")
Back to Archived Journals » Research and Reports in Biochemistry » Volume 6
Synonymous codons influencing gene expression in organisms
Authors Mitra S, Ray SK, Banerjee R
Received 4 March 2016
Accepted for publication 8 July 2016
Published 13 December 2016 Volume 2016:6 Pages 57—65
DOI https://doi.org/10.2147/RRBC.S83483
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Nikolay Dokholyan
Sutanuka Mitra,1 Suvendra Kumar Ray,2 Rajat Banerjee1
1Department of Biotechnology, University of Calcutta, Kolkata, West Bengal, 2Department of Molecular Biology and Biotechnology, Tezpur University, Napaam, Tezpur, Assam, India
Abstract: Nowadays, it is beyond doubt that synonymous codons are not the same with respect to expression of a gene. In favor of this, ribosome profiling experiments in vivo and in vitro have suggested that ribosome occupancy time is not the same for different synonymous codons. Therefore, synonymous codons influence differently the speed of translation elongation, which guides further cotranslational folding kinetics of a protein. It is now realized that the position of each codon in a coding sequence is important. The effect of synonymous codons on protein structure is an exciting field of research nowadays. This review discusses the recent developments in this field.
Keywords: codon usage bias, synonymous codons, ribosome profiling, cotranslational protein folding, protein structure
Introduction
In the standard genetic code table, of the 64 triplets or codons, 61 codons correspond to the 20 amino acids. While Met and Trp are encoded by one codon each, the other 18 amino acids are encoded by two to six different codons. This is called codon degeneracy. Different codons that encode the same amino acid are known as synonymous codons. Even though synonymous codons encode the same amino acid, it has been shown for all organisms that the distribution of these codons in a genome is not random. Certain synonymous codons are preferred over other synonymous codons, leading to different frequencies of occurrence of synonymous codons within a genome. This phenomenon has been termed as codon usage bias.1,2
Codon usage bias is observed in genomes of all organisms including viruses.1–6 Hence, there has been a long-standing interest among biologists to understand the evolutionary significance of this phenomenon. During the 1980s and 1990s, comparative analysis among coding sequences at both intragenomic and intergenomic levels supported the role of both selection and mutation in codon usage bias phenomenon.7–17 In these periods, much attention was paid toward understanding the relation between codon usage bias and gene expression in organisms.8,9,15,18,19 Scientists were keen to find out the contributions from genome composition,11 gene expression,8,9 and transfer RNA (tRNA) gene copy number7,10,14 toward codon usage bias in a genome.
During the last decade, several articles have been published that suggests the impact of synonymous changes or single nucleotide polymorphisms (SNPs) in coding sequences on gene function and the structure of the coded protein.20,21 In fact, synonymous SNPs have been linked to different diseases in human, which has increased the research impact in this field.20 This is an unbelievable discovery, which has now changed our earlier perception regarding the physical location of synonymous codons along a gene sequence. There may be selection along the entire coding sequence for determining the synonymous codon and the position in a gene for proper expression of genes in organisms.22
Figure 1 outlines how a single nucleotide substitution in a coding sequence may influence gene function without affecting amino acid sequence of a protein. A codon is a sequence of three nucleotides in messenger RNA (mRNA) that are read simultaneously by the anticodon sequence of tRNA within a ribosome during translation. Apart from translation, the sequence of nucleotides in codons also has impact on the structure and stability of mRNA that is important for the expression level of a gene.23,24 Therefore, the impact of different synonymous codons at a specific locus on mRNA structure and stability may not be the same. Further, the efficiency of decoding of different synonymous codons by anticodons might not be the same by virtue of being different in their nucleotide sequences. Apart from this, the association rate of ternary complex formation between an anticodon, the A-site of ribosome, and the mRNA may be dissimilar for different synonymous codons.25 In addition, the impact of codon context during translation26 and the effect of certain sequences in mRNA on ribosome movement during translation27 are attributes of synonymous codons. Therefore, synonymous codons can influence gene expression at both the posttranscriptional and translational levels.
