")
Back to Archived Journals » Research and Reports in Medicinal Chemistry » Volume 6
Synthesis, characterization, antitubercular and antibacterial activity, and molecular docking of 2,3-disubstituted quinazolinone derivatives
Authors Komarla R, Nizamuddin ND , Surur A, Mekonnen YT
Received 30 June 2015
Accepted for publication 23 January 2016
Published 11 May 2016 Volume 2016:6 Pages 15—26
DOI https://doi.org/10.2147/RRMC.S91474
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Xing-Cong Li
Video abstract presented by Abdrrahman Shemsu Surur.
Views: 923
KK Rajasekhar,1 ND Nizamuddin,1 Abdrrahman Shemsu Surur,2 Yenus Tadesse Mekonnen2
1Department of Pharmaceutical Chemistry, Sri Padmavathi School of Pharmacy, Tiruchanur, India; 2Department of Pharmaceutical Chemistry, School of Pharmacy, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
Abstract: Quinazolinone derivatives, which are known for their versatile biological activities, have been reported to show significant antibacterial and antitubercular activities. Fourteen compounds that belong to either 2-methyl substituted quinazolinone or 2-phenyl substituted quinazolinones were synthesized. Compounds 5a–e and 8a–c showed a minimum inhibitory concentration value between 6.25 and 100 µg/mL against Mycobacterium tuberculosis. Compounds 5g and 8d, on the other hand, showed significant antibacterial activity against Staphylococcus albus and Streptococcus pyogenes. The use of amido, thioamido, imidamido, N,N-dimethyl guanidinyl, or N-pyridoyl substituents at 3-position of quinazolinone was found to increase antitubercular activity. A binding affinity prediction by autodock vina was higher for the 2-phenyl series, which may be due to increased hydrophobic interactions within the binding site of enoyl-acyl carrier protein reductase.
Keywords: quinazolinones, antitubercular activity, antibacterial activity, autodock vina
Introduction
Multidrug-resistant bacteria represent a significant health threat with many of the last-line treatment options losing their effectiveness. Despite this rising concern, only 22 new antibiotics were launched worldwide between 2000 and 2013. For drugs targeting Gram-negative pathogens, there has not been a compound approved with a novel mechanism in nearly half a century.1
Tuberculosis (TB) ranks as the second leading cause of death from a single infectious agent, after human immunodeficiency virus. The TB mortality rate has decreased by 45% since 1990. In 2013, however, 1.5 million people died from TB. This is partly due to the increased resistance by Mycobacterium tuberculosis. The number of people diagnosed with multidrug-resistant TB tripled between 2009 and 2013, with an estimated 210,000 people dying from multidrug-resistant TB in 2013. To make the conditions even worse, an estimated 9% of people with multidrug-resistant TB have extensive drug-resistant TB.2 This report by World Health Organization necessitates increased efforts in the discovery and development of antitubercular agents.
Literature survey revealed the versatile biological activities of quinazolinone derivatives.3,4 It has been established that quinazolinones possess antiviral,4 antifungal,5 antiallergic,6 antitumor,7 and antidiabetic activities.8 In the recent past, quinazolinones were reported to exhibit pronounced coronary vasodilatory9 and histamine receptor type 3 inverse agonism.10
Various researchers have reported the antibacterial activity of quinazolinone derivatives.11–14 Subramaniam et al15 also evaluated the antibacterial and antitubercular activity of some quinazolinone derivatives. In the present study, 14 2,3-disubstitued quinazoline-4-one compounds were synthesized and their antibacterial and antitubercular activities were evaluated. The compounds were synthesized by varying the substitution pattern at the second and third positions of 1,3-benzoxazin-4-one and their in vitro antibacterial and antitubercular activities were evaluated.
Keeping this in view, the synthesized small molecules were docked against enoyl-acyl carrier protein reductase (InhA) of M. tuberculosis, which catalyzes the NADH-dependent reduction of the trans double bond between positions C2 and C3 of fatty acyl substrates. In addition, InhA prefers fatty acyl substrates of C16 or greater, consistent with it being a member of the mycobacterial FAS-II system.16 The docking was performed to predict the binding affinity of the synthesized quinazolinone derivatives against this enzyme. This will help to identify if there exists a relation between the binding affinity to InhA and minimum inhibitory concentration (MIC) for quinazolinone-based antitubercular drugs. The docking can also generate useful information for further studies on the structure-based drug design of quinazolinone-based antitubercular drugs.
Experimental
All chemicals were purchased from Sigma Aldrich Co., (St Louis, MO, USA), Merck (Whitehouse Station, NJ, USA), Qualigens Fine Chemicals (Mumbai, India), Loba Chemie Pvt. Ltd (Mumbai, India), and Himedia Laboratories Pvt. Ltd (Mumbai, India). The melting points of the synthesized compounds were determined in an open capillary tube using digital melting point apparatus and are uncorrected. The homogeneity and purity of the compounds were ascertained by thin layer chromatography (TLC) on silica gel G-plates using cyclohexane:ethyl acetate (2:1) and the spots were visualized in UV chamber. Infrared spectra (υ cm−1) were recorded on a SHIMADZU FT-IR 6000 using KBr disks. CHNO elemental analysis was carried out by a Perkin Elmer Series II 2400 CHNS/O Elemental analyzer. Mass spectra were obtained on a JEOL GC mate II GC- Mass spectrometer at 70 eV using direct insertion probe method. 1H NMR spectra were taken on a BRUKER AVIII-500MHz FT–NMR spectrometer by using tetramethylsilane (TMS) as an internal standard and the solvent used was dimethyl sulfoxide (DMSO).
Retrosynthetic analysis
The target structures were subjected to disconnection, which corresponds to the reverse of synthetic reaction, so as to convert the target structure to simpler precursor structures (synthons) without any assumptions regarding the starting materials. Each of the precursors so generated was then examined in the same way, and the process was repeated until simple or commercially available structures were obtained.
General synthetic procedures
Synthesis of 2-methyl-(4H)-benzo[1,3]oxazin-4-one
A mixture of anthranilic acid 1 (0.02 mol, 2.7242 g) in acetic anhydride 2 (2 mL) was heated for 1 hour; the excess solvent was then distilled off under reduced pressure. The reaction mixture was cooled, filtered, washed with petroleum ether, dried, and recrystallized with absolute ethanol to get 2-methyl-(4H)-benzo[1,3]oxazin-4-one 3 (Figure 2A). Completion of the reaction was determined by TLC using cyclohexane:ethyl acetate (2:1) as mobile phase.
