007;323(1):277–284. | ||
Porras G, Berthet A, Dehay B, et al. PSD-95 expression controls L-DOPA dyskinesia through dopamine D1 receptor trafficking. J Clin Invest. 2012;122(11):3977–3989. | ||
Lindenbach D, Ostock CY, Eskow Jaunarajs KL, et al. Behavioral and cellular modulation of L-DOPA-induced dyskinesia by beta-adrenoceptor blockade in the 6-hydroxydopamine-lesioned rat. J Pharmacol Exp Ther. 2011;337(3):755–765. | ||
Mabrouk OS, Mela F, Calcagno M, et al. GluN2A and GluN2B NMDA receptor subunits differentially modulate striatal output pathways and contribute to levodopa-induced abnormal involuntary movements in dyskinetic rats. ACS Chem Neurosci. 2013;4(5):808–816. | ||
Santini E, Valjent E, Usiello A, et al. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci. 2007;27(26):6995–7005. | ||
Santini E, Sgambato-Faure V, Li Q, et al. Distinct changes in cAMP and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. PLoS One. 2010;5(8):e12322. | ||
Brown AM, Deutch AY, Colbran RJ. Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. Eur J Neurosci. 2005;22(1):247–256. | ||
Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285(5433):1569–1572. | ||
Nai Q, Li S, Wang SH, et al. Uncoupling the D1-N-methyl-D-aspartate (NMDA) receptor complex promotes NMDA-dependent long-term potentiation and working memory. Biol Psychiatry. 2010;67(3):246–254. | ||
Ladepeche L, Yang L, Bouchet D, Groc L. Regulation of dopamine D1 receptor dynamics within the postsynaptic density of hippocampal glutamate synapses. PLoS One. 2013;8(9):e74512. | ||
Chase TN, Oh JD. Striatal dopamine- and glutamate-mediated dysregulation in experimental parkinsonism. Trends Neurosci. 2000;23(Suppl 10):S86–S91. | ||
Picconi B, Centonze D, Hakansson K, et al. Loss of bidirectional striatal plasticity in L-DOPA-induced dyskinesia. Nat Neurosci. 2003;6(5):501–506. | ||
Dekundy A, Lundblad M, Danysz W, Cenci MA. Modulation of L-DOPA-induced abnormal involuntary movements by clinically tested compounds: further validation of the rat dyskinesia model. Behav Brain Res. 2007;179(1):76–89. | ||
Pascoli V, Turiault M, Lüscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2011;481(7379):71–75. | ||
Cahill E, Pascoli V, Trifilieff P, et al. D1R/GluN1 complexes in the striatum integrate dopamine and glutamate signalling to control synaptic plasticity and cocaine-induced responses. Mol Psychiatry. 2014;19(12):1295–1304. | ||
Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294(5544):1024–1030. | ||
Bateup HS, Santini E, Shen W, et al. Distinct subclasses of medium spiny neurons differentially regulated striatal motor behaviors. Proc Natl Acad Sci U S A. 2010;107(33):14845–14850. | ||
Fiorentini C, Rizzetti MC, Busi C, et al. Loss of synaptic D1 dopamine/N-methyl-D-aspartate glutamate receptor complexes in L-DOPA-induced dyskinesia in the rat. Mol Pharmacol. 2006;69(3):805–812. | ||
Ahmed I, Bose SK, Pavese N, et al. Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain. 2011;134(4):979–986. | ||
Hallett PJ, Dunah AW, Ravenscroft P, et al. Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology. 2005;48(4):503–516. | ||
Nishi A1, Watanabe Y, Higashi H, Tanaka M, Nairn AC, Greengard P. Glutamate regulation of DARPP-32 phosphorylation in neostriatal neurons involves activation of multiple signaling cascades. Proc Natl Acad Sci U S A. 2005;102(4):1199–1204. | ||
Ramsey AJ, Laakso A, Cyr M, Sotnikova TD, Salahpour A, Medvedev IO. Genetic NMDA receptor deficiency disrupts acute and chronic effects of cocaine but not amphetamine. Neuropsychopharmacology. 2008;33(11):2701– 2714. | ||
Fan X, Jin WY, Wang YT. The NMDA receptor complex: a multifunctional machine at the glutamatergic synapse. Front Cell Neurosci. 2014;8:160. | ||
Zhang J, Xu TX, Hallett PJ, et al. PSD-95 uncouples dopamine-glutamate interaction in the D1/PSD-95/NMDA receptor complex. J Neurosci. 2009;29(9):2948–2960. | ||
Wang M, Wong AH, Liu F. Interactions between NMDA and dopamine receptors: a potential therapeutic target. Brain Res. 2012;1476: 154–163. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.