")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Synthesis, antimicrobial and in vitro antitumor activities of a series of 1,2,3-thiadiazole and
1,2,3-selenadiazole derivatives
Authors Mhaidat N, Al-Smadi ML, Al-momani F, Alzoubi KH , Mansi I, Al-Balas Q
Received 3 April 2015
Accepted for publication 26 May 2015
Published 16 July 2015 Volume 2015:9 Pages 3645—3652
DOI https://doi.org/10.2147/DDDT.S86054
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Nizar M Mhaidat,1,2 Mousa Al-Smadi,3 Fouad Al-Momani,4 Karem H Alzoubi,1 Iman Mansi,2 Qosay Al-Balas5
1Department of Clinical Pharmacy, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, 2Faculty of Pharmaceutical Sciences, Hashemite University, Zarqa, 3Department of Applied Chemical Sciences, 4Department of Applied Biological Sciences, Faculty of Science and Arts, 5Department of Medicinal Chemistry and Pharmacognosy, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan
Abstract: Three derivatives of substituted 1,2,3-thia- or 1,2,3-selenadiazole (4a–c) were prepared and characterized by different chemical techniques. These compounds were evaluated for their antimicrobial and antitumor activities. Compounds 4a (propenoxide derivative), 4b (carbaldehyde derivative), and 4c (benzene derivative) were active against the yeast-like fungi Candida albicans. Compound 4a was active against gram-negative Escherichia coli, and compound 4c was active against the gram-positive Staphylococcus aureus. For the antitumor activity, both compounds 4b and 4c were active against all tested tumor cell lines, namely, SW480, HCT116, C32, MV3, HMT3522, and MCF-7. The activity of compound 4c was greater than that of compound 4b and more than that of the reference antitumor 5-flourouracil against the SW480, HCT116, and MCF-7 tumor cell lines. In conclusion, a number of the prepared 1,2,3-thia- or 1,2,3-selenadiazole compounds showed promising antifungal, antibacterial, and in vitro antitumor activities. Further investigations are required to explore the mechanism by which active compound are inducing their cytotoxicity.
Keywords: thiadiazole, selenadiazole, cell lines, antimicrobial activity
Introduction
Microbes are highly diverse, and their genetic makeup is subjected to continuous wavering in response to environmental settings.1 Human pathogens, both nosocomial and enterobacter species, are the most common infectious pathogens.2,3 The induction of highly resistant pathogens urges researchers to develop new antimicrobial agents from natural sources.3 Heterocyclic compounds containing sulfur and selenium are attractive due to their interesting biological and synthetic applications. 1,2,3-Thia- and 1,2,3-selenadiazole derivatives are well-known, and the organic synthesis of their intermediates have attracted the attention of researchers.4 Numerous substituted 1,2,3-thia- and 1,2,3-selenadiazole derivatives have been prepared, and most of them have showed noticeable antibacterial activities against Bacillus subtilis and Escherichia coli. The antifungal activity of 1,2,3-thia- and 1,2,3-selenadiazole against Aspergillus niger, Cryptococcus neoformans, and Candida albicans has also been determined.5–7 It has been found that the introduction of the 1,2,3-selenadiazole ring to known molecules with biological activity will modify and even improve their biological activity. Other heterocyclic compounds containing triazole, oxazole, benzoxazole, quinazoline, pyridazoline, pyridazine, pyrazole, and thiazole have been found to be biologically active substances.8–10
Heterocyclic ring systems are present in numerous antiparasitic, antifungal, anthelmintic, and anti-inflammatory drugs.11 The beta-lactam antibiotics derivatized with a 1,2,3-thiadiazole-5-mercapto moiety have been found to be active against gram-negative bacteria, such as Pseudomonas aeruginosa. 4-Methyl-1,2,3-selenadiazole-5-carboxamides have been described to inhibit tumor cell colony formation.12 In the area of antibacterial therapeutics, resistance to currently available drugs has been progressively limiting their utility in treating bacterial infections. Discovering novel pharmaceutical agents has become a necessity to combat the increased resistance of microbes to the current antimicrobial arsenal.13 Advances in molecular microbiology and genomics have led to the identification of numerous bacterial genes that code for novel druggable proteins and could potentially serve as antibacterial targets.13 Regulatory proteins, such as the two-component histidine kinases involved in bacterial signal transduction, have recently gained considerable attention as one such class of potential targets.14,15 New synthetic compounds and natural products from plants are showing promising antitumor and antimicrobial products.16 It was reported that some 1,2,3-selenadiazole derivatives show antimicrobial activity against highly resistance reference and local nosocomial pathogens isolated from the clinical environment.17 As a continuation of our previous work on the synthesis of heterocyclic compounds containing 1,2,3-thi- or 1,2,3-selenadiazole rings,18 in this study, substituted 1,2,3-thiadiazole (Figure 1) or 1,2,3-selenadiazole (Figure 2) were synthesized, and their antitumor and antimicrobial activities were investigated in comparison with control drugs.
