")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Synthetic biology for pharmaceutical drug discovery
Authors Trosset J, Carbonell P
Received 21 May 2015
Accepted for publication 24 September 2015
Published 3 December 2015 Volume 2015:9 Pages 6285—6302
DOI https://doi.org/10.2147/DDDT.S58049
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Wei Duan
Jean-Yves Trosset,1 Pablo Carbonell2,3
1Bioinformation Research Laboratory, Sup’Biotech, Villejuif, France; 2Faculty of Life Sciences, SYNBIOCHEM Centre, Manchester Institute of Biotechnology, University of Manchester, Manchester, UK; 3Department of Experimental and Health Sciences (DCEXS), Research Programme on Biomedical Informatics (GRIB), Hospital del Mar Medical Research Institute (IMIM), Universitat Pompeu Fabra (UPF), Barcelona, Spain
Abstract: Synthetic biology (SB) is an emerging discipline, which is slowly reorienting the field of drug discovery. For thousands of years, living organisms such as plants were the major source of human medicines. The difficulty in resynthesizing natural products, however, often turned pharmaceutical industries away from this rich source for human medicine. More recently, progress on transformation through genetic manipulation of biosynthetic units in microorganisms has opened the possibility of in-depth exploration of the large chemical space of natural products derivatives. Success of SB in drug synthesis culminated with the bioproduction of artemisinin by microorganisms, a tour de force in protein and metabolic engineering. Today, synthetic cells are not only used as biofactories but also used as cell-based screening platforms for both target-based and phenotypic-based approaches. Engineered genetic circuits in synthetic cells are also used to decipher disease mechanisms or drug mechanism of actions and to study cell–cell communication within bacteria consortia. This review presents latest developments of SB in the field of drug discovery, including some challenging issues such as drug resistance and drug toxicity.
Keywords: metabolic engineering, plant synthetic biology, natural products, synthetic quorum sensing, drug resistance
Introduction
The new area of synthetic biology (SB) is arguably reorienting the field of drug discovery (DD) in the same way as one century ago the field of organic chemistry was at the center of innovation in the pharmaceutical industries. Today, the increasing drug attrition rate, with 95% of drugs tested in Phase I not reaching approval,1 testifies the difficulty to innovate for safe medicines with the current approaches of medicinal chemistry.
SB brings the engineer’s view into biology, which transforms a biological cell into an industrial biofactory. Nature has been the source of human medicines for thousands of years, but the difficulty of large-scale production of natural products (NPs) made pharmaceutical industries to abandon this source of natural medicinal compounds. As such, their therapeutic advantages (eg, biocompatibility) were sacrificed to turn toward simpler chemistry at the risk of increased cross-reactivity with secondary therapeutic targets and even unwanted off-targets as confirmed by recent studies in system chemical biology.2–4 Such target promiscuity is often responsible for observed toxicity issues that can jeopardize a project at clinical stage.5
A breakthrough discovery in the 1990s made the rational-based genetic design a potential strategy for DD. Microorganisms (as well as plants and others) produce secondary metabolites using gigantic biosynthetic units.6 These enzymatic modules can be manipulated in combinatorial fashion in synthetic cells to produce new NPs derivatives.7 The first application of SB in DD was to boost innovation in creating new chemical scaffolds that have properties similar to well-known NP-derived human medicines, increasing the chance of being bioactive with the right pharmacological properties.8
With the recent advanced genome editing, molecular biology, and protein engineering tools, SB has focused its aim at creating biological devices that can produce controlled phenotypes from a given input, such as a molecular or light switch (Figure 1 for a description of the concepts in SB). The design of genetic circuits in SB is used in pharmaceutical research not only for bioproduction9 of drugs by microorganisms but also to support the different steps of drug development.10
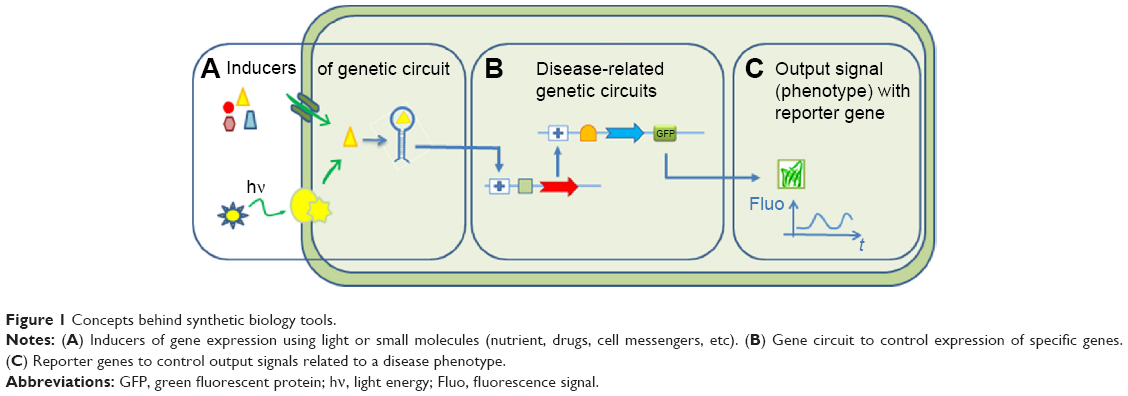 | Figure 1 Concepts behind synthetic biology tools. |
The first section of this review presents an overview of the basic concept of SB followed by an historical evolution of the concepts used in pharmaceutical research and how the application of SB in DD naturally emerged from modern poly-pharmacology. The third section presents the impact of SB in the field of NPs. The fourth section shows the latest development of metabolic engineering in the large-scale production of drugs by microorganisms. Synthetic cellular models can be constructed to identify or validate drug target (fifth section) as well as to create target-based or phenotypic-based drug screening platform (sixth section).11–15 The following section describes the construction of disease models with the help of optogenetics to decipher disease mechanisms with examples in cancer and neuronal diseases. Finally, the last two sections discuss some other challenging topics in DD, such as toxicity and drug resistance.
Some basic concepts of synthetic biology
The basic concepts of SB tools in use for DD are summarized in Figure 1. A typical synthetic cell is composed of the following three elements: an inducer represented by a small molecule, a ligand of a membrane receptor, or lights (Figure 1A) that triggers a de novo-designed genetic circuit (Figure 1B). Inducing this circuit produces an output signal that can be followed by a light-emitting reporter gene (Figure 1C). These three basic constituents can be integrated in different manners according to the type of applications in DD. Figure 2 presents some of these applications. Gene circuits from secondary metabolisms or cryptic biosynthetic units of microorganism can be integrated into host microorganisms to facilitate gene expression of targeted compound (first row of Figure 2) or to shuffle modules within biosynthetic units for combinatorial exploration of chemical space of NPs (second row of Figure 2). Light may induce activation of expression of specific receptor (eg, the bacterial enzymatic light-emission system, encoded by the lux operon [Lux], protein photo sensors such as LOV [light, oxygen or voltage] domains, or green fluorescent protein [GFP]). These biosensors can be used in many ways to, eg, validate drug targets, understand drug’s mechanism of action through a designed disease model, and induce a drug delivery mechanism at a specific site or under specific condition (third row of Figure 2). Synthetic quorum sensing (QS) can be used to study antibiotics persistence or resistance mechanisms in population of bacteria by altering the cell–cell communication system (fourth row of Figure 2). Protein engineering is another tool of SB. Site-directed mutagenesis can increase the regio- or stereospecificity of an enzyme (Figure 3A), increase the binding constant of a chosen ligand (Figure 3B), or choose between enzyme isoforms (Figure 3C). Other approaches not shown in the figure include directed precursor biosynthesis approach,7,16 in which enzyme is mutated through selection pressure with imposed substrates, or using mutational biosynthesis or mutasynthesis,17 in which wild-type enzymatic path is shut off by mutation forcing supplemented substrates analogs to be processed by the enzyme through selective evolution.
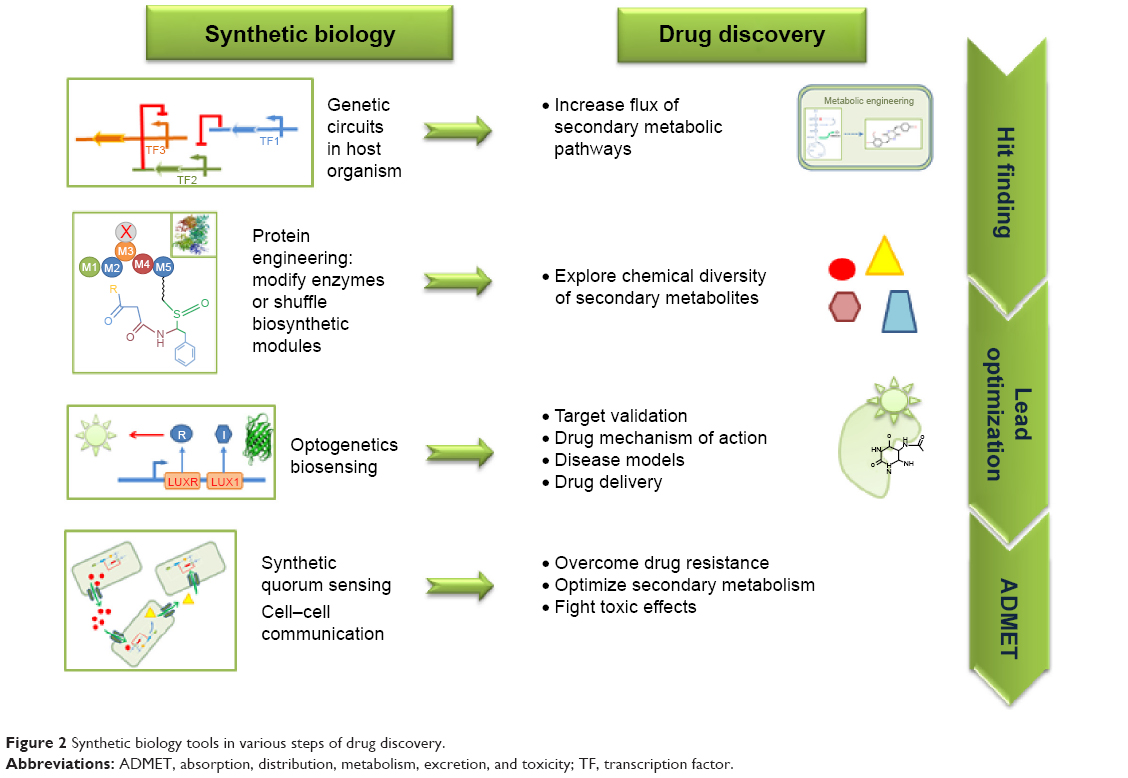 | Figure 2 Synthetic biology tools in various steps of drug discovery. |
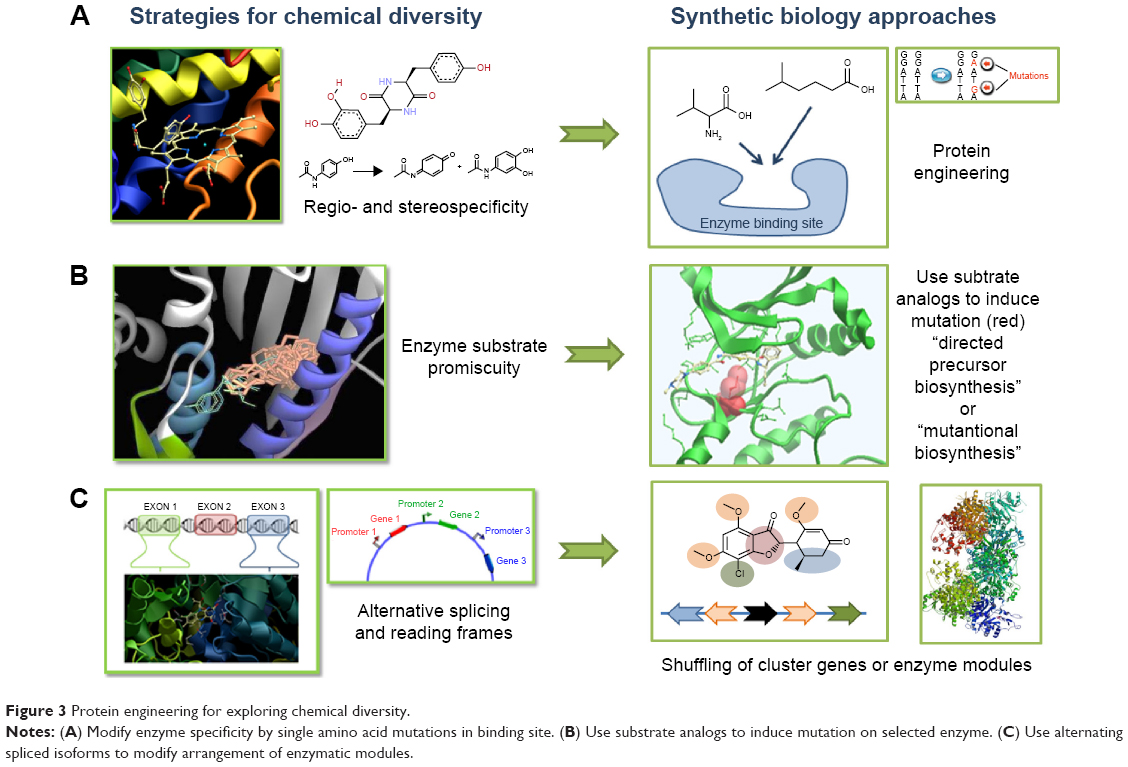 | Figure 3 Protein engineering for exploring chemical diversity. |
Emergence of SB in DD: a historical perspective
One hundred years ago, Paul Ehrlich launched the concept of “One drug – One Target – One disease” in which a drug would cure a disease by targeting a specific component of our body.18 Such “magic bullet” was to be found in nature through pharmacologically active compounds extracted from plants or microorganisms. “Tour de force” was achieved by chemists to reproduce these molecules by total synthesis.19 New cost-effective approaches such as function-oriented synthesis emerged in order to mimic complex NPs with simpler products while retaining similar pharmacophore pattern of interactions. NPs and NPs biomimetism marked the golden age of chemistry and established the success of the pharmaceutical industries for the next century.20
A culminant period was observed at the turn of the last century with the advances in solid- and liquid-phase syntheses. Combinatorial chemistry opened the possibility of exploring unknown regions of the chemical space by systematic decoration of predefined chemical scaffolds.21,22 Together with the miniaturization of biochemical assays, large-scale chemical libraries could be screened for binding affinity with a chosen biological target.23 Before validating the proof of success of this approach, pharmaceutical industries abandoned most of their research programs on NPs and bet on the high-throughput screening (HTS) approach to find initial hits for the therapeutic target under investigation.24
Success of this approach depends heavily on the quality of the input compound libraries. To increase the chance of success, selection of compound libraries was based on chemical diversity and optimization of physicochemical properties (eg, solubility and permeability). With the addition of structural knowledge on the target, new constraints were included in the optimization process to produce target-focused compound libraries that are specifically designed for a selected set of targets.25
This raises another problem of compound specificity, ie, its capacity to bind a unique biological target.26 Cross-reactivity with structurally similar and unwanted biological targets would induce toxicity. This usually arises when targeting a specific member of a large protein family (eg, kinases). To control the selectivity of drug candidates, industries and academic structural genomics research centers developed high-throughput biochemical assay platforms to screen compounds’ libraries on a representative set of a protein target family members (eg, kinases27).This approach led to the accumulation of chemical proteomics data that showed that even selective compounds bind to more than one biological target.28,29 In that way, the systemic view of pharmacology has moved the Paul Ehrlich’s concept (“single drug – single target”) toward a more subtle point of view involving interactions with multiple targets (poly-pharmacology) which are modulated by synergistic drug combinations.30–33
