")
Back to Archived Journals » Research and Reports in Medicinal Chemistry » Volume 4
Synthesis molecular modeling and anticonvulsant activity of some hydrazone, semicarbazone, and thiosemicarbazone derivatives of benzylidene camphor
Authors Agrawal S, Jain J, Kumar A, Gupta P, Garg V
Received 13 April 2014
Accepted for publication 2 July 2014
Published 22 September 2014 Volume 2014:4 Pages 47—58
DOI https://doi.org/10.2147/RRMC.S66115
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Saurabh Agrawal,1 Jainendra Jain,2 Ankit Kumar,3 Pratibha Gupta,4 Vikas Garg5
1Meerut Institute of Engineering and Technology, Meerut, Uttar Pradesh, India; 2Ram–Eesh Institute of Vocational and Technical Education, Greater Noida, Uttar Pradesh, India; 3Kharvel Subharti College of Pharmacy, Swami Vivekanand Subharti University, Meerut, Uttar Pradesh, India; 4Atarra Degree College, Atarra, Banda, India; 5Manipal College of Pharmaceutical Sciences, Manipal University, Manipal, Karnataka, India
Abstract: Four series of 20 novel derivatives of benzylidene camphor with hydrazones, semicarbazones, and thiosemicarbazones were designed and synthesized. The newly synthesized compounds were evaluated for their anticonvulsant activity by maximal electroshock seizure model. Compounds showed varying degrees of anticonvulsant activity, most marked effect was observed for compounds 2f and 4d with lesser neurotoxicity. Molecular docking studies of most active compounds (2f and 4d) of the series revealed that they interact with LYS329A, GLN 301A, and THR 353B residues of 1OHV protein via hydrogen bonding and Pi interaction.
Keywords: camphor, semicarbazone, hydrazone, thiosemicarbazone, anticonvulsant
Introduction
Epilepsy is an agelong disease that often involves convulsive seizures. Epilepsy threatens approximately 50 million clinical cases worldwide annually.1 Approximately 20%–30% of the patients have seizures that are resistant to the available medical therapies. Despite the major medical need for a new chemotherapeutic agent, drug discovery for anticonvulsants is very challenging.2 With the help of novel drug discovery methods like computer added drug design we can design high quality leads which are more likely to succeed in clinical trials.3
Derivatives of hydrazone, semicarbazone, and thiosemicarbazone have already been approved as effective anticonvulsants.4–6 Camphor is a ketone found to have good anticonvulsant and nicotinic receptor-blocking properties.7,8 Herein, camphor moiety also plays an important role as it imparts lipophilic character to the synthesized compounds, which is essential to cross the blood–brain barrier.9
Gamma amino butyric acid (GABA) has been proposed as a validated target for antiepileptic drugs, because its selective inhibition raises GABA concentration in the brain.10 It is a pyridoxal phosphate (PLP)-dependent homodimeric enzyme, catalyzing reversible transfer of the amino group of GABA to α-ketoglutarate to yield succinic semialdehyde and L-glutamate. GABA–amino transferase (AT) is a homodimer with each subunit containing an active site PLP, covalently bound to LYS329 of chain A via a Schiff base.11 When the concentration of GABA diminishes below a threshold level in the brain, convulsion results while raising the brain GABA level terminates the seizure. So, due to these features pig GABA-AT [Protein Data Bank code: 1OHV] was taken for interaction studies.
Biological targets do not recognize a drug’s structural makeup. They respond instead to the properties around the drug that are generated by the structure. We can now generate and compare these property fields to indicate similar biological action.12 In order to design potent anticonvulsants molecular field mapping and alignment studies of camphor derivatives of hydrazone, semicarbazone and thiosemicarbazones were performed and compared with the molecular field of gabapentin. Field mapping results showed that all the derivatives have field similarity with gabapentin with a field similarity score ≥0.5 (Figure 1).
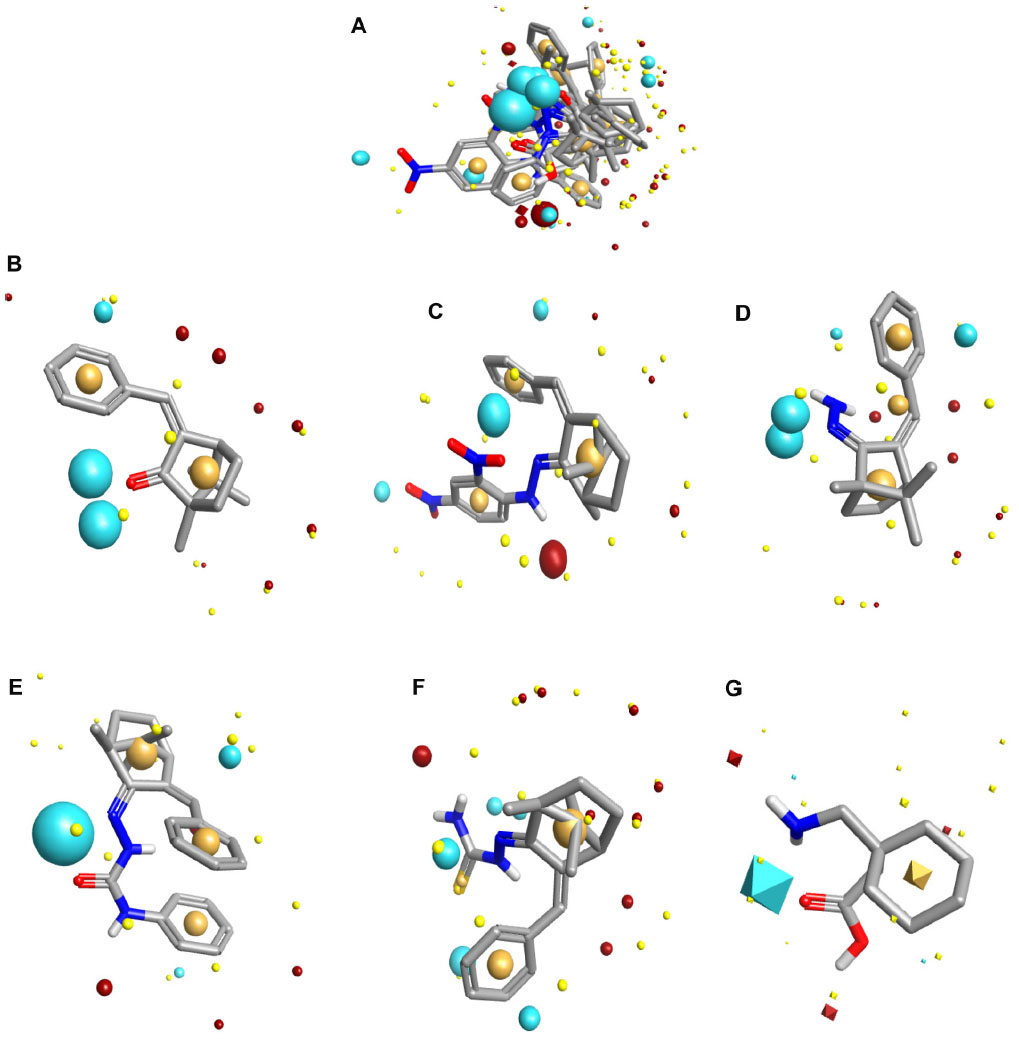 | Figure 1 Field aligning study (A) with gabapentin. Field mapping study of benzylidene camphor (B) benzylidene derivatives of camphor with hydrazone (C and D) semicarbazone (E) thiosemicarbazones (F) and gabapentin (G). The size of the point indicates the potential strength of the interaction. Round-shaped field points are of test compounds. Diamond-shaped field points are of reference compound (gabapentin). Sky blue color, negative ionic fields. Magenta color, positive ionic fields. Light yellow color, van der Waals interactions. Dark yellow color, hydrophobic fields. Field similarity score: ≥0.5. |
These findings encouraged us to synthesize novel derivatives of camphor with hydrazones, semicarbazones, and thiosemicarbazones. The newly synthesized derivatives were evaluated for their anticonvulsant activity by maximal electroshock seizure (MES) model and their neurotoxicity was assessed by rotarod test.