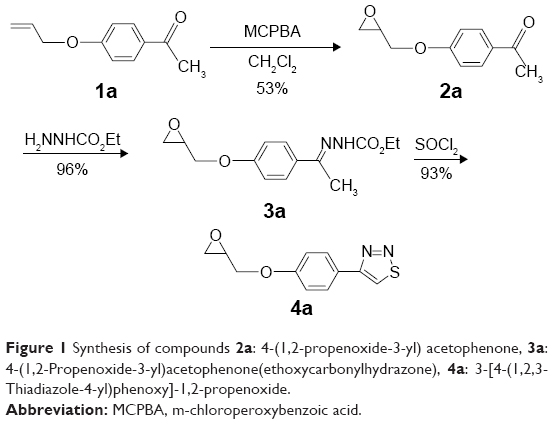 | Figure 1 Synthesis of compounds 2a: 4-(1,2-propenoxide-3-yl) acetophenone, 3a: 4-(1,2-Propenoxide-3-yl)acetophenone(ethoxycarbonylhydrazone), 4a: 3-[4-(1,2,3-Thiadiazole-4-yl)phenoxy]-1,2-propenoxide. |
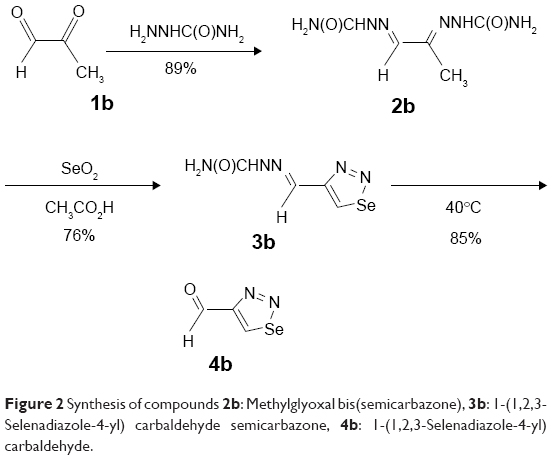 | Figure 2 Synthesis of compounds 2b: Methylglyoxal bis(semicarbazone), 3b: 1-(1,2,3-Selenadiazole-4-yl) carbaldehyde semicarbazone, 4b: 1-(1,2,3-Selenadiazole-4-yl) carbaldehyde. |
Materials and methods
Chemistry
The melting points (mps) were determined on an electrothermal digital melting point apparatus. The solvents were purified by standard procedures. Infrared (IR) spectra were recorded (in cm−1) using a Nicolet Impact 410 FTIR Spectrometer (Nicolet Instrument Corp, Madison, WI, USA). The IR spectra of pure substances were measured as KBr pellets. Proton nuclear magnetic resonance (1H-NMR) spectra were recorded on a 400 MHz Fourier transform nuclear magnetic resonance (FT-NMR) system (JNM-ECP400; JEOL, Ltd, Peabody, MA, USA) with a 300 MHz Bruker AC 200 spectrometer Bruker Corporation, Billerica, MA, USA). Tetramethylsilane was used as internal reference. The spectral data were reported in delta (δ) units relative to the tetramethylsilane reference line. Carbon-13 nuclear magnetic resonance (13C-NMR) spectra were recorded on the 100 MHz JEOL FT-NMR system, (75 MHz). Mass spectroscopy was carried out by using the Finnigan MAT95XP mass spectrometer (Thermo Fisher Scientific Inc, Waltham, MA, USA) (field desorption [FD]: 5 kV ionizing energy).
4-(1,2-Propenoxide-3-yl)acetophenone (2a)
A solution of (3.20 mmol) 4-(2-propenoxy)acetophenone(1a) in (12 mL) dry dichloromethane cooled in an ice bath to 0°C was added dropwise into a solution of (4.32 mmol) m-chloroperoxybenzoic acid in (12 mL dry dichloromethane) at 0°C under vigorous stirring. After complete addition, the whole solution was left stirring at room temperature for 6 hours and under reflux for further 3 hours. The precipitated m-chlorobenzoic acid was removed from solution by simple filtration. The filtrate was washed many times with 10% NaOH followed by distilled water and then dried over magnesium sulfate. After removing the solvent, through evaporation, to dryness, the residue was purified on silica gel column chromatography using ethanol/dichloromethane (1:30) as eluent. This compound was obtained as light brown viscous oil in 53% yield (2.1 g); 1H NMR (CDCl3): δ =2.27 (s, 3H, CH3), 2.61 (q, 3Jcis =4.3 Hz, 1H, CH2-epoxide), 2.92 (t, 3Jtrans =8.5 Hz, 1H, CH2-epoxide), 3.43 (m, 3Jcis =4.3 Hz, 1H, CH-epoxide), 3.95 (q, 3Jtrans =11.2 Hz, 1H, CH2O), 4.27 (q, 3Jcis =4.3 Hz, 1H, CH2O), 7.13 (d, 2H, CH-phenyl), 7.87 (d, 2H, CH-phenyl) ppm; 13C NMR (CDCl3): δ =25.9 (1C, CH3), 44.7 (1C, CH2-epoxide), 50.4 (1C, CH-epoxide), 69.3 (1C, CH2O), 115.9 (2C, CH-phenyl), 127.6 (1C, CC-phenyl), 128.9 (2C, CH-phenyl), 159.9 (1C, CO-phenyl), 195.9 (1C, CO) ppm; IR (KBr): ν =3,067, 2,889, 1,662, 1,460, 1,251, 1,221, 933 cm−1; MS: (5 kV, FD) m/z (%) 192 (100%). Analytically calculated For C11H12O3: C, 68.75; H, 6.25. Found: C, 68.79; H, 6.29.