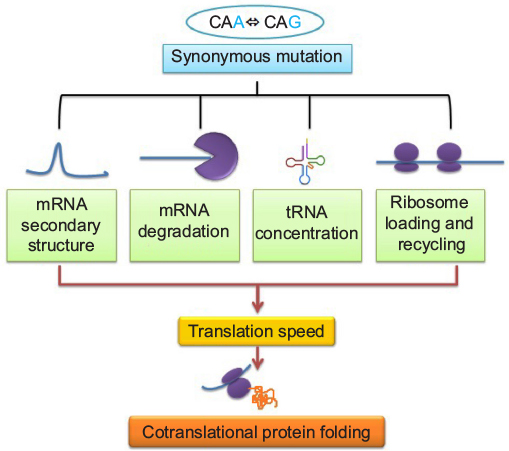 | Figure 1 Relation between synonymous mutation and cotranslational protein folding: its effects on various factors controlling translation speed. Abbreviations: mRNA, messenger RNA; tRNA, transfer RNA. |
There are several ways of removing defective mRNA to prevent formation of defective proteins containing aberrant sequences, and recent studies clearly indicated that removal of mRNA variants due to synonymous substitutions can also add an additional layer of quality control during cotranslation protein folding process. In eukaryotes, three major mechanisms of mRNA surveillance are operational, which are nonsense-mediated mRNA decay found in all eukaryotes that senses and degrades transcripts that contain premature stop codons,28,29 nonstop mRNA decay activation when the ribosome reaches 3′-end of the mRNA molecule without encountering a stop codon.30 Cotranslational quality control is also activated when ribosome stalled abnormally. This is known as no-go mRNA decay. Thus, mRNA structural features, such as stable secondary structure and slower translational elongation, contribute directly to a range of abnormalities resulting in low translation efficiency, mediated by codon usage.31
One of the recent studies demonstrated that variations in mRNA secondary structure surprisingly affect posttranslational modifications due to difference in translation speed. This is another level of protein regulation that is believed to be unrelated with the RNA-level regulation. Recently, it was shown for actins whose posttranslational arginylation is regulated by translation speed.32
The most intriguing effect of nonsynonymous substitution is probably alteration of target site for microRNA binding that may be implicated in disease development. One such example is synonymous polymorphism in the human IRGM gene that affects the binding site for miR-196 and leads to tissue-specific deregulation of the IRGM-dependent xenophagy that causes a predisposition to Crohn’s disease.33–35
Similarly, RNA splicing is an important component for maintenance of several essential cellular functions in eukaryotes. Zafrir and Tuller showed that pre-mRNA folding is weak in both intronic donor and acceptor sites, which correlates with splicing efficiency. Thus, it may be assumed that any synonymous change that promotes mRNA secondary structure around these regions would impact splicing and adversely affect the protein production.36
Several review articles have described the influence of synonymous codons on gene expression.34,37–40 In this review, we give an account of the recent evidences from ribosome profiling, in vivo translation, and heterologous expression experiments on the influence of synonymous codons on gene expression.
Folding energy landscape, cotranslational folding, and role of ribosome
In his famous in vitro experiment, Anfinsen41 (in 1973) observed the successful refolding of an unfolded ribonuclease A (RNase A). The experimental observation led him to propose that the three-dimensional structure of a protein is solely dependent upon its primary structure, which is nothing but the amino acid sequence of the protein. This theory as proposed by Anfinsen not only explained the puzzle behind protein structure but also suggested that synonymous codons in a gene sequence are redundant with respect to protein structure and function. Subsequent studies proposed many protein folding model such as “framework model”, “nucleation–condensation model”, and “hydrophobic collapse model”.
The more recent idea that gains popularity is “energy landscape theory” that describes the idea of folding funnels followed by a nascent polypeptide chain from its random coil to the globular form. According to this theory, a protein can fold via multiple routes going downhill rather than through a single pathway. The ground state of the folded protein is assumed to be composed of several degenerate states, and the energy of the protein is thought to be a function of the topological arrangement of the atoms. Thus, the protein sequences have been selected in evolution to assist folding by choosing residues that stabilize the final folded structure. This choice creates the funnel-shaped protein energy landscape and helps the proteins to fold on a biologically relevant timescale.42
The major question is how cotranslational folding of polypeptides on the ribosome modulates the free-energy landscape of folding. The relatively slower rate of translation (~4−20 amino acids s−1) makes the partially folded nascent chains aggregation prone. Furthermore, many nonnative interactions with the highly charged ribosomal surface also delay the folding process during vectorial synthesis of polypeptide chains allowing more time to partially folded states to aggregate. Under these circumstances, ribosome-bound chaperones are believed to interact cotranslationally with the nascent chains to avoid premature folding and misfolding and preserve the nascent chain in a nonaggregated, folding-competent state. The problem is acute for multidomain proteins that undergo domain-wise cotranslational folding and emerge sequentially from the ribosome. Thus, ribosome-bound chaperones protect them from nonnative interdomain contacts, hence smoothing the folding energy landscape for large proteins. Any synonymous substitutions that either slow down or speed up the cotranslational folding process due to differential ribosomal speed will presumably experience perturbed interactions between nascent aggregation-prone polypeptide with folding machinery. Thus, the overall concentration of misfolded proteins may increase within the cytosol triggering unfolded protein response and ubiquitination leading to protein degradation. Additionally, translation pausing at rare codon may fine-tune the cotranslational folding process that is highly optimized through evolution, ensuring proficient folding for the majority of newly synthesized proteins43,44
Synonymous changes are not redundant: synonymous changes influence protein structure and function
The observations of codon usage bias difference between the high-expression gene and low-expression genes in genomes were perceived as translational selection on synonymous codons for gene expression.45,46 Synonymous codons that were more abundant in the high-expression genes than the low-expression genes were called as preferred or optimal codons.47,48 Similarly, the reverse was named as nonoptimal codons. It was hypothesized that the decoding of these optimal codons are faster than the nonoptimal codons for which the former types are enriched in the high-expression genes.49,50 Therefore, during the 1980s and 1990s, much emphasis was given to the study of the relation between codon usage bias and gene expression. Thus, the research in these periods was mainly aimed to understand the optimal codons in organisms,51,52 to develop different measures to quantify codon usage bias,53,54 and also to predict gene expression from these values in different genomes.18,53,54 As more genome sequences were available in the beginning of the 21st century, later scientists could relate growth rate46,55 and lifestyle of organisms56 with genome-level selection on codon usage bias. Still, there was no clear experimental demonstration to give a mechanistic explanation of differential codon functions that were speculated for the high-expression genes.