Compound 3 (0.01 mol) and amino reagent 4 (0.02 mol) in ethanol (30 mL) were heated under reflux for 3 hours. Then, the reaction mixture was concentrated and the separated solid was dried and recrystallized with ethanol to get 2-methyl-4H-3-substituted quinazolin-4-one 5. The homogeneity and purity of the compounds were ascertained by TLC on silica gel G-plates using cyclohexane:ethyl acetate (2:1) and the spots were visualized using a UV chamber.
Synthesis of 2-phenyl-(4H)-benzo[1,3]oxazin-4-one
Anthranilic acid 1 (0.1 mol) was dissolved in pyridine (60 mL) and benzoyl chloride 6 (0.2 mol) was added. The mixture was stirred for 30 minutes followed by treatment with 5% NaHCO3 (15 mL). The solid obtained was recrystallized with ethanol to get 2-phenyl-(4H)-benzo[1,3]oxazin-4-one 7 (Figure 2B). Completion of the reaction was determined by TLC using cyclohexane:ethyl acetate (2:1) as mobile phase.
Compound 7 and amino reagent 4 (0.02 mol) were refluxed for 3–4 hours in the presence of glacial acetic acid. The reaction mixture was kept overnight and the product obtained was recrystallized using ethanol to get 2-phenyl-4H-3-substituted quinazolin-4-one 8. The homogeneity and purity of the compounds were ascertained by TLC on silica gel G-plates using cyclohexane:ethyl acetate (2:1) and the spots were visualized using UV chamber.
By adopting the above synthetic procedures, compounds 5a, 5b, 5c, 5d, 5e, 5f, 5g, 8a, 8b, 8c, 8d, 8e, 8f, and 8g were also synthesized using different amino reagents. The seven amino reagents used are urea, thiourea, guanidine, metformin, isoniazid, phenyl hydrazine, and dinitrophenyl hydrazine.
The IUPAC and spectra details of the synthesized quinazolinones
2-Methyl-4-oxoquinazoline-3(4H)-carboxamide (5a)
Yield, 79%: mp 178°C–185°C; FT-IR (KBr): 2,978.4 (–CH3), 1,689.8 (O=C–NH2), 1,647.4 (C=N), 1,310 (30 N), 1,294.3 (10 N) cm−1; 1H NMR (DMSO): δ 7.00 (m, 4H, Ar-H), 3.0 (s, 3H, CH3), 2.00 (s, 2H, NH2) ppm; MS (m/z,%): 203.198 (M+).
2-Methyl-4-oxoquinazoline-3(4H)-carbothioamide (5b)
Yield, 92%: mp 189°C–196°C; FT-IR (KBr): 2,968.8 (–CH3), 1,267.3 (C=S), 1,684.8 (O=C–NH2), 1,647.4 (C=N), 1,310 (30 N), 1,294.3 (10 N) cm−1; 1H NMR (DMSO): δ 7.00 (m, 4H, Ar-H), 3.0 (s, 3H, CH3), 2.00 (s, 2H, NH2) ppm; MS (m/z,%): 219.262 (M+).
2-Methyl-4-oxoquinazoline-3(4H)-carboximidamide (5c)
Yield, 74%: mp 184°C–189°C; FT-IR (KBr): 2,972.67 (–CH3), 1,676.3 (O=C–NH2), 1,649.4 (C=N), 1,313.68 (30 N), 1,302.11 (10 N) cm−1; 1H NMR (DMSO): δ 7.00 (m, 4H, Ar-H), 3.0 (s, 3H, CH3), 2.00 (s, 2H, NH2), 2.47 (s, 1H, NH) ppm; MS (m/z,%): 202.246 (M+).
N-(dimethylcarbamimidoyl)-2- methyl-4-oxoquinazoline-3(4H)- carboximidamide (5d)
Yield, 89%: mp 195°C–202°C; FT-IR (KBr): 2,951.4 (–CH3), 1,684.06 (O=C–NH2), 1,647.4 (C=N), 1,270.7 (30 N), 1,298.25 (20 N) cm−1; 1H NMR (DMSO): δ 7.00 (m, 4H, Ar-H), 5.5 (s, 1H, NH), 3.2 (s, 9H, CH3), 2.47 (m, 2H, NH) ppm; MS (m/z,%): 272.305 (M+).
N-(2-methyl-4-oxoquinazolin-3(4H)-yl)pyridine-4-carboxamide (5e)
Yield, 72%: mp 162°C–167°C; FT-IR (KBr): 2,988.8 (–CH3), 1,682.13 (O=C–NH2), 1,633.13 (C=N), 1,309.98 (30 N), 1,275.10 (20 N) cm−1; 1H NMR (DMSO): δ 7.28 (m, 8H, Ar-H), 3.0 (s, 3H, CH3), 2.0 (s, 1H, NH) ppm; MS (m/z,%): 280.281 (M+).
2-Methyl-3-(phenylamino)quinazolin-4(3H)-one (5f)
Yield, 88%: mp 156°C–160°C; FT-IR (KBr): 2,972.67 (–CH3), 1,689.8 (O=C–NH2), 1,657.4 (C=N), 1,300.18 (30 N), 1,325.26 (20 N) cm−1; 1H NMR (DMSO): δ 6.5 (m, 9H, Ar-H), 3.0 (s, 3H, CH3), 4.0 (s, 1H, Ar-NH) ppm; MS (m/z,%): 265.309 (M+).
3-[(2,4-Dinitrophenyl)amino]-2-methylquinazolin-4(3H)-one (5g)
Yield, 84%: mp 162°C–165°C; FT-IR (KBr): 2,953.38 (–CH3), 1,508.52 (NO2), 1,684.8 (O=C–NH2), 1,647.41 (C=N), 1,310 (30 N), 1,294.3 (20 N) cm−1; 1H NMR (DMSO): δ 8.4 (m, 7H, Ar-H), 3.0 (s, 3H, CH3), 4.0 (s, 1H, Ar-NH) ppm; MS (m/z,%): 355.504 (M+).
4-Oxo-2-phenylquinazoline-3(4H)-carboxamide (8a)
Yield, 78%: mp 275°C–280°C; FT-IR (KBr): 3,053.15 (C–H), 1,689.8 (O=C–NH2), 1,647.4 (C=N), 1,310 (30 N), 1,294.3 (10 N) cm−1; 1H NMR (DMSO): δ 7.64 (m, 9H, Ar-H), 2.00 (s, 2H, NH2) ppm; MS (m/z,%): 265.225 (M+).