4-(1,2-Propenoxide-3-yl)acetophenone(ethoxycarbonylhydrazone) (3a)
A solution of 4-(1,2-propenoxide-3-yl)acetophenone (2a) (2.50 mmol) and ethoxycarbonyl hydrazine (2.60 mmol) in dry chloroform (120 mL) was heated under reflux and under dean-stark for 13 hours. When no more water was generated, the solvent was removed using rotary evaporator. The residue was washed several times with diethyl ether, and then the remaining ether was concentrated using the rotatory evaporator. This compound was obtained as colorless solid in 96% yield, decomposed at 143°C. 1H NMR (CDCl3): δ =1.98 (t, 3H, CH3), 2.21 (s, 3H, CH3), 2.59 (q,3Jcis =4.2 Hz, 1H, CH2-epoxide), 2.92 (t, 3Jtrans =8.6 Hz, 1H, CH2-epoxide), 3.43 (m, 3Jcis =4.2 Hz, 1H, CH-epoxide), 3.95 (q, 3Jtrans =11.1 Hz, 1H, CH2O), 4.21 (q, 2H, OCH2), 4.27 (q, 3Jcis =4.2 Hz, 1H, CH2O), 7.13 (d, 2H, CH-phenyl), 7.87 (d, 2H, CH-phenyl), 8.86 (s, 1H, NH) ppm; 13C (CDCl3): δ =18.9 (1C, CH3), 25.9 (1C, CH3), 44.7 (1C, CH2-epoxide), 50.4 (1C, CH-epoxide), 60.3 (1C, OCH2), 69.4 (1C, CH2O), 115.6 (2C, CH-phenyl), 127.2 (1C, CC-phenyl), 128.5 (2C, CH-phenyl), 151.3 (1C, CN), 159.8 (1C, CO-phenyl), 195.7 (1C, CO) ppm; IR (KBr): ν =3,223, 3,041, 2,989, 1,608, 1,523, 1,463, 1,251, 1,241 cm−1; MS: (5 kV, FD) m/z (%) 250 (100%). Anal Calcd For C14H18O4: C, 67.25; H, 7.24. Found: C, 67.59; H, 7.28.
3-[4-(1,2,3-Thiadiazole-4-yl)phenoxy]-1,2-propenoxide (4a)19
The reaction of 4-(1,2-propenoxide-3-yl)acetophenone(etho-xycarbonylhydrazone) (3a) (5.20 mmol) with excess SOCl2 (121 mmol) was carried out in an ice bath under vigorous stirring for 8 hours. Then the solution was left to warm to room temperature for a further 3 hours. The excess SOCl2 was removed by distillation. The residue was washed several times with ether and concentrated. This compound was obtained as a colorless solid in 93% yield with mp 76°C–78°C; 1H NMR (CDCl3): δ =2.75 (q, 3Jcis =4.2 Hz, 1H, CH2-epoxide), 2.94 (t, 3Jtrans =8.6 Hz, 1H, CH2-epoxide), 3.35 (m, 3Jcis =4.2 Hz, 1H, CH-epoxide), 3.97 (q, 3Jtrans =11.1 Hz, 1H, CH2O), 4.27 (q, 3Jcis =4.2 Hz, 1H, CH2O), 7.03 (d, 2H, CH-phenyl), 7.92 (d, 2H, CH-phenyl), 8.53 (s, 1H, CHS) ppm.
Methylglyoxal bis(semicarbazone) (2b)
A solution of semicarbazidhydrochloride (1.0 g, 10.0 mmol) and sodium acetate (1.0 g, 12.0 mmol) in absolute ethanol was heated for 20 minutes under reflux. The product was filtered while hot to remove precipitated sodium chloride salt. Then one equivalent of methylglyoxal (0.35 g, 4.86 mmol) was added to the product solution. This mixture was refluxed for 35 minutes and followed by thin layer chromatography (TLC) (chloroform). When the reaction was completed, ethanol was removed and the residue was washed with diethyl ether. The colorless solid of this compound was obtained in 89% yield with mp132°C (decomposition); 1H NMR (dimethyl sulfoxide [DMSO]-d6): δ =1.46 (s, 3H, CH3), 6.35 (s, 4H, NH2), 8.83 (s, 2H, NH), 9.78 (s, 1H, NCH); 13C NMR (DMSO-d6): δ =18.7 (CH3), 147.9 (CH3C=N), 148.2 (HC=N), 156.8–157.3 (2C, CO); IR (KBr): ν =3,476, 3,148, 2,937, 2,863, 1,755, 1,682, 1,254 cm−1; MS: (5 kV, FD): m/z=186 (M+). Anal Calcd for C5H10N6O2: C 32.26, H 5.38, N 45.16. Found: C, 32.53; H, 5.60; N, 45.37.
