")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Targeted and synergistic therapy for hepatocellular carcinoma: monosaccharide modified lipid nanoparticles for the co-delivery of doxorubicin and sorafenib
Received 23 February 2018
Accepted for publication 31 May 2018
Published 11 July 2018 Volume 2018:12 Pages 2149—2161
DOI https://doi.org/10.2147/DDDT.S166402
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jianbo Sun
Wendu Duan, Yan Liu
Department of Hepatobiliary Surgery, Affiliated Hospital of Hebei University, Baoding, Hebei Province 071000, People’s Republic of China
Purpose: Targeted hepatocellular carcinoma therapy was carried out to improve the efficacy of liver cancer treatment. The purpose of this study was to design an N-acetylgalactosamine (NAcGal) modified and pH sensitive doxorubicin (DOX) prodrug (NAcGal-DOX) for the construction of lipid nanoparticles (LNPs).
Methods: NAcGal-DOX and sorafenib (SOR) co-loaded LNPs were designed and the synergistic effects were evaluated on human hepatic carcinoma (HepG2) cells in vitro and anti-hepatic carcinoma mice model in vivo.
Results: Cellular uptake efficiency of NAcGal modified LNPs was significantly higher than unmodified LNPs. NAcGal modified LNPs showed the most significant inhibition effect among all the samples tested. The results revealed that the LNPs system achieved significant synergistic effects, best tumor inhibition ability and the lowest systemic toxicity.
Conclusion: These results proved that the NAcGal conjugated and pH sensitive co-delivery nano-system could be a promising strategy for treatment of hepatocellular carcinoma.
Keywords: hepatocellular carcinoma, asialoglycoprotein receptor, N-acetylgalactosamine, pH sensitive, prodrug
Introduction
Hepatocellular carcinoma (HCC) remains a major global health problem.1 It is the fifth most common cancer and the second leading cause of cancer-related death worldwide.2 Currently, due to the majority of patients diagnosed with HCC that are not eligible for surgery, systemic therapy has often been another treatment option for those patients with advanced disease including common therapeutic regimens based on doxorubicin and sorafenib.3–5 However, various clinical studies have demonstrated that conventional cytotoxic chemotherapy had low response rates and lacked overall survival benefit.6
In HCC patients, doxorubicin (DOX) has the highest activity among chemotherapy agents including gemcitabine and capecitabine, with a response rate of 20% and a median survival of 4 months.7,8 However, the lower response rate and severe side effects like cardiotoxicity and cytotoxicity to normal tissues have hindered its clinical application.9 Therefore, targeted drug delivery strategies and targeted drugs have emerged as the most promising fields, and have become research hotspots globally. Current strategies to deliver DOX include using nanoparticles (NPs) with long-circulative polyethylene glycol (PEG) and stimuli-responsive materials.10,11
Prodrug-based targeted nanoparticulate drug delivery systems have many advantages as platforms for cancer therapy.12 Thus, our strategy to address the limitations of DOX is to synthesize N-acetylgalactosamine modified and pH sensitive DOX prodrug (NAcGal-DOX). N-acetylgalactosamine (NAcGal) targets hepatic cancer cells where asialoglycoprotein receptors (ASGPR) are highly expressed. The pH sensitivity allows rapid drug release at the tumor site where pH is lower than 7.4.13,14 Recently, studies have designed monosaccharide (galactosamine, NAcGal) and disaccharide (pullulan) decorated nanocarriers for DOX delivery, and achieved liver target in vitro and in vivo based on animal models.14–18 However, there are no studies about NAcGal conjugated and pH sensitive (hydrazine linked) DOX prodrugs.
Targeted drugs for HCC therapy are other effective choices. Sorafenib (SOR) is the only US Food and Drug Administration approved treatment for advanced HCC, achieving modest response rates.19–21 It is an oral multikinase inhibitor that suppresses tumor cell proliferation and angiogenesis.22 It inhibits various receptors, namely VEGFR1-3, PDGFR-B, c-KIT and Fms-related tyrosine kinase-3, and exerts cytostatic effects to achieve its anticancer activities.23,24 In a randomized phase II trial, outcomes have revealed that the combination of SOR and DOX had synergistic effect; specifically, the combination arm had an overall survival of 13.7 months compared to 6.5 months in the DOX group, and progression free survival (PFS) of 6 months in the combination arm vs 2.7 months in the DOX arm.25 Therefore, lipid nanoparticles (LNPs) were designed for co-delivery of SOR and NAcGal-DOX to improve their synergy and reduce the side effects.
In this present study, we report the development of multifunctionalized LNPs for HCC targeted delivery of NAcGal-DOX and SOR. NAcGal-DOX conjugates were synthesized with NAcGal decoration, PEG and acid-labile hydrazine links. The LNPs were fabricated by single-step nanoprecipitation. pH responsiveness of the LNPs was evaluated in vitro by drug release behavior with different pH mediums. Finally, in vitro cell viability assays were carried out on HCC cells (HepG2 cells); and in vivo antitumor activity of drugs loaded LNPs was evaluated in HepG2 tumor xenograft mouse model.
Materials and methods
Materials
SOR was purchased from LC Laboratories (Woburn, MA, USA). NAcGal, DOX, polysorbate 80, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), dimethyl sulfoxide (DMSO), fetal bovine serum (FBS), Dulbecco’s Modified Eagle’s Medium (DMEM), and 3-[4,5-dimethylthiazol-2yl]–2,5 diphenyltetrazolium (MTT) were purchased from Sigma Aldrich (St Louis, MO, USA). Compritol® 888 ATO was obtained from Gattefossé (Saint-Priest, Lyon, France). Injectable soya lecithin was obtained from Lipoid GmbH (Ludwigshafen, Germany). All other chemicals and reagents were of analytical grade or high performance liquid chromatography (HPLC) grade and used without further purification.
Synthesis and characterization of NAcGal-DOX
NAcGal (1 mmol) was dissolved in 10 mL of dimethylformamide (DMF) and NaH (1 mmol) was added as a solid followed by the addition of tertbutyl bromoacetate (1 mmol) in 2 mL of DMF to get mixture 1.17 After stirring at room temperature for 48 hours, hydrazine (1 mmol) and EDC (1 mmol) was added into mixture 1 and stirred for 4 hours in an ice bath to form mixture 2. DOX (1 mmol) was dissolved in DMF (10 mL) and added into mixture 2 and stirred for another 24 hours, dialyzed against excess ultrapure water for 48 hours to produce NAcGal-DOX (65% yield). Hydrogen nuclear magnetic resonance (1H NMR) of NAcGal-DOX in dimethyl sulfoxide-d6 (600 mHz) is presented, and each proton peak is identified according to the structural formula: δ 1.21 (1, −CH3); 2.05 (2, −C−OH); 2.82 (3, −CH2−); 3.11 (4, −CH2−O); 3.84 (5, −O−CH3); 4.19 (6, −CH−); 4.93 (7, −CH2−C=O); 5.38 (8, −O−CH−C−); 5.96 (9, −C−CH−C−); 6.91 (10, −NH−CO−); 8.11 (11, −NHAc) (Figure 1).