4-Oxo-2-phenylquinazoline-3(4H)-carbothioamide (8b)
Yield, 92%: mp 282°C–286°C; FT-IR (KBr): 3,029.6 (C–H), 1,267.3 (C=S), 1,684.8 (O=C–NH2), 1,647.4 (C=N), 1,310 (30 N), 1,294.3 (10 N) cm−1; 1H NMR (DMSO): δ 7.00 (m, 9H, Ar-H), 2.00 (s, 2H, NH2) ppm; MS (m/z,%): 281.262 (M+).
4-Oxo-2-phenylquinazoline-3(4H)-carboximidamide (8c)
Yield, 64%: mp 187°C–190°C; FT-IR (KBr): 3,047.90 (C–H), 1,653.20 (O=C–NH2), 1,639.5 (C=N), 1,344.35 (30 N), 1,327.19 (10 N) cm−1; 1H NMR (DMSO): δ 7.00 (m, 9H, Ar-H), 2.00 (s, 2H, NH2), 2.47 (s, 1H, NH) ppm; MS (m/z,%): 264.28 (M+).
N-(dimethylcarbamimidoyl)-4-oxo-2-phenylquinazoline-3(4H)-carboximidamide (8d)
Yield, 88%: mp 218°C–221°C; FT-IR (KBr): 3,037.95 (C–H), 1,684.06 (O=C–NH2), 1,647.4 (C=N), 1,325.20 (30 N), 1,300.18 (20 N) cm−1; 1H NMR (DMSO): δ 7.45 (m, 9H, Ar-H), 2.95 (s, 6H, CH3), 5.5 (s, 1H, NH), 2.45 (m, 2H, NH) ppm; MS (m/z,%): 334.375 (M+).
N-(4-oxo-2-phenylquinazolin-3(4H)-yl)pyridine-4-carboxamide (8e)
Yield, 72%: mp 162°C–167°C; FT-IR (KBr): 3,048.8 (C–H), 1,682.13 (O=C–NH2), 1,633.13 (C=N), 1,359.98 (30 N), 1,275.10 (20 N) cm−1; 1H NMR: δ 8.2 (m, 13H, Ar-H), 2(s, 1H, NH) ppm; MS (m/z,%): 342.375 (M+).
2-Phenyl-3-(phenylamino)quinazolin-4(3H)-one (8f)
Yield, 68%: mp 198°C–201°C; FT-IR (KBr): 3,027.44 (C–H), 1,689.8 (O=C–NH2), 1,657.4 (C=N), 1,300.18 (30 N) 1,325.26 (20 N) cm−1; 1H NMR: δ 6.8 (m, 14H, Ar-H), 4.0 (s, 1H, Ar-NH) ppm; MS (m/z,%): 313.352 (M+).
3-[(2,4-Dinitrophenyl)amino]-2-phenylquinazolin-4(3H)-one (8g)
Yield, 74%: mp 169°–172 °C; FT-IR (KBr): 3,030.44 (C–H), 1,508.52 (–NO2), 1,684.8 (O=C–NH2), 1,647.41 (C=N), 1,310 (30 N), 1,294.3 (20 N) cm−1; 1H NMR: δ 8.4 (m, 12H, Ar-H), 4.0 (s, 1H, Ar-NH) ppm; MS (m/z,%): 403.347 (M+).
Antitubercular activity
The synthesized quinazolinones were screened for antitubercular activity using the microplate Alamar blue assay method. Accordingly, each quinazolinone was screened against M. tuberculosis H37 RV strain in the Middlebrook 7H9 (MB 7H9) broth using streptomycin and pyrazinamide as standard drugs at concentrations of 6.25 and 3.125 μg/mL, respectively.
An amount of 200 μL of sterile deionized water was added to all outer perimeter wells of a sterile 96-well plate to minimize evaporation of the medium in the test wells during incubation. The 96-well plate received 100 μL of the MB 7H9 broth and serial dilution of compounds was made directly on plate. The final drug concentrations tested were 0.2, 0.4, 0.8, 1.6, 3.125, 6.25, 12.5, 25, 50, and 100 μg/mL. Plates were covered and sealed with Parafilm and incubated at 37°C for 5 days. After this time, 25 μL of freshly prepared 1:1 mixture of Alamar blue reagent and 10% Tween 80 was added to the plate and incubated for 24 hours. A blue color in the well was interpreted as no bacterial growth, and pink color was scored as growth. The MIC, which is the required concentration to inhibit 90% of the standardized bacterial inoculums, was defined as the lowest drug concentration which prevented the color change from blue to pink.17
Antibacterial activity
The antibacterial activity of the synthesized quinazolinones was evaluated using agar cup plate method. Accordingly, the compounds were screened against Gram-negative organisms, namely, E. coli and Klebsiella and Gram-positive organisms S. albus and Streptococcus pyogenes using the MIC method. Ciprofloxacin was employed as a reference standard to compare the results.
Brain heart infusion agar was used at room temperature. The required colonies were transferred to the plates and the turbidity was adjusted visually with broth to equal that of a 0.5 McFarland turbidity standard that has been vortexed. The entire surface of agar plate was swabbed three times, rotating plates ~60° between streaking to ensure even distribution. The inoculated plate was allowed to stand for at least 5 minutes before applying disks.
A 5 mm hollow tube was heated, pressed on the inoculated agar plate, and removed immediately five times by making five wells in the plate. Subsequently, 75, 50, 25, 10, and 5 μL of the synthesized compounds were added into the respective wells on each plate. The plates were incubated within 15 minutes of compounds application for 24 hours at 37°C in incubator. The diameter of inhibition zone was measured to the nearest whole millimeter by holding the measuring device. According to the MIC procedure, the serial dilution was repeated up to 10−9 dilution for each synthesized quinazolinone.18,19
Molecular docking
Preparation of the macromolecule
The 3D crystal structure of M. tuberculosis InhA in complex with 1-cyclohexyl-N-(3,5-dichlorophenyl)-5-oxopyrrolidine-3-carboxamide (PDB code: 4TZK) was retrieved from the RCSB protein data bank.20 The dock prep tool of UCSF chimera version 1.10.1 for Mac was used to prepare the enzyme for docking.21 Eventually, Python Prescription (PyRx) 0.8 for Mac was used to save the macromolecule in pdbqt format, which contains hydrogen atoms in all polar residues.22
Ligand preparation
The 2D chemical structures of the ligands were prepared using ChemBioDraw 14.0.0.117 for Mac (Cambridge, MA, USA).23 The 2D chemical structures were converted into the respective 3D structures using the Open Babel of PyRx 0.8 virtual screening tool for Mac. After energy minimization of each ligand using Uff force field of the Open Babel, the ligands were converted into autodock ligands.
