")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Structure-based virtual screening and characterization of a novel IL-6 antagonistic compound from synthetic compound database
Authors Wang J, Qiao C, Xiao H, Lin Z, Li Y, Zhang J, Shen B, Fu T, Feng J
Received 29 July 2016
Accepted for publication 8 November 2016
Published 15 December 2016 Volume 2016:10 Pages 4091—4100
DOI https://doi.org/10.2147/DDDT.S118457
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Manfred Ogris
Jing Wang,1,* Chunxia Qiao,1,* He Xiao,1 Zhou Lin,1 Yan Li,1 Jiyan Zhang,1 Beifen Shen,1 Tinghuan Fu,2 Jiannan Feng1
1Department of Molecular Immunology, Beijing Institute of Basic Medical Sciences, 2First Affiliated Hospital of PLA General Hospital, Beijing, People’s Republic of China
*These authors contributed equally to this work
Abstract: According to the three-dimensional (3D) complex structure of (hIL-6·hIL-6R·gp 130)2 and the binding orientation of hIL-6, three compounds with high affinity to hIL-6R and bioactivity to block hIL-6 in vitro were screened theoretically from the chemical databases, including 3D-Available Chemicals Directory (ACD) and MDL Drug Data Report (MDDR), by means of the computer-guided virtual screening method. Using distance geometry, molecular modeling and molecular dynamics trajectory analysis methods, the binding mode and binding energy of the three compounds were evaluated theoretically. Enzyme-linked immunosorbent assay analysis demonstrated that all the three compounds could block IL-6 binding to IL-6R specifically. However, only compound 1 could effectively antagonize the function of hIL-6 and inhibit the proliferation of XG-7 cells in a dose-dependent manner, whereas it showed no cytotoxicity to SP2/0 or L929 cells. These data demonstrated that the compound 1 could be a promising candidate of hIL-6 antagonist.
Keywords: virtual screening, structural optimization, human interlukin-6, small molecular antagonist, XG-7 cells, apoptosis
Introduction
IL-6 is a pleiotropic cytokine involved in the regulation of a multitude of cellular functions, including cell proliferation, apoptosis, and differentiation.1 In addition, it plays a role in the modulation of immune responses, hematogenesis, acute immune reaction, etc.2–4 IL-6 can be expressed by various kinds of cells, such as monocytes, lymphocytes, mechanocyte, and marrow stroma cell (MSC). Abnormal expression of IL-6 or its receptor IL-6R correlates closely with cancer, inflammation diseases or autoimmune diseases such as multiple myeloma (MM), Castleman disease, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and hypercalcemia.5–9
hIL-6 was discovered in 1980s. It belongs to cytokine superfamily and is composed of 184 amino acids with two disulfide bonds (Cys44–Cys50 and Cys73–Cys83).10 X-ray crystal diffraction showed that IL-6 contained four alpha helices (helices A, B, C, and D), which were linked with loops. The receptor-binding domain was located at the C-terminus (175–181),11 in which Arg179 was the key residue.12 AB loop and helices A and D were important in receptor binding and signal transduction.13–18
hIL-6R is composed of 468 amino acids, including 19 residues of signal peptide, 339 residues of extracellular domain, 28 residues of transmembrane sequence and 82 residues of intracellular domain. The extracellular domain of IL-6R consists of three domains: D1 (1–93), D2 (94–149), and D3 (195–299). D1 on the N-terminus belongs to Ig superfamily, which is composed of irregular β-sheet. It influences not only the ligand identification and signal transduction but also the stability of protein.19 D2 and D3 are the cytokine-binding domains (CBDs). D2 has four conserved Cys residues and redundant prolines, meanwhile D3 contains a “Tyr–Arg” ladder, which plays a key role in stabilizing the structure of D3.20 Furthermore, this ladder contains a conserved “WSXWS” motif (284–288) at the C-terminus of D3. Three-dimensional (3D) crystal structure of hIL-6R showed that the extracellular domain has eight antiparallel β-sheet at the N-terminus, four antiparallel β-sheet and one α-helix at the C-terminus.21,22
gp130 (CD130) belongs to hematopoietic factor superfamily, which functions as a signal transducer in various pathways, including hIL-6.23 It can also be activated in response to IL-6-related cytokines, such as LIF and IL-11. It is a glycoprotein with a molecular weight of 130 kDa, which also contains a extracellular domain (597 amino acids), a transmembrane domain (22 amino acids) and a intracellular domain (277 amino acids). The extracellular domain contains an Ig-like domain and six type III fibronectin structure, in which a CBD is conformed with four conserved Cys residues and a WSXWS motif between the second and the third fibronectin.21,22,24 IL-6 signals through membrane receptor that is composed of the ligand-binding subunit and the signal transduction subunit gp130. IL-6 receptors are expressed in a variety of benign or malignant cells. Following homodimerization of gp130, there is a formation of a high-affinity-binding hexameric complex consisting of two molecules each of IL-6, IL-6R, and gp130.
In the present study, a virtual screening approach was developed for discovering novel blockers of hIL-6. According to the 3D crystal structure of (hIL-6·hIL-6R·gp 130)2 complex, three small molecular antagonistic compounds against IL-6R (compounds 1, 2, and 3) targeting hIL-6 were screened out, optimized and evaluated theoretically using the computer-aided molecular docking-based virtual screening methods. Furthermore, the bioactivities of these compounds were analyzed with IL-6-dependent MM cell line (XG-7). The results suggested that compound 1 acted as a potential specific antagonist of IL-6 and could be a lead compound for treating various diseases caused by excess IL-6 production, such as MM.