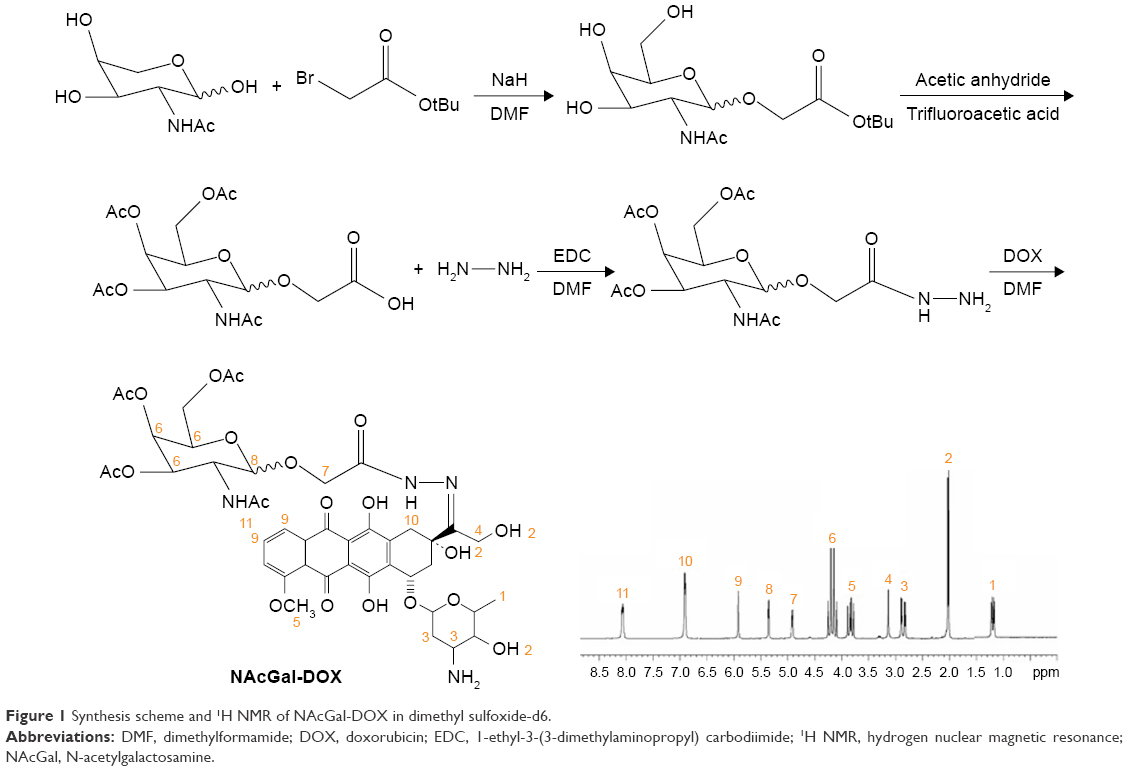 | Figure 1 Synthesis scheme and 1H NMR of NAcGal-DOX in dimethyl sulfoxide-d6. |
Preparation of LNPs
NAcGal-DOX and SOR loaded LNPs (NAcGal-DOX/SOR LNPs) were prepared via single-step nanoprecipitation (Figure 2).26 Briefly, NAcGal-DOX (100 mg), SOR (100 mg), and injectable soya lecithin (1 g) were dissolved in DMSO (5 mL) as the oil phase. Polysorbate 80 (200 mg) was dispersed in deionized water (20 mL) to form the water phase. The oil phase was added to the water phase dropwise under gentle stirring (300 rpm). The NAcGal-DOX/SOR LNPs were self-assembled with continuous stirring for 30 minutes at room temperature. For purification, the solution was dialyzed against excess ultrapure water for 2 days (moelcular weight cut off, 3,500 Da).
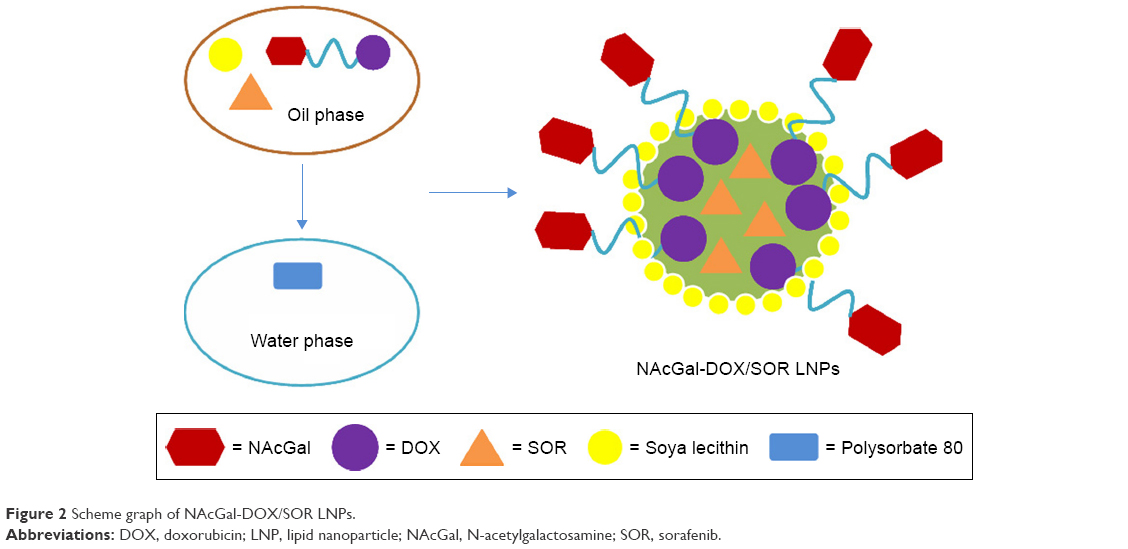 | Figure 2 Scheme graph of NAcGal-DOX/SOR LNPs. |
DOX and SOR loaded LNPs (DOX/SOR LNPs) were prepared by the same procedure using DOX instead of NAcGal-DOX. NAcGal-DOX loaded LNPs (NAcGal-DOX LNPs) were prepared by the same procedure without SOR. SOR loaded LNPs (SOR LNPs) were prepared by the same procedure without NAcGal-DOX. Blank LNPs were prepared by the same procedure without NAcGal-DOX and SOR.
Characterization of LNPs
The particle size and zeta potential were measured by the Nano-ZS apparatus (Malvern Instruments, Malvern, UK).27 To evaluate the drug contents and loading efficiency, 5 mg of LNPs were distributed into the mobile phase (acetonitrile/methanol/1% acetic acid in a ratio of 35:38:27) and stirred overnight. DOX concentration was determined with UV spectrophotometry (Shimadzu, Kyoto, Japan) at 478 nm and SOR concentration was evaluated by high-performance liquid chromatography (HPLC). The percentage drug content was calculated using the following equations:
|
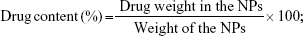
|

In vitro drug release of LNPs
LNPs was dissolved in 1 mL of phosphate buffered saline (PBS) in the presence or absence of 10% mouse serum, and placed in a sealed dialysis tube (molecular weight cut off, 8–10 kDa).28 The dialysis bag was then submerged in 50 mL tubes (Falcon, BD Labware, NJ, USA) containing 20 mL sodium phosphate buffer (50 mM) of different pH (ie, 7.4 and 5.5), and incubated in water-bath at 37°C with a shaking rate of 100 rpm. Drugs released from the dialysis bags were collected at scheduled time intervals and their amounts were quantified by HPLC as described above.
Cells and animals
Human liver carcinoma cells (HepG2 and HuH7 cells) were from the American Type Culture Collection (ATCC; Manassas, VA, USA), and the cells were cultured in DMEM with 10% FBS, at 37°C under atmosphere of 5% CO2/95% air. BALB/c nude mice (4–6 weeks old, 18–22 g weight) were purchased from Beijing Vital River Experimental Animal Technical Co., Ltd (Beijing, China), and were maintained under specific pathogen-free conditions. All animal experiments complied with the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No 8023, revised 1978) and approved by the Medical Ethics Committee of Hebei University (HBU-No 1020170607002).