Docking validation
The ligand from the active site of the crystal structure of InhA was removed using UCSF Chimera for Mac. After the ligand was redocked, the alignment between the docked ligand and the ligand from the crystal structure was done using MacPyMOL (New York, NY, USA).24
Molecular docking
Docking was performed using PyRx autodock vina.25 The results were quantified in terms of free binding energy (ΔG). The highest binding energy values corresponding to the RMSD value of zero were considered as the binding affinity value of the ligands. The postdock analysis was made using PyMOL and UCSF chimera.
Results and discussion
Synthesis
Applying retrosynthetic analysis suggested the use of substituted primary amines, anthranilic acid, and acetic anhydride for the synthesis of 2-methyl-(4H)3-substituted quinazolin-4-one. The same strategy suggested the use of substituted primary amine, anthranilic acid, and benzoyl chloride for the synthesis of 2-phenyl-(4H)3-substituted quinazolin-4-one (Figure 1).
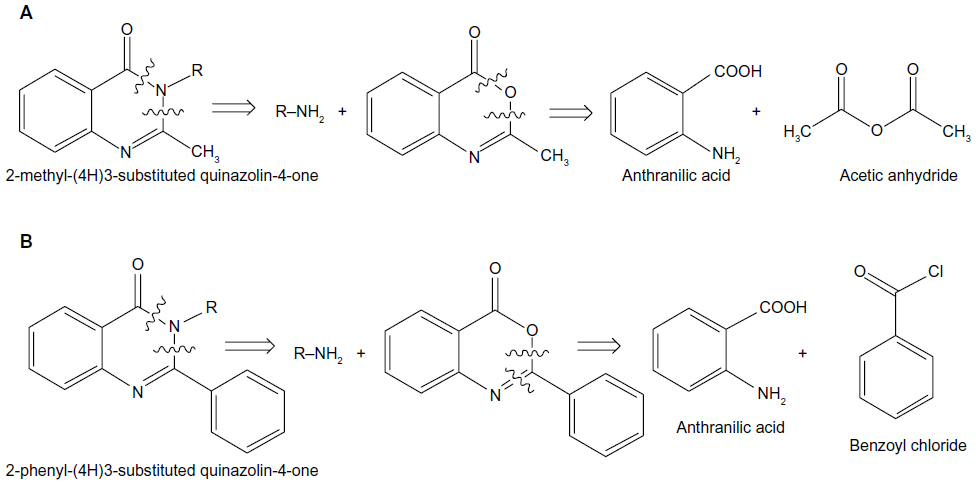 | Figure 1 Retrosynthetic analysis of (A) 2-methyl-(4H)3-substituted quinazolin-4-one and (B) 2-phenyl-(4H)3-substituted quinazolin-4-one. |
Compounds 5a–g were synthesized by condensation of 2-methyl-4H-benzo[d][1.3]oxazin-4-one with primary amines (4a–g) in alcohol (Figure 2A). Compounds 8a–g, on the other hand, were synthesized by condensation of 2-phenyl-4H-benzo[d][1,3]oxazin-4-one with primary amines (4a–g) in the presence of glacial acetic acid (Figure 2B). The method required mild experimental conditions and the yields were satisfactory.
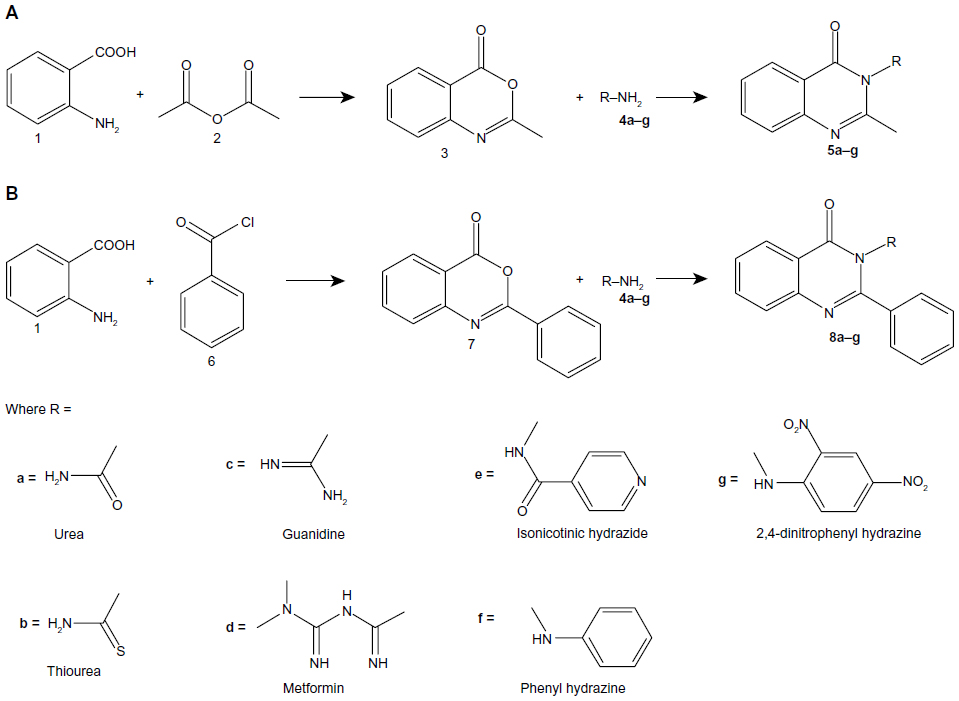 | Figure 2 General synthetic route for (A) 2-methyl(4H)3-substituted quinazolin-4-one and (B) 2-phenyl(4H)3-substituted quinazolin-4-one. |
The proposed scheme led to compounds which are in conformity with the structure envisioned. Structural confirmation was done by using the spectral and analytical data from Fourier transform infrared spectroscopy (FT-IR), proton nuclear magnetic resonance (1H NMR), and mass spectrometry (MS). In all cases, the products were obtained in pure form. Moreover, they were purified by recrystallization from ethanol.
Different synthetic routes for compounds 5c,26 5e,27 5g,28 and 8c26 are available. Moreover, synthesis and biological activities other than antibacterial and antitubercular activity of compounds 5a,29,30 5b,30 5e,31 5f,32 8a,30 8b,30 and 8e31,33 have been previously reported.