1-(1,2,3-Selenadiazole-4-yl)carbaldehyde semicarbazone (3b)
Methylglyoxal bis(semicarbazone) (2b) (0.30 mmol) was mixed with selenium dioxide powder (0.60 mmol) and sodium sulfate (2.5 g) in a solution of 1,4-dioxane (15 mL) under vigorous stirring at room temperature. The solution was kept in dark, and the reaction progress was followed up with TLC that showed reaction completion in approximately 14 hours. After reducing the volume of the reaction mixture to half its original volume under vacuum, distilled water (10 mL) was added, and chloroform (3×15 mL) was used for extraction. The combined chloroform layers were dried using magnesium sulfate. After removing the solvent in vacuum, the product was obtained as a light brown solid in 76% yield with mp 126°C (decomposed); 1H NMR (DMSO-d6): δ =6.31 (s, 2H, NH2), 8.76 (s, 1H, NH), 8.94 (s, 1H, CHSe), 9.81 (s, 1H, NCH) ppm; 13C NMR (DMSO-d6): δ =148.5 (HC=N), 157.1 (1C, CO), 136.4 (1C, C-Se), 158.6 (1C, C=N) ppm; IR (KBr): ν =3,466, 3,134, 2,926, 2,848, 1,743, 1,659, 1,213, 941 cm−1. MS: (5 kV, FD) m/z (%) 218 (100%). Anal Calcd For C4H5N5OSe: C, 22.02; H, 2.29; N, 32.11; Se, 36.24. Found: C, 22.07; H, 2.35; N, 32.05.
1-(1,2,3-Selenadiazole-4-yl)carbaldehyde (4b)
1-(1,2,3-Selenadiazole-4-yl)carbaldehydesemicarbazone (3b) (0.36 mmol) was added to 1,4-dioxane (20 mL) under vigorous stirring. The solution was kept in dark, and the reaction progress was followed up with TLC that showed reaction completion in approximately 20 hours. Then, 1M H2SO4 (10 mL) was added and the solution was gently heated to 40°C for 5 hours. After reducing the volume of the reaction mixture to two-thirds of its original volume under vacuum, distilled water (30 mL) was added, and chloroform (3×50 mL) was used for extraction. The combined chloroform layers were dried using magnesium sulfate. The crude product was obtained after solvent removal under vacuum. This compound was obtained as dark brown solid in 85% yield with mp 112°C (decomposition); 1H NMR (CDCl3): δ =9.83 (s, 1H, CHO), 8.98 (s, 1H, CHSe) ppm; 13C NMR (CDCl3): δ =136.7.2 (1C, C-Se), 158.3 (1C, C=N), 192.3 (1C, CHO) ppm; IR (KBr): ν =3,057, 1,628, 1,476, 1,412, 1,263, 1,218, 959 cm−1; MS: (5 kV, FD) m/z (%) 161 (100%). Anal Calcd For C3H2N2OSe: C, 22.37; H, 1.24; N, 17.39; Se, 49.04. Found: C, 22.27; H, 1.75; N, 17.41.
1,3,5-Tris(1,2,3-thiadiazole-4-yl)benzene (4c)
This compound (Figure 3) was prepared according to the procedure described,20 and obtained as pale yellow powder in 93% yield with mp 227°C (decomposition); 1H NMR (DMSO-d6): δ =8.92 (s, 3H, CH-phenyl), 9.83 (s, 3H, CHS) ppm.
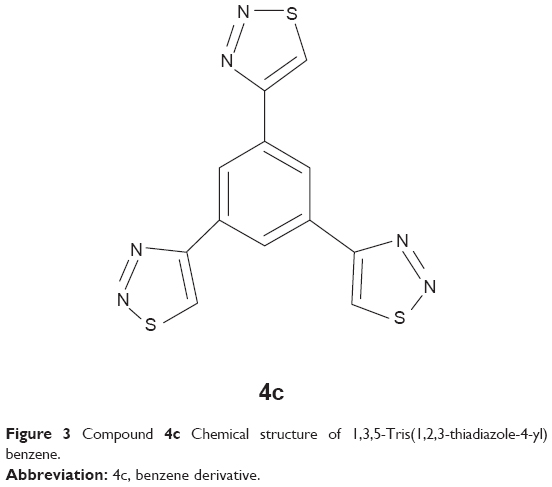 | Figure 3 Compound 4c Chemical structure of 1,3,5-Tris(1,2,3-thiadiazole-4-yl) benzene. |
Microbiology
All bacterial strains were obtained from American Type Culture Collection (ATCC) (Manassas, VA, USA). The filter paper disk and the well diffusion methods were applied to measure the inhibitory activity as indicated by the diameter of the inhibition zone.21,22 The well method was carried out as follows: a 100 μL of the heterocyclic compounds at each concentration were placed in 5 mm-diameter wells with nutrient agar that was inoculated spontaneously with the tested pathogens. The plates were incubated at 37°C for 48 hours. The clear zones around the wells were measured as inhibition zones. No clear zone around the well was indicated as a negative activity. The filter paper disk method was carried out as follows: Whatman® III filter paper puncture disks (5 mm) were saturated with different concentrations of the tested heterocyclic compounds. They were dried in an oven at 60°C for 12 hours. The dried disks were placed on already cultured nutrient agar plates with the different pathogens. The diameter of clear zone around the disks was measured after 48 hours of incubation at 37°C. No clear zone around the disks indicated negative activity. In both methods, dimethyl sulfoxide (DMSO) was used as a solvent to prepare the heterocyclic compound solutions.