Materials and methods
Reagents
rhIL-6R and hIL-6 were purchased from R&D Systems, Inc. (Minneapolis, MN, USA). 2-Mercaptoethanol, Giemsa, dimethyl sulfoxide (DMSO), and MTT were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). 3H-tritiated thymidine and ATPlite kit were purchased from PerkinElmer Inc. (Waltham, MA, USA). Genomic DNA Purification Kit was purchased from Promega Corporation, (Fitchburg, WI, USA).
Rational design of antagonist compounds
Based on the 3D complex crystal structure of hIL-6 and hIL-6R X-ray crystallography1 and the interaction mode of hIL-6 and its antagonistic peptides,25–28 the character of pharmacophore, such as specific chemical group (eg, aliphatic series), hydrogen bond donor/receptor, groups with positive or negative electricity and hydrophobic groups, was confirmed in virtue of distant geometry and intermolecular hydrogen-bond theory. Considering the surrounding range (the radius was defined as 0.5 nm) of the binding residues in hIL-6R, the matching molecular fragments were selected from the standard fragment library offered by the program Ludi, which had ~10,000 candidate compounds available. The rationality of these selected fragments was determined using distance geometry and root-mean-square deviation analysis; meanwhile, the distance of these fragments from other sites of hIL-6R and the static effect were also optimized. After evaluation of these fragments offered by Ludi, the mode of primitive molecule candidates was obtained by expanding these fragments. Finally, potential antagonistic compounds were screened out from 3D-Available Chemicals Directory (ACD) and MDL Drug Data Report (MDDR) libraries.29,30 All the molecules were visualized and analyzed using the InsightII (2000) software (Accelrys lnc., San Diego, CA, USA) running on an Octane2 R12000 Silicon Graphics workstation.
Preparation of sample solution
Compounds purchased from Specs Inc. (Zoetermeer, the Netherlands) were dissolved separately in DMSO to a final stock concentration of 3 mM. Then, they were diluted to appropriate concentrations with 10 mmol/L phosphate-buffered saline (PBS; pH 7.2) or cell culture medium.
Cell culture
Human myeloma cell line XG-7 (a kind gift from Professor Xue-guang Zhang of Suzhou University, China), mouse fibrosarcoma L929 cells and SP2/0 cells (purchased from American Type Culture Collection [ATCC], Manassas, VA, USA) were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% fetal bovine serum (FBS) and penicillin–streptomycin (50 IU/mL and 50 g/mL, respectively) at 37°C. Moreover, 50 μmol/L 2-mercaptoethanol and 2 ng/mL rhIL-6 were added to the media in XG-7 cells.31,32 The study was approved by the Ethics Review Boards at Beijing Institute of Basic Medical Sciences.
Competition binding analysis
Polystyrene plates were coated with 2 μg/mL of hIL-6R (R&D Systems) in 0.05 M bicarbonate buffer (pH 9.6) at 4°C overnight and blocked with 5% (w/v) low-fat milk for 2 hours at room temperature. Various dilutions of hIL-6 antagonist compounds were added to the wells and incubated overnight at 4°C. PBS and 0.1% DMSO were set as negative controls. Then, the plates were incubated with 4 μg/mL hIL-6 at 37°C for 2 hours. After washing three times with PBS, the plates were added with mouse anti-hIL-6 antibody (1:5000, BD Biosciences, San Jose, CA, USA) for 2 hours at 37°C, followed by incubation with the HRP-conjugated GAM IgG for 40 minutes at room temperature. After washing, the substrate o-phenylenediamine dihydrochloride was added to develop the color, and the absorbance at 492 nm was measured.
Antagonist compounds treatment
In order to test the ability of the compounds to block hIL-6, IL-6-dependent XG-7 cells were washed three times with RPMI-1640 medium and seeded at 1×105 cells/mL in the RPMI-1640 medium supplemented with 2% FBS for 12 hours (without rhIL-6). Then, 1×104 XG-7 cells/well in 0.1 mL of complete medium were dispensed into each well of 96-well microtiter plate, and compounds were added at the concentration of 0.003 μM, 0.03 μM, 0.3 μM, and 3 μM. After incubation for 5 hours, rhIL-6 was added for another 72 hours cultivation period to achieve a final concentration of 2 ng/mL.33 Cell growth was measured by MTT assay, ATP-based assay and 3H-TdR method.28 Triplicate wells were set for each group.
Morphological analysis
Cells were starved, resuspended and treated with or without compounds and hIL-6R as mentioned earlier. Then, they were washed with PBS and spread onto clean glass slides. After being fixed with methanol and acetic acid mixture for 10 minutes, the air-dried glass slides were stained with Giemsa solution (pH 6.8) for 10–15 minutes. The apoptotic cells were identified under an inverted microscope.26,27
DNA fragmentation analysis
Cells were treated with compounds and hIL-R as mentioned earlier. Then, they were collected and washed twice with PBS. Genomic DNA Purification Kit was used for extracting DNA, which was electrophoresed on 2% agarose gel to evaluate the internucleosomal DNA fragmentation.
Cytotoxic assay
L929 cells and SP2/0 cells were washed three times and cultured in the RPMI-640 medium supplemented with 2% FBS for 8 hours (without rhIL-6 for cell starvation). Then, the cells were harvested and diluted to a final concentration of 105 cells/mL in the RPMI-1640 medium containing 10% FBS, and 100 μL of cell suspension per well was cultivated in 96-well plates; meanwhile, diluted compound 1 was added to each well. After incubation for another 72 hours, cell growth was measured using the MTT method.
Statistical analysis
The mean values from different treatments were compared using one-way analysis of variance or Student’s t-test. Statistical significance was set at P<0.05.