Antibacterial and antitubercular activity
All the synthesized quinazolinones were screened for their antitubercular activity according to microplate Alamar blue assay method (Table 1). It is interesting to note that synthesized compounds 5a–e and 8a–c exhibited MICs at a concentration ranging from 6.25 to 100 μg/mL. The rest of the compounds 5f–g and 8d–g exhibited no antitubercular activity at the studied concentration interval. Accordingly, the presence of amido, thioamido, and guanidino groups at 3-position of quinazolinone nucleus, as in compounds 5a–c and 8a–c, may be necessary for the antitubercular activity. In particular, a thioamido or guanidino group can greatly increase the antitubercular activity of quinazolinones, as noted by 6.25 μg/mL MIC value for compounds 5b and 8c.
 | Table 1 The antitubercular activity (minimum inhibitory concentration [MIC] values and the binding affinities to InhA) for quinazolinone derivatives |
In addition, all the synthesized quinazolinones were screened for their antibacterial activity using agar cup plate method. Accordingly, ciprofloxacin sensitive at 10 μg/mL for Staphylococcus albus and Streptococcus pyogenes are 26 and 24 mm of zone of inhibition, respectively. Ciprofloxacin sensitive at 10 μg/mL for Escherichia coli and Klebsiella are 32 and 30 mm of zone of inhibition, respectively. The antibacterial activity of the synthesized quinazolinones in comparison with ciprofloxacin is shown in Figures 3–6. Compounds 5g (13 mm) and 8d (11 mm) exhibited significant antibacterial activity against Gram-positive bacteria at 10 μg/mL concentration. This result showed the importance of 2,4-dinitrophenyl hydrazine or N,N–dimethyl guanidinyl group at 3-position of quinazolinones for activity against Gram-positive bacteria. However, the general activity of the synthesized quinazolinones against Gram-negative bacteria was found to be low.