During the 1990s, it was experimentally confirmed that many proteins fold during their synthesis,57,58 termed as cotranslational protein folding. The discovery of cotranslational protein folding and its relation with translational kinetics stimulated scientists to rethink the role of synonymous mutations on the functions of a protein, which were not investigated previously. In 2007, in a remarkable study, Kimchi-Sarfaty et al59 reported the impact of the synonymous SNP in the multidrug resistance 1 (MDR1) gene product P-glycoprotein with respect to altered drug and inhibitor concentrations. The authors clearly demonstrated that the impact of the synonymous SNP was due to the anomaly in the protein structure and not due to any abnormality of the mRNA expression and stability, which perceived to be the role of synonymous mutation impact on gene function. Unlike the earlier views regarding the role of synonymous mutation on gene function, in this review, the hypothesis was given that the synonymous change that resulted into a rare synonymous codon where the translation kinetic was slow is likely to have an impact of protein structure. As synthesis of the protein is slower due to the rare codon, it has likely influenced the cotranslational folding rate of the nascent protein, which in turn affected the proper folding of the mature protein. So the translation kinetics is important for protein folding, and the rate of translation is not the same for all synonymous codons (Figure 2).60,61 This discovery was a paradigm shift for the scientists in the way synonymous mutations were perceived in genes in terms of protein structure and function. Now, several different synonymous mutations of several genes are known to be related to human diseases that affect the protein structure and function.20,21
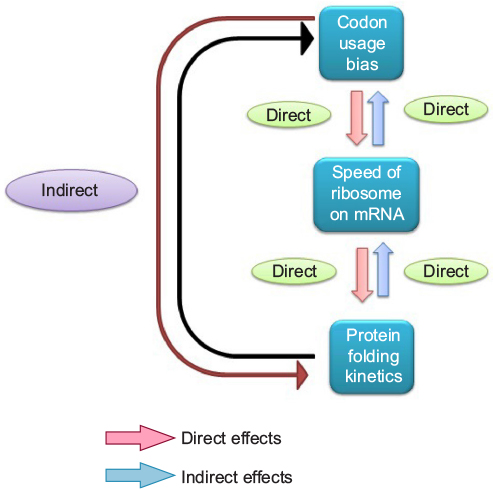 | Figure 2 Relation between codon usage bias, speed of ribosome on mRNA, and protein folding kinetics. Abbreviation: mRNA, messenger RNA. |
If synonymous mutations are not the same, heterologous expression of a gene with synonymous mutations is likely to produce proteins of variable structure and activities. Kudla et al62 did an experiment to express the same green fluorescent protein but with different synonymous sites in Escherichia coli. They constructed a synthetic library of 154 genes that varied randomly at synonymous sites but are the same with respect to their coded protein sequence. The amount of green fluorescent protein produced from different constructs in E. coli strains was variable. The authors concluded that the role of mRNA secondary structure near the Shine–Dalgarno sequence required for ribosome access to initiate translation is very important for gene expression. The authors were more biased to study the impact of synonymous codons on mRNA structure and, therefore, did not provide any information regarding the structure and folding of the green fluorescent protein in E. coli. In this context, Hu et al63 studied the heterologous expression of anti-IgE single chains (scFv) in E. coli. They constructed the synonymous codon library of the gene. In this study, it was demonstrated that scFv encoded by genes with different synonymous codons could be synthesized in E. coli with different solubility and antigen-binding ability.63 The result in this experiment clearly suggested that the variation in protein structure and function can be brought by varying the synonymous sites in the coding sequence. These experiments clearly demonstrated that synonymous changes are not redundant.