Materials and methods
All the chemicals used were of laboratory grade and procured from Thermo Fisher Scientific, (Waltham, MA, USA), S D Fine–Chem Limited (Mumbai, India), and Central Drug House (P) Ltd (New Delhi, India). Melting points were determined by the open tube capillary method and are uncorrected. Thin layer chromatography plates (silica gel G) were used to confirm the purity of commercial reagents used, compounds synthesized, and to monitor the reactions as well. Two different solvent systems – chloroform, methanol 5:1 (for compounds of Figures 2 and 3) and hexane, ethyl acetate in the ratio 6:4 (for compounds of Figures 4 and 5) – were used to run the thin layer chromatography. The spots were visualized under iodine vapors/ultraviolet light (254 nm and 365 nm). Infrared spectra were obtained on a Shimadzu 8400S FT-IR spectrometer (Shizmadzu Corporation, Kyoto, Japan) (KBr pellets). The 1H nuclear magnetic resonance (NMR) spectra were recorded on a 300 MHz DPX spectrometer in deuterated dimethyl sulfoxide (DMSOd6) using tetramethylsilane as the internal standard. The 13C NMR spectra were recorded at 100 MHz on Bruker AMX 400 (Bruker Corporation, Billerica, MA, USA) in DMSOd6. Mass spectra were recorded on an API 3000 liquid chromatography-tandem-mass spectrometry (LC/MS/MS) Q3 spectrometer (Shimadzu). The elemental analysis was performed on an Elemental Combustion System 4010 from Costech Analytical Technologies, Inc., Valencia, CA, USA.
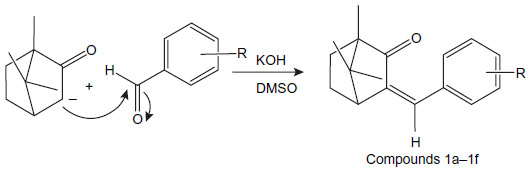 | Figure 2 Synthesis of benzylidene derivatives of camphor. |
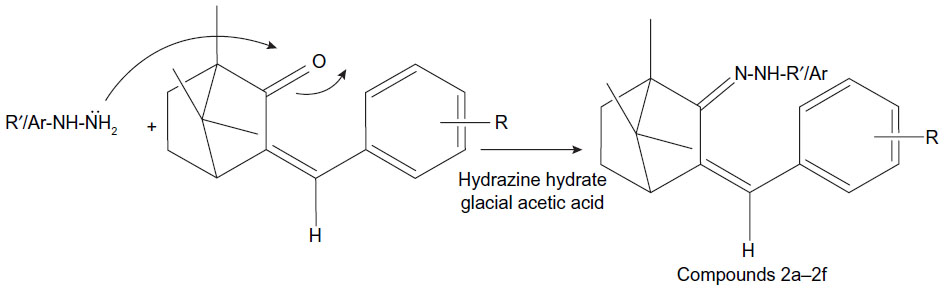 | Figure 3 Synthesis route of hydrazones of benzylidene derivatives of camphor. |
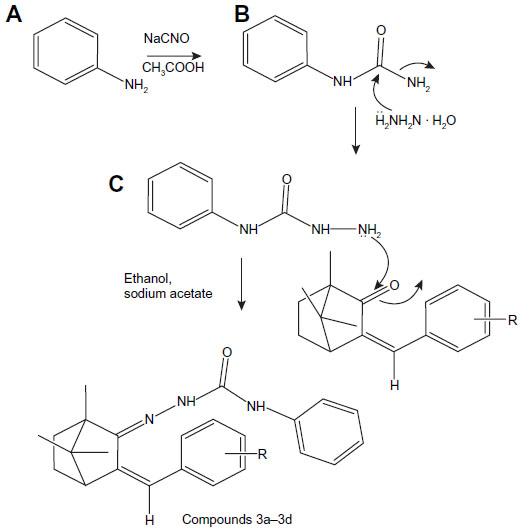 | Figure 4 Synthesis route of semicarbazones of benzylidene derivatives of camphor. |
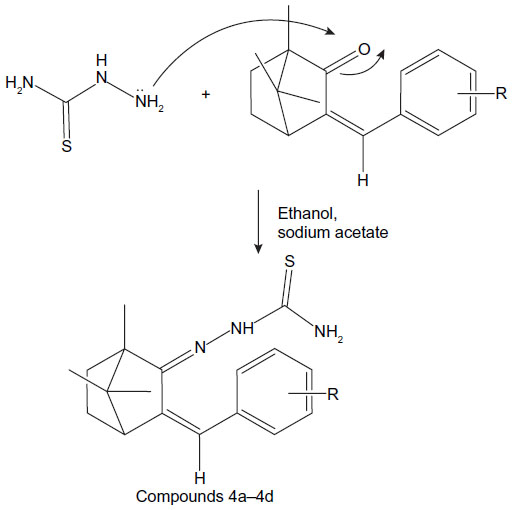 | Figure 5 Synthesis route of thiosemicarbazones of benzylidene camphor. |
General procedure for synthesis of benzylidene derivatives of camphor
Benzylidene derivatives of camphor were synthesized by condensing camphor with substituted aromatic aldehydes in anhydrous dimethyl sulfoxide (DMSO) in the presence of potassium hydroxide. Camphor (0.2 mol) and substituted benzaldehydes (0.26 mol) were dissolved in 25 mL of anhydrous DMSO. To the reaction mixture, potassium hydroxide (0.1 mol) in 10 mL anhydrous DMSO, was added dropwise, and the mixture was stirred at 20°C for 42 hours. The reaction mixture was cooled to 0°C, and the mixture was acidified with glacial acetic acid. The precipitate was filtered, washed with ice-cold water, and dried at room temperature (37°C). The colorless residue was recrystallized by using ethanol (Figure 2).