Results
Virtual screening of small molecule antagonistic compounds against hIL-6
Based on the hIL-6/hIL-6R complex structure, the key residues in hIL-6 to bind hIL-6R within the distance of 0.4 nm were identified according to computer graphics and distant geometry (Figure 1A), which showed that residues in loops A and B (green balls) and helix D (red balls) in hIL-6 participated in hIL-6R binding. Then, the contact surface area of hIL-6 and hIL-6R to bind each other was calculated as 523.29 A2 and 1,244.69 A2 by the Ludi program, respectively. Meanwhile, the spherical surface of hIL-6 to bind hIL-6R was displayed also by Ludi (Figure 1B). Considering the size of the interactive area and small molecular compound, the active spherical surface was empirically separated into three parts: Q1, Q2, and Q3 (Figure 1C), by which 3,000 molecular fragments were screened from 3D-ACD or MDDR library according to their unique structure and pharmacophore character (1,050, 1,020, and 930 fragments targeting Q1, Q2, and Q3, respectively). Molecular docking was performed to preliminary evaluate the binding energy of those fragments and hIL-6R, suggesting that compound 1 (phthalimide like), 2 (uracil like), and 3 (aminocytosine like) located in Q1, Q2, or Q3 region were the best (Figure 1D).
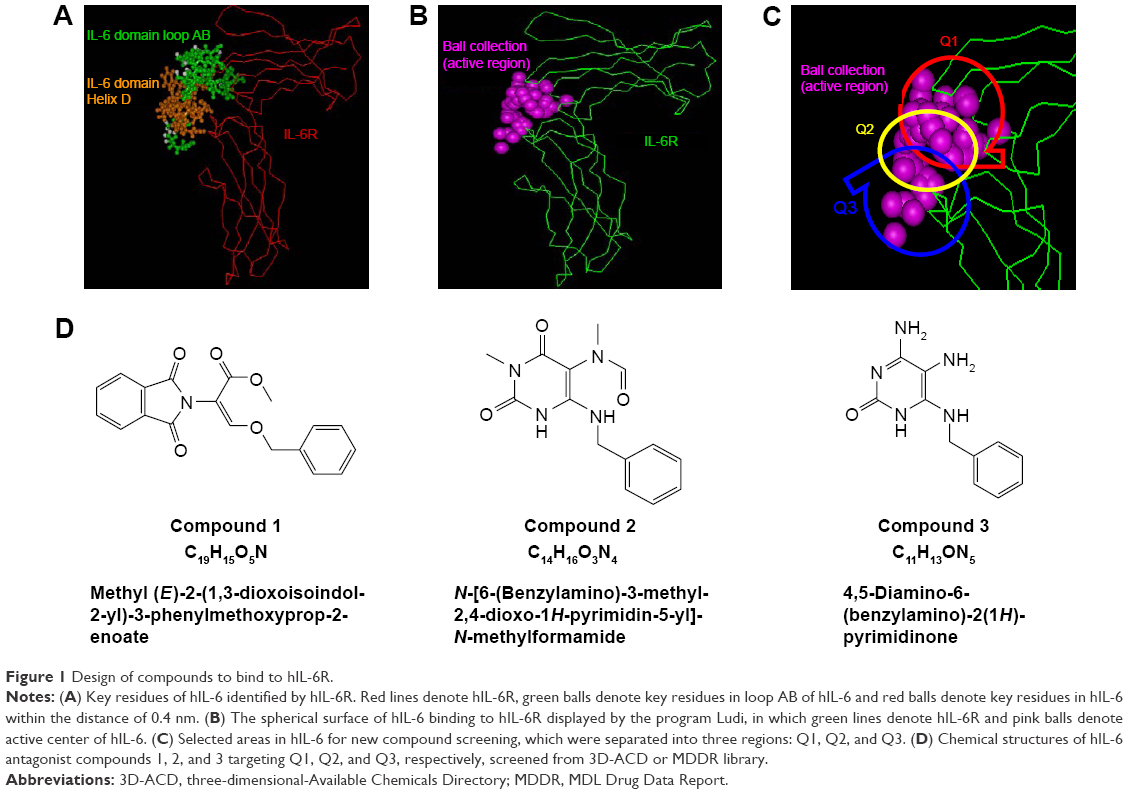 | Figure 1 Design of compounds to bind to hIL-6R. |
Theoretical analysis of candidate compounds binding to hIL-6R
After structural optimization of these compounds by semi-experienced AM1 method of quantum chemistry, hIL-6R-binding region of these compounds was defined theoretically by molecular docking and dynamic optimization. The stable model of compound/hIL-6R complex is displayed in Figure 2; meanwhile, the interacted key residues and the reaction energy of compounds binding to hIL-6R were calculated. As shown in Table 1, the binding energies of three compounds were −18.73 kJ/mol (compound 1), −26.46 kJ/mol (compound 2), and −36.28 kJ/mol (compound 3). The data denote that compound 3 possessed the best binding capacity among the three, while compound 1 had the weakest.