Discrepancy among synonymous codons with respect to translation speed: evidence from ribosome profiling experiments
How synonymous variation can influence protein structure and function? There are two mechanistic views regarding the difference among synonymous codons. One school of thoughts support that synonymous codons are different from each other with respect to the speed of decoding a codon. This view argues that the optimal codons are decoded faster than the nonoptimal codons.60,61 As high-expression genes are needed to be translated rapidly, these codons occur more frequently in these genes to favor the high abundance of these proteins. The other schools of thought support that synonymous codons are different with respect to accuracy of translation.64 Some codons are more prone to misincorporation of an amino acid, and these codons are avoided in the high-expression genes. So arguing from the point of accuracy, it can be said that synonymous codon-induced malfunction of a protein might be due to mistranslation-induced misfolding.65 Till date, no protein has been sequenced individually to find out if the malfunction of the protein is due to misincorporation of the amino acid or not. So both speed and accuracy can be argued to affect the protein structure due to synonymous change.
The following two experiments demonstrated that influence of synonymous codons is more due to difference in speed of translation rather than the accuracy. The argument is that if there will be difference in speed of translation, then both lower and higher speed of decoding will be important for protein folding depending upon its folding kinetics. In that case, changing optimal codon to nonoptimal codon or the vice versa will affect protein function since the rate of decoding of two synonymous codons is different. But in case of accuracy, the protein function will only be affected by changing optimal to nonoptimal but not the reverse. An elegant experiment was done in Neurospora crassa. In this fungus, frq gene controls the circadian clock function and exhibits nonoptimal codon usage. The changing of the nonoptimal codon to optimal codon in frq gene increased the expression level of Frq as expected but led to defects in folding of this protein and affected the circadian clock.66 So in this fungus, the nonoptimal codons have been selected in frq gene for proper structure and function of the protein. Similarly, the kaiB and kaiC genes in the cold-adapted cyanobacterium Synechococcus elongatus are also enriched in nonoptimal codons. In this organism, kaiB and kaiC genes are critical for regulation of circadian rhythms, and codon optimization increased the protein levels of KaiB and KaiC but disturbed the circadian rhythm in the cyanobacterium.67 These two experiments supported the notion that synonymous codons are indeed different with regard to the speed of translation elongation.
It is equally important to demonstrate that the speed of translation for optimal codons is faster than the nonoptimal codon in an organism. Very recently, Yu et al68 conducted an in vitro translation experiment, which demonstrated that optimal codons are indeed translated faster than nonoptimal codons. They studied translation kinetics of fire fly luciferase gene (luc gene) in the cell-free extract of N. crassa and of yeast. The luc gene constructs they used were the wild type from fire fly, optimized with synonymous optimal codons of N. crassa and de-optimized with nonoptimal synonymous codons of N. crassa. When translation was done in N. crassa cell-free translation system, the protein folded to produce the fluorescence earliest in case of the optimized construct. This indicated that the completion of the translation was fastest in case of optimized construct. The fluorescence was observed to be slowest in case of the de-optimized construct, indicating that the translation was the slowest. The observed difference in fluorescence timing between the different constructs was not due to any influence of mRNA secondary structure, which was eliminated by performing translation after substituting first ten codons of optimal mRNA construct with wild-type sequence and the same difference in time of fluorescence appearance was observed. The difference in the timing of fluorescence between the different gene constructs was not due to any influence of mRNA secondary structure because the possibility of formation of mRNA secondary structures was ruled out by doing translation using additional constructs from different regions of the gene, namely, N-terminal (2–223) and middle region (223–423). The sooner appearance of fluorescence signal for optimized codon constructs than the WT construct undoubtedly indicated that the difference in elongation rate is due to cumulative codon usage, not due to change in mRNA structure. Yu et al68 also did 35S labeling experiment to confirm that optimal codons are translated faster than the nonoptimal codons.
Mechanistically, faster rate of translation in case of optimal codons suggests that decoding of these codons occurs faster in translation inside ribosome than that of nonoptimal codons. Subsequently, the ribosome retention time during translation of the optimal codons will be shorter than that of the nonoptimal codons. Ribosome profiling is an elegant technique to map ribosome-bound mRNA in a cell.69,70 This is an emerging technique to study dynamics of ribosome on mRNA during translation. The technique uses deep sequencing of that part of mRNA that is protected by the ribosome. The underlying principle is that a translating ribosome protects 28–30 nucleotides of the transcript during translation from nuclease digestion by shielding this region. These protected fragments or “footprints” indicate the exact position of the ribosomes on the mRNA. A footprint is roughly centered on the A-site of the ribosome. Now, as said earlier, optimal codons are assumed to be translated faster than nonoptimal ones due to abundance of its cognate tRNAs. Therefore, ribosome should take a longer time on encountering a nonoptimal codon at the A-site. Hence, footprints generated from ribosomes at nonoptimal codons should be greater in number than those generated from ribosomes at optimal codons.69,70
In order to probe this phenomenon, in vivo translational speeds for all sense codons from Saccharomyces cerevisiae were analyzed70 using genome-wide ribosome profiling data. Similar translational speeds among synonymous codons were found, suggesting that preferentially used codons in highly expressed proteins are not translated faster than nonpreferred ones. The finding in this study did not support the notion that ribosome occupancy is more in the nonoptimal codon than optimal codons. It is pertinent to note that there are several limitations of the ribosome profiling experiment of which noise is the most important.