Cellular uptake of LNPs
Intracellular accumulation assay was used on HepG2 cells to quantitatively determine the cellular uptake of the LNPs.29 Coumarin 6 (C6) was applied as a model fluorescent molecule and was loaded into the LNPs by adding C6 to the oil phase during the preparation of the LNPs. After cells were equilibrated with Hank’s buffered salt solution (HBSS) at 37°C for 1 hour, C6 loaded LNPs were added at concentrations of 200 mg/mL, respectively. The medium was removed after incubation for the determined time, the fluorescence intensity was measured by inversion fluorescence microscope and the picture was captured. Then the cells were washed three times with cold PBS solution and detached with trypsin/EDTA. The cells were centrifuged at 1,500 rpm, 4°C for 5 minutes, the supernatant was discarded, 300 μL of PBS was added to re-suspend the cells and injected to a FACSCalibur flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
In vitro cytotoxicity of LNPs
The in vitro cytotoxicity of LNPs towards HepG2 and HuH7 cells was evaluated by MTT assay.30 Cells were seeded in 96-well plates at a density of 6,000 cells per well in 0.1 mL DMEM solution and incubated in 5% CO2 atmosphere at 37°C for 24 hours, followed by removing culture medium and then adding 0.1 mL of NAcGal-DOX/SOR LNPs, DOX/SOR LNPs, NAcGal-DOX LNPs, SOR LNPs, free DOX and SOR mixture (free DOX/SOR), free DOX, free SOR, and blank LNPs. Cells incubated with 0.1 mL of PBS were used as control. After 24 hours’ incubation, the medium was discarded and 20 μL of MTT solution was added to the cells for another 4 hours. The absorbency of the medium solution was measured on a microplate reader at 570 nm. The cell viability was expressed using the following equation:
|
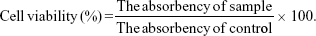
In vitro synergistic effects of LNPs
Half-maximal inhibitory concentration (IC50) was then calculated for each sample according to the in vitro cytotoxicity results. Synergistic effects of the systems containing dual drugs can be evaluated by a combination index (CI) analysis based on the Chou and Talalay’s method.31,32 CI values for DOX and SOR were calculated using the following equation:
|
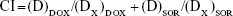
where (D)DOX and (D)SOR are the concentrations of DOX and SOR in the combination system at the ICX value; (DX)DOX and (DX)SOR are ICX value of DOX alone and SOR alone.
CIX< 1 represents synergism and CIX> 1 represents antagonism. In this study, CI50 values were applied and the IC50 values were used for calculation.
In vivo tissue distribution of LNPs
The hepatocellular carcinoma bearing mice model was induced by subcutaneous injection of HepG2 cells (107 cells suspended in 100 μL normal saline) into the right and left flanks on the dorsal side of the BALB/c nude mice.33 The mice were divided into three groups (n=6) and NAcGal-DOX/SOR LNPs, DOX/SOR LNPs, and free DOX/SOR were administered through the tail vein, separately. At predetermined time intervals, mice were sacrificed and the heart, liver, spleen, lung, kidney, stomach, colon, and tumor of mice were collected. The tissues were cut into small pieces and homogenized with physiological saline. After appropriate dilution of supernatants, the content of DOX and SOR was quantified by HPLC as described above.
In vivo therapeutic efficacy of LNPs
The hepatocellular carcinoma bearing mice were randomly divided into nine groups (n=6) and administered with NAcGal-DOX/SOR LNPs, DOX/SOR LNPs, NAcGal-DOX LNPs, SOR LNPs, free DOX/SOR, free DOX, free SOR, blank LNPs, and 0.9% normal saline, respectively, through the tail vein on Days 0, 3, and 6.34 Tumor volumes were calculated according to the following equation:
|
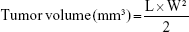
where L is the longest and W is the shortest tumor diameter (mm).
Then mice were sacrificed and tumors were harvested at the completion of the experiment. Tumor weights were measured, and tumor growth inhibition ratio was calculated using the following equation:
|
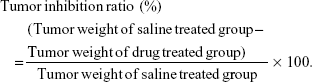
Blood collection and analysis
The hepatocellular carcinoma bearing mice were divided into three groups (n=6) and NAcGal-DOX/SOR LNPs, DOX/SOR LNPs, free DOX/SOR, and 0.9% normal saline were administered through the tail vein, separately.35 On Day 7 after injection, the blood was collected into heparinized tubes through cardiac puncture under anesthesia and animals were euthanized via CO2 overdose. Blood was centrifuged at 15,000 rpm for 20 minutes at 4°C to isolate plasma, which was immediately flash frozen on liquid nitrogen until processing. Plasma was assayed for blood enzymes as biomarkers for different tissue toxicity using assay kits, namely, lactate dehydrogenase (LDH), alanine transaminase (ALT), and creatine phosphokinase (CPK).
Evaluation of toxicity during the repeated treatment
The hepatocellular carcinoma bearing mice were divided into three groups (n=6) and NAcGal-DOX/SOR LNPs, DOX/SOR LNPs, free DOX/SOR, and 0.9% normal saline were administered through the tail vein, separately. The physical conditions and body weight change of mice were monitored for 3 weeks.30
Results
Characterization of LNPs
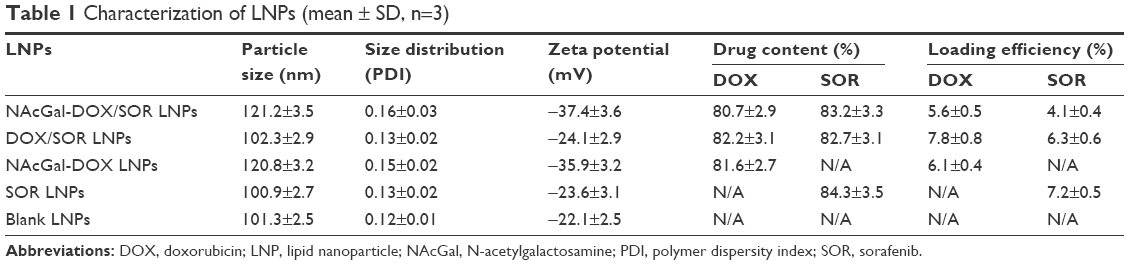
The particle size, zeta potential, drug content and loading efficiency of LNPs were measured (Table 1). The size of NAcGal-DOX/SOR LNPs is 121 nm, which is larger than that of DOX/SOR LNPs (102 nm). Sizes of NAcGal-DOX/SOR LNPs and NAcGal-DOX LNPs are the same. DOX/SOR LNPs, SOR LNPs and blank LNPs have similar sizes. The polymer dispersity index (PDI) of all LNPs was below 0.2. Drug contents of drugs loaded LNPs are over 80%.
 | Table 1 Characterization of LNPs (mean ± SD, n=3) |
In vitro drug release
DOX releases from NAcGal-DOX contained LNPs at pH 5.5 are faster than pH 7.4 (Figure 3A). For NAcGal-DOX/SOR, over 80% release of DOX was achieved at 36 hours in acidic condition (pH 5.5). In contrast, in a neutral environment the release reached 80% at 48 hours. The SOR releases were not affected by the pH conditions (Figure 3B). The release curves of the same system in pH 7.4 and 5.5 followed the same behavior. The SOR release from NAcGal-DOX/SOR LNPs was slower than that from DOX/SOR LNPs and SOR LNPs.