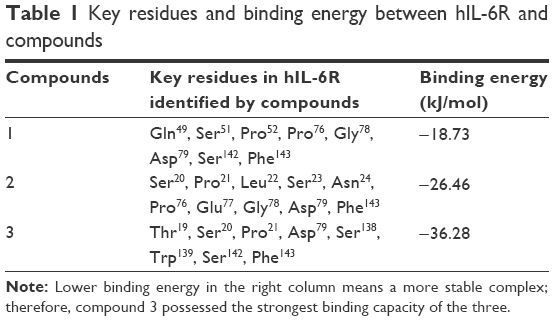 | Table 1 Key residues and binding energy between hIL-6R and compounds |
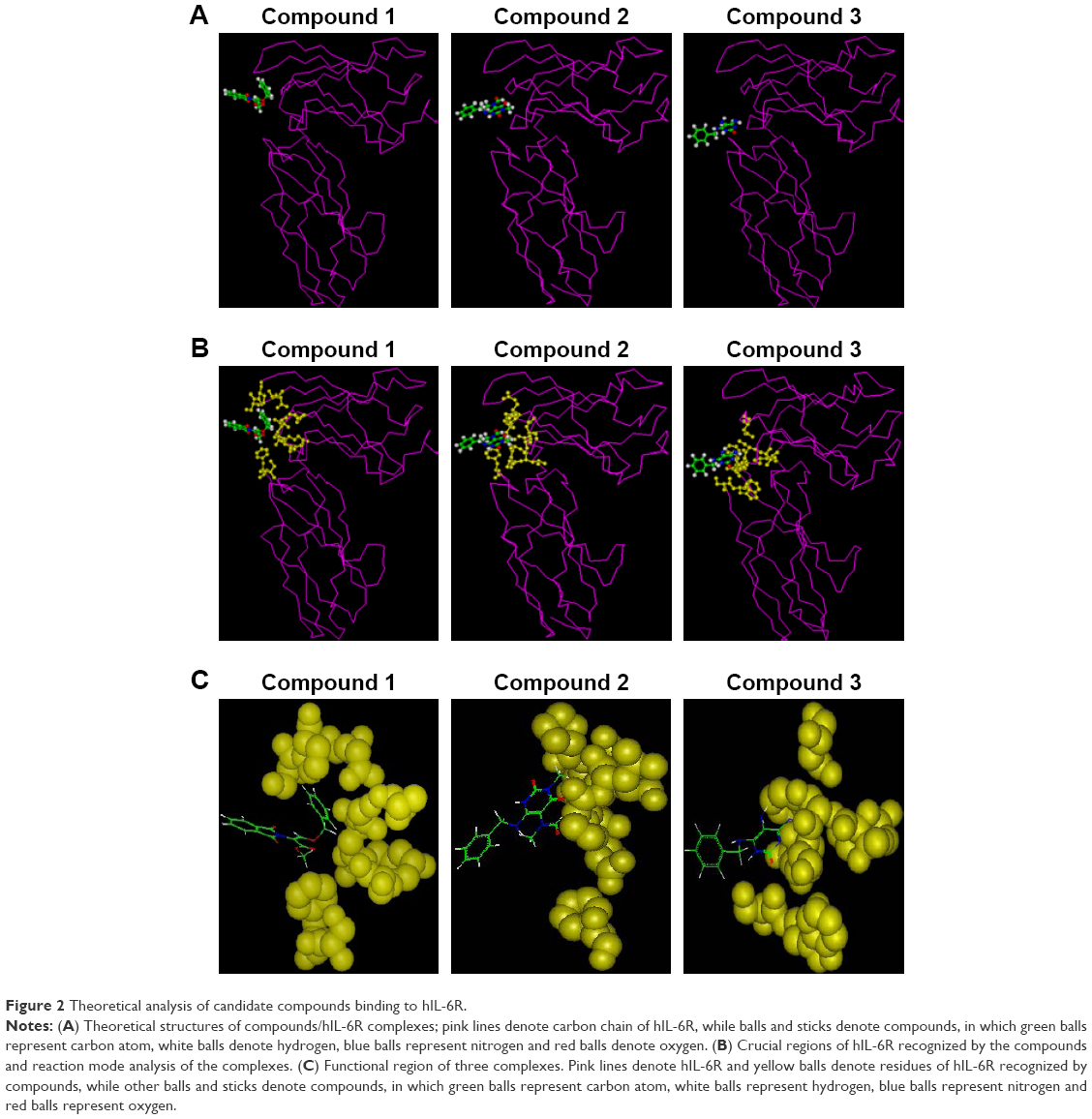 | Figure 2 Theoretical analysis of candidate compounds binding to hIL-6R. |
IL-6R binding activity of the compounds to compete with IL-6
Competing enzyme-linked immunosorbent assay (ELISA) analysis was carried out to evaluate the inhibitory potential of compound candidates. As shown in Figure 3A, compounds 1, 2, and 3 were able to compete with hIL-6 to bind hIL-6R in a dose-dependent manner, while negative controls (PBS and DMSO) did not inhibit the formation of hIL-6/hIL-6R complex. At the concentration of 3 μM, the inhibition ratio was 11.8% (compound 1), 24.8% (compound 2), and 25.6% (compound 3; Figure 3B), which supports the theoretical conclusion mentioned earlier.