Yu et al68 further did in vivo and in vitro experiments exploiting ribosome profiling, and their data clearly showed higher ribosome occupancy around the nonoptimal codons and lower ribosome occupancy around the optimal codons. This experiment unequivocally proved that the speed of translation elongation due to optimal codons is higher than that due to nonoptimal codons. In this pioneering study, Yu et al also brought a new revolution in ribosome profiling experiment: profiling of reporters in translationally active N. crassa and S. cerevisiae (yeast) lysates.68,71 The above-mentioned study thus clearly showed that the choice of codon has a profound effect on both protein expression and folding, and the genetic code may influence more processes in the cell beyond translation.71
Translation kinetics guides cotranslational protein folding
If synonymous codons differentially influence the translational kinetics, which further guides cotranslational protein folding, it may be possible that two coding sequences different in synonymous sites may produce proteins that are structurally distinct. Though all the previous experiments were suggestive of this, experimental evidences demonstrating variation in cotranslation folding due to translation kinetics in a time-resolved manner were lacking. Very recently, Buhr et al72 demonstrated how synonymous codons guide the real kinetics of translation and cotranslational folding. They analyzed the in vivo expression of bovine eye lens protein gamma B crystallin in E. coli cells and the in vitro translation system of E. coli. They monitored the kinetics of synthesis and cotranslational folding of the protein in real time by fluorescence and Foster resonance energy transfer. Buhr et al72 used an alternative optimization strategy, where the expression of original unoptimized Bos taurus sequence (here, U) was compared with that of a “harmonized” variant (here, H). Harmonization is optimizing translation by using those synonymous codons that have the most similar usage frequencies in the native and the host organisms.72,73 Harmonization of gamma B crystallin resulted in codon usage profile more similar to native B. taurus than the original unoptimized B. taurus sequence as well as increase in the number of optimal codons in E. coli. In E. coli, expression of the H variant yielded ~1.5–1.6 times more full-length protein than expression of the U variant, despite identical mRNA levels. Expression of the H variant also yielded more soluble protein and fewer truncated peptides compared to expression of the U variant. Western blotting followed by mass spectrometry analysis established the fact that the products of the two variants have different degradation patterns, thereby suggesting adoption of different conformations. Most notably, the amino acid sequences were found to be identical. Gamma B crystallin contains seven cysteines, six of which are located in the N-terminal domain (NTD). They also found that the two variants differ with respect to the oxidation state of cysteine residues in the NTD. The oxidation state of at least two cysteine residues within the NTD is a major cause of overall structural changes. It is often observed that when a protein is expressed in a heterologous system, it either folds differently forming insoluble aggregates or in few occasions even if they are soluble,74 the underlying mechanism was not demonstrated till Buhr et al proved it in this study. Another exciting finding of this study is that from the same mRNA sequence, proteins of different conformations are formed.
Then they used BOF and BOP (Bodipy FL and Bodipy 567/589, respectively) to assay translation kinetics as well as folding of the nascent polypeptide chains using E. coli translation system in vitro. They observed that the protein from the H-mRNA was translated faster as well as folded faster (43 seconds) than that of the U-mRNA (57 seconds). Also, more translational pauses were found to occur in case of synthesis of NTD of U variant as compared to the H variant, thereby indicating abundance of rare codons in the corresponding region of U-mRNA. These findings were further supported from the protease digestion of the proteins. To summarize, this experiment proved that codon variation may alter the translation kinetics, thereby changing the folding kinetics, conformation, and stability of a certain protein (Figure 3).