1,7,7-trimethyl-3-(2-nitrobenzylidene)bicyclo[2.2.1]heptan-2-one (compound 1a)
Colorless solid: Melting point (mp) 102°C–104°C; mass spectrometry (MS), m/z: 285.124, 286.140, 287.143 (C17H19NO3 calculated [calcd] 285.338); Fourier Transform infared (FTIR) (KBr) (cm−1): 2,927 (C–H stretching [str]), 1,720 (cyclic C=O str), 1,637 (C=C str), 3,150–3,050 (ring C–H str), 1,550 (NO2 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.89 (6H, s), 1.35 (3H, s), 1.60–1.65 (4H, m), 7.79–8.20 (4H, m), 7.60 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 9.30, 17.5 (2C), 26.7, 47.2, 30.5, 45.6, 58.2, 127.1 (2C), 134.5, 135.4, 128.2, 123.4, 147.5, 143.5, 208.1. Analysis (anal) calcd C, 71.56; H, 6.71; N, 4.91; O, 16.82. Found: C, 71.53; H, 6.69; N, 4.12; O, 16.80.
1,7,7-trimethyl-3-(4-nitrobenzylidene)bicyclo[2.2.1]heptan-2-one (compound 1b)
Colorless solid; mp 98°C–102°C; MS, m/z: 285.124, 286.140, 287.143 (C17H19NO3 calcd 285.338); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,720 (cyclic C=O str), 1,637 (C=C str), 3,150–3,050 (ring C-H str), 1,550 (NO2 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.89 (6H, s), 1.35 (3H, s), 1.60–1.65 (4H, m), 8.02–8.21 (4H, m), 7.20 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 9.30, 17.5 (2C), 26.7, 47.2, 30.5, 45.6, 58.2, 123.6 (2C), 134.5, 135.4, 129.4 (2), 147.5, 141.5, 208.6. Anal calcd C, 71.56; H, 6.71; N, 4.91; O, 16.82. Found: C, 71.52; H, 6.68; N, 4.10; O, 16.80.
3-(2-fluorobenzylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-one (compound 1c)
Colorless solid; mp 107°C–109°C; MS, m/z: 258.124, 259.140, 260.143 (C17H19FO calcd 258.331); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,250 (C−F str), 1,720 (cyclic C=O str), 1,637 (C=C str), 3,150–3,050 (ring C–H str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.89 (6H, s), 1.35 (3H, s), 1.60–1.65 (4H, m), 7.12–7.31 (4H, m), 7.15 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 9.30, 17.5 (2C), 26.7, 30.5, 45.5, 58.2, 135.6, 115.4, 129.2, 124.4, 128.4, 123.5, 143.9, 147.5, 161.5, 208.6. Anal calcd C, 79.04; H, 7.41; F, 7.35; O, 6.19. Found: C, 79.01; H, 7.38; F, 7.32; O, 6.21.
3-(4-fluorobenzylidene)-1,7,7- trimethylbicyclo[2.2.1]heptan-2-one (compound 1d)
Colorless solid; mp 107°C–109°C; MS, m/z: 258.110, 259.140, 260.131 (C17H19FO calcd 258.331); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,250 (C−F str), 1,720 (cyclic C=O str), 1,637 (C=C str), 3,150–3,050 (ring C–H str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.89 (6H, s), 1.35 (3H, s), 1.60–1.65 (4H, m), 7.12–7.69 (4H, m), 7.15 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 9.30, 17.5 (2C), 26.7, 30.5, 45.5, 58.2, 135.6, 115.4 (2), 129.9 (2), 130.4, 143.9, 147.5, 161.5, 208.6. Anal calcd C, 78.66; H, 7.01; F, 7.78; O, 6.55. Found: C, 78.64; H, 7.05; F, 7.74; O, 6.50.
3-(4-methoxybenzylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-one (compound 1e)
Colorless solid: mp 79°C–85°C; MS, m/z: 270.160, 271.162, 272.161 (C18H2202 calcd 270.366); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,250 (O−CH3 str), 1,720 (cyclic C=O str), 1,637 (C=C str), 3,150–3,050 (ring C–H str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.89 (6H, s), 3.68 (3H), 1.35 (3H, s), 1.60–1.65 (4H, m), 7.12–7.69 (4H, m), 7.15 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 9.30, 17.5 (2C), 26.7, 30.5, 45.5, 47.0, 58.2, 55.6, 130.6 (2), 114.4 (2), 127.9, 135.4, 143.5, 159.6, 208.6. Anal calcd C, 79.96; H, 8.20; O, 11.84. Found: C, 79.93; H, 8.25; O, 11.81.
3-benzylidene-1,7,7-trimethylbicyclo[2.2.1]heptan-2-one (compound 1f)
Colorless solid: mp 85°C–90°C; MS, m/z: 240.160, 240.152, (C17H200 calcd 240.151); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,720 (cyclic C=O str), 1637 (C=C str), 3,150–3,050 (ring C–H str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.89 (6H, s), 3.68 (3H), 1.35 (3H, s), 1.60–1.65 (4H, m), 7.33–7.69 (5H, m), 7.15 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 9.30, 17.5 (2C), 26.7, 30.5, 45.5, 47.0, 58.2, 55.6, 135.6, 127.7, 128.5 (2), 128.6 (2), 143.5, 208.6. Anal calcd C, 84.96; H, 8.39; O, 6.66. Found: C, 84.95; H, 8.32; O, 6.60.
General procedure for synthesis of derivatives of hydrazone with camphor
Hydrazones of benzylidene camphor were prepared by condensing of benzylidene camphor derivatives (Figure 2) and hydrazine hydrate. Equimolar quantity of benzylidene camphor and hydrazine hydrate were dissolved in ethanol and transferred to a round bottom flask. The reaction mixture was made acidic with glacial acetic acid and resulting mixture was refluxed on a water bath for 5 hours. The mixture was cooled and residue was separated, filtered, dried and recrystallized from benzene and hexane (1:1) to afford colorless crystalline powder (Figure 3).
3-benzylidene-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene hydrazine (compound 2a)
Colorless solid: mp 75°C−79°C; MS, m/z: 254.12, 256.14 (C17H22N2 calcd 254.18); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH2 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.35 (3H, s), 1.60–1.65 (4H, m), 2.17 (1H, t), 7.22–7.51 (5H, m), 6.68 (1H, s), 7.0 (2H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.50, 17.5 (2C), 26.7, 50.3, 52.3, 57.9, 136.1, 131.7, 127.2, 128.5 (2C), 130.1 (2C), 135.2. Anal calcd C, 80.27; H, 8.72; N, 11.01. Found: C, 80.20; H, 8.61; N, 10.90.
3-(2-fluorobenzylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene hydrazine (compound 2b)
Colorless solid: mp 85°C−89°C; MS, m/z: 272.12, 273.12 (C17H21N2F calcd 272.36); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,250 (C−F str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH2 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.35 (3H, s), 1.60–1.65 (4H, m), 2.17 (1H, t), 7.17–7.63 (4H, m), 6.68 (1H, s), 7.0 (2H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.50, 17.5 (2C), 26.7, 32.5, 50.3, 52.3, 57.9, 136.1, 131.7, 123.2, 128.0, 129.5, 124.1, 115.1, 161.3, 155.6. Anal calcd C, 74.97; H, 7.72; F, 6.98; N, 10.29. Found: C, 74.92; H, 7.78; F, 6.93; N, 10.69.