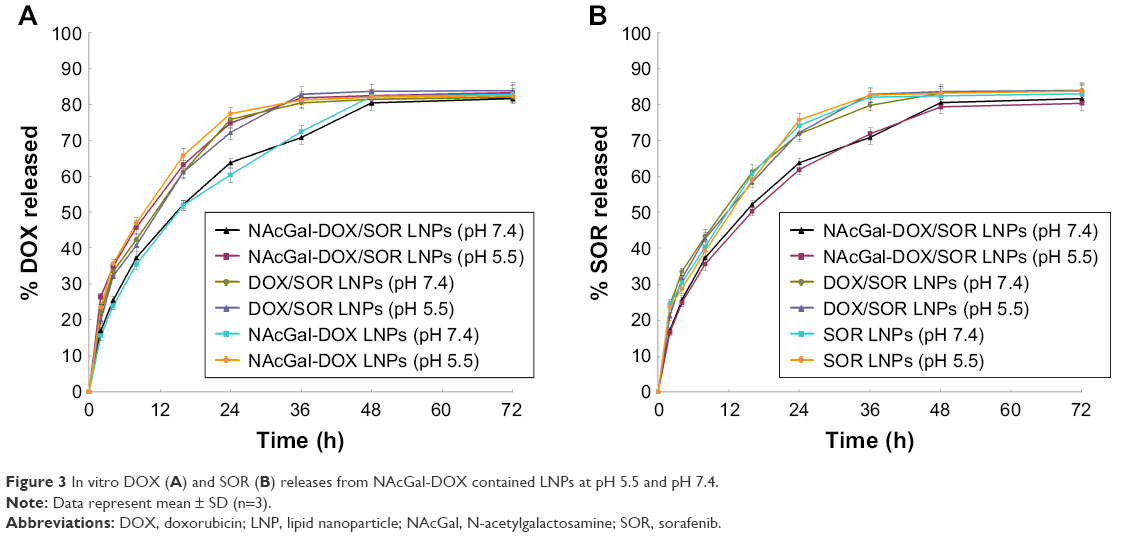 | Figure 3 In vitro DOX (A) and SOR (B) releases from NAcGal-DOX contained LNPs at pH 5.5 and pH 7.4. |
Cellular uptake of LNPs
Cellular uptake of the LNPs increased with time (Figure 4). Cellular uptake efficiency of NAcGal modified LNPs was significantly higher than unmodified LNPs (P<0.05). After 9 hours of incubation, over 70% of cell uptake was achieved by NAcGal-DOX/SOR LNPs.
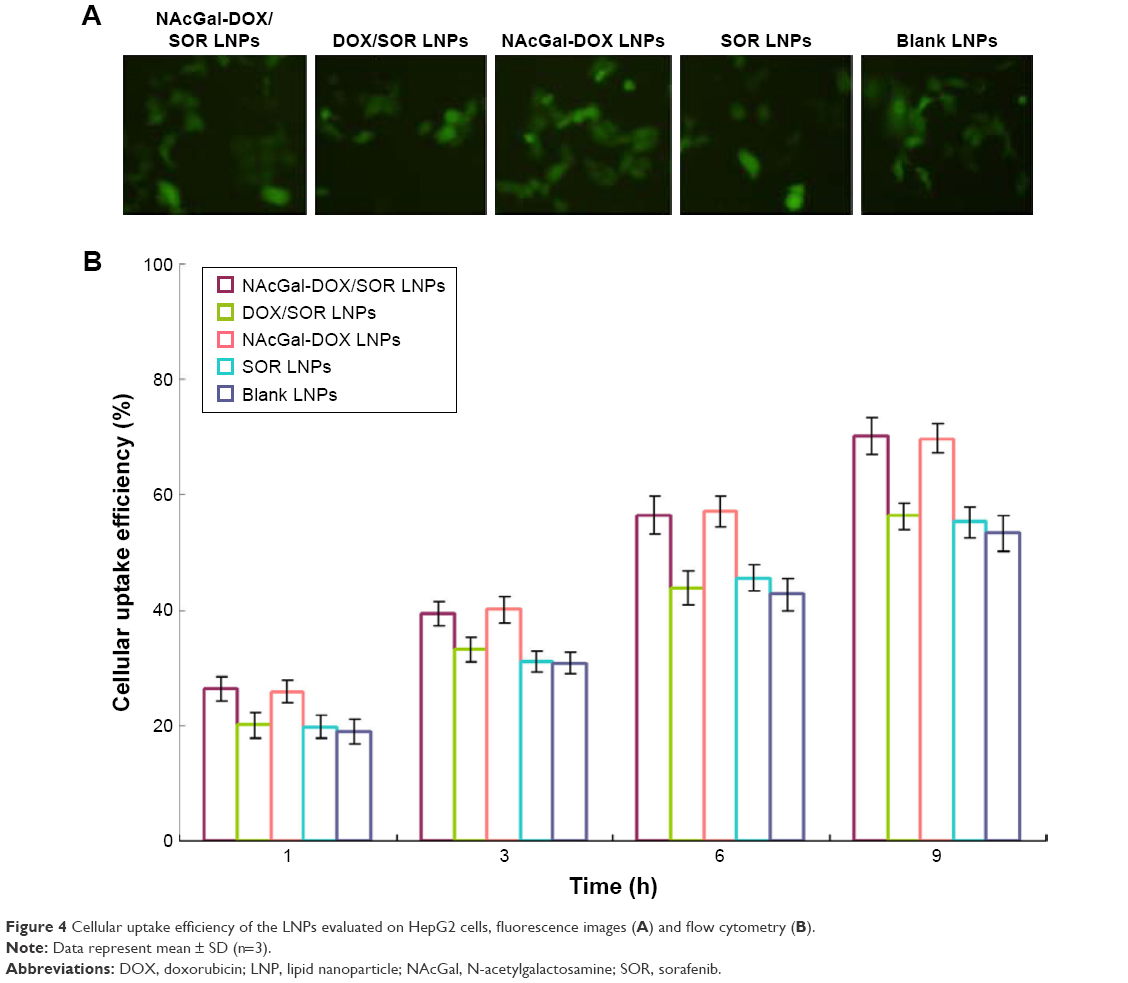 | Figure 4 Cellular uptake efficiency of the LNPs evaluated on HepG2 cells, fluorescence images (A) and flow cytometry (B). |
In vitro cytotoxicity of LNPs
NAcGal modified LNPs showed the most significant inhibition effect among all the samples tested (P<0.05). Dual drug loaded LNPs exhibited higher efficiency than single drug loaded LNPs (P<0.05). The LNP samples had better performances than their free drug counterparts (P<0.05). In addition, data revealed blank NPs that do not contain drugs showed negligible toxicity (Figure 5). The in vitro cytotoxicity results indicated that DOX and SOR released from LNPs could play an enhanced anti-tumor effect.
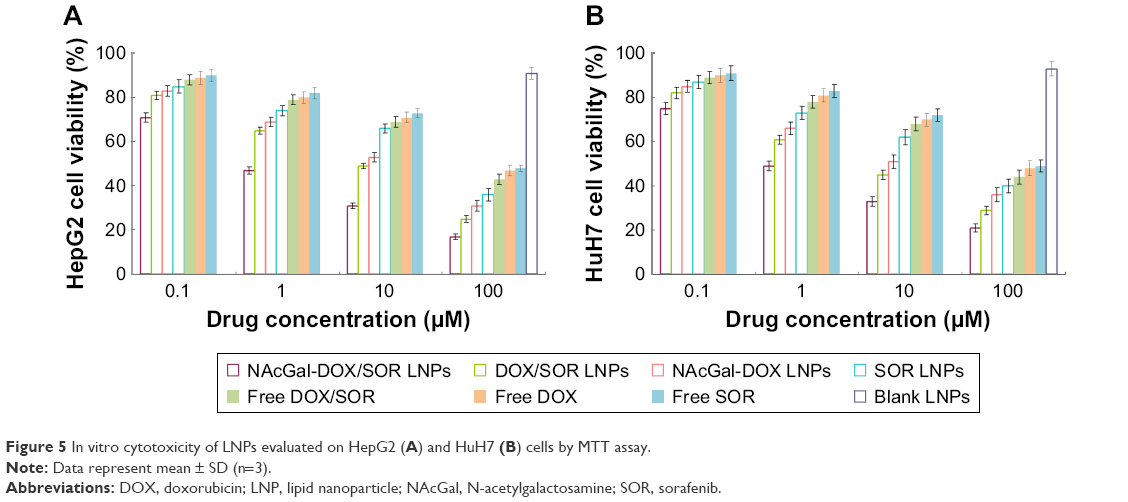 | Figure 5 In vitro cytotoxicity of LNPs evaluated on HepG2 (A) and HuH7 (B) cells by MTT assay. |
In vitro synergistic effects of LNPs
To select the suitable DOX and SOR amount loaded in the LNPs to get the best synergism effect, CI50 values were calculated according to the IC50 values of NAcGal-DOX/SOR LNPs, NAcGal-DOX LNPs, and SOR LNPs (Table 2). When DOX:SOR ratios were between 10:1 and 1:1, NAcGal-DOX/SOR LNPs showed synergistic effects. The best synergistic effect was achieved at the weight ratio of 2:1 (DOX:SOR). So the DOX and SOR loaded in LNPs is determined as 2:1.