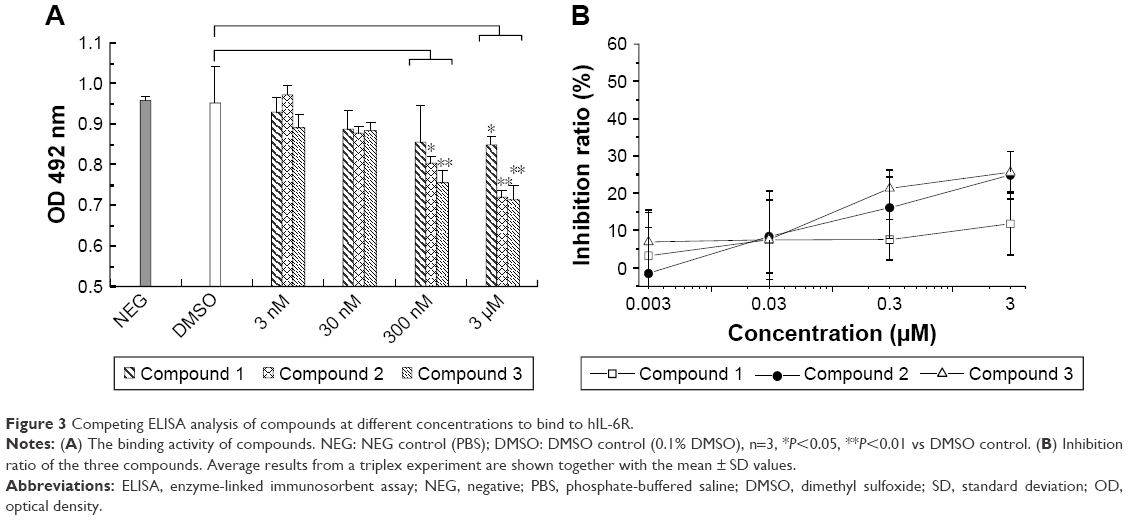 | Figure 3 Competing ELISA analysis of compounds at different concentrations to bind to hIL-6R. |
Effects of the compounds on IL-6-dependent cell growth
The survival and growth of XG-7 cells were dependent on hIL-6, and the cells underwent rapid growth arrest and apoptosis with IL-6 withdrawal.31 Therefore, XG-7 cells represented an ideal model for evaluating the biological activity of candidate IL-6 inhibitors.25,28 The inhibitory effect of antagonists was investigated at various concentrations, while XG-7 cells with and without hIL-6 were taken as negative and positive controls. In this study, three wildly used proliferation assays (MTT, 3H-TdR, and ATPlite) were carried out to evaluate the inhibitory activity of these compounds (Figure 4). Similar results were obtained with the abovementioned three methods, even though the 3H-TdR and ATPlite methods were much more sensitive than MTT.34 Among all the three compounds, only compound 1 could inhibit XG-7 proliferation through antagonizing hIL-6 in a dose-dependent manner, and the inhibitory ratio reached up to 24.5% at the concentration of 3 μM when compared to the negative controls. The results indicated that the Q1 region may be the key biological activate area for hIL-6/hIL-6R interaction, although the compound 3 showed the best binding activity.
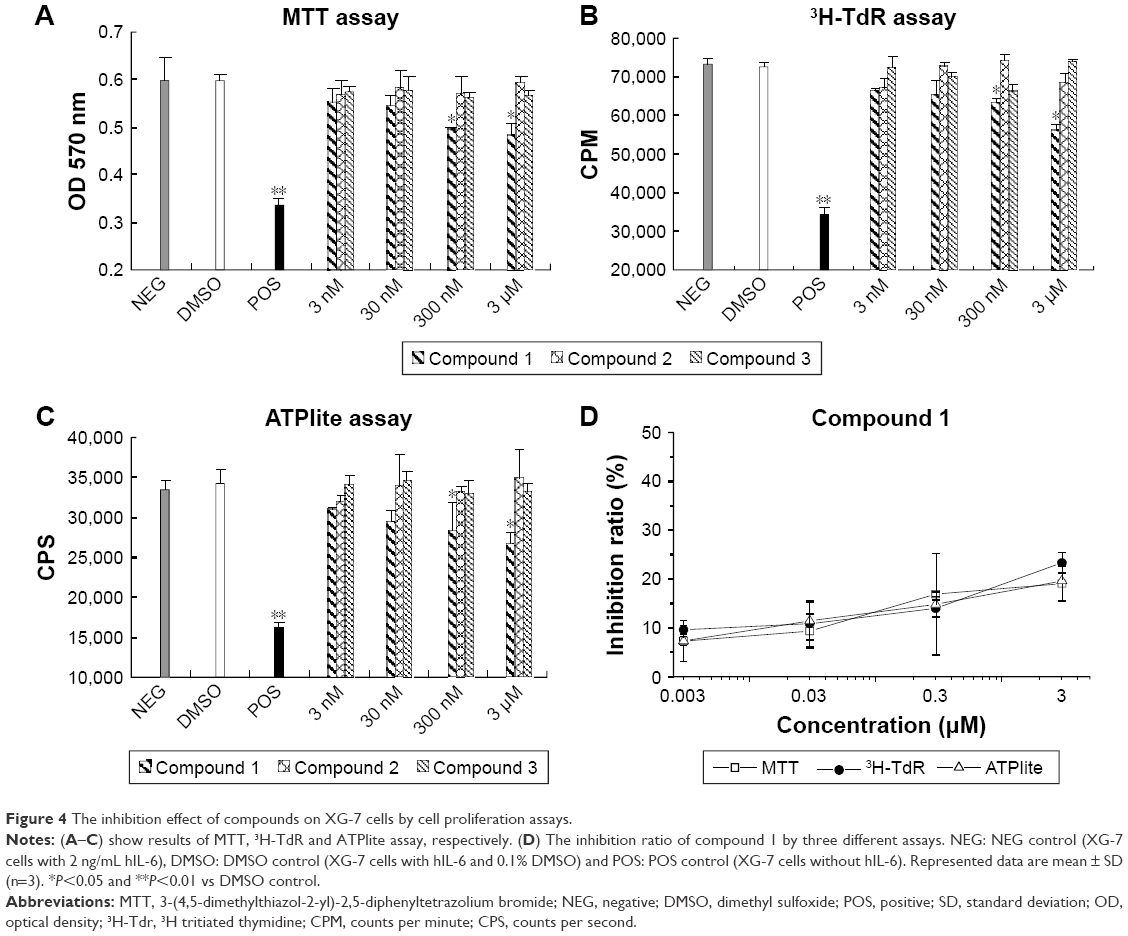 | Figure 4 The inhibition effect of compounds on XG-7 cells by cell proliferation assays. |
Compound 1-induced apoptosis examined by morphological and DNA fragmentation analyses
Compound-induced apoptosis in XG-7 cells was also identified by morphological analysis. XG-7 cells were treated with or without drugs, then stained with Giemsa assay and observed with a light microscope. A marked enhancement of apoptotic change in decreased cell size, pyknosis formation of apoptotic bodies and karyorrhexis could be observed in the samples treated with 0.3 μM and 3 μM of compound 1 (Figure 5A and B), while the negative control and DMSO-treated cells were not affected. XG-7 cells were treated with 0.3 μM and 3 μM of compound 1, and then, the genomic DNA was isolated and examined using agarose gel electrophoresis. Fragmented DNA ladder pattern existed obviously in DNA samples extracted from compound 1-treated cells (Figure 5B), while the genomic DNA of negative or DMSO-treated cells did not present DNA fragmentation.