3-(4-methoxybenzylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene hydrazine (compound 2c)
Colorless solid: mp 75°C−79°C; MS, m/z: 284.184, 285.192 (C18H24ON2 calcd 284.88); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,250 (C−OCH3 str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH2 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.35 (3H, s), 1.60–1.65 (4H, m), 2.17 (1H, t), 3.83 (3H), 6.97–7.63 (4H, m), 6.68 (1H, s), 7.0 (2H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.50, 17.5 (2C), 26.7, 32.5, 50.3, 52.3, 55.8, 57.9, 136.1, 131.7, 128.0, 130.5 (2), 115.1 (2), 159.8, 155.6. Anal calcd C, 76.02; H, 8.5; O, 5.63; N, 9.85. Found: C, 76.05; H, 8.1; O, 5.60; N, 9.82.
1-(3-benzylidene-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)-2-(2,4-dinitrophenyl)-hydrazine (compound 2d)
Dark brown solid: mp 79°C−86°C; MS, m/z: 420.19, 422.14 (C23H24N4O4 calcd 420.18); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH2 str) 1,550 (NO2 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.15 (3H, s), 1.61–1.65 (4H, m), 2.17 (1H, t), 7.52–7.61 (5H, m), 7.97–8.82 (3H, m), 6.68 (1H, s), 7.0 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.20, 17.5 (2C), 26.7, 32.9, 50.3, 52.3, 57.9, 136.0, 131.7, 127.9, 128.5 (2C), 130.1 (2C), 135.2, 145.2, 116.6, 130.8, 138.5, 123.5, 129.1, 155.6. Anal calcd C, 80.27; H, 8.72; N, 11.01. Found: C, 80.20; H, 8.61; N, 10.90.
1-(2,4-dinitrophenyl)-2-(3-(2-fluorobenzylidene) 1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)hydrazine (compound 2e)
Dark brown sticky: mp 79°C−87°C; MS, m/z: 438.19, 439.14 (C23H23N4O4F calcd 438.450); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,250 (C−F str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH2 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.15 (3H, s), 1.61–1.65 (4H, m), 2.17 (1H, t), 7.17–7.61 (4H, m), 7.97–8.80 (3H, m), 6.68 (1H, s), 7.0 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.20, 17.5 (2C), 26.7, 32.9, 50.3, 52.3, 57.9, 124.0, 131.7, 123.9, 128.5, 130.1, 135.2, 145.2, 115.4, 116.6, 130.8, 138.5, 123.5, 129.1, 155.6, 161.0. Anal calcd C, 63.27; H, 5.29; F, 4.33, N, 12.78, O, 14.60. Found: C, 63.21; H, 5.21; F, 4.38, N, 12.72, O, 14.50.
1-(2,4-dinitrophenyl)-2-(3-(4-methoxybenzylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene) hydrazine (compound 2f)
Dark brown solid: mp 85°C−95°C; MS, m/z: 450.19, 451.198 (C24H26N4O5 calcd 458.47); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH2 str) 1,250 (O−CH3 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.15 (3H, s), 1.61–1.65 (4H, m), 2.17 (1H, t), 3.85 (3H), 7.52–7.61 (5H, m), 7.97–8.82 (3H, m), 6.68 (1H, s), 7.0 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.20, 17.5 (2C), 26.7, 32.9, 50.3, 52.3, 57.9, 55.5 136.0, 131.7, 127.9, 114.5 (2C), 130.1 (2C), 135.9, 145.2, 116.6, 130.8, 132.5, 119.5, 139.1, 159.6. Anal calcd C, 80.27; H, 8.72; N, 11.01. Found: C, 80.20; H, 8.61; N, 10.90.
General method of synthesis of semicarbazone of benzylidene camphor derivatives
Semicarbazone of benzylidene camphor derivatives were synthesized in three steps.
Synthesis of aryl urea
Aryl amine (0.1 mol, 10.7 mL) was dissolved in 20 mL of glacial acetic acid and 10 mL of water. To this, 0.1 mol of sodium cyanate (6.5 g) in 80 mL of warm water was added with stirring. Allowed to stand for 30 minutes, it was then cooled in ice and filtered with suction and dried, recrystallized from boiling water to yield aryl urea.
Synthesis of aryl semicarbazide
Equimolar quantities (0.1 mol) of phenyl urea (9.2 g) and hydrazine hydrate (2.5 mL) in ethanol were refluxed for 24 hours with stirring. The two-thirds volume of alcohol was distilled by the vacuum distillation unit and then poured into ice. The resultant precipitate was filtered, washed with water, and dried. The solid was recrystallized from 50 mL of 90% alcohol to which 25 mL of concentrated hydrochloric acid was added. The precipitate of semicarbazide hydrochloride was filtered by vacuum and dried.
Synthesis of semicarbazones of benzylidene camphor
Equimolar quantities (0.5 mol) of benzylidene camphor and semicarbazide hydrochloride were dissolved in 25 mL of ethanol. The pH of the reaction mixture was made alkaline by using sodium acetate and refluxed for 6 hours. The resultant precipitate was filtered, washed with water, and dried. The solid was recrystallized from 50 mL of 90% hot ethanol (Figure 4).
2-(3-benzylidene-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)-N-phenylhydrazine carboxamide (compound 3a)
Light yellowish solid: mp 110°C−112°C; MS, m/z: 373.19, 374.14, 375.16 (C24H27N3O calcd 373.22); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH str), 1,625 (NH bend). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.15 (3H, s), 1.61–1.65 (4H, m), 2.17 (1H, t), 7.42–7.61 (10H, m), 6.68 (1H, s), 5.8 (1H,s), 7.0 (1H, s). 13C NMR (100 MHz) DMSO d6, δ (ppm): 16.20, 17.8 (2C), 26.7, 32.9, 50.3, 52.3, 57.9, 136.0, 131.7, 127.9, 128.5 (2C), 129.1 (2C), 135.2, 152.2, 139.6, 121.8 (2C), 128.5 (2C), 121.5, 129.1, 155.6. Anal calcd C, 77.18; H, 7.29; N, 11.25; O, 4.28. Found: C, 76.99; H, 7.19; N, 11.20; O, 4.21.
2-3-(2-fluorobenzylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene- N-phenylhydrazine carboxamide (compound 3b)
Light yellowish solid: mp 115°C−119°C; MS, m/z: 391.209, 392.216, (C24H26N3OF calcd 391.481); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,250 (C−F str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH str), 1,625 (NH bend). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.15 (3H, s), 1.61–1.65 (4H, m), 2.17 (1H, t), 7.12–7.61 (9H, m), 6.68 (1H, s), 5.8 (1H,s), 7.0 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.20, 17.8 (2C), 26.7, 32.9, 50.3, 52.3, 57.9, 136.0, 131.7, 115.5 (2C), 129.1 (3C), 135.2, 152.2, 121.8 (2C), 128.5 (2C), 121.5, 129.1, 162.1. Anal calcd C, 73.63; H, 6.69; F, 4.85, N, 10.75; O, 4.08. Found: C, 73.60; H, 6.65; F, 4.80, N, 10.75; O, 4.03.