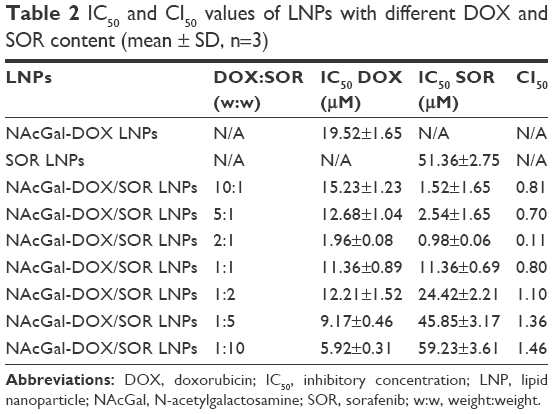 | Table 2 IC50 and CI50 values of LNPs with different DOX and SOR content (mean ± SD, n=3) |
In vivo tissue distribution of LNPs
In vivo tissue distribution of NAcGal-DOX/SOR LNPs, DOX/SOR LNPs, and free DOX/SOR was investigated in hepatocellular carcinoma bearing mice model (Figure 6). After 48 hours of administration, DOX and SOR distributions of NAcGal-DOX/SOR LNPs were higher in the tumor tissue than DOX/SOR LNPs (P<0.05). DOX/SOR LNPs showed better tumor tissue distribution than free DOX/SOR (P<0.05). At 2 hours post injection, free DOX/SOR showed higher accumulation in heart and kidney than LNP formulas (P<0.05).
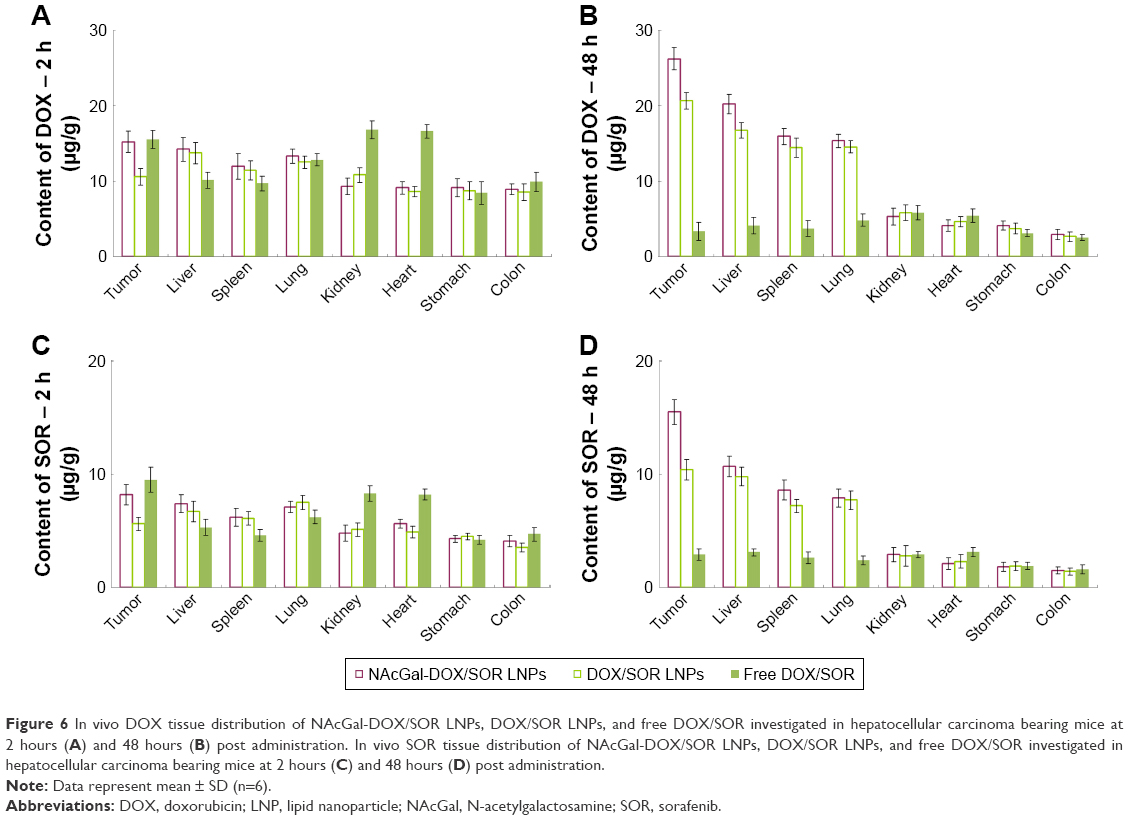 | Figure 6 In vivo DOX tissue distribution of NAcGal-DOX/SOR LNPs, DOX/SOR LNPs, and free DOX/SOR investigated in hepatocellular carcinoma bearing mice at 2 hours (A) and 48 hours (B) post administration. In vivo SOR tissue distribution of NAcGal-DOX/SOR LNPs, DOX/SOR LNPs, and free DOX/SOR investigated in hepatocellular carcinoma bearing mice at 2 hours (C) and 48 hours (D) post administration. |
In vivo therapeutic efficacy of LNPs
In vivo antitumor efficiency of LNPs evaluated in hepatocellular carcinoma bearing BALB/c mice (Figure 7) was stronger than free drug(s), which showed modest anti-cancer activity with tumor growth inhibition ratios of 17%–44% (Table 3). Saline-treated mice displayed a rapid increase in tumor size during the experiment. Among all the groups, NAcGal-DOX/SOR LNPs exhibited the most potent anti-tumor activity. The tumor growth was inhibited with a tumor growth inhibition ratio as high as 87%.