SB is a rational attempt to understand the basic concepts of this apparent complexity. Using an engineer’s view in biology, SB designs biological devices (synthetic cells or cell-free system) to trigger a biological response with respect to input controlled signal (Figure 1). In DD, such devices would be used to activate gene expression of biosynthetic units to explore NP-like chemical space. In-cell synthesis has the advantage to make use of natural evolution to create compounds compliant to biological environment, which is part of the lead optimization process. Genome editing tools give the possibility of following through reporter genes the action of a particular output signal, which is very useful to validate drug target or disease models as well as merging constraints from both DD and drug production. This “rational-based biosynthetic drug design”34 approach is somehow the other side of the de novo rational-based drug design of the last century.35
Mining NPs space
Chemistry in a cell makes use of biosynthetic machinery of plants, microorganisms, and fungi36 to produce NPs derivatives with therapeutic interest. NPs have already provided myriad of human medicines, including antibiotics, antifungals, antitumors, immunosuppressants, and cholesterol-lowering agents.37–39 Major classes of NPs include polyketides,40 nonribosomal peptides (NRPs),41–43 terpenoids,44 isoprenoids, alkaloids,45,46 and flavonoids.47
The idea of transforming cells into a synthetic chemical biofactory takes place after the discovery of Katz and Leadlay at the beginning of 1990s, which showed independently that the antibiotic erythromycin used in the defense system of Actinomycetes is synthesized by a giant biosynthetic unit made of 28 protein modules from a unique gene cluster.48–51 Such biosynthetic units could then easily be isolated and implemented into host organism to further modify at the genetic level to produce NPs derivatives.37,52
Large-scale genome or metagenomes sequencing of microorganisms with the help of bioinformatics tools, such as Secondary Metabolite Unknown Regions Finder (SMURF) and antibiotics & Secondary Metabolite Analysis Shell (antiSMASH),53–56 extend the discovery of such biosynthetic gene clusters.55,57 However, most biosynthetic gene clusters remain cryptic or silent under culturing conditions. Lots of effort have been made to boost or reactivate silent gene expression through the design of synthetic transcription factors (TFs),58 ligand-controlled aptamers, or riboswitches59 “knock-in” promoter replacement strategy.60,61 Early reviews present these or similar approaches.62–64
The major interest of these biosynthetic units is modularity.51,65 Each unit can be modified individually by site-specific mutation to increase the catalytic efficiency or substrate preference66 (Figure 3). Mimicking the combinatorial chemistry approach, chemical diversity can be explored by genetic shuffling or individual modification of the corresponding biosynthetic modules.67–70 Xu et al engineered random pairs of sequentially acting iterative polyketide synthases subunits in a yeast heterologous host to create a diverse library of benzendiol lactone polyketides derivatives, including a polyketide with an unnatural skeleton and heat shock response-inducing activity.71
Another example concerns erythromycin A, one of the most studied antibiotics for treating human infections. Analogs have been created through combinatorial precursor-directed biosynthesis, leading to a new class of alkynyl- and alkenyl-substituted macrolides.72 Applications of these combinatorial approaches have been reviewed for various classes of NPs.63,73 For instance, Smanski et al optimized the function of gene clusters through combinatorial assembly of well-characterized bioparts.74 This enables the exploration of the very important classes of therapeutically derived NPs, such as terpenoids, flavonoids, and alkaloids.75,76 Klein et al reported a series of 74 novel compounds belonging to various classes, including type III polyketide and flavonoids.77 These small-molecular-weight scaffolds (200–300 MW) were produced using a combinatorial genetic approach in baker’s yeast. They made use of the concept of coevolution with the presence of a target protein in an intracellular primary survival assay as a screening strategy to evaluate hits. SB uses cells as a chemical factory (NP derivatives) and a screening platform against therapeutic targets even difficult ones (eg, protein–protein interactions).
Plants represent a rich source of secondary metabolites with human therapeutic activity. One of the greatest successes of SB is to incorporate pathways or biosynthetic units of plants secondary metabolites into microorganisms or algae.78–80 Specific alkaloids or terpenoids only seen in plants can be synthesized in highly growing organisms, such as cyanobacteria or yeasts. However, the integration of biosynthetic units into host organisms is not an easy task as it requires the synchronization of multiple components on different temporal and spatial scales81,82 as well as the incorporation of additional regulating element to achieve efficient production yield.83–85
The independence (orthogonality) of the manufactured gene circuits with respect to the host organism is an important criterion in order to integrate them easily using a plug-and-play approach.59,83,86–88 Cell-free systems are also emerging as powerful tools to avoid many of the pitfalls of in vivo SB. This in vitro approach allows the direct control of regulatory elements, the addition of cofactors, and enzymes, as well as in situ monitoring.89
Plant’s synthetic biology
Plants have been used for thousands of years not only for human medicine and insecticide but also for dyes, flavors, and fragrance. The impossibility to change location made plants to develop an extensive defense system and adaptation capabilities. They therefore synthesize a tremendous amount of secondary metabolites that they use as defense systems against pathogens, herbivores, or external stress, such as ultra violet (UV) radiation.90
Plants have interesting features for SB.91 The first one is the use of a photosynthetic system to convert sunlight energy into organic compounds. This feature (shared by other organisms, such as cyanobacteria and algae) is a major interest in the economy of production. As they use minerals as nutriment and gases (O2 and CO2) for respiration and photosynthesis, plants have developed an extremely rich arsenal of enzymes (eg, cytochrome P450s) to carry out a broad range of regio- and stereospecific chemical reactions. This explains in part the versatile panel of plant secondary metabolites. P450s are also involved in the biosynthetic pathways of chemicals involved in the defense system against hostile organisms, which are often retrieved in animals as part of their detoxication pathways. Opposite coevolution with respect to animals might also explain the large amounts of P450 isoforms found in plants and the link with human medicine.92–94
Of particular interest for plant SB is the presence of various cellular compartments, including chloroplast, vacuole, nucleus, endoplasmic reticulum, and cytosol. Individual biochemical steps may take place in particular subcellular locations to benefit from specific enzymes and reactive conditions. Large-scale production of such compounds is a major challenge in plant SB. This supposes the coexpression or sequential expression of specific genes. Plant compartments help very much in this respect by decomposing the full pathway in independent parts while optimizing reactions and precursor compounds conditions in each cellular compartment.95,96 Combinatorial approach used in microorganisms for genome shuffling has also been adapted for plants using the multiplex hybridization technique.97
Metabolic engineering and SB for drug production
Production of NP drugs as the ones reviewed in previous sections in their natural producers is often expensive resulting in low-titer levels. Notably, the application of SB techniques can help to expand the variety of pharmaceutical products that can be efficiently produced.98 As we increase our understanding of the complexity of biosynthetic pathways and their regulatory circuitry, more successful applications of SB to metabolic engineering of pharmaceuticals are becoming possible.99–102 Chemicals of pharmaceutical interest are often complex molecules with chirality, which are difficult to synthesize chemically. Metabolic engineering, combined with SB, provides innovative solutions for red biotechnology through modification and by importing enzymatic pathways into industrial organisms103 for conversion of natural feedstock (Figure 4A) and/or enzymatic bioprocessing of supplemented chemical precursors (Figure 4B). Microbial bioproduction is often preferred, as it is relatively easier to manipulate, fermentation can be better controlled, it has fast growth, and inexpensive substrates can be used in many cases.104 Even if it is a promising solution for pharmaceutical production, the number of compounds with pharmaceutical interest having reached industrial scale of bioproduction (>50 g/L) is still limited to some amino acids and isoprenoids, while medium scale (5–50 g/L) has been reached for artemisinin and several antibiotics.105 Increasing the number of pharmaceuticals that can be bioproduced at industrial scale is, thus, a challenge at present. SB addresses the challenge and contributes to the delivery of pharmaceuticals to market by providing an engineering approach that combines modeling and simulation of metabolic pathways with design, build, test, and optimization of host strains.106
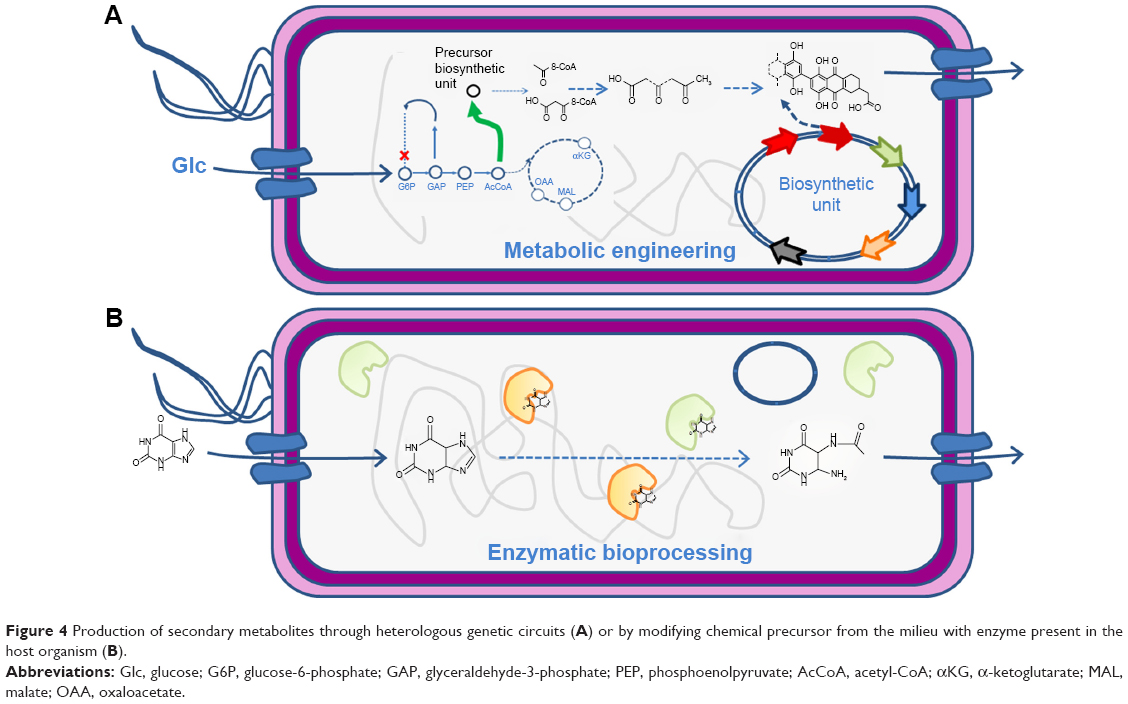 | Figure 4 Production of secondary metabolites through heterologous genetic circuits (A) or by modifying chemical precursor from the milieu with enzyme present in the host organism (B). |
Major classes of NP drugs that are bioproduced are isoprenoids, polyketides, NRPs, and other naturally produced polyphenols (flavonoids, stilbenoids, etc). Isoprenoids are a large class of NPs (>40,000 structurally unique compounds) that include many pharmaceutical relevant compounds, such as antioxidants, anticancer, or antimalarial drugs. The isoprenoid pathway has been expressed in several hosts and assembled from genes imported from multiple sources.107 Several SB techniques have been used to increase the performance of the isoprenoid pathways, such as modular tuning of gene expression,108 increasing the flux,109 controlling substrate toxicity,110 or co-localization by synthetic protein scaffolds.111 Perhaps the most well-known case is the semisynthetic production of the isoprenoid artemisinin, an antimalarial drug, achieved through the combined use of metabolic engineering and SB.112 Identification of limiting enzymes and gene expression balance through plasmid copy number and promoter strength was possible to arrive at a production of the precursor amorphadiene of 25 g/L in Escherichia coli and of 40 g/L in yeast.112