2-3-(4-fluorobenzylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene- N-phenylhydrazine carboxamide (compound 3c)
Light yellowish solid: mp 115°C−119°C; MS, m/z: 391.209, 392.216, (C24H26N3OF calcd 391.48); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,250 (C−F str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH str), 1,625 (NH bend). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.15 (3H, s), 1.61–1.65 (4H, m), 2.17 (1H, t), 7.12–7.61 (9H, m), 6.68 (1H, s), 5.8 (1H, s), 7.0 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.20, 17.8 (2C), 26.7, 32.9, 50.3, 52.3, 57.9, 136.0, 131.7, 115.5 (2C), 129.1 (3C), 135.2, 152.2, 121.8 (2C), 128.5 (2C), 121.5, 129.1, 162.1. Anal calcd C, 73.63; H, 6.69; F, 4.85, N, 10.75; O, 4.08. Found: C, 73.60; H, 6.65; F, 4.80, N, 10.75; O, 4.03.
2-3-(4-methoxybenzylidene)-1,7,7- trimethylbicyclo-[2.2.1]heptan-2-ylidene- N-phenylhydrazine carboxamide (compound 3d)
Dark yellowish solid: mp 125°C−129°C; MS, m/z: 402.225, 404.226, (C25H29N3O2 calcd 403.517); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,250 (O−CH3 str), 1,680 (cyclic C=N str), 1,250 (C−F str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH str), 1,625 (NH bend). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.15 (3H, s), 1.61–1.65 (4H, m), 2.17 (1H, t), 3.83 (3H), 7.12–7.61 (8H, m), 6.68 (1H, s), 5.8 (1H, s), 7.0 (1H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.20, 17.8 (2C), 26.7, 32.9, 50.3, 52.3, 55.8, 57.9, 127.0, 131.7, 115.5 (2C), 129.1 (3C), 135.2, 152.2, 121.8 (2C), 128.5 (2C), 129.1, 159.5, 155.1. Anal calcd C, 74.41; H, 7.24; N, 10.45; O, 7.98. Found: C, 74.40; H, 7.25; N, 10.55; O, 7.93.
Synthesis of thiosemicarbazones of benzylidene camphor
Equimolar quantities (0.5 mol) of benzylidene camphor and thiosemicarbazide hydrochloride were dissolved in 25 mL of ethanol. The pH was made alkaline by using sodium acetate. The reaction mixture was refluxed for 6 hours. The resultant precipitate was filtered, washed with water, and dried. The solid was recrystallized from 50 mL of 90% hot ethanol (Figure 5).
2-(3-benzylidene-1,7,7-trimethyl bicyclo[2.2.1]heptan-2-ylidene) hydrazine carbothioamide (compound 4a)
Yellowish solid: mp 104°C−108°C; MS, m/z: 314.16 (C18H23N3S calcd 313.16); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH2 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.35 (3H, s), 1.60–1.65 (4H, m), 2.17 (1H, t), 7.22–7.51 (5H, m), 6.68 (1H, s), 7.0 (1H, s) 8.52 (2H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.50, 17.5 (2C), 26.7, 50.3, 52.3, 57.9, 1362.1, 131.7, 127.2, 128.5 (2C), 130.1 (2C), 135.2, 181.1. Anal calcd C, 68.97; H, 7.40; N, 13.41; S, 10.23. Found: C, 68.90; H, 7.10; N, 13.11; S, 9.98.
2-3-(2-fluorobenzylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene hydrazine carbothioamide (compound 4b)
Yellowish solid: mp 104°C−108°C; MS, m/z: 331.16, 332.159 (C18H22N3SF calcd 331.451); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,250 (C−F str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH2 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.35 (3H, s), 1.60–1.65 (4H, m), 2.17 (1H, t), 7.12–7.63 (4H, m), 6.98 (1H, s), 7.0 (1H, s) 8.52 (2H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.50, 17.5 (2C), 26.7, 32.2, 50.3, 52.3, 57.9, 115.1, 161.3, 124.7, 123.2, 128.5 (2C), 131.1, 136.2, 155.0, 181.1. Anal calcd C, 65.23; H, 6.69; F, 5.73, N, 12.68; S, 9.67. Found: C, 65.20; H, 6.63; F, 5.78, N, 12.60; S, 9.61.
2-3-(4-fluorobenzylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene hydrazine carbothioamide (compound 4c)
Yellowish solid: mp 104°C−110°C; MS, m/z: 331.16, 332.159 (C18H22N3SF calcd 331.451); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,250 (C−F str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH2 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.35 (3H, s), 1.60–1.65 (4H, m), 2.17 (1H, t), 7.29–7.72 (4H, m), 6.68 (1H, s), 7.0 (1H, s) 8.52 (2H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.50, 17.5 (2C), 26.7, 32.2, 50.3, 52.3, 57.9, 115.1 (2C), 161.3, 130.7, 129.5 (2C), 131.1, 136.2, 155.0, 181.1. Anal calcd C, 65.23; H, 6.69; F, 5.73, N, 12.68; S, 9.67. Found: C, 65.20; H, 6.63; F, 5.78, N, 12.60; S, 9.61.
2-3-(4-methoxybenzylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene) hydrazine carbothioamide (compound 4d)
Dark yellowish solid: mp 104°C−110°C; MS, m/z: 343.170, 344.179 (C19H25N3SO calcd 343.481); FTIR (KBr) (cm−1): 2,927 (C–H str), 1,680 (cyclic C=N str), 1,250 (O−CH3 str), 1,637 (C=C str), 3,100 (ring C–H str), 3,350 (NH2 str). 1H NMR (300 MHz) DMSOd6, δ (ppm): 0.99 (6H, s), 1.35 (3H, s), 1.60–1.65 (4H, m), 2.17 (1H, t), 3.83 (3H), 6.99–7.72 (4H, m), 6.68 (1H, s), 7.0 (1H, s) 8.52 (2H, s). 13C NMR (100 MHz) DMSOd6, δ (ppm): 16.50, 17.5 (2C), 26.7, 32.2, 50.3, 52.3, 55.8, 57.9, 114.1, 161.3, 127.7, 159.5, 129.5 (2C), 131.1, 136.2, 155.0, 181.1. Anal calcd C, 66.44; H, 7.34; N, 12.23; S, 9.34; O, 4.66. Found: C, 66.40; H, 7.31; N, 12.20; S, 9.39; O, 4.60.