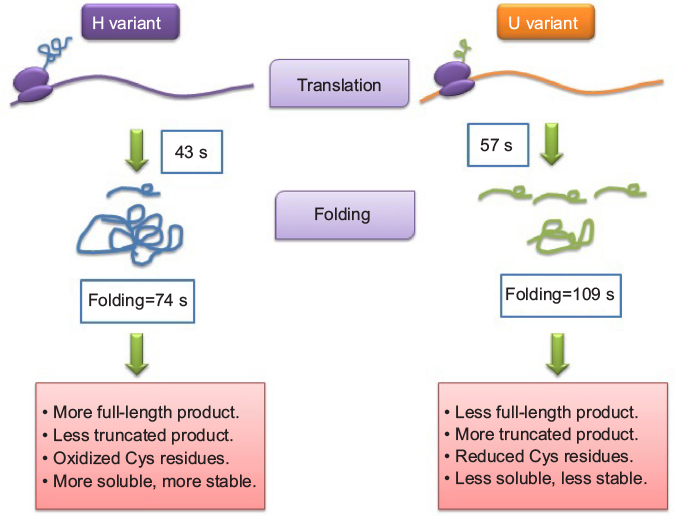 | Figure 3 A simplified model based on recent experimental data. Notes: Adapted from Molecular Cell, Vol 61/edition 3, Buhr F, Jha S, Thommen M, et al, Synonymous codons direct cotranslational folding toward different protein conformations, Pages 341–351, Copyright 2016, with permission from Elsevier.72 Abbreviations: Cys, cysteine; s, seconds. |
In silico studies have been performed in support of cotranslational protein folding and synonymous codon usage. There is evidence of computational analysis that fast translation speed can increase the probability of cotranslational protein folding.75–77 It is now important to consider the coding sequence while performing protein folding in silico. In this regard, the recently developed CSandS database is worth to consider while designing protein three-dimensional structure.78
Conclusion
When the degeneracy in the genetic code table was discovered, several scientists were thought of redundancy in the table among the synonymous codons. However, the differential influence of synonymous codons on protein structure and function has revealed that synonymous codons are not the same in all aspects. Therefore, the genetic code table is no more believed to be redundant. The long-standing Anfinsen’s theory on protein three-dimensional structure has now been challenged. In near future, it will be interesting to explore if codon degeneracy and evolution of a protein structure are related.
The discovery of cotranslational protein folding and its linkage with codon usage in coding sequences has now suggested a few cautious notes for scientists studying protein folding and heterologous gene expression: 1) it is now a challenge for computational biologist or protein modeler who tries to fold protein taking the whole protein into account and do not consider the rate of the protein synthesis. Consideration of the coding sequences while performing protein folding will be important in in silico studies; 2) it is now proved that in vitro protein folding experiments may not mimic the in vivo condition. The most fundamental is that in the former, the whole protein is allowed to refold after unfolding, while inside the cell, folding is occurring along the synthesis; 3) homologous proteins in a genome with nonidentical coding sequences may not be redundant. There may be a possibility that both may carry the same function with different efficiencies or may carry different functions in extreme conditions; 4) if protein folding is dependent upon the rate at which it is translated and not on the amino acid sequence of the protein, once it is not folded well, it is difficult to fold it back. Posttranslational folding to its natural conformation will not be easy; 5) heterologous expression to produce recombinant proteins will not be easy. Rate of elongation is dependent upon different physical and chemical factors such as temperature and tRNA availability. Any change in these factors is likely to affect cotranslational fielding.
By the discovery of synonymous codon influencing cotranslational folding, the field of protein folding has got new impetus. It is now a challenge that lies in front of evolutionary biologists to find out which protein structures are influenced and which protein structures are not influenced by synonymous codons, which might link with synonymous diversity of a gene.
Finally, it may be noted that differential usage of synonymous mRNA variants due to variable speed of decoding on ribosome may be a sign of a “code” within the genetic code for guiding accurate cotranslational protein folding and subsequently maintenance of cellular functions.71
Acknowledgments
RB and SKR are grateful to the Department of Biotechnology, India, for funding our collaborative project under DBT-NE Twinning Project (DBT-Twinning NER Project No BT/511/NE/TBP/2013). We are also grateful to two anonymous reviewers for their valuable comments and suggestions.
Disclosure
The authors report no conflicts of interest in this work.
References
Grantham R, Gautier C, Gouy M, Mercier R, Pave A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980;8(1):r49–r62. | ||
Ermolaeva MD. Synonymous codon usage in bacteria. Curr Issues Mol Biol. 2001;3(4):91–97. | ||
Baruah VJ, Satapathy SS, Powdel BR, Konwarh R, Buragohain AK, Ray SK. Comparative analysis of codon usage bias in Crenarchaea and Euryarchaea genome reveals differential preference of synonymous codons to encode highly expressed ribosomal and RNA polymerase proteins. J Genet. (2016). doi:10.1007/s12041-016-0667-5. | ||
Satapathy SS, Ray SK, Sahoo AK, Begum T, Ghosh TC. Codon usage bias is not significantly different between the high and the low expression genes in human. Int J Mol Genet Gene Ther. 2015;1(1):1–6. | ||
Belalov IS, Lukashev AN. Causes and implications of codon usage bias in RNA viruses. PLoS One. 2013;8(2):e56642. | ||
Subramanian S. Nearly neutrality and the evolution of codon usage bias in eukaryotic genomes. Genetics. 2008;178(4):2429–2432. | ||