Well diffusion assay
Antibacterial activity of the chemical compounds was tested against several gram-positive and gram-negative bacteria, and Candida by well diffusion method. The antibacterial susceptibility tests were performed on Muller–Hinton agar medium. Briefly, 20 mL agar medium was poured into the plates to obtain uniform depth. The standard inoculum suspensions (106 cfu/mL) were streaked over the surface of the media using a sterile cotton swab to ensure the confluent growth of the organism. Each compound was dissolved in 5% DMSO to obtain stock solution concentrations. Then, 5 mm-diameter wells were prepared in the medium and filled with 100 μL of each compound. Chloramphenicol (100 μg/mL) and cyclohexamide (100 μg/mL) were used as positive references against the bacteria and Candida, respectively, and 5% DMSO was used as negative control. Finally, the inoculated plates were incubated at 37°C for 24 hours, and the inhibition zones were observed, including the diameter of the well (5 mm). All the experiments were carried out in triplicate.
Determination of minimum inhibitory concentration (MIC)
The MIC values were determined for the bacterial strains that were sensitive to the compounds in the well diffusion assay. The MIC of the compounds was tested in Mueller–Hinton broth by broth macrodilution method.23 The inoculation of the bacterial strains was prepared from 4-hour-old broth cultures, and the suspensions were adjusted to standard turbidity (107 cfu/mL). The compounds were dissolved in 5% DMSO to obtain 20 mg/mL stock solution. The stock solution was diluted with Muller–Hinton broth to give different concentrations (12, 25, and, 50 μg/mL). Finally, bacteria were incorporated with the chemical compounds and Muller–Hinton broth to give a final concentration of 107 cfu/mL. The control tubes containing bacteria with chloramphenicol (30 μg/mL), cyclohexamide (10 μg/mL), and 5% DMSO were used as control. The culture tubes were incubated at 37°C for 24 hours. The lowest concentrations that did not show any growth of the test organism on the culture medium after macroscopic evaluation was determined as MIC.
Antitumor activity
The acute cytotoxic effects of the compounds on a panel of cancer cell lines, namely, breast (HMT3522, MCF7), colorectal (SW480, HCT116), and melanoma (MV3, C32) were determined using 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay.24 Briefly, cells were cultured in phenol red–free Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (Bio Whittaker, Verviers, Belgium) containing the antimicrobial agents amphotericin B and penicillin/streptomycin (PAA Laboratories, GmbH, Pasching, Austria). Cells were seeded at 5,000 per well onto flat-bottomed 96-well culture plates and allowed to grow for 24 hours before the desired treatment. Cells were then incubated with a wide range of the compounds concentrations (0–250 μg/mL) for 72 hours. The antimetabolite anticancer agent 5-fluorouracil (5-FU), at 50 μg/mL, was used as a control. Cells were then labeled with MTT (at a final concentration of 1.2 mM) from the Vybrant MTT Cell Proliferation Assay Kit (Molecular Probes®; Grand Island, NY, USA) according to the manufacturer’s instruction, and resulting formazan was solubilized with DMSO. Absorbance was read on a microplate reader (ELx800, Bio-tek instruments, plate reader, Highland Park, VT, USA) at 540 nm.
Statistical analysis
Data were analyzed using GraphPad PRISM version 5 (La Jolla, CA, USA). All data points were presented as (mean ± standard deviation) of three independent experiments. One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test were used for statistical analysis. P<0.05 was considered significant.
Results
1,2,3-Thiadiazole derivative 4a was prepared from the corresponding ketone 1a, as described previously19 but with a different methodology, as shown in Figure 1. The synthetic procedure started from ketone 1a that was prepared following a specific procedure.19 Ketone 1a was transformed into the corresponding epoxide derivative 2a through oxidation with m-chloroperbenzoic acid. The hydrazone derivative 3a was obtained from the condensation reaction between compound 2a and ethyl carbazate. The 1,2,3-thiadiazole derivative 4a was obtained from the reaction of the hydrazone 3a with thionyl chloride, as described by Li.25 1,2,3-Selenadiazole derivative 4b was prepared from the corresponding methylglyoxal 1b, which was transformed into the corresponding methylglyoxal bis(semicarbazone) (2b) through condensation with semicarbazide. The 1,2,3-selenadiazole derivative 3b was obtained from the reaction of the methylglyoxal bis(semicarbazone) (2b) with selenium dioxide in glacial acetic acid, as previously described26 and shown in Figure 2.