Similarly, SB techniques have been applied to the production of the isoprenoid paclitaxel (known as taxol), a cancer chemotherapy drug. Taxol is difficult to synthesize chemically and its extraction from its natural producer, the Pacific yew, is very inefficient. By using a modular approach, the optimal combination of expression levels of the different parts of the taxol precursor pathway was determined, achieving a titer of 1 g/L in E. coli.113 Similarly, a modular approach was used as well in the production of the nutraceutical resveratrol in E. coli.114,115
SB can serve to devise combinatorial biosynthesis of pharmaceutical compounds, such as NRPs such as cyclic lipopeptide antibiotics,116 and to help improving the properties of drugs. For instance, by implementing pathways that incorporated fluorine into the polyketide backbone result in fluorinated NPs with improved the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties.117 SB for DD has also been applied to the combinatorial biosynthesis in plants97 (Figure 4). Plants offer several advantages, for instance, plant cytochrome P450 enzymes are often found in the metabolic pathways. In some cases, plants with a fast, photosynthesis-driven accumulation of biomass, such as tobacco species, may serve as an alternative bioreactor to microbial production of pharmaceutical compounds. Cyanobacteria are prolific producers of NPs, such as NRPs and polyketides.118 Blue green algae (microalgae) and red algae (macroalgae) phyla are recognized as a rich source of diverse and novel bioactive compounds from marine resources. They live on water and sun as a source for nutriments and energy and can therefore be easily set up for large-scale production of molecules of pharmaceutical importance.119,120
Synthetic bioproduction, however, is a complex process whose success not only depends on the appropriate choice of the pathway but also on many other factors, such as cofactor and redox balance,121 thermodynamic feasibility, flux coupling,122 or regulatory elements.123 Synthetic pathways are often inefficient and need to be improved through protein engineering124–127 and directed evolution, which can be applied not only to enzymes but also to full pathways.128 Automated computer-aided design of metabolic pathways can help to cope with the complexity of finding the most efficient pathway among the large number of potential routes.129–132 Based on that strategy, a fully automated framework for pathway design based on a retrosynthetic approach has been applied to the production of flavonoid compounds.133 Flavonoids possess pharmaceutical potential due to their health-promoting activities, with naringenin and pinocembrin as the key flavonoid scaffolds and precursors.134 Beyond these proofs of concept applications, the integration of computer-aided design with automated DNA assembly and genome compilation methods and robotized manufacturing will accelerate pharmaceutical DD in the future.
One of the most promising contributions of SB to pharmaceutical bioproduction is in the use of biological devices built from well characterized and standardized genetic parts to exploit dynamics pathway regulation and metabolic control.135–137 Metabolite-responsive transcription regulators and riboswitches are two types of genetic parts that can be used to engineer synthetic, dynamically regulated metabolic pathways.138 Zhang et al devised a fatty acid synthesis pathway that is regulated by a transcription repressor that becomes deactivated when bound to fatty acids.139 This feedback loop increased the yield of fatty acid ethyl ester by threefold and improved the stability of pathway genes, facilitating the high-yield production of other malonyl-CoA-derived compounds.135
Another domain of SB for bioproduction is cell-free metabolic engineering140 consisting of in vitro ensembles of enzymes. Some groups have been able to reconstruct biosynthetic pathways in vitro, for instance, for isoprenoids141 or for protein production.142
Target identification and validation
Drug target identification and validation are essential in the DD process to minimize risk of future failure at later stages of drug development. Standard knock-out and knock-in experiments in cell or in mice are the common strategies for studying the influence of gene in cell-based or animal models. By using SB tools, expression of essential genes can be modulated by small molecules making synthetic cells a tool to identify drug–target interactions as presented, for example, by Firman et al.143 Today, the recent application of clustered regularly interspaced short palindromic repeats CRISPR/Cas9 genome editing technique (discovered for the first time in 1987 by Ishino et al144 in E. coli) enables us to insert mutations or incorporate a gene at any specified sites of the genome. CRISPR/Cas9 is a programmable DNA cleaving enzyme that uses guided RNA to identify the targeted region of the DNA. “CRISPy”, a user-friendly bioinformatics tool has been developed to identify rapidly the “small-guided” sgRNA target sequences in the CHO-K1 genome.145
The introduction of site-specific mutations that confer drug resistance in cells is an ultimate way for validating a target and estimating the selectivity of a drug. This approach was not straightforward in mammalian cells before the arrival of CRISPR/Cas9 technology. Neggers et al used this technique recently to validate the interaction of the anticancer drug selinexor with its primary target exportin-1 (CRM1/XPO1).146 The authors created a mutant drug target XPO1C528S that conferred >250-fold resistance to selinexor. The effect of the mutation on cell viability, apoptosis, and cell cycle progression in the presence of the drug-validated XPO1 as the prime and selective target of selinexor.
Kasap et al integrated recently this “gold standard” (or “genetic”) proof of a drug’s target into a genomic platform DrugTargetSeqR.147 This platform is based on high-throughput sequencing of drug resistance-conferring mutations in human cancer cells and uses the CRISPR/Cas9 “nickase” system and homology-directed repair to check whether the observed mutations were sufficient to confer resistance to the drug. This platform was tested to analyze ispinesib, an anticancer agent that inhibits drug target kinesin-5 in human cancer cells. CRISPR/cas9 technique was also used by Zheng et al to assess the target candidate leucine aminopeptidase to treat Plasmodium, Babesia, and Trypanosoma pathogens.148
Citorik et al used the CRISPR/Cas9 technology to create antimicrobials with spectrum of activity specified by design. The RNA-guided nucleases were delivered to microbial populations through transmissible plasmids carried by bacteriophage. These RNA-guided nucleases targets DNA genes that induce antibiotic resistance or virulence determinants in carbapenem-resistant Enterobacteriaceae (CRE) and entero-hemorrhagic E. coli.149 Such an approach induces selective pressure at the DNA level and can be used as a tool to modulate the composition of complex microbial communities in particular, the programmable remodeling of microbiota.
Phenotypic cell-based screening
With the help of structure-based drug design and human genome sequencing, the target-based approach has overwhelmed modern DD since 30 years or so. The major advantage of this approach is to jumpstart therapeutic project with a predefined drug mechanism of action, ie, the selected therapeutic target, which can be validated through a chemical series that present structure–activity relationship. The counterpart of this approach is that the long and iterative process of drug target validation is made on the road, and experimental evidence for the poor choice of the target may come at late stage of DD jeopardizing the whole project (refer Table 1 of Zheng et al for the advantages and inconveniences of the target- and phenotypic-based approaches150).
Today, the increasing difficulty of drug approval and the resurgence of a systemic view of poly-pharmacology lead to a resurgence of the phenotypic-based approach. This is a drug screening strategy in which disease phenotype is incorporated into a cell-based assay. Multiple targets or pathways may be altered by the drug, and no assumption on the mechanism of action is made at this level.20
There are three types of cell assays that are commonly performed in DD. 1) Cell viability assays, which measure the capability of a compound to kill a cell (eg, cancer cell, bacteria, fungi, protozoa, and parasites); 2) cell signaling pathway assays in which compound activates or inhibits a cell signaling or metabolic pathways; and finally 3) the disease-related phenotypic assays in which the compound triggers phenotypic responses with respect to apoptosis, immune-response, induction of cell cycle, motility, and nuclear translocation leading to neurite outgrowth. For example, neurite outgrowth assays are used to study Alzheimer’s and Parkinson’s diseases, aberration of cytoskeletal structure in myopathy, and CNS pathologies.150
One of the objectives of SB in the field of screening for therapeutic research is to construct a minimal synthetic cell containing the genes that are essential for cell survival and proliferation together with a disease-related genetic circuit, which aligns very much with this phenotypic-based strategy. A general review of the synthetic construction of phenotypic cell-based screening assay has been presented by Chiba et al.151
So far, those screening cell-based assays are made to identify small molecules that modulate gene expression by direct interaction with repressors or inducers, such as aptamers, riboswitchs, or TFs. Genetic circuit integrates luminescence or fluorescence reporter gene (eg, GFP) to monitor output signal, ie, the binding interactions with those gene regulators. The implementation of target- and phenotypic-based drug screening approaches in synthetic cell is illustrated in Figure 5. This conceptual workflow is inspired from the work of Duportet et al on the construction of modular and combinatorial assembly of functional gene expression vectors.152 It incorporates various components that could illustrate an ideal synthetic cell-based assay as connection between certain elements remains theoretical.