Biological activity
Maximal electroshock-induced seizure model
Maximal electroshock induced seizure model was used for evaluation of anticonvulsant activity. Swiss albino mice were used for the activity. Mice deprived of food and water overnight were randomly distributed in to ten groups of six animals each. Group I served as control (vehicle treated); group II served as standard (phenytoin sodium 25 mg/kg body weight); group III–group X were treated with test drugs 30, 100, and 300 mg/kg body weight, respectively. The control, standard, and test drugs were administered intraperitoneally (ip) in 2% volume/volume Tween 80 solution, 0.5 hours prior to induce the convulsion, and standard drug was administered ip 30 minutes before. Electroconvulsive shock (50 mA for 0.2 seconds) was delivered through corneal electrodes to induce convulsions. The various phases of convulsion are flexion, extension, clonus, and stupor. After the electric stimulation occurrence, the duration phases were noted, and the hind limb tonic extension phase was compared with the control group. The decrease in the duration of the hind limb extension was considered as protective action.3,13–16
Neurotoxicity screening
The minimal motor impairment was measured in mice by the rotarod test. The mice were trained to stay on an accelerating rotarod of diameter 3.2 cm that rotates at 10 rpm. Trained animals were given the ip injection of the test compounds 30, 100, and 300 mg/kg. Neurotoxicity was indicated by the inability of the animal to maintain equilibrium on the rod for at least 1 minute in each of the trials.17
Molecular docking
Molecular field mapping and alignment studies were performed using Torch software (Cresset, Cambridgeshire, UK). The receptor model was built by using AutoDock Tools 1.4.6 (The Scripps Research Institute, La Jolla, CA, USA) and MGL Tools 1.5.4 (The Scripps Research Institute) packages. Gasteiger partial charges were added to the ligand atoms. Nonpolar hydrogen atoms were merged, and rotatable bonds were defined. Docking calculations were carried out on 1OHV protein model.10 Essential hydrogen atoms, Kollman united atom type charges, and solvation parameters were added with the aid of AutoDock tools.18 The affinity (grid) maps of 20 A° grid points and 0.375 A° spacing were generated using the AutoGrid program (AutoGrid Systems, Redwood Shores, CA, USA).19
The AutoDock parameter set and distance-dependent dielectric functions were used in the calculation of the van der Waals and the electrostatic terms, respectively. Docking simulations were performed, using the Lamarckian genetic algorithm and the Solis and Wets local search method. Initial position, orientation, and torsions of the ligand molecules were set randomly. All rotatable torsions were released during docking. Each docking experiment was set to terminate after a maximum of 2,500,000 energy evaluations. The population size was set to 150. During the search, a translational step of 0.2 A°, and quaternion and torsion steps of 5 were applied.20,21
Results and discussion
Preparation of compounds
The synthesis of benzylidene derivatives of camphor and their hydrazones, semicarbazones, and thiosemicarbazones (compounds 1a–1f, 2a–2f, 3a–3d, and 4a–4d) are outlined in Figures 2–5. The chemical structure and physical data of these compounds are presented in Table 1. In Figure 2 benzylidene derivatives of camphor (compounds 1a–1f) were synthesized by condensing camphor with substituted aromatic aldehydes, in anhydrous DMSO, in the presence of potassium hydroxide. In Figure 3, hydrazones of benzylidene derivatives of camphor were synthesized. Hydrazones were prepared by condensing benzylidene derivatives of camphor (1c, 1e, and 1f) with substituted hydrazine hydrates.
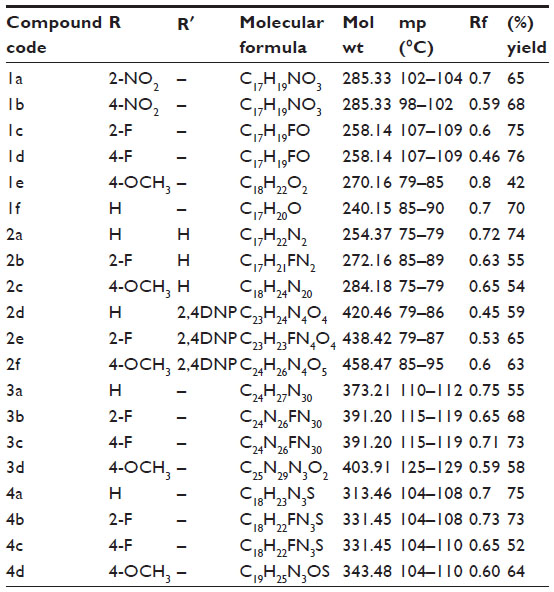 | Table 1 Physical data of the benzylidene derivatives of camphor |
In Figures 4 and 5, semicarbazones and thiosemicarbazones of benzylidene derivatives of camphor were prepared respectively. For their synthesis, first aryl urea derivatives were prepared by treating aryl amine with sodium cyanate in the presence of glacial acetic acid. Synthesized aryl urea derivatives were than refluxed with hydrazine hydrate to yield hydrazine carboxamides. The final compounds were synthesized by the reaction of hydrazine carboxamides with the appropriate ketone group of benzylidene derivatives of camphor. Thiosemicarbazones were prepared by condensing benzylidene derivatives of camphor with thiosemicarbazide as per given protocol. Analytical and spectral data of all the synthesized compounds were found in agreement with the composition of synthesized compounds. The data of physicochemical properties of all the compounds is given in Table 1.
Biological activity
The synthesized compounds were tested for anticonvulsant activity and neurotoxicity by the MES method and rotarod method, respectively. Compounds 2f and 4d showed activity at dose 30 mg/kg after 0.5 hours of injection. Compounds 1a, 1b, 2a, 2b, 2c, 3a, 3b, 3d, and 4b showed activity at dose 100 mg/kg after 0.5 hours of injection. Compounds 2f and 4d showed activity at dose 100 mg/kg after 4 hours of injection. Compounds 1a, 1b, 2b, and 3a showed activity at dose 300 mg/kg after 4 hours of injection. Other compounds did not show activity at the given doses. Compounds 3c and 4c showed neurotoxicity at dose 300 mg/kg after 0.5 hours of injection. Compounds 1c, 1d, and 2e showed neurotoxicity at dose 100 mg/kg after 0.5 hours of injection. Compounds 1d and 2e showed neurotoxicity at dose 300 mg/kg after 4 hours of injection. Among the tested compounds, 2f and 4d were found most active, displaying significant activity at 30 mg/kg dose level comparable to phenytoin 25 mg/kg. Further, these two compounds (2f and 4d) did not show neurotoxicity at the treated doses (Table 2).
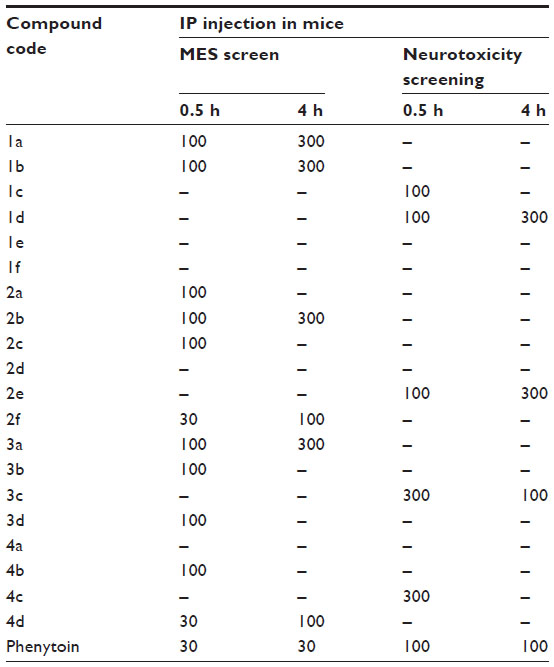 | Table 2 Anticonvulsant activity and minimal motor impairment of benzylidene derivatives of camphor |
Molecular modeling study
Molecular modeling and field mapping studies of most active derivatives (compounds 2f and 4d) of the four series were carried out to find the probable mechanism of the action of their derivatives and to develop a model for anticonvulsant drugs. The results of the molecular modeling study of most active derivatives are summarized in Table 3. Compounds 2f and 4d showed maximum docking score −137.71 and −105.34, respectively. Compound 2f showed an interaction with protein 1OHV by hydrogen bonding with residues: lysine 329 residue in chain A (LYS329A, 2.061 and 2.349); threonine 353 residue in chain B (THR353B, 2.223 and 2.045); glutamine 301 residue in chain A (GLN301A, 2.386); and Pi stacking interaction with phenyl alanine residue in chain A (PHE189A, 5.077). Compound 4d was shown to interact with 1OHV protein by hydrogen bonding with residues LYS329A (2.505, 2.495, and 2.545), THR353B (2.291), and GLN301A (2.488). Compound 4d also found to show Pi-stacking interactions with residue PHE189A (4.683 and 5.473) (Table 3 and Figure 6). It means these groups play an important role for anticonvulsant property of these compounds. The field aligning study of compounds 2f and 4d was performed, and it was found that both compounds have field similarity with a score of 0.702 (Figure 7). Further, it was found that the introduction of the 2, 4-dinitrophenyl group in hydrazones and thiosemicarbazones formation from the benzylidene camphor increase van der Waals attraction (Figure 8). Both compounds 2f and 4d have strong hydrophobic, H donor, H acceptor and van der Waals attraction regions which forms electrostatic and hydrophobic interactions with the receptor thus resulting in anticonvulsant activity.