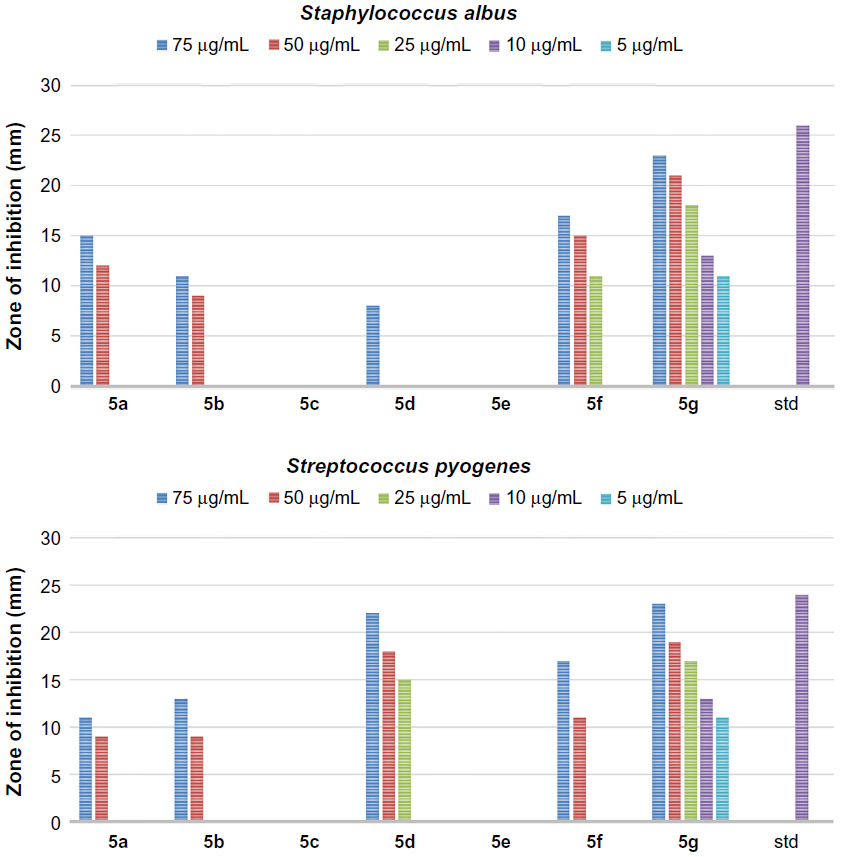 | Figure 3 The antibacterial activity of compounds 5a–g against Gram-positive bacteria, Staphylococcus albus and Streptococcus pyogenes. |
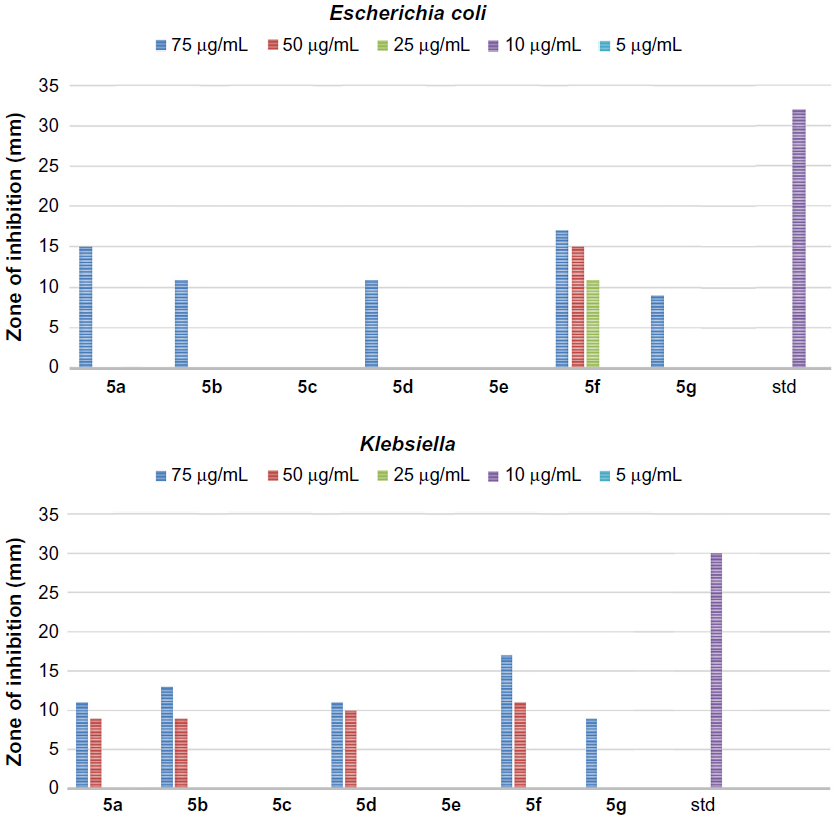 | Figure 4 The antibacterial activity of compounds 5a–g against Gram-negative bacteria, Escherichia coli and Klebsiella. |
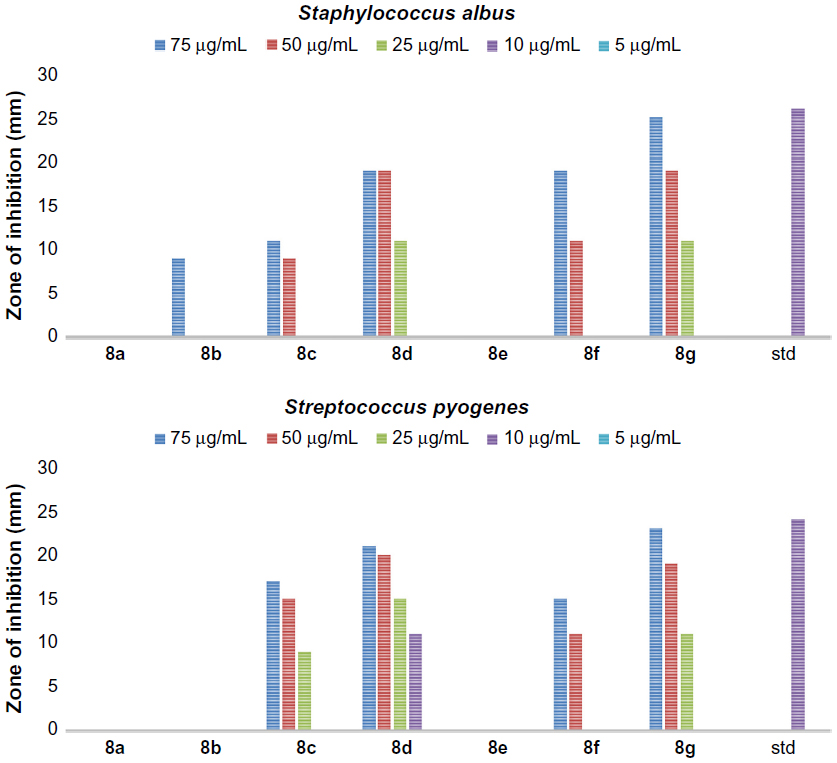 | Figure 5 The antibacterial activity of compounds 8a–g against Gram-positive bacteria, Staphylococcus albus and Streptococcus pyogenes. |
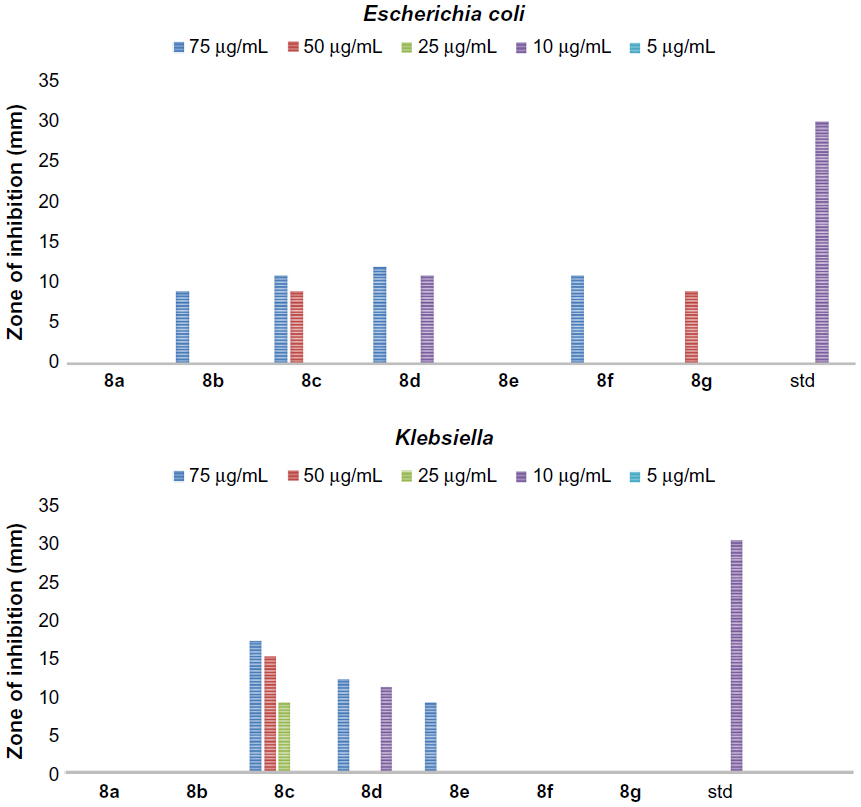 | Figure 6 The antibacterial activity of compounds 8a–g against Gram-negative bacteria, Escherichia coli and Klebsiella. |
Molecular docking
The quality of prepared macromolecule
Ramachandran plot, which is a commonly used indicator of quality of a model, of prepared macromolecule was obtained using RAMPAGE.34 The percentage of residues in the favored region and allowed region was 96.6% and 3.4%, respectively. Moreover, the number of residues in outlier region was zero (Figure 7).
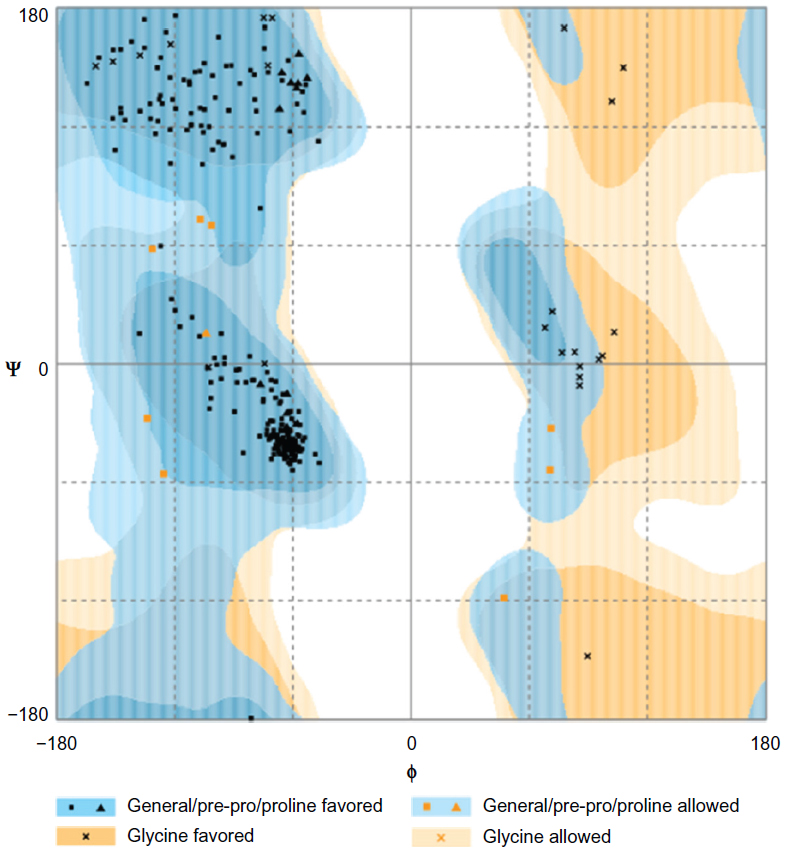 | Figure 7 Ramachandran plot for the analysis of ψ and φ torsion angles for all residues on the macromolecule prepared for docking. |
The Structural Analysis and Verification Server was also used for checking and validation of the protein structure.35 The ERRAT analysis gave an overall quality factor of 96.54%, which is acceptable as high resolution structures generally produce values around 95% or higher. In VERIFY3D result, 96.64% of the residues had an averaged 3D-1D score ≥0.2. This value is higher than the pass value, which is at least 80% of the amino acids with a score ≥0.2 in the 3D-1D profile.