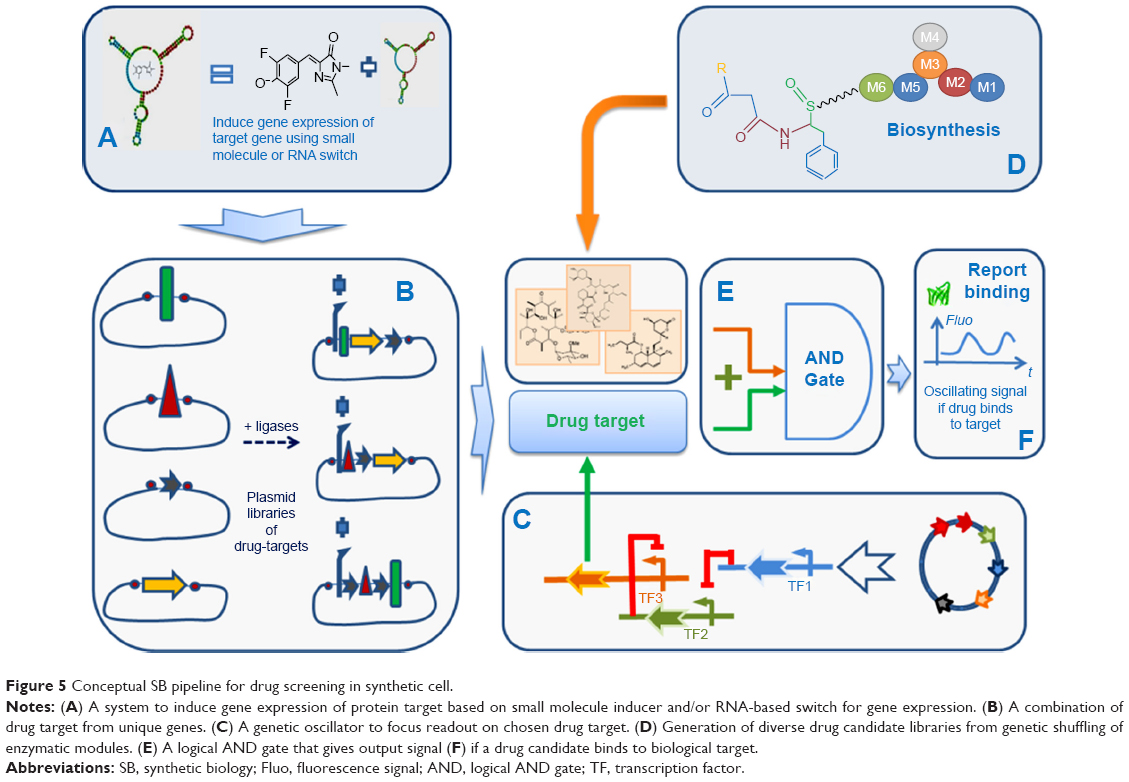 | Figure 5 Conceptual SB pipeline for drug screening in synthetic cell. |
The starting point is a library of plasmids for producing a set of drug targets by combinatorial shuffling of basic elements (Figure 5A). The expression of a given target is stimulated by external small-molecule binders of TFs, RNA-based switches, such as aptamers, which can be controlled through green fluorescence with a suitable fluorophore (Figure 5B). An oscillatory genetic circuit controls gene expression of the selected drug target. Shuffling of the biosynthetic modules produce a combinatorial library of compounds (Figure 5D) that enter the “AND gate” (logical AND gate) with the drug target. This AND gate is also a genetic circuit (Figure 5E). The output signal is monitored by measuring variation in oscillation if drug binds to the selected drug-target using fluorescent reporter gene such as GFP (Figure 5F).
Phenotypic screening using synthetic TFs
Synthetic TFs, transcription activators, and transcription activators-like effectors can be designed to bind to specified regions of the genome to modulate gene expression.153 Other TFs can be engineered to sense small metabolites or small-molecular-weight drugs, such as isopropyl β-D-1-thiogalactopyranoside (IPTG), doxycycline, 4-hydroxy-tamoxifen, uric acid, rapamycin, macrolides, and streptogramin.84 Such TFs can be fused with fluorescent reporter genes (eg, GFP) to serve as drug screening assay. That approach was developed by Fusseneger’s group to identify new antibiotics of the streptogramin group. Candidate antibiotics activate the expression of a reporter gene, such as GFP, or gene secreted alkaline phosphatase by inhibiting the repressor of the pristinamycin-induced protein. This repressor is observed in the streptogramin-resistance operon and is used as a target against drug resistance.154
Bhattacharjee developed another transcription assay to identify toxic endocrine disruptive chemicals that modulate hormone signaling and cause developmental and reproductive anomalies. They used mammalian and yeast cell systems together with various reporter genes, such as bacterial luciferase (Lux) and GFP, which showed improved speed and sensitivity of detection.155 Quantitative readout from graded response to inducer was obtained by McIsaac et al.156 They used artificial TF to activate a unique target gene (human estrogen receptor) in the presence of the inducer (β-estradiol-like chemicals). Eggelin et al reviewed development and screening applications of such TF-based biosensors.157
Phenotypic screening using aptamers
In recent years, RNA, DNA, and peptide aptamers have emerged as novel molecular tools for biosensing, drug delivery, disease diagnosis, and therapy.158 Aptamers can bind with high affinity and high specificity to a wide range of targets, from large proteins to small molecules, such as glucose. Aptamer can be isolated using SELEX (Systematic Evolution of Ligands by Exponential Enrichment) technology159 in which specific aptamers can be produced at large amount158,160 and optimized for nearly any kind of protein targets, especially undruggable targets, including transiently structured, intrinsically disordered proteins,161 partners of protein–protein interactions.162 A recent study shows that they can also be used as sensors of a molecular chaperone and to modulate its folding state and lifetime and decrease the antiapoptotic and tumorigenic activities of cancer cells.163
An aptamer-based sorting system capable of extracting and releasing target biomolecules from a solution mixture has been designed by Shastri et al.164 Noncoding RNAs are also used in epigenetic regulation165 or splicing events regulation. The use of aptamers for the treatment of cancer has been reviewed by Sun et al.166 Guo et al developed a method for the rapid selection of looped DNA aptamers against eIF4e oncology target. Selection and purification were combined in a single step followed by high-throughput sequencing to characterize aptamer candidate.167
Peptide aptamers are well adapted to bind protein targets with very high affinity and specificity and for this reason are called chemical antibodies. They are used in immunotherapies to target cell-specific receptors.166 Peptide aptamers can be fused to a stable protein scaffold to search for partners of protein–protein interaction or to optimize the biochemical function of multidomain proteins.160 Miller et al developed a scaffold protein by fusing three protein domains, FKBP12, FRB, and GST, which bind only in the absence of the small-molecule rapamycin. Miller et al developed a ligand-regulated peptide aptamer that interact with and inhibit the 5′-AMP-activated protein kinase. The LiRPs interact with the substrate peptide binding region of both AMP-activated protein kinase α1 and α2.168 Reverdatto et al developed a yeast two-hybrid screen to identify peptide aptamers that bind to various domains of the receptor for advanced glycation end products using a combinatorial library of improved peptide aptamers.169
Combinatorial selection of peptide aptamers is also a promising approach to screen for inhibitors of protein–protein interactions. AptaScreen is based on the generation of combinatorial libraries of peptide sequences that typically target the surface loops of a scaffold protein. This HTS assay identifies small molecules that displace interactions between proteins and their cognate peptide aptamers.170 Yeh described the peptide-aptamer interference approach to identify modulators of protein–protein interaction.171
Phenotypic screening using riboswitches or ribozymes
Riboswitches and ribozymes are RNA-based gene regulatory devices that change conformation or autocatalyze while binding with small metabolites,172–173 resulting in an on or off switch of gene expression. Their importance in controlling central metabolism makes them an attractive target for antibiotics or antiparasitics.
Thiamine pyrophosphate riboswitches, for example, are involved in the regulation of thiamine metabolism in numerous bacteria and are examples of promising antibacterial targets.174 Prommana et al described a glucosamine-induced ribozyme activation system to modulate a drug target dihydrofolate reductase-thymidylate synthase in Plasmodium falciparum.175
Ligand sensing riboswitch in fusion with a reporter gene can be used as a screening strategy to identify new chemical scaffolds that trigger riboswitch-mediated gene regulation. Nelson et al developed an efficient HTS using a fluoride riboswitch reporter fusion gene, which led to the identification of compounds that enhance the innate toxicity of fluoride in toxicity in E. coli and Streptococcus mutants.176 These synthetic riboregulators can respond to multiple orthogonal input signals regulating multiple genes inside a cell and represent programmable kill switches for pathological microorganisms.177
Synthetic cellular models of disease
SB tools are also used in DD to understand disease mechanisms in order to help the development of new diagnostics tools and treatments.83,84,178,179 Disease models are engineered genetic circuits to study cellular response with respect to input stimulating signals to either control molecular mechanisms under input light stimuli (eg, optogenetics) or study pathogenic-related situations, such as UV stress, growth factor, or drug action using a reporter gene (eg, fluorescent protein).
A major application of synthetic cellular models is in the field of immuno-oncology.180–184 Such models aim at studying the regulation of the B-cell antigen receptor (BCR) signaling from various cell surface receptors in order to understand the pathological mechanisms of B-cell immunodeficiency. Harumiya et al reconstructed such BCR-signaling pathway within J558L myeloma cell lines.185 Other disease models have been made to study host–pathogen interactions, immune disorders, metabolic disorders, neurodegenerative diseases,12,186–189 or cancer.190,191
The design principles and generation of novel signaling properties have been reviewed by Furukawa and Hohmann in the context of engineered yeast mitogen-activated protein kinase pathways.192 Moreover, metabolic network modeling is of growing interest in therapeutic research, especially in cancer research to understand cell proliferation signaling mediated by Tyr-kinase receptors.193
Neural science and brain research is another field of application of SB tools. An optical genetic sensor is integrated into synthetic cells to follow cell signaling in vivo. Optogenetics is an approach to switch protein on and off using either light or synthetic photoswitches ligands.194–196 Coupled with pharmacogenetics and chemogenetics, optogenetics is a powerful tool to study and dissect the neural circuits and study neuropathological conditions, such as sleep disorders and other psychiatric diseases.197–200
Antimicrobial and drug resistance
Overcoming the emergence of drug-resistance or combatting persistence cells (persisters) is a major challenge in DD.201–204 Kohanski et al discussed the different drug resistant mechanisms from a biological network viewpoint.205 Liu et al made an in silico investigation to study intrinsic and induced drug resistance mechanism in solid tumor by bridging the gap between cell and tissues.206
Manipulation of biosynthetic units as presented in second section can be applied to synthesize new antibiotics against resistant bacteria207 Saxena et al used genetic–synthetic strategies to target multidrug resistant Mycobacterium tuberculosis.208 Zakeri and Lu reviewed similar SB approaches for phage engineering.204
Cheng et al used high-throughput technology to identify genetic combinations that could potentiate antibiotics activity against CRE.209 They developed the Combinatorial Genetics en Masse technology to identify genetic combinations that enhance the effectiveness of antibiotics against CRE.
Another SB strategy is to design synthetic cell–cell communication network as a model to study persistence in bacteria and how to sensitize cells or to use it as a screening platform to target QS in bacterial communities.210–213 QS is a cell–cell communication system that allows bacteria or microorganisms to synchronize expression of a particular gene in a cell density-dependent manner (Figure 6).