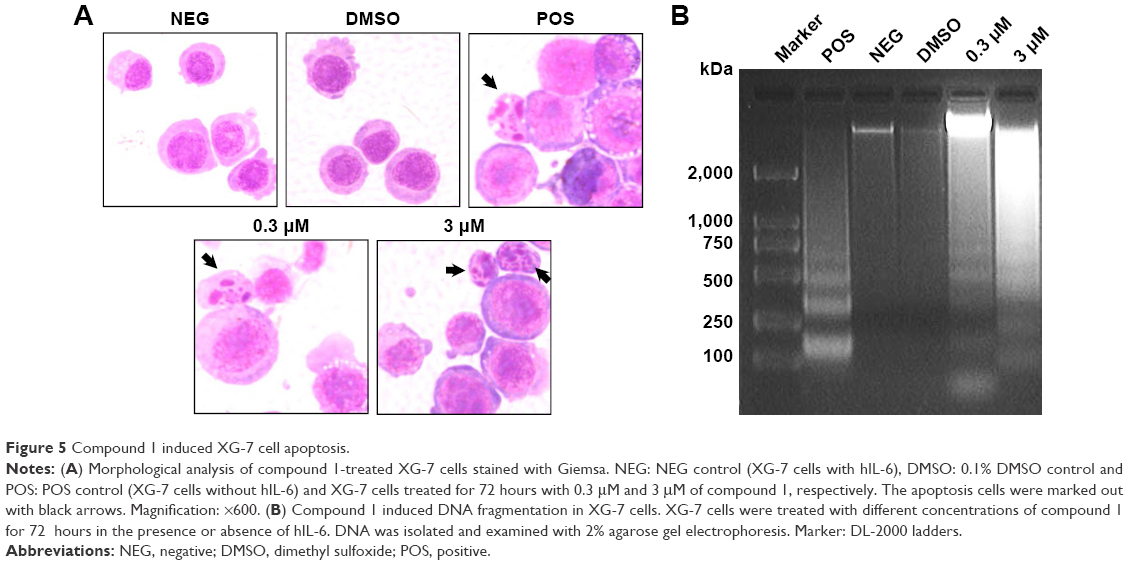 | Figure 5 Compound 1 induced XG-7 cell apoptosis. |
Compound cytotoxicity evaluation
To estimate the cytotoxic effect of compound 1, IL-6-nonresponsive L929 (mouse fibroblast cell) and SP2/0 (mouse myeloma cell) cells were chosen and cultivated in RPMI-1640 complete media supplemented with different doses of compound 1 (Figure 6). Compared to the DMSO control group, compound 1 exhibited no significant cytotoxic effect on L929 cells (Figure 6A) or SP2/0 cells (Figure 6B) within 72 hours in the dose range.
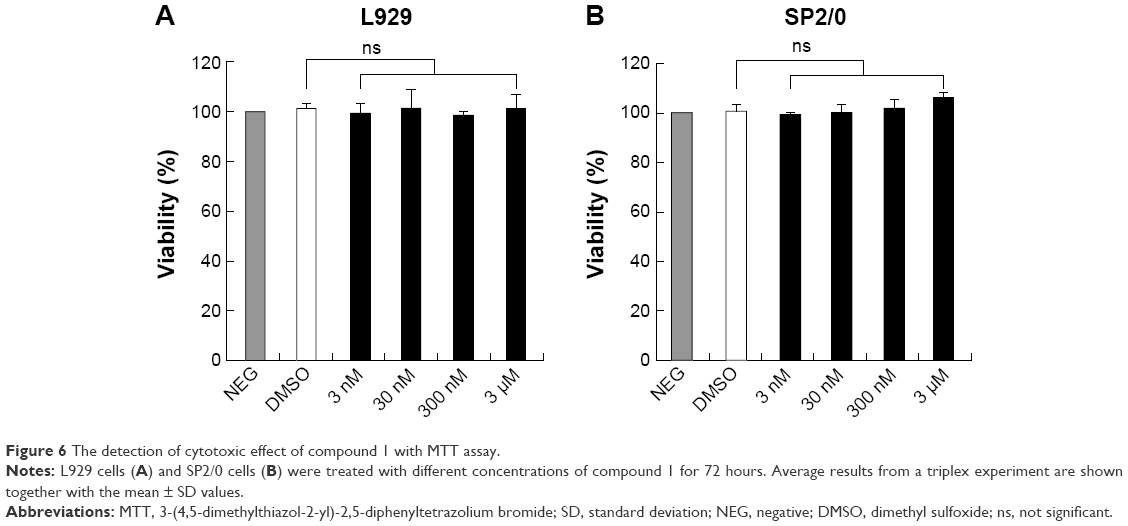 | Figure 6 The detection of cytotoxic effect of compound 1 with MTT assay. |
Discussion
It was shown that hIL-6 was relevant to the development and progression of tumors of various organs, particularly for MM, renal and prostate cancers, and melanoma. Research results suggested the possible involvement of the cytokine in the pathogenesis of these neoplasias as an autocrine and paracrine growth factor. Therefore, the inhibition or modulation of hIL-6 could have profound therapeutic benefits in MM and in several other diseases.35 Extensive investigation of the interaction mode between hIL-6 and hIL-6R has formed the basis for the development of highly specific receptor antagonists.33 Now, the researchers are focusing on generating monoclonal antibody or its mutants to block the bioactivity of hIL-6. High-molecular-weight biocompounds, such as various IL-6 variants,13 bifacial mutants,14 stable IL-6 mutant,15 or humanized antibodies,16 have suggested the possibility of selectively controlling the activity or production of IL-6. Development of a low-molecular-weight antagonist is desirable for clinical application due to its superiority for oral intake, antigenicity and other qualities. However, a very limited research on the low-molecular-weight antagonists targeted to hIL-6 has been reported. In 2003, the 3D structures of hIL-6, hIL-6R, and gp130 complex have been determined by X-ray crystallography.1 The contact regions of hIL-6 binding with hIL-6R are constituted of three distinct sites. Site I is formed by the C-terminal parts of helices A and D and the AB loop interacted with domains 2 and 3 of non-signaling receptor (hIL-6R).28 The interaction is the prerequisite for the engagement of the signaling gp130 receptor, which was recruited by site II and site III. Site II was formed by a limited number of exposed residues on helix A and helix C, while site III was formed by the residues at the amino-terminal end of helix D, spatially flanked by residues in the initial part of the AB loop.21,36
Recently, computational techniques for docking potential ligands based on the shape of protein receptors had been developed dramatically. The docking methods used in structure-based virtual database screening offer the ability to quickly and cost-efficiently estimate the affinity and binding mode of a ligand for the protein receptor of interest, such as a drug target.37 In this study, based on the 3D crystal structure of (hIL-6·hIL-6R·gp 130)2 complex, the computer virtual screen approach was used to discover highly selective, novel hIL-6 inhibitors. From the 3D-ACD and MDDR chemical databases, three candidate compounds were screened out. Theoretical analysis showed that these compounds offered a practical means of imposing long-term blockade of hIL-6 activity and possessed very high affinity to hIL-6R in silico. The biological functions of candidate compounds were analyzed using competition ELISA, which demonstrated that the three compounds all could block IL-6 to bind with IL-6R specifically. Then, IL-6 growth-dependent cell line XG-7 was chosen to evaluate the inhibitory ability on cell proliferation and enhanced effect on cell apoptosis, which showed that only compound 1 could affect the hIL-6-dependent biological character and antagonize hIL-6 in a dose-dependent manner in XG-7 cells. In addition, DNA fragmentation and morphological analysis of compound 1-treated XG-7 cells also suggested the antagonistic effect of compound 1 against hIL-6. Furthermore, compound 1 did not have cytotoxic effect on hIL-6-unrelated cell lines in the engaged dose range.