Docking
The docking validation showed that the X-ray crystallographic conformer was nearly identical with the docked conformer, as deducted from the alignment of the two structures with an RMSD value of 0.057 Å (Figure 8).
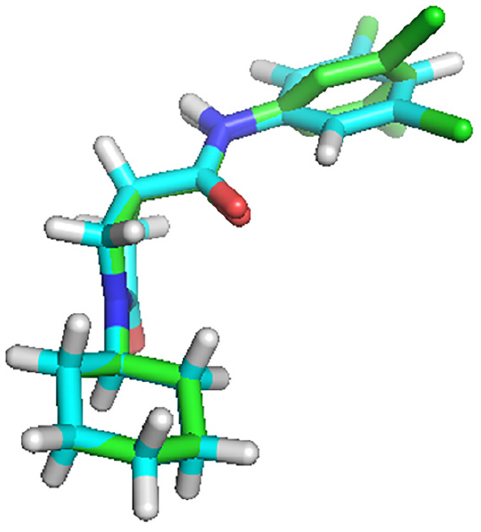 | Figure 8 The alignment of the docked ligand on the X-ray crystal ligand in the active site of InhA. |
The binding energy of each ligand against InhA macromolecule was predicted using autodock vina, which is one of the most commonly used docking software. In the docking procedure, eight binding poses were obtained, and the binding pose with the highest binding energy corresponding to the RMSD value of zero was considered as the binding affinity value of the ligand. From 2-methyl quinazolinone series, compound 5g (−9.3 kcal/mol) showed the highest binding energy. The 2-phenyl quinazolinone series, however, showed better binding energy than the 2-methyl quinazolinone series. In particular, compounds 8e (−9.8 kcal/mol) and 8g (−9.6 kcal/mol) showed the highest binding energy among all synthesized quinazolinones.
The amino acid residues interacting with the quinazolinone derivatives are given in Table 2. With the exception of compounds 5d and 5f, all derivatives in the 2-methyl series showed hydrogen bond interactions. Compound 5g, the most active in 2-methyl series, showed two hydrogen bond interactions (Figure 9A) and many hydrophobic contacts with different amino acid residues in the binding site.
 | Table 2 Summary of the residues interacting with quinazolinone derivatives |
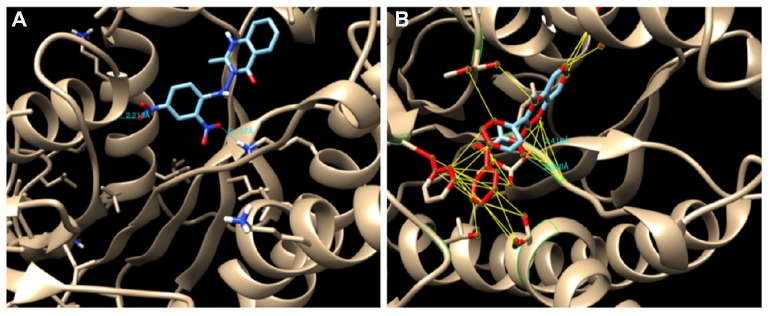 | Figure 9 (A) Hydrogen bond (HB) interactions of compound 5g and (B) HB interactions and hydrophobic contact of compound 8c in the binding site of InhA. |
In general, the 2-phenyl series of quinazolinones were found to have more binding affinity than the 2-methyl series. This is due in part to an increased number of hydrophobic contacts with the amino acids of the binding site or stronger hydrophobic interaction of a 2-phenyl moiety with I20, F40, F148, and G191. In addition, the benzene ring of quinazolinone is also involved in a more hydrophobic interaction with Y157, M198, and I214 residues than the same moiety in the 2-methyl series. The most active compound was compound 8e, which showed two hydrogen bonding interactions and more hydrophobic contacts with amino acid residues, such as F40 and G95 (Figure 9B).
Conclusion and recommendations
The results of antitubercular activity testing revealed that quinazolinone compounds 5a–e and 8a–c exhibited significant activity. Amido, thioamido, imidamido, N,N-dimethyl guanidinyl, and N-pyridoyl moieties at 3-position of quinazolinone were found to increase antitubercular activity. A further synthesis and evaluation of compounds having these moieties may lead to a much more potent antitubercular agent.
The quinazolinone compounds 5g and 8d exhibited significant activity against Gram-positive bacteria, which may be due to the presence of N-phenyl (5g) and N,N-dimethyl guanidinyl moieties at 3-position. However, the activity of the synthesized quinazolinones against Gram-negative bacteria was low. A further synthetic work on derivatives with potential to be ionized is important, so as to increase activity against Gram-negative bacteria.
Acknowledgments
The authors are grateful for the material support received from Sri Padmavathi School of Pharmacy and University of Gondar. The authors are also grateful for the authorities of UCSF chimera, PyRx, and PyMOL for providing free software.
Disclosure
The authors report no conflicts of interest in this work.