The antimicrobial activity of the three synthetic compounds was evaluated against gram-positive Enterococcus faecalis and Staphylococcus aureus, and gram-negative Salmonella typhi, E. coli, and P. aeruginosa, as well as against C. albicans, a fungal pathogen. The sensitivity test of the three compounds indicated their highly antagonistic activity against the selected microbes except P. aeruginosa. At 10 μg/mL concentration of each of the three compounds, the antimicrobial activity was higher than 5 μg/mL, as indicated in Table 1. At 10 μg/mL, compound 4B inhibited the growth of C. albicans, E. coli, E. faecalis, and S. typhi, with 25, 21, 16 and 12 mm inhibition zones, respectively; S. aureus was resistant to compound 4b. Compound 4c had inhibitory activity only against S. aureus. Compound 4a was active against C. albicans, and E. Coli with 11 and 16 mm inhibition zones, respectively. The antimicrobial activity of these compounds at 10 μg/mL was similar to antimicrobial activity of 5 μg/mL of chloramphenicol or 5 μg/mL of cyclohexamide and the antifungal reference compounds. These results proved the promise of these synthetic compounds as antimicrobial agents. The MIC of the three synthetic compounds against reference microbes varied from 0.625 to 6.25 μg/mL, as shown in Table 1 and Figure 4.
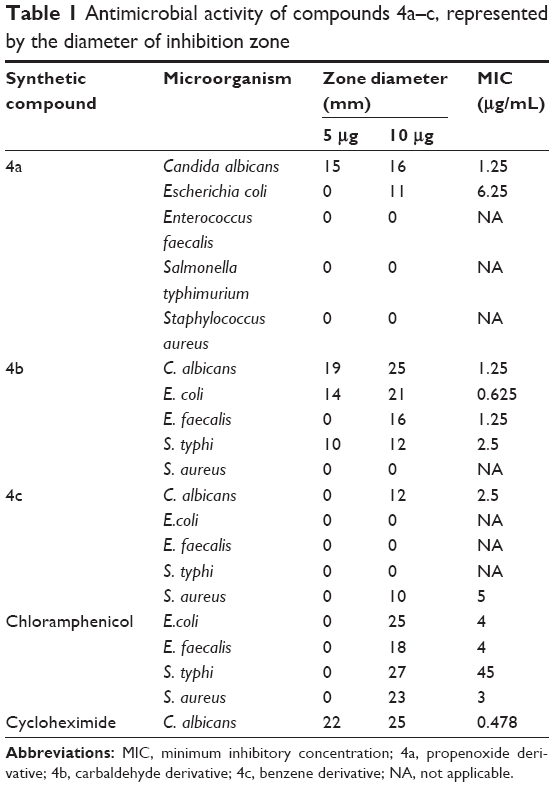 | Table 1 Antimicrobial activity of compounds 4a–c, represented by the diameter of inhibition zone |
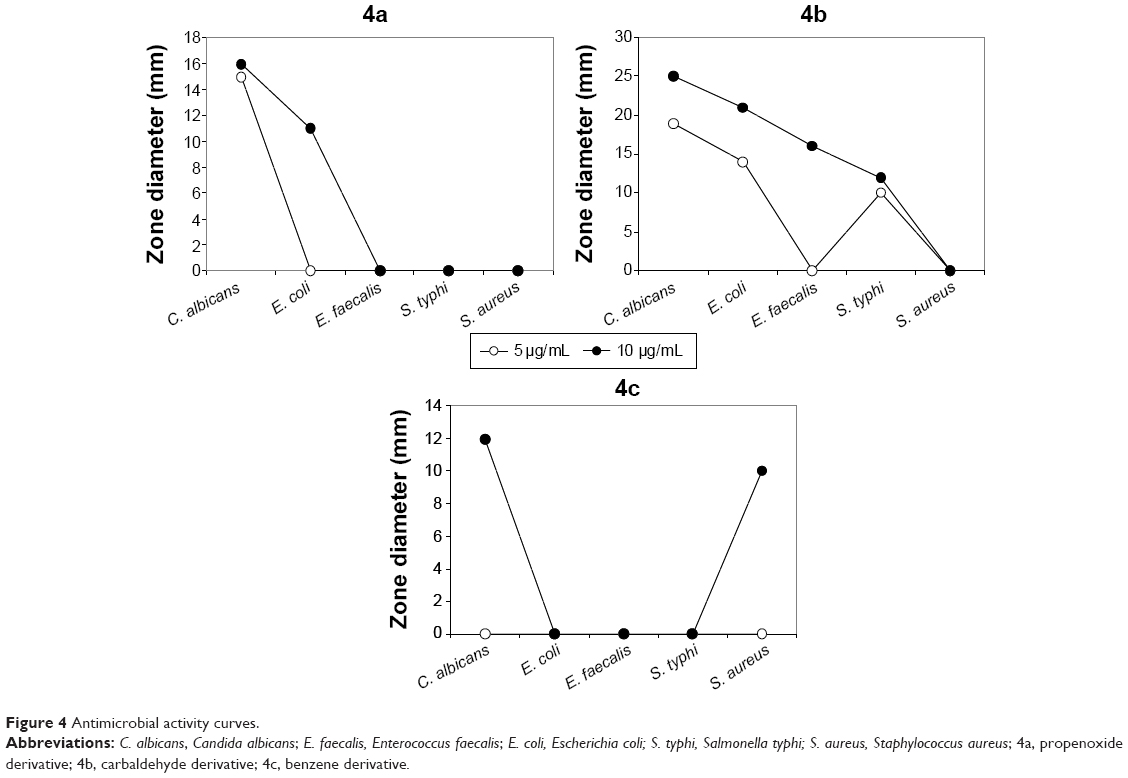 | Figure 4 Antimicrobial activity curves. |
The antitumor activity of the three synthetic compounds against SW480, HCT116, C32, MV3, HMT3522, and MCF-7 cell lines showed that compound 4b was active against all tumor cell lines, with variable half-maximal inhibitory concentration (IC50) values ranging from 52.17 to 114.79 μg/mL. Compound 4c was more potent than compound 4b against all tumor cell lines, as shown in Table 2. The IC50 of compound 4c varied from 31.91 to 528.10 μg/mL. These results showed that compound 4c had higher antitumor activity than did compound 4B. Compound 4a had no antitumor activity against any of the selected tumor cell lines. Comparing the IC50 of compound 4c with the reference antitumor compound 5-FU, the IC50 against SW480, HCT116, and MCF-7 was lower, indicating its higher potential to inhibit tumor cell proliferation. In addition, results showed that HMT3522 breast cancer cells were highly resistant to both compounds 4b and 4c. The rest of the tumor cell lines were more sensitive to compound 4c than to compound 4b.