Ikemura T. Correlation between the abundance of yeast tRNAs and the occurrence of the respective codons in its protein genes. J Mol Biol. 1982;158(4):573–597. | ||
Bennetzen JL, Hall BD. Codon selection in yeast. J Biol Chem. 1982;257(6):3026–3031. | ||
Gouy M, Gautier C. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 1982;10(22):7055–7074. | ||
Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 1985;2(1):13–34. | ||
Muto A, Osawa S. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc Natl Acad Sci U S A. 1987;84(1):166–169. | ||
Marfn A, Bertranpetit’ J, Oliver JL, Medina JR. Variation in G+C-content and codon choice: differences among synonymous codon groups in vertebrate genes. Nucleic Acids Res. 1989;17(15):6181–6189. | ||
Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129(3):897–907. | ||
Dong H, Nilsson L, Kurland CG. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J Mol Biol. 1996;260(5):649–663. | ||
Duret L, Mouchiroud D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc Natl Acad Sci USA. 1999;96(8):4482–4487. | ||
McInerney JO. Replicational and transcriptional selection on codon usage in Borrelia burgdorferi. Proc Natl Acad Sci USA. 1998;95:10698–10703. | ||
Morton BR. Strand asymmetry and codon usage bias in the chloroplast genome of Euglena gracilis. Proc Natl Acad Sci USA. 1999;96(9):5123–5128. | ||
Sharp PM, Li WH. The codon adaptation index- a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15(3):1281–1295. | ||
Sharp PM, Li WH. The rate of synonymous substitution in enterobacterial genes is inversely related to codon usage bias. Mol Biol Evol. 1987;4(3):222–230. | ||
Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 2011;12(10):683–691. | ||
Chaney JL, Clark PL. Roles for synonymous codon usage in protein biogenesis. Annu Rev Biophys. 2015;44:143–166. | ||
Ray SK, Baruah VJ, Satapathy SS, Banerjee R. Cotranslational protein folding reveals the selective use of synonymous codons along the coding sequence of a low expression gene. J Genet. 2014;93(3):613–617. | ||
Park C, Chen X, Yang J-R, Zhang J. Differential requirements for mRNA folding partially explain why highly expressed proteins evolve slowly. Proc Natl Acad Sci USA. 2013;110(8):E678–E686. | ||
Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505(7485):701–705. | ||
Varenne S, Buc J, Lloubes R, Lazdunski C. Translation is a non-uniform process. Effect of tRNA availability on the rate of elongation of nascent polypeptide chains. J Mol Biol. 1984;180(3):549–576. | ||
Chevance FF, Le Guyon S, Hughes KT. The effects of codon context on in vivo translation speed. PLoS Genet. 2014;10(6):e1004392. | ||
Li G-W, Oh E, Weissman JS. The anti Shine–Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. | ||
Gatfield D, Unterholzner L, Ciccarelli FD, Bork P, Izaurralde E. Nonsense-mediated mRNA decay in Drosophila: at the intersection of the yeast and mammalian pathways. EMBO J. 2003;22(15):3960–3970. | ||
Hwang J, Maquat LE. Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: to die or not to die, that is the question. Curr Opin Genet Dev. 2011;21(4):422–430. | ||
Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. | ||
Zhang Z, Zhou L, Hu L, et al. Nonsense-mediated decay targets have multiple sequence-related features that can inhibit translation. Mol Syst Biol. 2010;6:442. | ||
Zhang F, Saha S, Shabalina SA, Kashina A. Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science. 2010;329(5998):1534–1537. | ||
Brest P, Lapaquette P, Souidi M, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43(3):242–245. | ||
Shabalina SA, Spiridonov NA, Kashina A. Sounds of silence: synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res. 2013;41(4):2073–2094. | ||
Venetianer P. Are synonymous codons indeed synonymous? Biomol Concepts. 2012;3(1):21–28. | ||
Zafrir Z, Tuller T. Nucleotide sequence composition adjacent to intronic splice sites improves splicing efficiency via its effect on pre-mRNA local folding in fungi. RNA. 2015;21(10):1704–1718. | ||
Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011;12(1):32–42. | ||
Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. 2008;42:287–299. | ||
Bali V, Bebok Z. Decoding mechanisms by which silent codon changes influence protein biogenesis and function. Int J Biochem Cell Biol. 2015;64:58–74. | ||
López D, Pazos F. Protein functional features are reflected in the patterns of mRNA translation speed. BMC Genomics. 2015;16:513. | ||
Anfinsen CB. The formation and stabilization of protein structure. Biochem J. 1972;128(4):737–749. | ||
Giri Rao VH, Gosavi S. Using the folding landscapes of proteins to understand protein function. Curr Opin Struct Biol. 2016;36:67–74. | ||
Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475(7356):324–332. | ||
Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16(6):574–581. | ||
Sharp PM, Emery LR, Zeng K. Forces that influence the evolution of codon bias. Philos Trans R Soc Lond B Biol Sci. 2010;365(1544):1203–1212. | ||