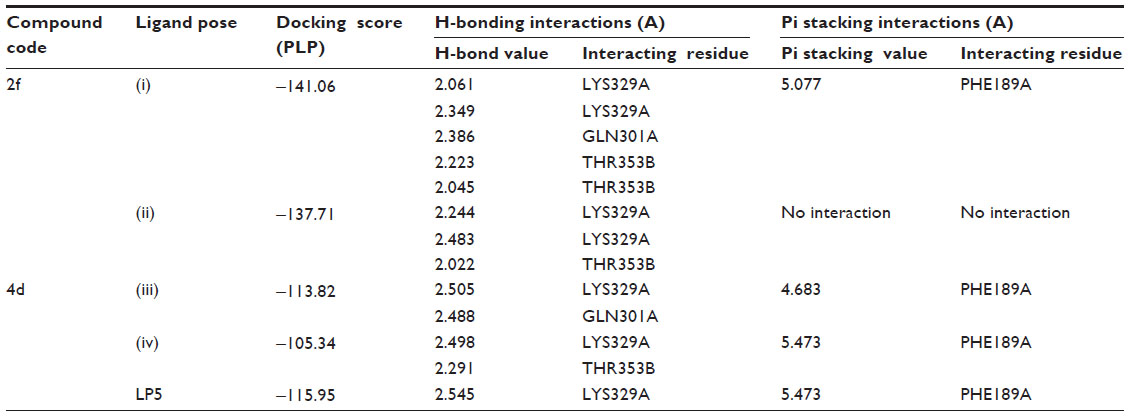 | Table 3 1OHV interaction table of active benzylidene derivatives of camphor |
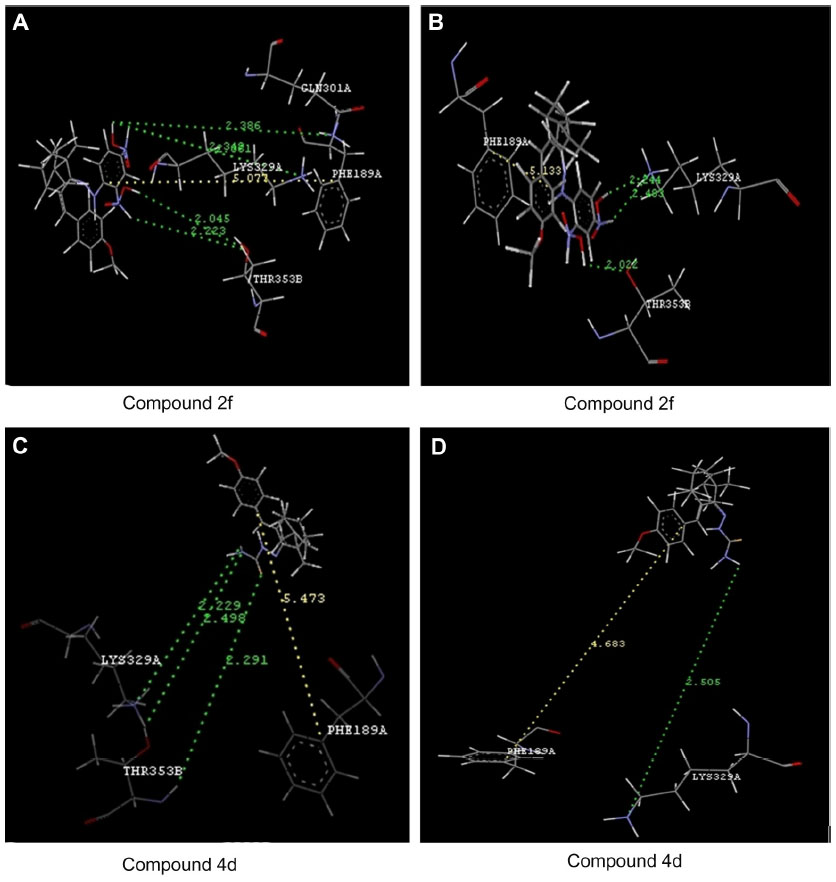 | Figure 6 Docking of compounds into the active site of 1OHV protein. |
 ), hydrogen bonding interaction. Yellow line (
), hydrogen bonding interaction. Yellow line ( ), Pi interaction. (
), Pi interaction. (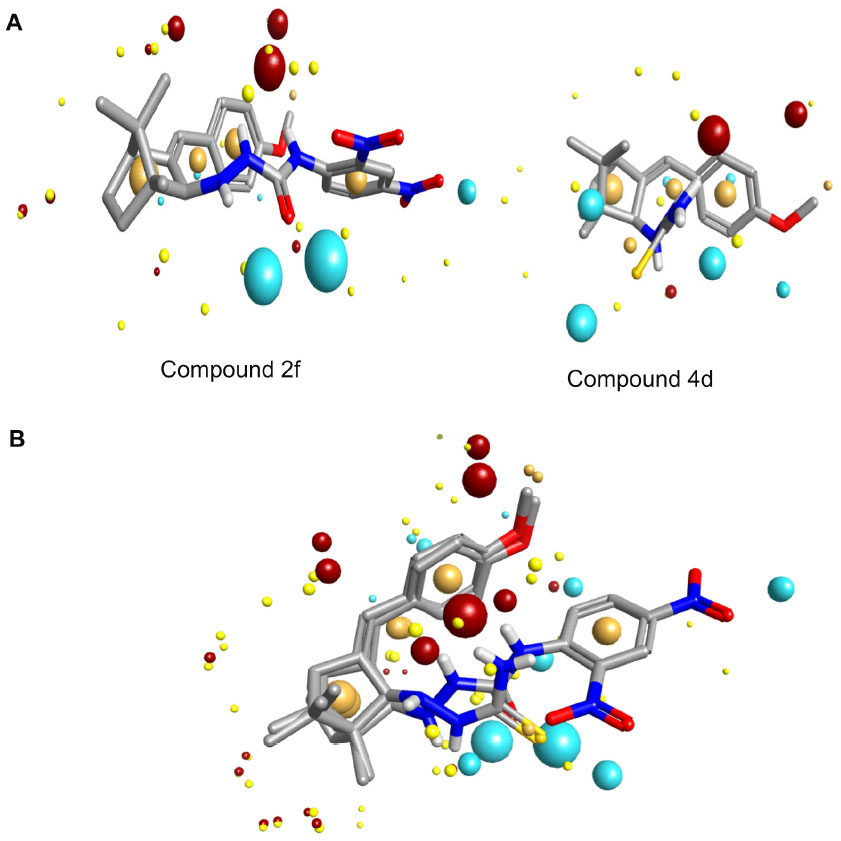 | Figure 7 Model development from active compounds. |
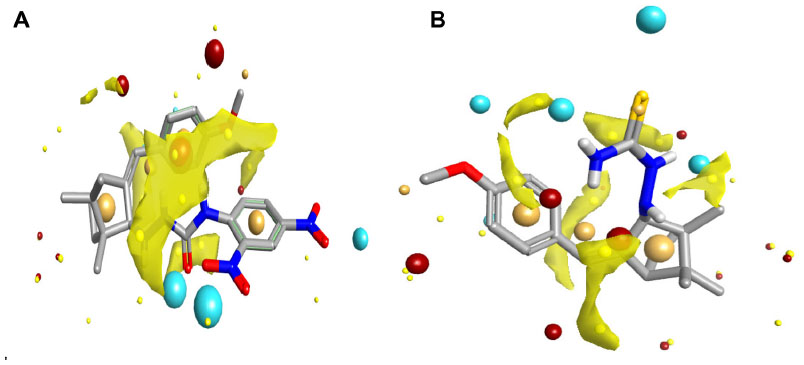 | Figure 8 Van der Waals attraction regions (in yellow) of compounds 2f (left) and 4d (right). |
Conclusion
In the present study, a series of 20 novel derivatives of benzylidene with camphor were synthesized. The newly synthesized compounds were evaluated for their anticonvulsant activity by the MES model. Compounds 2f and 4d found to be most active at a dose of 30 mg/kg comparable to phenytoin. Further these compounds did not show any neurotoxicity at the tested doses. Molecular docking studies of most active compounds (2f and 4d) of the series revealed that they interact with LYS329A, GLN 301A, and THR 353B residues of 1OHV protein, present in GABA-AT receptor of the brain of a pig via hydrogen bonding and Pi interaction. Field alignment studies of compounds 2f and 4d showed that compounds have strong hydrophobic regions, H donor and acceptor regions, and van der Waals attraction regions that formed electrostatic regions and steric regions responsible for interaction with GABA-AT receptor and anticonvulsant activity. These interaction data of synthesized compounds with active residues like LYS329A, GLN301A, and THR 353B of GABA-AT have suggested its possible mechanism of anticonvulsant action. Inhibition of GABA-AT results in an increased level of GABA in the glial cells and, thereby, suppresses seizure spread.
Acknowledgments
The authors are thankful to Meerut Institute of Engineering and Technology, Meerut, and Institute of Technology and Science Paramedical College, Muradnagar, Uttar Pradesh, India, for the support during the study. The authors are also very thankful to the Cresset Group for providing Torch V 10 Next Generation Chemistry Software.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization [homepage on the Internet]. Epilepsy. World Health Organization; 2012. Available from: http://www.who.int/mediacentre/factsheets/fs999/en/. Accessed February 15, 2014. | |
Ratti E, Trist D. Continuing evolution of the drug discovery process in the pharmaceutical industry. Pure and Applied Chemistry. 2001;73(1):67–75. | |
Dimmock JR, Sidhu KK, Tumber SD, et al. Some aryl semicarbazones possessing anticonvulsant activities. Eur J Med Chem. 1995;30(4):287–301. | |
Leppik LE. Antiepileptic drugs in development: prospects for the near future. Epilepsia. 1994;35 Suppl 4:S29–S40. | |