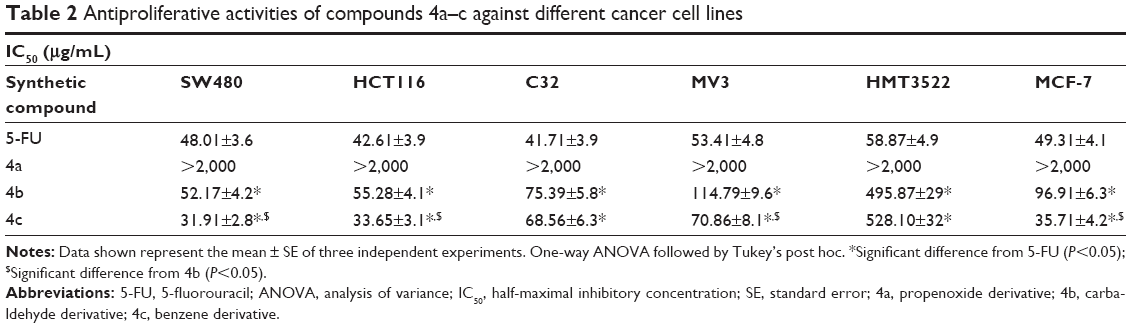 | Table 2 Antiproliferative activities of compounds 4a–c against different cancer cell lines |
Discussion
The results of the present study showed the antimicrobial and antitumor activities of the synthetic derivatives (4a, 4b, and 4c), with promising potential in comparison with the active reference chemicals. Variation in antimicrobial activity, as indicated by the diameters of the inhibition zones of the three compounds, could have been due to variation in the genetic makeup of the microbial reference strains used in this study, variation in compounds metabolic activity, or compounds with different resistance or different sensitivity. Compound 4b showed higher antimicrobial activity of over compound 4c but lower antitumor or cytotoxicity in comparison with compound 4c. This could have been due to their active sites on the living cells, especially the genetic makeup, or could have been due to the different molecular weight and different solubility in water and hence, differences in diffusion rate in the culture medium.
In summary, new derivatives of substituted 1,2,3-thia- or 1,2,3-selenadiazole (4a–c) were prepared and characterized. These compounds were evaluated for their antimicrobial and antitumor activity. Compounds 4a–c were found to be active against the yeast-like fungi C. albicans. Compound 4a was active against gram-negative E. coli, and compound 4c was very active against the gram-positive S. aureus. The antitumor activity of the three synthetic compounds against different tumor cell lines (SW480, HCT116, C32, MV3, HMT3522, and MCF-7) showed that both compounds 4b and 4c had an antitumor activity against all the tumor cell lines. Furthermore, the activity of compound 4c was greater than those of compound 4b and the reference antitumor 5-FU, against SW480, HCT116, and MCF-7 tumor cell lines.
Notably, investigations of the specific mechanism of cytoxicity of the tested compounds (4a–c) are beyond the scope of the current study, and it is certainly the subject of our future work. This study was carried out with the rationale of investigating the antibacterial and antitumor activities of synthesized 1,2,3-thia- and 1,2,3-selenadiazole derivatives. The results have showed various activities of the tested compounds, as described earlier.
Acknowledgments
We are grateful to the Deanship of Scientific Research of the Jordan University of Science and Technology, for financial support (grant number 226/2009).
Disclosure
The authors report no conflicts of interest in this work.