Sharp PM, Bailes E, Grocock RJ, Peden JF, Sockett RE. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 2005;33(4):1141–1153. | ||
Satapathy SS, Dutta M, Buragohain AK, Ray SK. Transfer RNA gene numbers may not be completely responsible for the codon usage bias in asparagine, isoleucine, phenylalanine, and tyrosine in the high expression genes in bacteria. J Mol Evol. 2012;75(3–4):151–153. | ||
Satapathy SS, Powdel BR, Dutta M, Buragohain AK, Ray SK. Selection on GGU and CGU codons in the high expression genes in bacteria. J Mol Evol. 2014;78(1):13–23. | ||
Higgs PG, Ran W. Coevolution of codon usage and tRNA genes leads to alternative stable states of biased codon usage. Mol Biol Evol. 2008;25(11):2279–2291. | ||
Ran W, Higgs PG. The influence of anticodon-codon interactions and modified bases on codon usage bias in bacteria. Mol Biol Evol. 2010;27(9):2129–2140. | ||
Hershberg R, Petrov DA. General rules for optimal codon choice. PLoS Genet. 2009;5(7):e1000556. | ||
Wang B, Shao Z-Q, Xu Y, et al. Optimal codon identities in bacteria: implications from the conflicting results of two different methods. PLoS One. 2011;6(7):e22714. | ||
Wrigh F. The ‘effective number of codons’ used in a gene. Gene. 1990;87(1):23–29. | ||
Novembre JA. Accounting for background nucleotide composition when measuring codon usage bias. Mol Biol Evol. 2002;19(8):1390–1394. | ||
Rocha EP. Codon usage bias from tRNA’s point of view: redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 2004;14(11):2279–2286. | ||
Wald N, Alroy M, Botzman M, Margalit H. Codon usage bias in prokaryotic pyrimidine-ending codons is associated with the degeneracy of the encoded amino acids. Nucleic Acids Res. 2012;40(15):7074–7083. | ||
Komar AA. A pause for thought along the co-translational folding pathway. Trends Biochem Sci. 2009;34(1):16–24. | ||
Komar AA, Lesnik T, Reiss C. Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett. 1999;462(3):387–391. | ||
Kimchi-Sarfaty C, Oh JM, Kim IW, et al. A ‘silent’ polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315(5811):525–528. | ||
Sørensen M, Kurland CG, Pedersen S. Codon usage determines translation rate in Escherichia coli. J Mol Biol. 1989;207(2):365–377. | ||
Sørensen MA, Pedersen S. Absolute in vivo translation rates of individual codons in Escherichia coli. The two glutamic acid codons GAA and GAG are translated with a threefold difference in rate. J Mol Biol. 1991;222(2):265–280. | ||
Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324(5924):255–258. | ||
Hu S, Wang M, Cai G, He M. Genetic code-guided protein synthesis and folding in Escherichia coli. J Biol Chem. 2013;288(43):30855–30861. | ||
Akashi H. Synonymous codon usage in Drosophila melanogaster: natural selection and translational accuracy. Genetics. 1994;136(3):927–935. | ||
Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134(2):341–352. | ||
Zhou M, Guo J, Cha J, et al. Non-optimal codon usage affects expression, structure and function of FRQ clock protein. Nature. 2013;495(7439):111–115. | ||
Xu Y, Ma PJ, Shah P, Rokas A, Liu Y, Johnson CH. Non-optimal codon usage is a mechanism to achieve circadian clock conditionality. Nature. 2013;495(7439):116–120. | ||
Yu CH, Dang Y, Zhou Z, et al. Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol Cell. 2015;59(5):744–754. | ||
Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet. 2014;15(3):205–213. | ||
Qian W, Yang JR, Pearson NM, Maclean C, Zhang J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet. 2012;8(3):e1002603. | ||
Koutmou KS, Radhakrishnan A, Green R. Synthesis at the speed of codons. Trends Biochem Sci. 2015;40(12):717–718. | ||
Buhr F, Jha S, Thommen M, et al. Synonymous codons direct cotranslational folding toward different protein conformations. Mol Cell. 2016;61(3):341–351. | ||
Angov E, Hillier CJ, Kincaid RL, Lyon JA. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS One. 2008;3(5):e2189. | ||
de Marco A, Vigh L, Diamant S, Goloubinoff P. Native folding of aggregation-prone recombinant proteins in Escherichia coli by osmolytes, plasmid- or benzyl alcohol-overexpressed molecular chaperones. Cell Stress Chaperones. 2005;10(4):329–339. | ||
Wang E, Wang J, Chen C, Xiao Y. Computational evidence that fast translation seed can increase the probability of cotranslational protein folding. Sci Rep. 2015;5:15316. | ||
Nissley DA, Sharma AK, Ahmed N, et al. Accurate prediction of cellular co-translational folding indicates proteins can switch from post- to co-translational folding. Nature Commun. 2015;7:10341. | ||
Sharma AK, Bukau B, O’Brien EP. Physical origins of codon positions that strongly influence co-translational folding: a framework for controlling nascent protein folding. J Am Chem Soc. 2015;138(4):1180–1195. | ||
Saunders R, Deane CM. Synonymous codon usage influences the local protein structure observed. Nucleic Acids Res. 2010;38(19):6719–6728. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.