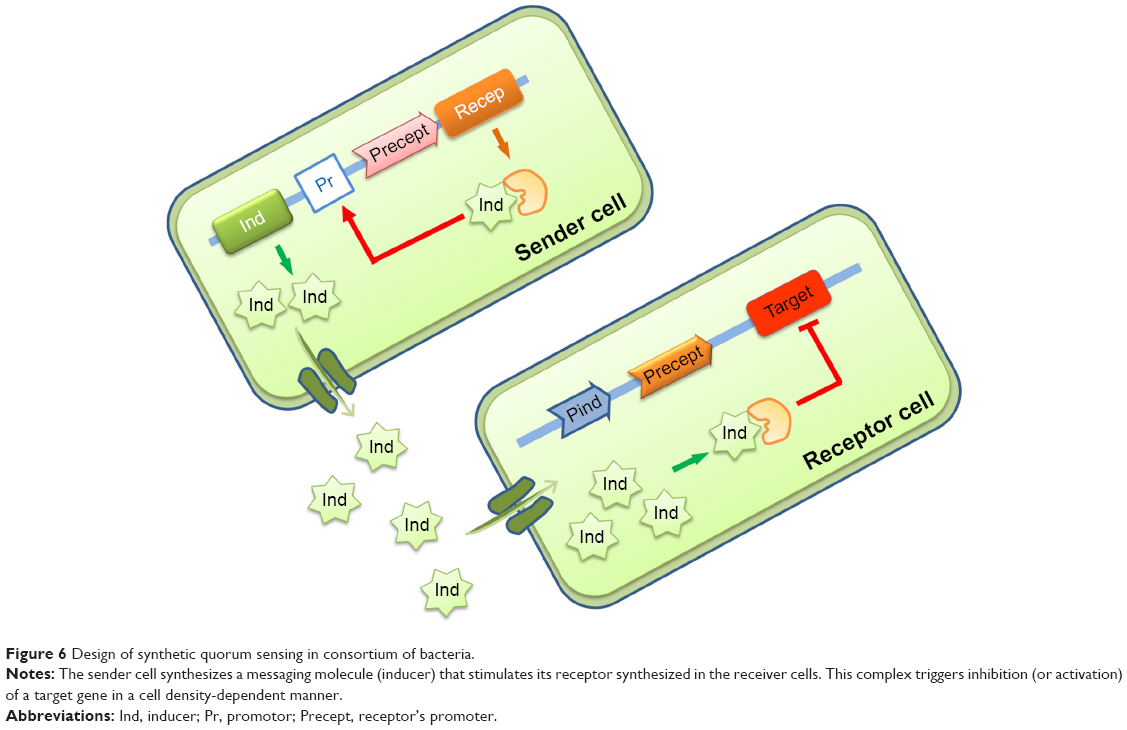 | Figure 6 Design of synthetic quorum sensing in consortium of bacteria. |
Holm and Vikström recently reviewed how bacteria use QS to interact with human cells and in particular, how they communicate information concerning population density to trigger the production of virulence factors, biofilm formation, or to develop drug resistance.214 Many antibiotic biosynthesis routes are regulated by small signaling molecules such as acyl homoserine lactones in Pseudomonas215 or γ-butyrolactones (GBLs) in Streptomyces.216 The small-molecule GBLs bind to cytoplasmic proteins receptors that are themselves repressor of gene TF of the antibiotics biosynthetic units. Very often, these biosynthetic units contain genes encoding transporters for antibiotic efflux to avoid the accumulation of toxic compounds in the cytoplasm. The stimulation of efflux pumps expression by signal molecules, such as GBL, also induces resistance to the antibiotics. Targeting the acyl homoserine lactone or GBL-based QS is a strategy that has gained much attention recently as it can inhibit the production of virulence factors as well as lower the impact of resistant mechanisms, such as drug-efflux pump systems.217 Three distinct signaling pathways, such as Lux, Las, and Rhl, are used to engineer synthetic communication systems as reviewed by Davis et al.218 Scutera et al reviewed the latest strategies in designing QS inhibitors219 and Singh and Ray reviewed the role of QS in Staphylococcus aureus and Staphylococcus epidermidis infections.220 They investigated in particular the role of accessory gene regulators in Staphylococci invasiveness through upregulation of secreted virulence factors or downregulating cell surface proteins.
A large body of evidence shows that reduced bacteria metabolism is linked to resistance and tolerance to many antibiotics, whereas enhanced metabolism induces drug sensitivity.221 Peng et al investigated the change in the metabolic states of resistant bacteria on treatment with antibiotic kanamycin and showed that resistant strains show greatest sensitivity in glucose and alanine deficiencies.222
Lee and Collins223 evoked four different consequences of the presence of antibiotics for which a SB targeting strategy can be defined. 1) Antibiotics stimulate the production of hydroxyl radicals, which can induce either cell death or resistant mutations if the antibiotics is at sublethal concentration; 2) antibiotics-resistant mutant induces indole formation by catabolizing L-tryptophan which through the QS network will stimulate drug-efflux pumps and oxidative stress detoxification pathways in the more sensible bacteria strain; 3) addition of metabolites such as glucose or alanine to the extracellular environment generates proton motive force that sensitizes persistent or dormant cells to aminoglycoside antibiotics; and finally 4) some bacteria use antibiotics as sole carbon source, which help microbial community to evade treatment by reducing the local concentration of antibiotics. Planson et al reviewed some strategies to counteract resistance to antibiotics from a combined metabolic engineering and SB standpoint.224
Toxicology
Drugs often modulate unwanted targets that induce toxic effects on human cells. Unraveling the components of the biological network that induce the toxicity pathways is an integral component of drug toxicity studies at preclinical stage. Cell-based dose–response assessments of toxicity rely on the detection of small cellular signals in response to perturbation of toxicity pathways. Zhang et al developed genetic circuitries, such as integral feedback, feedforward, and transcritical bifurcation, to obtain acceptable thresholds in response to changes in certain specific cellular states, such as levels of reactive oxygen species, oxygen molecules (O2), DNA damage, protein folding, metal ions, or osmolarity.225
Toxicity of drugs can also become an issue for engineered chassis organisms by limiting production titers. In order to reduce the toxicity effects, SB approaches have been proposed based on engineering export systems in the cell.226 A different approach consists of the development of models to estimate small-molecule toxicity in microorganism.227,228 Such type of models can help in order to select the most efficient biosynthesis constructs that avoid toxic intermediates when multiple pathways are available.
Conclusion
Application of SB in DD is at its infancy. However, it already plays a major role in reorienting pharmaceutical research. The abundance of experimental chemical proteomics data has revealed the existence of multiple biological targets for a given drug. This raises a systemic view of poly-pharmacology, which completely aligns with the cell-based phenotypic and holistic approaches of SB. The rational-based genetic design is to SB what rational-based drug design is to medicinal chemistry.
The great success of SB in the field of bioproduction with the success story of artemisinin will likely influence the early stages of DD. Next future interests are likely in the rational design of new biochemicals through genetic shuffling of biosynthetic modules in order to be compliant with large-scale production within microorganisms.
Synthetic cells have not only become a biofactory for producing added value compounds or innovating new NP-like derivatives but also a wet laboratory in which, therapeutic target or cell signaling pathway can be tested. It also provides a rational approach to engineer cell-based assays to screen for compounds that will trigger the designed disease phenotype. Such cell-based phenotypic assay has the advantage to study the action of the drug on the entire therapeutic pathway. Disruption of interactions between such pathways (including metabolism and cell signaling) can be studied through such synthetic cell-based models.
Synthetic cellular models can also be used to identify disease mechanisms or to dissect drug’s mechanism of action. The use of luminescent or fluorescent reporter genes is particularly useful to follow biomarkers of phenotypes with respect to physiological conditions. Optogenetics shows important applications in studying psychiatric disorders187 (eg, Alzheimer’s, Parkinson’s, and sleep disorders) or in immuno-oncology whose goals are to reactivate immunological response in tumors.181,229
Designing synthetic QS is a powerful tool to study cell–cell communication within bacteria consortium and in order to overcome drug-resistance and persistence mechanisms. Genetic circuits can reproduce artificial communication system and can be used to screen for compounds that disrupt QS or synergies between metabolism and drug resistance mechanisms. Indeed, sensitizing resistant or persistent bacteria using combination of antibiotics and metabolites is another promising approach to overcome drug resistance.
The holy grail of SB in the field of DD is to create a universal cell in which NP biosynthesis could be selectively triggered by sensed disease phenotypes. Lead optimization could be made through selective pressure (a precious optimization process offered by living systems) induced by chosen substrates. Modification of genome expression through environmental conditions (eg, nutriment or small molecular inducers or light) is another great property of living organisms, which can be modulated through SB and transformed into a therapeutic strategy to trigger molecular switches within pathological tissues.230
Acknowledgments
PC is supported by UPFellows program, which is funded in part by the Marie Curie COFUND program and by BBSRC/Engineering and Physical Sciences Research Council (EPSRC) grant BB/M017702/1.
Disclosure
The authors report no conflicts of interest in this work.
References
Moreno L, Pearson AD. How can attrition rates be reduced in cancer drug discovery? Expert Opin Drug Discov. 2013;8(4):363–368. | ||
Hu Y, Bajorath J. Monitoring drug promiscuity over time. F1000Res. 2014;3:218. | ||
Leil TA, Bertz R. Quantitative systems pharmacology can reduce attrition and improve productivity in pharmaceutical research and development. Front Pharmacol. 2014;5:247. | ||
Ryall KA, Tan AC. Systems biology approaches for advancing the discovery of effective drug combinations. J Cheminform. 2015;7:7. | ||
Srikanthan A, Ethier J-L, Ocana A, Seruga B, Krzyzanowska MK, Amir E. Cardiovascular toxicity of multi-tyrosine kinase inhibitors in advanced solid tumors: a population-based observational study. PLoS One. 2015;10(3):e0122735. | ||
Medema MH, Kottmann R, Yilmaz P, et al. Minimum information about a biosynthetic gene cluster. Nat Chem Biol. 2015;11(9):625–631. | ||
Sun H, Liu Z, Zhao H, Ang EL. Recent advances in combinatorial biosynthesis for drug discovery. Drug Des Devel Ther. 2015;9: 823–833. | ||
Atanasov AG, Waltenberger B, Pferschy-Wenzig E-M, et al. Discovery and resupply of pharmacologically active plant-derived natural products: a review. Biotechnol Adv. 2015;15:S0734–9750. | ||
Breitling R, Takano E. Synthetic biology advances for pharmaceutical production. Curr Opin Biotechnol. 2015;35C:46–51. | ||
Kis Z, Pereira HS, Homma T, Pedrigi RM, Krams R. Mammalian synthetic biology: emerging medical applications. J R Soc Interface. 2015;12(106):1000. | ||
Lee I, Lehner B, Crombie C, Wong W, Fraser AG, Marcotte EM. A single gene network accurately predicts phenotypic effects of gene perturbation in Caenorhabditis elegans. Nat Genet. 2008;40(2):181–188. | ||
Khalil AS, Collins JJ. Synthetic biology: applications come of age. Nat Rev Genet. 2010;11(5):367–379. | ||
Juhas M, Eberl L, Church GM. Essential genes as antimicrobial targets and cornerstones of synthetic biology. Trends Biotechnol. 2012;30(11):601–607. | ||
Bernardo D, Thompson MJ, Gardner TS, et al. Chemogenomic profiling on a genome-wide scale using reverse-engineered gene networks. Nat Biotechnol. 2005;23(3):377–383. | ||
Bayer TS. Using synthetic biology to understand the evolution of gene expression. Curr Biol. 2010;20(17):R772–R779. | ||
Harvey CJB, Puglisi JD, Pande VS, Cane DE, Khosla C. Precursor directed biosynthesis of an orthogonally functional erythromycin analogue: selectivity in the ribosome macrolide binding pocket. J Am Chem Soc. 2012;134(29):12259–12265. | ||
Weissman KJ. Mutasynthesis, uniting chemistry and genetics for drug discovery. Trends Biotech. 2007;25(4):139–142. | ||
Mulinari S. The specificity triad: notions of disease and therapeutic specificity in biomedical reasoning. Philos Ethics Humanit Med. 2014;9:14. | ||
Maruta H. From chemotherapy to signal therapy (1909–2009): a century pioneered by Paul Ehrlich. Drug Discov Ther. 2009;3(2):37–40. | ||