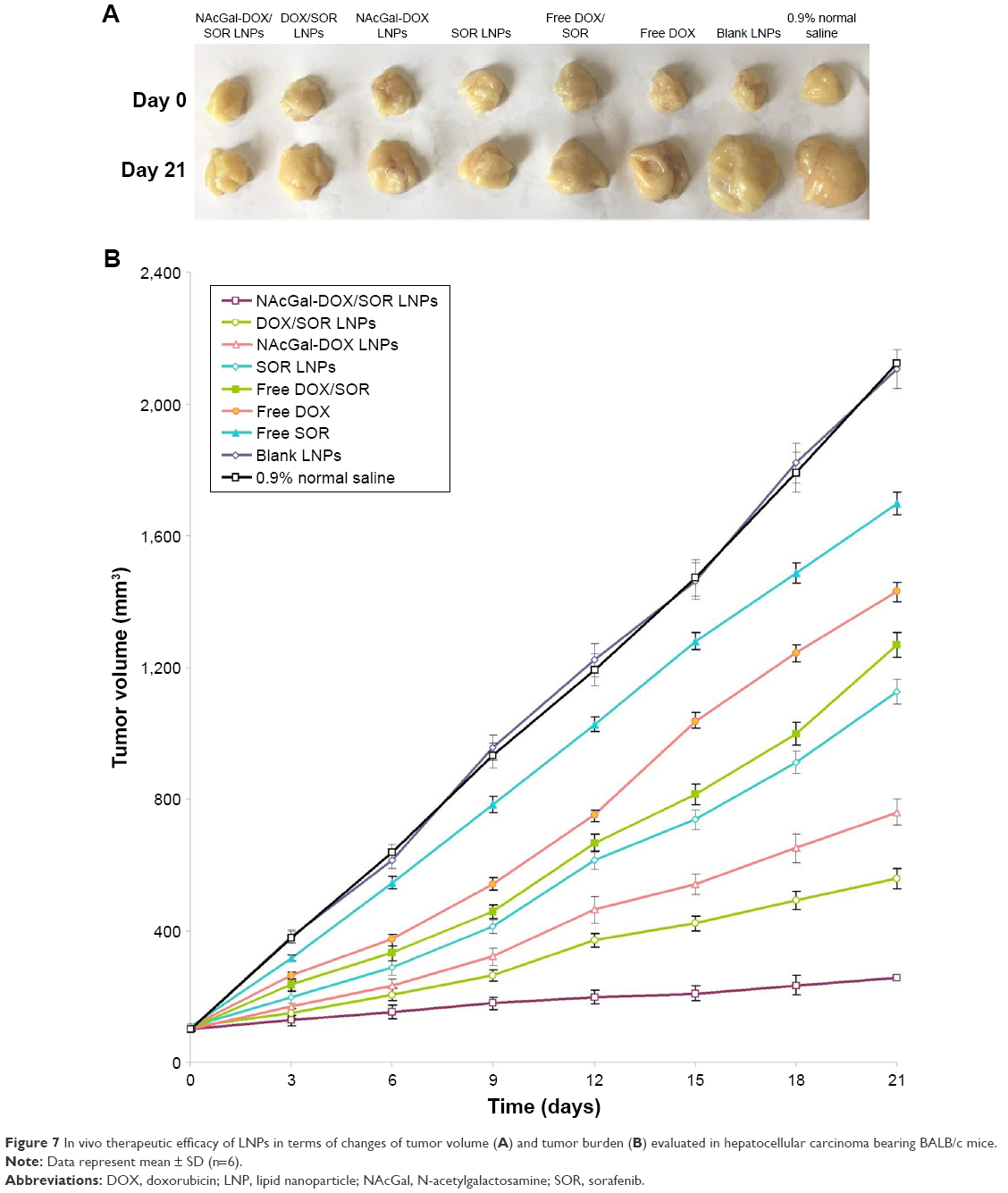 | Figure 7 In vivo therapeutic efficacy of LNPs in terms of changes of tumor volume (A) and tumor burden (B) evaluated in hepatocellular carcinoma bearing BALB/c mice. |
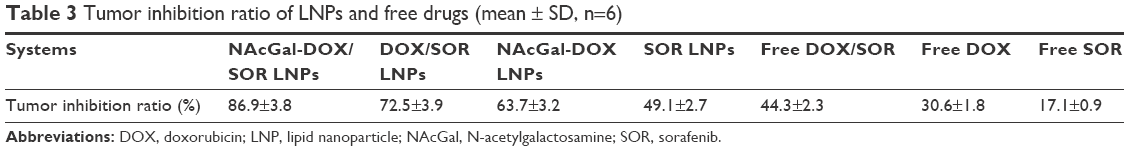 | Table 3 Tumor inhibition ratio of LNPs and free drugs (mean ± SD, n=6) |
Blood analysis
Blood enzyme levels were measured serially once a week over the 3-week course of treatment (Figure 8). Although the differences in blood enzyme activities between the treatment groups were not statistically significant (P>0.05), the trends in blood enzyme levels consistently suggested lower toxicity of LNP treatment relative to free DOX/SOR therapy in tumor models. Free DOX/SOR treatment resulted in an increase in LDH, ALT, and CPK.
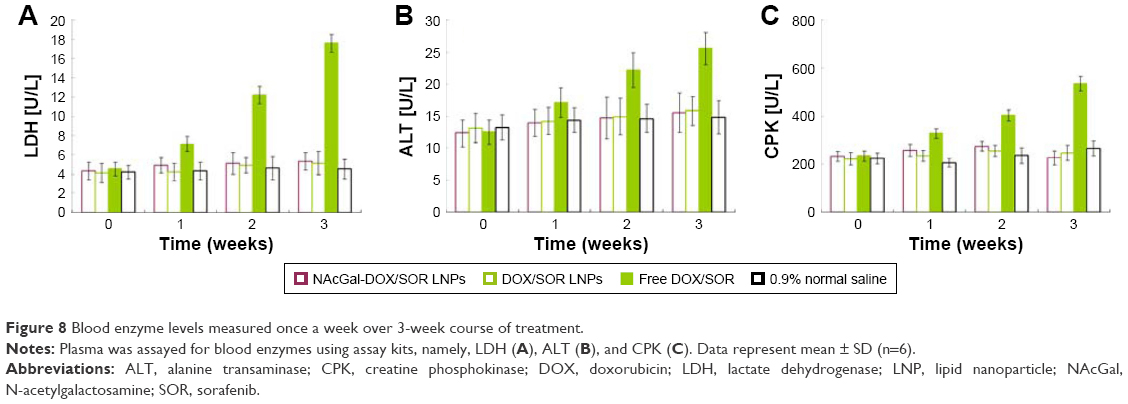 | Figure 8 Blood enzyme levels measured once a week over 3-week course of treatment. |
Toxicity during the repeated treatment
Toxicity of LNPs was studied in hepatocellular carcinoma bearing mice. As shown in Figure 9, the mice treated with free drugs led to over 20% body weight loss on Day 21. In contrast, the LNP administration group showed no obvious changes in body weight. This suggests that all of the LNP formulations were well tolerated. In contrast, decrease of body weights in free drugs treated mice could be the evidence of the toxicity of the systems.
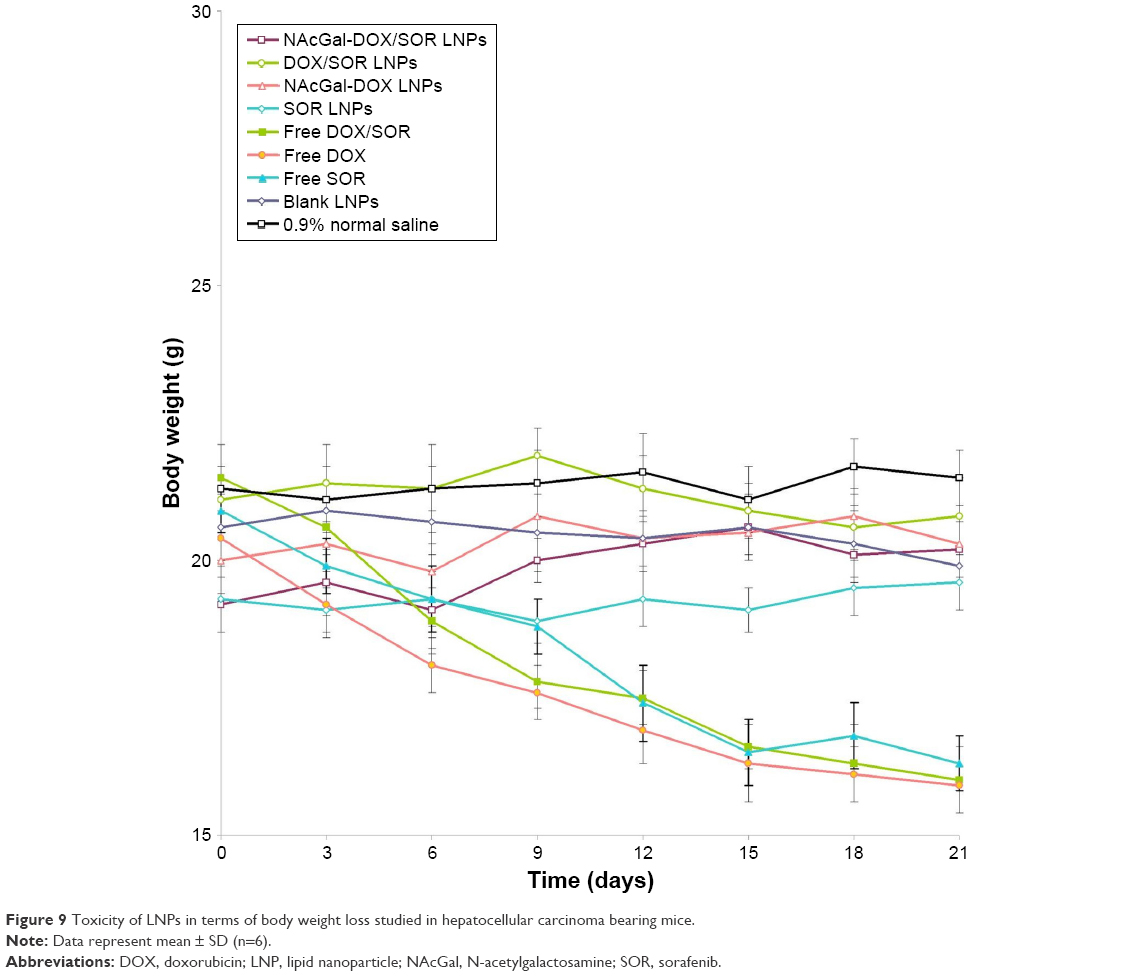 | Figure 9 Toxicity of LNPs in terms of body weight loss studied in hepatocellular carcinoma bearing mice. |
Discussion
In the present study, a novel NAcGal conjugated and pH sensitive LNP system is applied for treatment of hepatocellular carcinoma. At the beginning of this study, NAcGal modified and pH sensitive PEGylated DOX prodrug conjugate was synthesized. In order to achieve the synergistic effects of DOX and SOR on hepatocellular carcinoma, LNPs were successfully prepared by single-step nanoprecipitation.36 The size of blank LNPs and SOR LNPs was about 100 nm, with narrow polydispersity indexes lower than 0.2. This could be evidence that the loading of the drug has no obvious effect on the size of the LNPs. Particle size has a great impact on the in vitro and in vivo efficiency of the NPs, including prolonged blood circulation time and mediated targeted effect. Drug contents of all LNPs have no significant difference. These results indicated that LNPs had good drug entrapment capacity.