Yogeeswari P, Sriram D, Sunil Jit LR, Kumar SS, Stables JP. Anticonvulsant and neurotoxicity evaluation of some 6-chlorobenzothiazolyl-2-thiosemicarbazones. Eur J Med Chem. 2002;37(3):231–236. | |
Pandeya SN, Yogeeswari P, Stables JP. Synthesis and anticonvulsant activity of 4-bromophenyl substituted aryl semicarbazones. Eur J Med Chem. 2000;35(10):879–886. | |
Chatterjie N, Alexander GJ. Anticonvulsant properties of spirohydantoins derived from optical isomers of camphor. Neurochem Res. 1986;11(12):1669–1676. | |
Park TJ, Seo HK, Kang BJ, Kim KT. Noncompetitive inhibition by camphor of nicotinic acetylcholine receptors. Biochem Pharmacol. 2001;61(7):787–793. | |
Storici P, Capitani G, De Biase D, et al. Crystal structure of GABA-aminotransferase, a target for antiepileptic drug therapy. Biochemistry. 1999;38(27):8628–8634. | |
Bansal SK, Sinha BN, Khosa RL, Olson AJ. Novel GABA-AT inhibitors: QSAR and docking based virtual screening of phenyl substituted β-phenyl ethylidene hydrazine analogues. Medicinal Chemistry Research. 2010;20(9):1482–1489. | |
Rautio J, Laine K, Gynther M, Savolainen J. Prodrug approaches for CNS delivery. AAPS J. 2008;10(1):92–102. | |
Vinter A, Gardner S, Rose S. Overcoming the limitation of chemical structure. Chemical structure: Layout. 2009;13(37):58–63. | |
Pandeya SN, Ponnilavarasan I, Pandey A, Lakhan R, Stables JP. Evaluation of p-nitrophenyl substituted semicarbazones for anticonvulsant properties. Pharmazie. 1999;54(12):923–925. | |
Rollas S, Küçükgüzel SG. Biological activities of hydrazone derivatives. Molecules. 2007;12(8):1910–1939. | |
Tuncbilek M, Altanlar M. Synthesis of new 3-(substituted phenacyl)-5-[3′-(4H-4-oxo-1-benzopyran-2-yl)-benzylidene]-2,4-thiazolidinediones and their antimicrobial activity. Arch Pharm (Weinheim). 2006;339(4):213–216. | |
Krall RL, Penry JK, White BG, Kupferberg HJ, Swinyard EA. Antiepileptic drug development: II. Anticonvulsant drug screening. Epilepsia. 1978;19(4):409–428. | |
Kupferberg HJ. Antiepileptic drug development program: a cooperative effort of government and industry. Epilepsia. 1989;30 Suppl 1:S51–S56. | |
Halgren TA. Merck molecular force field. I. Basis, form, scope, parametrization, and performance of MMFF94. J Comput Chem. 1998;17(5–6):490–519. | |
Morris GM, Goodsell DS, Halliday RS, et al. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J Comput Chem. 1998;19(14):1639–1662. | |
Solis FJ, Wets RJB. Minimization by Random Search Techniques. Mathematics of Operations Research. 1981;6(1):19–30. | |
Storici P, De Biase D, Bossa S, et al. Structure of gamma-aminobutyric acid (GABA) aminotransferase a pyridoxal 5′-phosphate, and [2 Fe-2S] cluster-containing enzyme, complexed with gamma-ethynyl-GABA and with the antiepilepsy drug vigabatrin. J Biol Chem. 2004;279(1):363–373. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.