Eder J, Herrling PL. Trends in modern drug discovery. Handbook Experimental Pharmacology. 2015:1–20. | ||
Barot KP, Nikolova S, Ivanov I, Ghate MD. Liquid-phase combinatorial library synthesis: recent advances and future perspectives. Comb Chem High Throughput Screen. 2014;17(5):417–438. | ||
Sun X, Vilar S, Tatonetti NP. High-throughput methods for combinatorial drug discovery. Sci Transl Med. 2013;5(205):205rv1. | ||
Diller DJ. The synergy between combinatorial chemistry and high-throughput screening. Curr Opin Drug Discov Devel. 2008;11(3):346–355. | ||
Koehn FE, Carter GT. Rediscovering natural products as a source of new drugs. Discov Med. 2005;5(26):159–164. | ||
Harris CJ, Hill RD, Sheppard DW, Slater MJ, Stouten PFW. The design and application of target-focused compound libraries. Comb Chem High Throughput Screen. 2011;14(6):521–531. | ||
Miduturu CV, Deng X, Kwiatkowski N, et al. High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chem Biol. 2011;18:868–879. | ||
Fedorov O, Niesen FH, Knapp S. Kinase Inhibitor Selectivity Profiling Using Differential Scanning Fluorimetry. Totowa, NJ: Humana Press; 2012. | ||
Hu Y, Bajorath J. Quantifying the tendency of therapeutic target proteins to bind promiscuous or selective compounds. PLoS One. 2015;10(5):e0126838. | ||
Hu Y, Gupta-Ostermann D, Bajorath J. Exploring compound promiscuity patterns and multi-target activity spaces. Comput Struct Biotechnol J. 2014;9:e201401003. | ||
Boran ADW, Iyengar R. Systems approaches to polypharmacology and drug discovery. Curr Opin Drug Discov Devel. 2010;13(3):297–309. | ||
Tang J, Aittokallio T. Network pharmacology strategies toward multi-target anticancer therapies: from computational models to experimental design principles. Curr Pharm Des. 2014;20(1):23–36. | ||
Lehàr J, Krueger AS, Avery W, et al. Synergistic drug combinations improve therapeutic selectivity. Nat Biotechnol. 2010;27(7):659–666. | ||
Anighoro A, Bajorath J, Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem. 2014;57(19):7874–7887. | ||
Xiong A-S, Yao Q-H, Peng R-H, Cheng Z-M. Directed in vitro evolution of reporter genes based on semi-rational design and high-throughput screening. Methods Mol Biol. 2010;634:239–256. | ||
König H, Frank D, Heil R, Coenen C. Synthetic genomics and synthetic biology applications between hopes and concerns. Curr Genomics. 2013;14(1):11–24. | ||
Mattern DJ, Valiante V, Unkles SE, Brakhage AA. Synthetic biology of fungal natural products. Front Microbiol. 2015;6:775. | ||
Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75(3):311–335. | ||
Szychowski J, Truchon J-F, Bennani YL. Natural products in medicine: transformational outcome of synthetic chemistry. J Med Chem. 2014;57(22):9292–9308. | ||
Seyedsayamdost MR, Clardy J. Natural products and synthetic biology. ACS Synth Biol. 2014;3(10):745–747. | ||
Cummings M, Breitling R, Takano E. Steps towards the synthetic biology of polyketide biosynthesis. FEMS Microbiol Lett. 2014;351(2):116–125. | ||
Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106(8):3468–3496. | ||
Li J, Neubauer P. Escherichia coli as a cell factory for heterologous production of nonribosomal peptides and polyketides. N Biotechnol. 2014;31(6):579–585. | ||
Marahiel MA. Working outside the protein-synthesis rules: insights into non-ribosomal peptide synthesis. J Pept Sci. 2009;15(12):799–807. | ||
Moses T, Pollier J, Thevelein JM, Goossens A. Bioengineering of plant (tri)terpenoids: from metabolic engineering of plants to synthetic biology in vivo and in vitro. New Phytol. 2013;200(1):27–43. | ||
Minami H. Fermentative production of plant benzylisoquinoline alkaloids in microbes. Biosci Biotechnol Biochem. 2013;77(8): 1617–1622. | ||
Nakagawa A, Minami H, Kim J-S, et al. Bench-top fermentative production of plant benzylisoquinoline alkaloids using a bacterial platform. Bioeng Bugs. 2012;3(1):49–53. | ||
Trantas EA, Koffas MAG, Xu P, Ververidis F. When plants produce not enough or at all: metabolic engineering of flavonoids in microbial hosts. Front Plant Sci. 2015;6(7):1–16. | ||
Donadio S, Staver MJ, McAlpine JB, Swanson SJ, Katz L. Modular organization of genes required for complex polyketide biosynthesis. Science. 1991;252(5006):675–679. | ||
Caffrey P, Bevitt DJ, Staunton J, Leadlay PF. Identification of DEBS 1, DEBS 2 and DEBS 3, the multienzyme polypeptides of the erythromycin-producing polyketide synthase from Saccharopolyspora erythraea. FEBS Lett. 1992;304(2–3):225–228. | ||
Walsh CT, Fischbach MA. Natural products version 2.0: connecting genes to molecules. J Am Chem Soc. 2010;132(8):2469–2493. | ||
Hertweck C. Decoding and reprogramming complex polyketide assembly lines: prospects for synthetic biology. Trends Biochem Sci. 2015;40(4):189–199. | ||
Li JW-H, Vederas JC. Drug discovery and natural products: end of an era or an endless frontier? Science. 2009;325(5937):161–165. | ||
Medema MH, Blin K, Cimermancic P, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39(6):W339–W346. | ||
Seyedsayamdost MR. High-throughput platform for the discovery of elicitors of silent bacterial gene clusters. Proc Natl Acad Sci U S A. 2014;111(20):7266–7271. | ||
Wohlleben W, Mast Y, Muth G, Röttgen M, Stegmann E, Weber T. Synthetic biology of secondary metabolite biosynthesis in actinomycetes: engineering precursor supply as a way to optimize antibiotic production. FEBS Lett. 2012;586(15):2171–2176. | ||
Doroghazi JR, Albright JC, Goering AW, et al. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol. 2014;10(11):963–968. | ||
Cacho RA, Tang Y, Chooi Y-H. Next-generation sequencing approach for connecting secondary metabolites to biosynthetic gene clusters in fungi. Front Microbiol. 2015;5(774):1–16. | ||
Leavitt JM, Alper HS. Advances and current limitations in transcript-level control of gene expression. Curr Opin Biotechnol. 2015;34(3):98–104. | ||
Shao Z, Rao G, Li C, Abil Z, Luo Y, Zhao H. Refactoring the silent spectinabilin gene cluster using a plug and play scaffold. ACS Synth Biol. 2013;2(11):662–669. | ||
Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. 2015;14(2):111–129. | ||
Luo Y, Cobb RE, Zhao H. Recent advances in natural product discovery. Curr Opin Biotechnol. 2014;30(12):230–237. | ||
Chiang Y-M, Chang S-L, Oakley BR, Wang CCC. Recent advances in awakening silent biosynthetic gene clusters and linking orphan clusters to natural products in microorganisms. Curr Opin Chem Biol. 2011;15(1):137–143. | ||
Wu M, Law B, Wilkinson B, Micklefield J. Bioengineering natural product biosynthetic pathways for therapeutic applications. Curr Opin Biotechnol. 2012;23(6):931–940. | ||
Cobb RE, Wang Y, Zhao H. High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol. 2014;3(12):1–10. | ||
Road TC, Cb C. Polyketide biosynthesis: understanding and exploiting modularity. Philos Trans A Math Phys Eng Sci. 2004;362(1825):2671–2690. | ||
Mitchell W. Natural products from synthetic biology. Curr Opin Chem Biol. 2011;15(4):505–515. | ||
Zhang W, Tang Y. Combinatorial biosynthesis of natural products. J Med Chem. 2008;51(9):2629–2633. | ||
Menzella HG, Carney JR, Santi DV. Rational design and assembly of synthetic trimodular polyketide synthases. Chem Biol. 2007;14(2):143–151. | ||
Du Y-L, Ryan KS. Expansion of bisindole biosynthetic pathways by combinatorial construction. ACS Synth Biol. 2014;3(12):1–7. | ||
Schmidt EW, Heemstra JR. Assessing the combinatorial potential of the RiPP cyanobactin tru pathway. ACS Synth Biol. 2015;4(4):482–492. | ||
Xu Y, Zhou T, Zhang S, et al. Diversity-oriented combinatorial biosynthesis of benzenediol lactone scaffolds by subunit shuffling of fungal polyketide synthases. Proc Natl Acad Sci U S A. 2014;111(34):12354–12359. | ||
Harvey CJB, Puglisi JD, Pande VS, Cane DE, Khosla C. Precursor directed biosynthesis of an orthogonally functional erythromycin analogue: selectivity in the ribosome macrolide binding pocket. J Am Chem Soc. 2012;134(29):12259–12265. | ||
Winter JM, Tang Y. Synthetic biological approaches to natural product biosynthesis. Curr Opin Biotechnol. 2012;23(5):736–743. | ||
Smanski MJ, Bhatia S, Zhao D, et al. Functional optimization of gene clusters by combinatorial design and assembly. Nat Biotechnol. 2014;32(12):1241–1252. | ||
Xu P, Bhan N, Koffas MA. Engineering plant metabolism into microbes: from systems biology to synthetic biology. Curr Opin Biotechnol. 2012;24(2):291–299. | ||
Unkles SE, Valiante V, Mattern DJ, Brakhage AA. Synthetic biology tools for bioprospecting of natural products in eukaryotes. Chem Biol. 2014;21(4):502–508. | ||
Klein J, Heal JR, Hamilton WDO, et al. Yeast synthetic biology platform generates novel chemical structures as scaffolds for drug discovery. ACS Synth Biol. 2014;3(5):314–323. | ||
Kempinski C, Jiang Z, Bell S, Chappell J. Metabolic engineering of higher plants and algae for isoprenoid production. Adv Biochem Eng Biotechnol. 2015;148:161–199. | ||
Tholl D. Biosynthesis and biological functions of terpenoids in plants. Adv Biochem Eng Biotechnol. 2015;148:63–106. | ||
Engler C, Youles M, Gruetzner R, et al. A golden gate modular cloning toolbox for plants. ACS Synth Biol. 2014;3(11):839–843. | ||
Solé R, Macía J. Synthetic biology: biocircuits in synchrony. Nature. 2014;508(7496):326–327. | ||
Klavins E. Lightening the load in synthetic biology. Nat Biotechnol. 2014;32(12):1198–1200. | ||
Ye H, Fussenegger M. Synthetic therapeutic gene circuits in mammalian cells. FEBS Lett. 2014;588(15):2537–2544. | ||
Lienert F, Lohmueller JJ, Garg A, Silver PA. Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat Rev Mol Cell Biol. 2014;15(2):95–107. | ||
Lapique N, Benenson Y. Digital switching in a biosensor circuit via programmable timing of gene availability. Nat Chem Biol. 2014;10(12):1020–1027. | ||
Singh V. Recent advancements in synthetic biology: current status and challenges. Gene. 2014;535(1):1–11. | ||
Litcofsky KD, Afeyan RB, Krom RJ, Khalil AS, Collins JJ. Iterative plug-and-play methodology for constructing and modifying synthetic gene networks. Nat Methods. 2012;9(11):1077–1080. | ||
Brückner K, Schäfer P, Weber E, Grützner R, Marillonnet S, Tissier A. A library of synthetic transcription activator-like effector-activated promoters for coordinated orthogonal gene expression in plants. Plant J. 2015;82(4):707–716. | ||
Smith MT, Wilding KM, Hunt JM, Bennett AM, Bundy BC. The emerging age of cell-free synthetic biology. FEBS Lett. 2014;588(17):2755–2761. | ||
War AR, Paulraj MG, Ahmad T, et al. Mechanisms of plant defense against insect herbivores. Plant Signal Behav. 2012;7(10):1306–1320. | ||
Liu W, Stewart CN. Plant synthetic biology. Trends Plant Sci. 2015;20(5):309–317. | ||