References
Lode HM, Stahlmann R, Kresken M. [Multiresistant pathogens – a challenge for clinicians]. Zentralbl Chir. 2013;138(5):549–553. German. | ||
Lynch JP, Clark NM, Zhanel GG. Evolution of antimicrobial resistance among Enterobacteriaceae (focus on extended spectrum β-lactamases and carbapenemases). Expert Opin Pharmacother. 2013;14(2):199–210. | ||
Nordmann P. Carbapenemase-producing Enterobacteriaceae: overview of a major public health challenge. Med Mal Infect. 2014;44(2):51–56. | ||
Liu Y, Luo Y, Li X, Zheng W, Chen T. Rational design of selenadiazole derivatives to antagonize hyperglycemia-induced drug resistance in cancer cells. Chem Asian J. 2015;10(3):642–652. | ||
Chen JR, Wong JB, Kuo PY, Yang DY. Synthesis and characterization of coumarin-based spiropyran photochromic colorants. Org Lett. 2008;10(21):4823–4826. | ||
Jalilian AR, Sattari S, Bineshmarvasti M, Daneshtalab M, Shafiee A. Synthesis and in vitro antifungal and cytotoxicity evaluation of substituted 4,5-dihydronaphtho[1,2-d][1,2,3]thia(or selena)diazoles. Farmaco. 2003;58(1):63–68. | ||
Kandeel M, El-Meligie S, Omar R, Roshdy S, Youssef K. Synthesis of certain 1,2,3-selenadiazole, 1,2,3-thiadiazole and 1,2-oxazoline derivatives of anticipated antibacterial activity. Zagazig J Pharm Sci. 1994;3:197–205. | ||
Uto Y. 1,2-Benzisoxazole compounds: a patent review (2009–2014). Expert Opin Ther Pat. 2015;25(6):643–662. | ||
Asif M. Chemical characteristics, synthetic methods, and biological potential of quinazoline and quinazolinone derivatives. Int J Med Chem. 2014;2014:395637. | ||
Hroch L, Aitken L, Benek O, et al. Benzothiazoles – scaffold of interest for CNS targeted drugs. Curr Med Chem. 2015;22(6):730–747. | ||
Castelino PA, Naik P, Dasappa JP, et al. Synthesis of novel thiadiazolotriazin-4-ones and study of their mosquito-larvicidal and antibacterial properties. Eur J Med Chem. 2014;84:194–199. | ||
Güzeldemirci NU, Satana D, Küçükbasmaci O. Synthesis, characterization, and antimicrobial evaluation of some new hydrazinecarbothioamide, 1,2,4-triazole and 1,3,4-thiadiazole derivatives. J Enzyme Inhib Med Chem. 2013;28(5):968–973. | ||
Penesyan A, Gillings M, Paulsen IT. Antibiotic discovery: combatting bacterial resistance in cells and in biofilm communities. Molecules. 2015;20(4):5286–5298. | ||
Attwood PV. Histidine kinases from bacteria to humans. Biochem Soc Trans. 2013;41(4):1023–1028. | ||
Salazar ME, Laub MT. Temporal and evolutionary dynamics of two-component signaling pathways. Curr Opin Microbiol. 2015;24:7–14. | ||
Al Momani F, Alkofahi AS, Mhaidat NM. Altholactone displays promising antimicrobial activity. Molecules. 2011;16(6):4560–4566. | ||
Al-Smadi M, Al-Momani F. Synthesis, characterization and antimicrobial activity of new 1,2,3-selenadiazoles. Molecules. 2008;13(11):2740–2749. | ||
Al-Smadi M, Ratrout S. New 1,2,3-selenadiazole and 1,2,3-thiadiazole derivatives. Molecules. 2004;9(11):957–967. | ||
Lafi Al-Smadi M, Hanold M, Kalbitz H, Meier H. 1,2,3-Thiadiazoles with unsaturated side chains; synthesis, polymerization and photocrosslinking. Synthesis. 2009;15:2539–2546. | ||
Al-Smadi M, Meier H. Multi-arm 1,2,3-thiadiazole systems. European J Org Chem. 2006;1997(11):2357–2361. | ||
Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Disk Susceptibility Tests; Approved Standard – Eleventh Edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2012. | ||
Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, Nineteenth Informational Supplement. Wayne, PA: Clinical and Laboratory Standards Institute; 2012. | ||
Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard – Ninth Edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2012. | ||
Mhaidat NM, Qandil AM, Al-Balas QA, et al. Methoxyphenylcipro induces antitumor activity in human cancer cells. Arch Pharm Res. 2013;36(8):1023–1028. | ||
LI JJ. Hurd-Mori 1,2,3-thiadiazole synthesis. In: Li J, editor. Name Reaction. Berlin: Springer-Verlag Berlin Heidelberg; 2006:312–313. | ||
Lalezari I, Shafiee A, Yazdany S. Selenium heterocycles. X. Synthesis and antibacterial activity of pyridyl-1,2,3-thiadiazoles and pyridyl-1,2,3-selenadiazoles. J Pharm Sci. 1974;63(4):628–629. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.