Interestingly, even though the compounds 2 and 3 showed better binding activity than compound 1, only compound 1 had the inhibitory capacity on the hIL-6-dependent biological function. It provided an important hint that the Q1 region might be the IL-6R key biological activation area. In fact, structure-based drug design mainly depended on the biological process and the interface pattern of protein–protein interactions. The interfaces of protein–protein interactions were quite large (from 600 to >1,300 Å2), coupled with 10–30 side chains located on each side of the interfaces.26,27 Moreover, other important contact residues were even far away from the primary sequence. Thus, we may need to consider about combining the structural character of compound 3 (the binding activity advantage) and compound 1 (the biological activity advantage) together for the future optimization.
Conclusion
In this study, a combined computational approach, including computer virtual screening and molecular docking, has been applied to identify IL-6 antagonists from chemical databases, and three compound candidates were subject to a series of bioassay evaluations. As a result, one phthalimide-like compound 1 was identified to be a selective hIL-6 antagonist. During the present research work, the important functional group based on the compound 1 will be determined, and a series of the compounds with the same or similar functional group will be designed and synthesized. The designed compounds will be evaluated in a experiment. Thus, this strategy offers a feasible and effective approach to discover other bioactive compounds using in silico and in vitro screening.
Acknowledgments
The authors thank Professor Jian Sun (Tianjin University) and Renfeng Guo (University of Michigan) for helping revise the manuscript. This study was supported by National Sciences Fund (No 31070820 and 81672368), “863” Fund (No 2012AA02A), “973” Fund (No 2010CB833604) and China and Beijing Natural Science Fund (No 5162026).
Disclosure
The authors report no conflicts of interest in this work.
References
Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300(5628):2101–2104. | ||
Matsuda T, Yamasaki K, Taga T, Hirano T, Kishimoto T. Current concepts of B cell modulation. Int Rev Immunol. 1989;5(2):97–109. | ||
Eaves CJ, Cashman JD, Kay RJ, et al. Mechanisms that regulate the cell cycle status of very primitive hematopoietic cells in long-term human marrow cultures. II. Analysis of positive and negative regulators produced by stromal cells within the adherent layer. Blood. 1991;78(1):110–117. | ||
Dowton SB, Waggoner DJ, Mandl KD. Developmental regulation of expression of C-reactive protein and serum amyloid A in Syrian hamsters. Pediatr Res. 1991;30(5):444–449. | ||
Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood. 2004;104(3):607–618. | ||
Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627–2632. | ||
Smolen JS, Steiner G, Aringer M. Anti-cytokine therapy in systemic lupus erythematosus. Lupus. 2005;14(3):189–191. | ||
Takagi N, Mihara M, Moriya Y, et al. Blockage of interleukin-6 receptor ameliorates joint disease in murine collagen-induced arthritis. Arthritis Rheum. 1998;41(12):2117–2121. | ||
Schweitzer DH, Boxman IL, Lowik CW, et al. Parathyroid hormone related protein and interleukin-6 mRNA expression in larynx and renal cell carcinomas from normocalcaemic and hypercalcaemic patients. J Clin Pathol. 1995;48(10):896–900. | ||
Yamasaki K, Taga T, Hirata Y, et al. Cloning and expression of the human interleukin-6 (BSF-2/IFN beta 2) receptor. Science. 1988;241(4867):825–828. | ||
Somers W, Stahl M, Seehra JS. 1.9 A crystal structure of interleukin 6: implications for a novel mode of receptor dimerization and signaling. EMBO J. 1997;16(5):989–997. | ||
Fontaine V, Savino R, Arcone R, et al. Involvement of the Arg179 in the active site of human IL-6. Eur J Biochem. 1993;211(3):749–755. | ||
van Dam M, Mullberg J, Schooltink H, et al. Structure-function analysis of interleukin-6 utilizing human/murine chimeric molecules. Involvement of two separate domains in receptor binding. J Biol Chem. 1993;268(20):15285–15290. | ||
Savino R, Lahm A, Salvati AL, et al. Generation of interleukin-6 receptor antagonists by molecular-modeling guided mutagenesis of residues important for gp130 activation. EMBO J. 1994;13(6):1357–1367. | ||