In vitro drug release study of LNPs was investigated at pH 7.4 and 5.5.37 In acidic media, the release of DOX was faster than that in the neutral environment. This may be because the pH sensitive hydrazone bonds could cleave much more easily in the lower pH and the degradation of the LNPs occurs predominantly via hydrazone bond cleavage and released the DOX loaded in the LNPs. The pH value of the bloodstream is ~7.4, while the existing tumoral pH and that of endocytic compartments of the cells generally range from 4 to 6.38 This difference in pH value makes pH-triggered drug release possible. The DOX release rate from LNPs increased when pH was lowered. These findings suggest that the weak acidic condition of tumor tissue is favorable for release of DOX.28 Further, upon internalization into tumor cells via endocytosis, the low pH of the phago-lysosomal system will facilitate more rapid release of DOX due to lower pH. The SOR release from NAcGal-DOX/SOR LNPs was slower than that from DOX/SOR LNPs and SOR LNPs. In vitro drug release of the LNPs may be controlled by erosion, corrosion, and diffusion processes.39 Drug depot effects could be achieved by the carriers, which could lead to the sustained release of hydrophobic drugs. The more sustained SOR release behavior from NAcGal-DOX/SOR LNPs than from DOX/SOR LNPs might be explained by the modification of NAcGal ligands that decelerated the release of drugs. Once within the endosomal compartments, the drugs can be released from the LNPs and induce their toxic impacts. In addition, in the presence of serum, the release of drugs was not affected, indicating the stability of LNPs in circulation.
The in vitro cellular uptake research could provide some circumstantial evidence to display the advantages of the NP formulation entering the cancer cells. Cellular uptake efficiency of NAcGal modified LNPs was significantly higher than other LNPs. This could be attributed to enhanced cancer cell specific adherence of the NAcGal ligands to the liver cancer cell membrane. The improved activity and penetration of drugs delivered with NAcGal modified LNPs can be made use of to improve the efficacy of the standard drug dose, attenuate side effects, and overcome drug resistance.29
In order to verify the enhanced anticancer effect of the LNPs, the proliferation inhibition of LNPs was tested against HepG2 and HuH7 cells. Free drug and drug mixtures were used as the controls. The cell proliferation inhibition efficacy of all the samples exhibited a strongly dose-dependent pattern. Higher cytotoxicity of the drug-loaded LNPs than free drug indicated that LNP delivery systems can enhance cytotoxicity in vitro. It is encouraging that the anti-tumor activity of NAcGal-DOX/SOR LNPs was obviously higher than the other groups, especially in the range of higher drug concentration. To validate the synergistic effect of drugs co-loaded NPs on HepG2 cells, the CI was further determined using the isobologram equation of Chou and Talalay.40 NAcGal-DOX/SOR LNPs displayed an overall CI value <1 when the DOX:SOR ratio was between 10:1 and 1:1. In the ratio of 2:1, the combined anti-tumor effect of the dual drug-loaded LNPs was the best and could develop the ability of the drugs to the largest extent, suggesting the best DOX:SOR ratio in the LNP formulations.
In vivo drug distribution of LNPs was higher in tumor tissue and lower in the heart and kidney, which could decrease side effects during tumor therapy. Conversely, drug solution samples are mainly distributed in heart and kidney. This may lead to systemic toxicity. Higher distribution of drugs in LNPs in tumors than in other tissues, especially at 48 hours of testing, might be due to the sustained release behavior and targeted ability of the LNPs, thus prolonging blood circulation time and better targeting the tumor site. Higher accumulation of NAcGal-DOX/SOR LNPs than DOX/SOR LNPs was observed in the tumor tissue. This could be evidence that the NAcGal ligands would bring about the target ability to the LNP system. Toxicity of LNPs was studied in hepatocellular carcinoma bearing mice in terms of body weight changes. On Day 21, mice treated with LNPs showed no obvious changes in body weight, while free drugs led to over 20% body weight loss. The decrease of body weight in free drug treated mice is likely due to the toxicity caused by free DOX and SOR distributed to normal organs. Considering the results along with the blood enzyme level analysis and tissue distribution, these LNPs groups could be indicative of tumor cell death and not necessarily nonspecific tissue damage.
In vivo antitumor efficiency of LNPs was evaluated in BALB/c mice bearing hepatocellular carcinoma. The HepG2 cell line is an aggressive cancer cell line with a high proliferation rate, and saline-treated mice displayed a rapid increase in tumor size during the experiment. Free drug(s) showed modest anti-cancer activity, while stronger therapeutic responses were observed in LNP treated mice. NAcGal-DOX/SOR LNPs exhibited the most potent anti-tumor activity among all the groups; the tumor growth was almost completely inhibited. The results revealed that the higher anti-tumor efficiency of drugs after co-loading in NAcGal-DOX/SOR LNPs than in DOX/SOR LNPs is related to the targeted ability of NAcGal. Also, this high therapeutic efficacy is consistent with the strong synergy between DOX and SOR. The facilitated co-delivery of DOX and SOR by DOX/SOR LNPs also plays an important role as DOX/SOR LNPs were much more active than the free drug combination (free DOX/SOR) in antitumor activity.
Conclusion
In summary, this research provides a solution to formulate a specific NAcGal modified pH sensitive LNPs system for the co-delivery of DOX and SOR for combination hepatocellular carcinoma chemotherapy. The LNP system achieved significant synergistic effects, best tumor inhibition ability and the lowest systemic toxicity. These results proved that the NAcGal conjugated and pH sensitive co-delivery nano-system could be a promising strategy for treatment of hepatocellular carcinoma.
Disclosure
The authors report no conflicts of interest in this work.