Lassen LM, Nielsen AZ, Olsen CE, et al. Anchoring a plant cytochrome P450 via PsaM to the thylakoids in Synechococcus sp. PCC 7002: evidence for light-driven biosynthesis. PLoS One. 2014;9(7):e102184. | ||
Ohkawa H, Inui H. Metabolism of agrochemicals and related environmental chemicals based on cytochrome P450s in mammals and plants. Pest Manag Sci. 2015;71(6):824–828. | ||
Mao G, Seebeck T, Schrenker D, Yu O. CYP709B3, a cytochrome P450 monooxygenase gene involved in salt tolerance in Arabidopsis thaliana. BMC Plant Biol. 2013;13:169. | ||
Heinig U, Gutensohn M, Dudareva N, Aharoni A. The challenges of cellular compartmentalization in plant metabolic engineering. Curr Opin Biotechnol. 2013;24(2):239–246. | ||
Lau W, Fischbach MA, Osbourn A, Sattely ES. Key applications of plant metabolic engineering. PLoS Biol. 2014;12(6):e1001879. | ||
Pollier J, Moses T, Goossens A. Combinatorial biosynthesis in plants: a (p)review on its potential and future exploitation. Nat Prod Rep. 2011;28(12):1897–1916. | ||
Curran KA, Alper HS. Expanding the chemical palate of cells by combining systems biology and metabolic engineering. Metab Eng. 2012;14(4):289–297. | ||
Keasling JD. Synthetic biology and the development of tools for metabolic engineering. Metab Eng. 2012;14(3):189–195. | ||
Nielsen J, Keasling JD. Synergies between synthetic biology and metabolic engineering. Nat Biotechnol. 2011;29(8):693–695. | ||
Stephanopoulos G. Synthetic biology and metabolic engineering. ACS Synth Biol. 2012;1(11):514–525. | ||
Du J, Shao Z, Zhao H. Engineering microbial factories for synthesis of value-added products. J Ind Microbiol Biotechnol. 2011;38(8):873–890. | ||
Rajagopal R. Bio-based chemicals: in need of innovative strategies. Chem Wkly. 2012;2(4):195–200. | ||
Lee SYY, Kim HUU, Park JHH, Park JMM, Kim TYY. Metabolic engineering of microorganisms: general strategies and drug production. Drug Discov Today. 2009;14(1–2):78–88. | ||
Sun J, Alper HS. Metabolic engineering of strains: from industrial-scale to lab-scale chemical production. J Ind Microbiol Biotechnol. 2014;42(3):423–436. | ||
Hara KY, Araki M, Okai N, Wakai S, Hasunuma T, Kondo A. Development of bio-based fine chemical production through synthetic bioengineering. Microb Cell Fact. 2014;13(1):173–191. | ||
Immethun CM, Hoynes-O’Connor AG, Balassy A, Moon TSS. Microbial production of isoprenoids enabled by synthetic biology. Front Microbiol. 2013;4(75):1–8. | ||
Pfleger BF, Pitera DJ, Smolke CD, Keasling JD. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat Biotechnol. 2006;24(8):1027–1032. | ||
Shiba Y, Paradise EM, Kirby J, Ro D-K, Keasling JD. Engineering of the pyruvate dehydrogenase bypass in Saccharomyces cerevisiae for high-level production of isoprenoids. Metab Eng. 2007;9(2):160–168. | ||
Djordjevic M, Djordjevic M. A simple biosynthetic pathway for large product generation from small substrate amounts. Phys Biol. 2012;9(5):056004–056010. | ||
Dueber JE, Wu GC, Malmirchegini GR, et al. Synthetic protein scaffolds provide modular control over metabolic flux. Nat Biotechnol. 2009;27(8):5–8. | ||
Paddon CJ, Keasling JD. Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat Rev Microbiol. 2014;12(5):355–367. | ||
Ajikumar PK, Xiao W-H, Tyo KEJ, et al. Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science. 2010;330(6000):70–74. | ||
Lim CGG, Fowler ZL, Hueller T, Schaffer S, Koffas MA. High-yield resveratrol production in engineered Escherichia coli. Appl Environ Microbiol. 2011;77(10):3451–3460. | ||
Wu J, Liu P, Fan Y, et al. Multivariate modular metabolic engineering of Escherichia coli to produce resveratrol from L-tyrosine. J Biotechnol. 2013;167(4):404–411. | ||
Baltz RH. Combinatorial biosynthesis of cyclic lipopeptide antibiotics: a model for synthetic biology to accelerate the evolution of secondary metabolite biosynthetic pathways. ACS Synth Biol. 2012;3(10):748–758. | ||
Walker MC, Thuronyi BW, Charkoudian LK, Lowry B, Khosla C, Chang MCY. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways. Science. 2013;341(6150):1089–1094. | ||
Kehr J-CC, Picchi DG, Dittmann E. Natural product biosyntheses in Cyanobacteria: a treasure trove of unique enzymes. Beilstein J Org Chem. 2011;7(12):1622–1635. | ||
Uzair B, Tabassum S, Rasheed M, Rehman SF. Exploring marine cyanobacteria for lead compounds of pharmaceutical importance. ScientificWorldJournal. 2012;2012:179782. | ||
El Gamal AA. Biological importance of marine algae. Saudi Pharm J. 2010;18(1):1–25. | ||
Dhamankar H, Prather KLJ. Microbial chemical factories: recent advances in pathway engineering for synthesis of value added chemicals. Curr Opin Struct Biol. 2011;21(4):488–494. | ||
Reimers AC, Goldstein Y, Bockmayr A. Generic flux coupling analysis. Math Biosci. 2015;262(4):28–35. | ||
Porro D, Branduardi P, Sauer M, Mattanovich D. Old obstacles and new horizons for microbial chemical production. Curr Opin Biotechnol. 2014;30(12):101–106. | ||
Pleiss J. Protein design in metabolic engineering and synthetic biology. Curr Opin Biotechnol. 2011;22(5):611–617. | ||
Chen Z, Zeng A-P. Protein design in systems metabolic engineering for industrial strain development. Biotechnol J. 2013;8(5):523–533. | ||
Eriksen DT, Lian J, Zhao H. Protein design for pathway engineering. J Struct Biol. 2014;185(2):234–242. | ||
Carbonell P, Trosset J-Y. Computational protein design methods for synthetic biology. Methods Mol Biol. 2015;1244:3–21. | ||
Abatemarco J, Hill A, Alper HS. Expanding the metabolic engineering toolbox with directed evolution. Biotechnol J. 2013;8(12):1397–1441. | ||
Fernández-castané A, Fehér T, Carbonell P, Pauthenier C, Faulon J. Computer-aided design for metabolic engineering. J Biotechnol. 2014;192(12):302–313. | ||
Campodonico MA, Andrews BA, Asenjo JA, Palsson BO, Feist AM. Generation of an atlas for commodity chemical production in Escherichia coli and a novel pathway prediction algorithm, GEM-path. Metab Eng. 2014;25(10):140–158. | ||
Medema MH, van Raaphorst R, Takano E, Breitling R. Computational tools for the synthetic design of biochemical pathways. Nat Rev Microbiol. 2012;10(3):191–202. | ||
Carbonell P, Parutto P, Herisson J, Pandit SB, Faulon J-L. XTMS: pathway design in an eXTended metabolic space. Nucleic Acids Res. 2014;42(7):W389–W394. | ||
Feher T, Planson A-GG, Carbonell P, et al. Validation of RetroPath, a computer aided design tool for metabolic pathway engineering. Biotechnol J. 2014;9(11):1446–1457. | ||
Wu J, Du G, Zhou J, Chen J. Systems metabolic engineering of microorganisms to achieve large-scale production of flavonoid scaffolds. J Biotechnol. 2014;188(3):72–80. | ||
Xu P, Li L, Zhang F, Stephanopoulos G, Koffas M. Improving fatty acids production by engineering dynamic pathway regulation and metabolic control. Proc Natl Acad Sci U S A. 2014;111(31):11299–11304. | ||
Afroz T, Beisel CL. Understanding and exploiting feedback in synthetic biology. Chem Eng Sci. 2013;103(11):79–90. | ||
Fehér T, Libis V, Carbonell P, Faulon J-L. A sense of balance: experimental investigation and modeling of a malonyl-CoA sensor in Escherichia coli. Front Bioeng Biotechnol. 2015;3(4):46–60. | ||
Michener JK, Thodey K, Liang JC, Smolke CD. Applications of genetically-encoded biosensors for the construction and control of biosynthetic pathways. Metab Eng. 2012;14(3):212–222. | ||
Zhang QC, Petrey D, Deng L, et al. Structure-based prediction of protein–protein interactions on a genome-wide scale. Nature. 2012;490(7421):556–560. | ||
Hodgman CE, Jewett MC. Cell-free synthetic biology: thinking outside the cell. Metab Eng. 2012;14(3):261–269. | ||
Dudley QM, Karim AS, Jewett MC. Cell-free metabolic engineering: biomanufacturing beyond the cell. Biotechnol J. 2014;10(1):69–82. | ||
Jewett MC, Calhoun KA, Voloshin A, Wuu JJ, Swartz JR. An integrated cell-free metabolic platform for protein production and synthetic biology. Mol Syst Biol. 2008;4(10):220–230. | ||
Firman K, Evans L, Youell JA. Synthetic biology project – developing a single-molecule device for screening drug-target interactions. FEBS Lett. 2012;586(15):2157–2163. | ||
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169(12):5429–5433. | ||
Ronda C, Pedersen LE, Hansen HG, et al. Accelerating genome editing in CHO cells using CRISPR Cas9 and CRISPy, a web-based target finding tool. Biotechnol Bioeng. 2014;111(8):1604–1616. | ||
Neggers JE, Vercruysse T, Jacquemyn M, et al. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem Biol. 2015;22(1):107–116. | ||
Kasap C, Elemento O, Kapoor TM. DrugTargetSeqR: a genomics- and CRISPR-Cas9-based method to analyze drug targets. Nat Chem Biol. 2014;10(8):626–628. | ||
Zheng J, Jia H, Zheng Y. Knockout of leucine aminopeptidase in Toxoplasma gondii using CRISPR/Cas9. Int J Parasitol. 2015;45(2–3):141–148. | ||
Citorik RJ, Mimee M, Lu TK. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat Biotechnol. 2014;32(11):1141–1145. | ||
Zheng W, Thorne N, McKew JC. Phenotypic screens as a renewed approach for drug discovery. Drug Discov Today. 2013;18(21–22):1067–1073. | ||
Chiba T, Tsuchiya T, Mori R, Shimokawa I. Protein reporter bioassay systems for the phenotypic screening of candidate drugs: a mouse platform for anti-aging drug screening. Sensors (Basel). 2012;12(2):1648–1656. | ||
Duportet X, Wroblewska L, Guye P, et al. A platform for rapid prototyping of synthetic gene networks in mammalian cells. Nucleic Acids Research. 2014;42(21):13440–13451. | ||
Keung AJ, Joung JK, Khalil AS, Collins JJ. Chromatin regulation at the frontier of synthetic biology. Nat Rev Genet. 2015;16(3):159–171. | ||
Fussenegger M, Morris RP, Fux C, et al. Streptogramin-based gene regulation systems for mammalian cells. Nat Biotechnol. 2000;18(11):1203–1208. | ||
Bagchi Bhattacharjee G, Paul Khurana SM. In vitro reporter assays for screening of chemicals that disrupt androgen signaling. J Toxicol. 2014;2014(701752):1–7. | ||
McIsaac RS, Oakes BL, Botstein D, Noyes MB. Rapid synthesis and screening of chemically activated transcription factors with GFP-based reporters. J Vis Exp. 2013;26(81):e51153. | ||
Eggeling L, Bott M, Marienhagen J. Novel screening methods – biosensors. Curr Opin Biotechnol. 2015;35(3):30–36. | ||
Wu X, Chen J, Wu M, Zhao JX. Aptamers: active targeting ligands for cancer diagnosis and therapy. Theranostics. 2015;5(4):322–344. | ||
Mascini M, Palchetti I, Tombelli S. Nucleic acid and peptide aptamers: fundamentals and bioanalytical aspects. Angew Chem Int Ed Engl. 2012;51(6):1316–1332. | ||
Reverdatto S, Burz DS, Shekhtman A. Peptide aptamers: development and applications. Curr Top Med Chem. 2015;15(12):1082–1101. | ||
Cobbert JD, DeMott C, Majumder S, et al. Caught in action: selecting peptide aptamers against intrinsically disordered proteins in live cells. Sci Rep. 2015;5(3):9402–9411. | ||