Paonessa G, Graziani R, De Serio A, et al. Two distinct and independent sites on IL-6 trigger gp 130 dimer formation and signalling. EMBO J. 1995;14(9):1942–1951. | ||
Ehlers M, de Hon FD, Bos HK, et al. Combining two mutations of human interleukin-6 that affect gp130 activation results in a potent interleukin-6 receptor antagonist on human myeloma cells. J Biol Chem. 1995;270(14):8158–8163. | ||
Ehlers M, Grotzinger J, Fischer M, Bos HK, Brakenhoff JP, Rose-John S. Identification of single amino acid residues of human IL-6 involved in receptor binding and signal initiation. J Interferon Cytokine Res. 1996;16(8):569–576. | ||
Grotzinger J, Kurapkat G, Wollmer A, Kalai M, Rose-John S. The family of the IL-6-type cytokines: specificity and promiscuity of the receptor complexes. Proteins. 1997;27(1):96–109. | ||
Vollmer P, Oppmann B, Voltz N, Fischer M, Rose-John S. A role for the immunoglobulin-like domain of the human IL-6 receptor. Intracellular protein transport and shedding. Eur J Biochem. 1999;263(2):438–446. | ||
Bazan JF. Structural design and molecular evolution of a cytokine receptor superfamily. Proc Natl Acad Sci U S A. 1990;87(18):6934–6938. | ||
Varghese JN, Moritz RL, Lou MZ, et al. Structure of the extracellular domains of the human interleukin-6 receptor α -chain. Proc Natl Acad Sci U S A. 2002;99(25):15959–15964. | ||
Cole AR, Hall NE, Treutlein HR, et al. Disulfide bond structure and N-glycosylation sites of the extracellular domain of the human interleukin-6 receptor. J Biol Chem. 1999;274(11):7207–7215. | ||
Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol. 1997;15:797–819. | ||
Chow D, He X, Snow AL, Rose-John S, Garcia KC. Structure of an extracellular gp130 cytokine receptor signaling complex. Science. 2001;291(5511):2150–2155. | ||
Feng J, Li Y, Shen B. The design of antagonist peptide of hIL-6 based on the binding epitope of hIL-6 by computer-aided molecular modeling. Peptides. 2004;25(7):1123–1131. | ||
Yang Z, Feng J, Hu M, et al. A novel hIL-6 antagonist peptide from computer-aided design contributes to suppression of apoptosis in M1 cells. Biochem Biophys Res Commun. 2004;325(2):518–524. | ||
Yang Z, Feng J, Li Y, et al. Structure-based design and characterization of a Novel IL-6 antagonist peptide. Mol Immunol. 2005;42(9):1015–1021. | ||
Feng J, Yang Z, Li Y, et al. The rational designed antagonist derived from the complex structure of interleukin-6 and its receptor affectively blocking interleukin-6 might be a promising treatment in multiple myeloma. Biochimie. 2006;88(9):1265–1273. | ||
Ferrari S, Morandi F, Motiejunas D, et al. Virtual screening identification of nonfolate compounds, including a CNS drug, as antiparasitic agents inhibiting pteridine reductase. J Med Chem. 2010;54(1):211–221. | ||
Cummings MD, Schubert C, Parks DJ, et al. Substituted 1,4-benzodiazepine-2,5-diones as alpha-helix mimetic antagonists of the HDM2-p53 protein-protein interaction. Chem Biol Drug Des. 2006;67(3):201–205. | ||
Zhang XG, Gaillard JP, Robillard N, et al. Reproducible obtaining of human myeloma cell lines as a model for tumor stem cell study in human multiple myeloma. Blood. 1994;83(12):3654–3663. | ||
Song L, Li Y, Sun Y, Shen B. Mcl-1 mediates cytokine deprivation induced apoptosis of human myeloma cell line XG-7. Chin Med J (Engl). 2002;115(8):1241–1243. | ||
Manfredini R, Tenedini E, Siena M, et al. Development of an IL-6 antagonist peptide that induces apoptosis in 7TD1 cells. Peptides. 2003;24(8):1207–1220. | ||
Riss TL, Moravec RA, Niles AL, et al, editors. Cell viability assays. In: Sittampalam GS, Coussens NP, Nelson H, et al, editors. Assay Guidance Manual. Bethesda, MD: Eli Lilly & Company; 2004:1–30. | ||
Hitzler JK, Martinez-Valdez H, Bergsagel DB, Minden MD, Messner HA. Role of interleukin-6 in the proliferation of human multiple myeloma cell lines OCI-My 1 to 7 established from patients with advanced stage of the disease. Blood. 1991;78(8):1996–2004. | ||
Kurth I, Horsten U, Pflanz S, et al. Activation of the signal transducer glycoprotein 130 by both IL-6 and IL-11 requires two distinct binding epitopes. J Immunol. 1999;162(3):1480–1487. | ||
Grinter SZ, Zou X. Challenges, applications, and recent advances of protein-ligand docking in structure-based drug design. Molecules. 2014;19(7):10150–10176. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.