References
Butler MS, Blaskovich MA, Cooper MA. Antibiotics in the clinical pipeline in 2013. J Antibiot. 2013;66:571-591. | |
WHO. Global Tuberculosis Report 2014. Geneva: World Health Organization; 2014. Available form: http://www.who.int/tb/publications/global_report/en/. Accessed June 5, 2015. | |
Büyüktimkin S. Synthesis, pharmacology and structure-activity relationships of 2-methyl-3-(4-oxo-3-phenyl-thiazolidin-2-ylidenamino)-4(H)-quinazolinone derivatives. Arch Pharm. 1985;318:496–501. | |
Büyüktimkin S, Büyüktimkin N, Özdemir O, Rollas S. Synthesis of 3-[2-(2,3-dihydro-5-phenyl-4-substituted-3H-1,2,4-triazole-3-thione-2-yl)-acetylamino]-2-methyl-4(3H)-quinazolinones and their pharmacological activities. Arch Pharm. 1989;322:49–52. | |
Mukherji DD, Nintiyal SR, Prasad CR, Dhawan BN. Pharmacological studies on quinazolinones. Ind J Med Res. 1980;93:1426–1432. | |
Peet NP, Baugh LE, Sunder S, et al. 3-(1H-tetrazol-5-yl)-4(3H)-quinazolinone sodium salt (MDL 427): a new anti-allergic agent. J Med Chem. 1986;29:2403–2409. | |
Chaurasia MR, Sharma SK. Synthesis of some new 4(3H)-quinazolinones as a potent CNS depressant. Arch Pharm. 1982;315:377–381. | |
Li H, Wang JP, Yang F, et al. Design, synthesis and biological activity evaluation of 2-mercapto-4(3H) quinazolinone derivatives as a novel inhibitor of protein tyrosine phosphatase 1B. Heterocycles. 2012;85:1897–1911. | |
Chiou WF, Liao JF, Chen CF. Comparative study on the vasodilatory effects of three quinazoline alkaloids isolated from Evodia rutaecarpa. J Nat Prod. 1996;59:374–378. | |
Nagase T, Mizutani T, Ishikawa S, et al. Synthesis, structure-activity relationships, and biological profiles of a quinazolinone class of histamine H3 receptor inverse agonists. J Med Chem. 2008;51:4780–4789. | |
Nanthakumar R, Muthumani P, Girija K. Anti-inflammatory and antibacterial activity study of some novel quinazolinones. Arabian J Chem. 2014;7:1049–1054. | |
Khodarahmi GA, Khajouel MR, Hakimelahi GH, Abedi D, Jafari E, Hassanzadeh F. Antibacterial, antifungal and cytotoxic evaluation of some new 2,3-disubstituted 4(3H)-quinazolinone derivatives. Res Pharm Sci. 2012;7:151–158. | |
Hemalatha K, Girija K. Synthesis, characterization, preliminary QSAR studies and in vitro antioxidant activity of some novel 2,3-disubstituted quinazolinone derivatives. Int J Pharm Sci. 2012;4:99–103. | |
Panwar H, Chaudhary N, Singh S, Singh SH. Anti-inflammatory and antimicrobial activity of some new quinazolinones. Rasayan J Chem. 2011;4:498–505. | |
Subramaniam R, Rao G, Jaiswal PK. Synthesis of some 2,3-disubstituted quinazolinone derivatives as antitubercular and antibacterial activity. Der Pharmacia sinica. 2014;5:37–40. | |
Rozwarski D, Vilcheze C, Sugantino M, Bittman R, Sacchettini J. Crystal structure of the mycobacterium tuberculosis Enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J Biol Chem. 1999;274(22):15582–15589. | |
Franzblau SG, Witzig RS, Mclaughlin GC, et al. Rapid, low-technology MIC determination with clinical M. tuberculosis isolates by using the microplate Alamar blue assay. J Clin Microbiol. 1998;36:362–366. | |
Hanlon A, Taylor M, Dick JD. Agar dilution susceptibility testing. In: Schwalbe R, Moore LS, Goodwin AC, editors. Antimicrobial Susceptibility Testing Protocols. Volume 1. Boca Raton: CRC Press, Taylor and Francis group; 2007:91–104. | |
Garcia LS, Isenberg HD. Clinical Microbiology Procedures Handbook. Volume 3. Washington DC: ASM Press; 2010. | |
He X, Alian A, Stroud R, Montellano P. Pyrrolidine carboxamide as a novel class of inhibitors of enoyl acyl carrier protein reductase from M. tuberculosis. J Med Chem. 2006;49:6308–6323. | |
Pettersen E, Goddard T, Huang C, et al. UCSF chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;13:1605–1612. | |
Dallakyan S, Olson A. Small molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243–250. | |
Cambridgesoft corporation, a subsidiary of PerkinElmer, Inc. 2014. ChemBioDraw Ultra version 14.0.0.117 for Mac. Available from: www.cambridgesoft.com. | |
The PyMOL molecular graphics system, version 1.7.4 Schrodinger, LLC. Available from: www.schrodinger.com/pymol/. | |
Trott O, Olson A. Autodock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. | |
Kawadkar RK, Ghiya BJ. Synthesis of new quinazolin-4-one compounds of medicinal importance. Asian J Chem. 1999;11(2):388–391. | |
Babu RR, Naresh K, Ravi A, Reddy BM, Babu VH. Synthesis of novel isoniazid incorporated styryl quinazolinones as anti-tubercular agents against isoniazid sensitive and MDR M. tuberculosis strains. Med Chem Res. 2014; 23(10):4414–4419. | |
Nezhad AK, Haghighi, SM, Purkhosrow A, Panahi F. An efficient one-pot access to quinazolinone derivatives using TiO2 nanoparticles as catalyst: synthesis and vasorelaxant activity evaluation. Synlett. 2012;23(6):920–924. | |
Wilczkiewicz AM, Kalinowski DS, Musiol R, Finster J, Szurko A, Serafin K. Investigating the anti-proliferative activity of styrylazanaphthalenes and azanaphthalenediones. Bioorg Med Chem. 2010;18:2664–2671. | |
Babu YR, Bhagavanraju M, Reddy GD, Peters GJ, Prasad VV. Design and synthesis of quinazolinone tagged acridones as cytotoxic agents and their effects on EGFR tyrosine kinase. Arch Pharm (Weinheim). 2014;347(9):624–634. | |
Hemalatha K, Kumar MS, Girija K. Dihydrofolate reductase inhibitors: synthesis, characterization and biological evaluation of some novel 2,3-disubstituted quinazolinones. Indian J Heterocycl Chem. 2014; 24(1):35–40. | |
Srivastava VK, Palit G, Agarwal AK, Shanker K. Antiparkinsonian activity and behavioural effects of newer quinazolinones. Pharmacol Res Commun. 1987:19(9):617–628. | |
Hemalatha K, Girija K. Synthesis of some novel 2,3-disubstituted quinazolinone derivatives as analgesic and anti-inflammatory agents. Int J Pharm Pharm Sci. 2011;3(2):103–106. | |
Lovell S, Davis I, Arendall III W, et al. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2002;50:437–450. | |
Structural Analysis, Verification Server; 2011. Available from: http://nihserver.mbi.ucla.edu/SAVES/. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.