Chen L-C, Tzeng S-C, Peck K. Aptamer microarray as a novel bioassay for protein–protein interaction discovery and analysis. Biosens Bioelectron. 2013;42(4):248–255. | ||
Gibert B, Simon S, Dimitrova V, Diaz-Latoud C, Arrigo A-P. Peptide aptamers: tools to negatively or positively modulate HSPB1(27) function. Philos Trans R Soc Lond B Biol Sci. 2013;368(1617):20120075–20120083. | ||
Shastri A, McGregor LM, Liu Y, et al. An aptamer-functionalized chemomechanically modulated biomolecule catch-and-release system. Nat Chem. 2015;7(5):447–454. | ||
Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. 2013;20(3):300–307. | ||
Sun H, Zhu X, Lu PY, Rosato RR, Tan W, Zu Y. Oligonucleotide aptamers: new tools for targeted cancer therapy. Mol Ther Nucleic Acids. 2014;3(8):e182–e196. | ||
Guo WM, Kong KW, Brown CJ, et al. Identification and characterization of an eIF4e DNA aptamer that inhibits proliferation with high throughput sequencing. Mol Ther Nucleic Acids. 2014;3(12):e217–e227. | ||
Miller RA, Binkowski BF, Belshaw PJ. Ligand-regulated peptide aptamers that inhibit the 5′-AMP-activated protein kinase. J Mol Biol. 2007;365(4):945–957. | ||
Reverdatto S, Rai V, Xue J, Burz DS, Schmidt AM, Shekhtman A. Combinatorial library of improved peptide aptamers, CLIPs to inhibit RAGE signal transduction in mammalian cells. PLoS One. 2013;8(6):e65180–e65195. | ||
Bardou C, Borie C, Bickle M, Rudkin BB, Colas P. Peptide aptamers for small molecule drug discovery. Methods Mol Biol. 2009;535:373–388. | ||
Yeh JT-H, Binari R, Gocha T, Dasgupta R, Perrimon N. PAPTi: a peptide aptamer interference toolkit for perturbation of protein–protein interaction networks. Sci Rep. 2013;3(1):1156–1164. | ||
Berens C, Groher F, Suess B. RNA aptamers as genetic control devices: the potential of riboswitches as synthetic elements for regulating gene expression. Biotechnol J. 2015;10(2):246–257. | ||
Chappell J, Takahashi MK, Lucks JB. Creating small transcription activating RNAs. Nat Chem Biol. 2015;11(3):214–220. | ||
Lünse CE, Scott FJ, Suckling CJ, Mayer G. Novel TPP-riboswitch activators bypass metabolic enzyme dependency. Front Chem. 2014;2(53):1–8. | ||
Prommana P, Uthaipibull C, Wongsombat C, et al. Inducible knockdown of Plasmodium gene expression using the glmS ribozyme. PLoS One. 2013;8(8):e73783–e73793. | ||
Nelson JW, Plummer MS, Blount KF, Ames TD, Breaker RR. Small molecule fluoride toxicity agonists. Chem Biol. 2015;22(4):527–534. | ||
Callura JM, Dwyer DJ, Isaacs FJ, Cantor CR, Collins JJ. Tracking, tuning, and terminating microbial physiology using synthetic riboregulators. Proc Natl Acad Sci U S A. 2010;107(36):15898–15903. | ||
Ye H, Aubel D, Fussenegger M. Synthetic mammalian gene circuits for biomedical applications. Curr Opin Chem Biol. 2013;17(6):910–917. | ||
Liu Y, Zeng Y, Liu L, et al. Synthesizing and gate genetic circuits based on CRISPR-Cas9 for identification of bladder cancer cells. Nat Commun. 2014;5(5393):1–8. | ||
Kenderian SS, Ruella M, Gill S, Kalos M. Chimeric antigen receptor T-cell therapy to target hematologic malignancies. Cancer Res. 2014;74(22):6383–6389. | ||
June CH, Maus MV, Plesa G, et al. Engineered T cells for cancer therapy. Cancer Immunol Immunother. 2014;63(9):969–975. | ||
Abate-Daga D, Rosenberg SA, Morgan RA. Pancreatic cancer: hurdles in the engineering of CAR-based immunotherapies. Oncoimmunology. 2014;3(6):e29194–e29197. | ||
Huang C-T, Oyang Y-J, Huang H-C, Juan H-F. MicroRNA-mediated networks underlie immune response regulation in papillary thyroid carcinoma. Sci Rep. 2014;4(9):6495–6509. | ||
Geering B, Fussenegger M. Synthetic immunology: modulating the human immune system. Trends Biotechnol. 2014;33(2):65–79. | ||
Harumiya S, Yoshino A, Hayashizaki K, Mizuno K, Yakura H, Adachi T. A system for reconstructing B cell antigen receptor signaling in the mouse myeloma J558L cell line. Arch Biochem Biophys. 2013;533(1–2):18–24. | ||
Weber W, Fussenegger M. Emerging biomedical applications of synthetic biology. Nat Rev Genet. 2012;13(1):21–35. | ||
Agustín-Pavón C, Isalan M. Synthetic biology and therapeutic strategies for the degenerating brain: synthetic biology approaches can transform classical cell and gene therapies, to provide new cures for neurodegenerative diseases. Bioessays. 2014;36(10):979–990. | ||
Abil Z, Xiong X, Zhao H. Synthetic biology for therapeutic applications. Mol Pharm. 2015;12(2):322–331. | ||
Hörner M, Reischmann N, Weber W. Synthetic biology: programming cells for biomedical applications. Perspect Biol Med. 2012;55(4):490–502. | ||
Forbes NS. Engineering the perfect (bacterial) cancer therapy. Nat Rev Cancer. 2010;10(11):785–794. | ||
Hörner M, Weber W. Molecular switches in animal cells. FEBS Lett. 2012;586(15):2084–2096. | ||
Furukawa K, Hohmann S. Synthetic biology: lessons from engineering yeast MAPK signalling pathways. Mol Microbiol. 2013;88(1):5–19. | ||
Grusch M, Schelch K, Riedler R, et al. Spatio-temporally precise activation of engineered receptor tyrosine kinases by light. EMBO J. 2014;33(15):1713–1726. | ||
Reiner A, Isacoff EY. Photoswitching of cell surface receptors using tethered ligands. Methods Mol Biol. 2014;1148:45–68. | ||
Stuber GD, Mason AO. Integrating optogenetic and pharmacological approaches to study neural circuit function: current applications and future directions. Pharmacol Rev. 2013;65(1):156–170. | ||
Sidor MM. Psychiatry’s age of enlightenment: optogenetics and the discovery of novel targets for the treatment of psychiatric disorders. J Psychiatry Neurosci. 2012;37(1):4–6. | ||
Jeong J-W, McCall JG, Shin G, et al. Wireless optofluidic systems for programmable in vivo pharmacology and optogenetics. Cell. 2015;162(3):662–674. | ||
Rihel J, Schier AF. Sites of action of sleep and wake drugs: insights from model organisms. Curr Opin Neurobiol. 2013;23(5):831–840. | ||
Farrell MS, Roth BL. Pharmacosynthetics: reimagining the pharmacogenetic approach. Brain Res. 2013;1511:6–20. | ||
Whittle AJ, Walsh J, de Lecea L. Light and chemical control of neuronal circuits: possible applications in neurotherapy. Expert Rev Neurother. 2014;14(9):1007–1017. | ||
Tomasetti C. Drug resistance. Adv Exp Med Biol. 2014;844:303–316. | ||
Kwan BW, Chowdhury N, Wood TK. Combating bacterial infections by killing persister cells with mitomycin C. Environ Microbiol. In press, 2015. | ||
Wood TK, Knabel SJ, Kwan BW. Bacterial persister cell formation and dormancy. Appl Environ Microbiol. 2013;79(23):7116–7121. | ||
Zakeri B, Lu TK. Synthetic biology of antimicrobial discovery. ACS Synth Biol. 2013;2(7):358–372. | ||
Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol. 2010;8(6):423–435. | ||
Liu C, Krishnan J, Xu XY. Intrinsic and induced drug resistance mechanisms: in silico investigations at the cellular and tissue scales. Integr Biol (Camb). 2015;7(9):1044–1060. | ||
Thaker MN, Wright GD. Opportunities for synthetic biology in antibiotics: expanding glycopeptide chemical diversity. ACS Synth Biol. 2015;4(3):195–206. | ||
Saxena A, Mukherjee U, Kumari R, Singh P, Lal R. Synthetic biology in action: developing a drug against MDR-TB. Indian J Microbiol. 2014;54(4):369–375. | ||
Cheng AA, Ding H, Lu TK. Enhanced killing of antibiotic-resistant bacteria enabled by massively parallel combinatorial genetics. Proc Natl Acad Sci U S A. 2014;111(34):12462–12467. | ||
Jagmann N, Philipp B. Design of synthetic microbial communities for biotechnological production processes. J Biotechnol. 2014;184:209–218. | ||
Grosskopf T, Soyer OS. Synthetic microbial communities. Curr Opin Microbiol. 2014;18:72–77. | ||
Mee MT, Collins JJ, Church GM, Wang HH. Syntrophic exchange in synthetic microbial communities. Proc Natl Acad Sci U S A. 2014;111(20):E2149–E2156. | ||
Mahadevan R, Henson MA. Genome-based modeling and design of metabolic interactions in microbial communities. Comput Struct Biotechnol J. 2012;3:e201210008. | ||
Holm A, Vikström E. Quorum sensing communication between bacteria and human cells: signals, targets, and functions. Front Plant Sci. 2014;5(309):1–7. | ||
Saeidi N, Wong CK, Lo T-M, et al. Engineering microbes to sense and eradicate Pseudomonas aeruginosa, a human pathogen. Mol Syst Biol. 2011;7(521):1–11. | ||
Du Y-L, Shen X-L, Yu P, Bai L-Q, Li Y-Q. Gamma-butyrolactone regulatory system of Streptomyces chattanoogensis links nutrient utilization, metabolism, and development. Appl Environ Microbiol. 2011;77(23):8415–8426. | ||
Wang Y, Ma S. Small molecules modulating AHL-based quorum sensing to attenuate bacteria virulence and biofilms as promising antimicrobial drugs. Curr Med Chem. 2014;21(3):296–311. | ||
Davis RM, Muller RY, Haynes KA. Can the natural diversity of quorum-sensing advance synthetic biology? Front Bioeng Biotechnol. 2015;3(30):1–10. | ||
Scutera S, Zucca M, Savoia D. Novel approaches for the design and discovery of quorum-sensing inhibitors. Expert Opin Drug Discov. 2014;9(4):353–366. | ||
Singh R, Ray P. Quorum sensing-mediated regulation of staphylococcal virulence and antibiotic resistance. Future Microbiol. 2014;9(5):669–681. | ||
Bhargava P, Collins JJ. Boosting bacterial metabolism to combat antibiotic resistance. Cell Metab. 2015;21(2):154–155. | ||
Peng B, Su Y-B, Li H, et al. Exogenous alanine and/or glucose plus kanamycin kills antibiotic-resistant bacteria. Cell Metab. 2015;21(2):249–261. | ||
Lee HH, Collins JJ. Microbial environments confound antibiotic efficacy. Nat Chem Biol. 2012;8(1):6–9. | ||
Planson A-G, Carbonell P, Grigoras I, Faulon J-L. Engineering antibiotic production and overcoming bacterial resistance. Biotechnol J. 2011;6(7):812–825. | ||
Zhang Q, Bhattacharya S, Conolly RB, Clewell HJ, Kaminski NE, Andersen ME. Molecular signaling network motifs provide a mechanistic basis for cellular threshold responses. Environ Health Perspect. 2014;122(12):1261–1270. | ||
Dunlop MJ, Dossani ZY, Szmidt HL, et al. Engineering microbial biofuel tolerance and export using efflux pumps. Mol Syst Biol. 2011;7(487):1–7. | ||
Planson A-G, Carbonell P, Paillard E, Pollet N, Faulon J-L. Compound toxicity screening and structure–activity relationship modeling in Escherichia coli. Biotechnol Bioeng. 2012;109(3):846–850. | ||
Oberhardt MA, Yizhak K, Ruppin E. Metabolically re-modeling the drug pipeline. Curr Opin Pharmacol. 2013;13(5):778–785. | ||
Chakravarti D, Wong WW. Synthetic biology in cell-based cancer immunotherapy. Trends Biotechnol. 2015;33(8):449–461. | ||
Bacchus W, Aubel D, Fussenegger M. Biomedically relevant circuit-design strategies in mammalian synthetic biology. Mol Syst Biol. 2013;9:691. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.