References
Nordenstedt H, White DL, El-Serag HB. The changing pattern of epidemiology in hepatocellular carcinoma. Dig Liver Dis. 2010;42(Suppl 3):S206–S214. | ||
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Sj Y. A concise review of updated guidelines regarding the management of hepatocellular carcinoma around the world: 2010–2016. Clin Mol Hepatol. 2016;22(1):7–17. | ||
Yeo W, Mok TS, Zee B, et al. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst. 2005;97(20):1532–1538. | ||
Lencioni R, Llovet JM, Han G, et al. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: The SPACE trial. J Hepatol. 2016;64(5):1090–1098. | ||
Thomas MB, O’Beirne JP, Furuse J, Chan AT, Abou-Alfa G, Johnson P. Systemic therapy for hepatocellular carcinoma: cytotoxic chemotherapy, targeted therapy and immunotherapy. Ann Surg Oncol. 2008;15(4):1008–1014. | ||
Patt YZ, Hassan MM, Aguayo A, et al. Oral capecitabine for the treatment of hepatocellular carcinoma, cholangiocarcinoma, and gallbladder carcinoma. Cancer. 2004;101(3):578–586. | ||
Lombardi G, Zustovich F, Farinati F, et al. Pegylated liposomal doxorubicin and gemcitabine in patients with advanced hepatocellular carcinoma: results of a phase 2 study. Cancer. 2011;117(1):125–133. | ||
Dong L, Xia S, Wu K, et al. A pH/enzyme-responsive tumor-specific delivery system for doxorubicin. Biomaterials. 2010;31(24):6309–6316. | ||
Zhou H, Fan Z, Deng J, et al. Hyaluronidase embedded in nanocarrier PEG shell for enhanced tumor penetration and highly efficient antitumor efficacy. Nano Lett. 2016;16(5):3268–3277. | ||
Wang Y, Shim MS, Levinson NS, Sung HW, Xia Y. Stimuli-responsive materials for controlled release of theranostic agents. Adv Funct Mater. 2014;24(27):4206–4220. | ||
Luo C, Sun J, Sun B, He Z. Prodrug-based nanoparticulate drug delivery strategies for cancer therapy. Trends Pharmacol Sci. 2014;35(11):556–566. | ||
Yu W, Zhang N, Li C. Saccharide modified pharmaceutical nanocarriers for targeted drug and gene delivery. Curr Pharm Des. 2009;15(32):3826–3836. | ||
Guhagarkar SA, Majee SB, Samad A, Devarajan PV. Evaluation of pullulan-functionalized doxorubicin nanoparticles for asialoglycoprotein receptor-mediated uptake in Hep G2 cell line. Cancer Nanotechnol. 2011;2(1–6):49–55. | ||
Xu M, Qian J, Liu X, Liu T, Wang H. Stimuli-responsive PEGylated prodrugs for targeted doxorubicin delivery. Mater Sci Eng C Mater Biol Appl. 2015;50:341–347. | ||
Pranatharthiharan S, Patel MD, Malshe VC, et al. Asialoglycoprotein receptor targeted delivery of doxorubicin nanoparticles for hepatocellular carcinoma. Drug Deliv. 2017;24(1):20–29. | ||
Medina SH, Tekumalla V, Chevliakov MV, Shewach DS, Ensminger WD, El-Sayed ME. N-acetylgalactosamine-functionalized dendrimers as hepatic cancer cell-targeted carriers. Biomaterials. 2011;32(17):4118–4129. | ||
Kuruvilla SP, Tiruchinapally G, Elazzouny M, Elsayed ME, Elsayed ME. N-acetylgalactosamine-targeted delivery of dendrimer-doxorubicin conjugates influences doxorubicin cytotoxicity and metabolic profile in hepatic cancer cells. Adv Healthc Mater. 2017;6(5):1601046. | ||
Chong DQ, Tan IB, Choo SP, Toh HC. The evolving landscape of therapeutic drug development for hepatocellular carcinoma. Contemp Clin Trials. 2013;36(2):605–615. | ||
Kalyan A, Nimeiri H, Kulik L. Systemic therapy of hepatocellular carcinoma: current and promising. Clin Liver Dis. 2015;19(2):421–432. | ||
Zhu AX. Molecularly targeted therapy for advanced hepatocellular carcinoma in 2012: current status and future perspectives. Semin Oncol. 2012;39(4):493–502. | ||
Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5(10):835–844. | ||
Mcdermott U, Sharma SV, Dowell L, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci U S A. 2007;104(50):19936–19941. | ||
Tai WT, Cheng AL, Shiau CW, et al. Signal transducer and activator of transcription 3 is a major kinase-independent target of sorafenib in hepatocellular carcinoma. J Hepatol. 2011;55(5):1041–1048. | ||
Abou-Alfa GK, Johnson P, Knox JJ, et al. Doxorubicin plus sorafenib vs doxorubicin alone in patients with advanced hepatocellular carcinoma: a randomized trial. JAMA. 2010;304(19):2154–2160. | ||
Wang C, Su L, Wu C, Wu J, Zhu C, Yuan G. RGD peptide targeted lipid-coated nanoparticles for combinatorial delivery of sorafenib and quercetin against hepatocellular carcinoma. Drug Dev Ind Pharm. 2016;42(12):1938–1944. | ||
Kim DH, Kim MD, Choi CW, et al. Antitumor activity of sorafenib-incorporated nanoparticles of dextran/poly(dl-lactide-co-glycolide) block copolymer. Nanoscale Res Lett. 2012;7(1):91. | ||
Fan X, Zhao X, Qu X, Fang J. pH sensitive polymeric complex of cisplatin with hyaluronic acid exhibits tumor-targeted delivery and improved in vivo antitumor effect. Int J Pharm. 2015;496(2):644–653. | ||
Wang H, Sun G, Zhang Z, Ou Y. Transcription activator, hyaluronic acid and tocopheryl succinate multi-functionalized novel lipid carriers encapsulating etoposide for lymphoma therapy. Biomed Pharmacother. 2017;91:241–250. | ||
Cui T, Zhang S, Sun H. Co-delivery of doxorubicin and pH-sensitive curcumin prodrug by transferrin-targeted nanoparticles for breast cancer treatment. Oncol Rep. 2017;37(2):1253–1260. | ||
Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. | ||
Zhang R, Ru Y, Gao Y, Li J, Mao S. Layer-by-layer nanoparticles co-loading gemcitabine and platinum (IV) prodrugs for synergistic combination therapy of lung cancer. Drug Des Devel Ther. 2017;11:2631–2642. | ||
Gao Z, Li Z, Yan J, Wang P, Irinotecan WP. Irinotecan and 5-fluorouracil-co-loaded, hyaluronic acid-modified layer-by-layer nanoparticles for targeted gastric carcinoma therapy. Drug Des Devel Ther. 2017;11:2595–2604. | ||
Zhang P, Li J, Ghazwani M, et al. Effective co-delivery of doxorubicin and dasatinib using a PEG-Fmoc nanocarrier for combination cancer chemotherapy. Biomaterials. 2015;67:104–114. | ||
Shao M, Yang W, Han G. Protective effects on myocardial infarction model: delivery of schisandrin B using matrix metalloproteinase-sensitive peptide-modified, PEGylated lipid nanoparticles. Int J Nanomedicine. 2017;12:7121–7130. | ||
Lee ES, Gao Z, Kim D, Park K, Kwon IC, Bae YH. Super pH-sensitive multifunctional polymeric micelle for tumor pH(e) specific TAT exposure and multidrug resistance. J Control Release. 2008;129(3):228–236. | ||
Tan S, Wang G, Redox-Responsive WG. Redox-responsive and pH-sensitive nanoparticles enhanced stability and anticancer ability of erlotinib to treat lung cancer in vivo. Drug Des Devel Ther. 2017;11:3519–3529. | ||
Qu CY, Zhou M, Chen YW, Chen MM, Shen F, Xu LM. Engineering of lipid prodrug-based, hyaluronic acid-decorated nanostructured lipid carriers platform for 5-fluorouracil and cisplatin combination gastric cancer therapy. Int J Nanomedicine. 2015;10:3911–3920. | ||
Nazari-Vanani R, Moezi L, Heli H. In vivo evaluation of a self-nanoemulsifying drug delivery system for curcumin. Biomed Pharmacother. 2017;88:715–720. | ||
Chou TC, Talalay P. A simple generalized equation for the analysis of multiple inhibitions of Michaelis-Menten kinetic systems. J Biol Chem. 1977;252:6438–6442. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.