")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Spotlight on sirukumab for the treatment of rheumatoid arthritis: the evidence to date
Authors Lazzerini PE, Capecchi PL, Guidelli GM, Selvi E, Acampa M, Laghi-Pasini F
Received 13 June 2016
Accepted for publication 5 August 2016
Published 26 September 2016 Volume 2016:10 Pages 3083—3098
DOI https://doi.org/10.2147/DDDT.S99898
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Frank Boeckler
Pietro Enea Lazzerini,1 Pier Leopoldo Capecchi,1 Giacomo Maria Guidelli,1 Enrico Selvi,1 Maurizio Acampa,2 Franco Laghi-Pasini1
1Department of Medical Sciences, Surgery and Neurosciences, University of Siena, 2Stroke Unit, University Hospital of Siena, Siena, Italy
Abstract: Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory disease primarily affecting synovial joints and is characterized by persistent high-grade systemic inflammation. Proinflammatory cytokines, particularly interleukin-6 (IL-6), are of crucial importance in the pathogenesis of the disease, driving both joint inflammation and extra-articular comorbidities. Tocilizumab, a humanized IL-6 receptor-inhibiting monoclonal antibody, has been the first, and, to date, the only, IL-6 inhibitor approved for the treatment of RA. Many studies have demonstrated the potency and effectiveness of tocilizumab in controlling disease activity and radiological progression of RA. These successful results have encouraged the development of novel IL-6 inhibitors, among which a promising agent is sirukumab (SRK), a human anti-IL-6 monoclonal antibody currently under evaluation in Phase II/III studies in patients with RA, systemic lupus erythematosus, giant-cell arteritis, and major depressive disorder. The evidence to date indicates SRK as an effective and well-tolerated new therapeutic tool for patients with active RA, with some preliminary data suggesting a specific beneficial impact on relevant systemic complications associated with the disease, such as depression and cardiovascular disease. Conversely, although pathophysiological considerations make plausible the hypothesis that IL-6 blockade with SRK may also be beneficial in the treatment of many diseases other than RA (either autoimmune or not), available clinical data in patients with systemic lupus erythematosus do not seem to support this view, also giving rise to potentially relevant concerns about drug safety. If large Phase III clinical trials currently in progress in patients with RA confirm the efficacy and tolerability of SRK, then in the long term, this drug could, in the near future, occupy a place in the treatment of the disease, potentially also opening the doors to a more extended use of SRK in a wide range of disorders in which IL-6 plays a key pathogenic role.
Keywords: sirukumab, rheumatoid arthritis, interleukin-6, tocilizumab, systemic lupus erythematosus, cardiovascular disease, interleukin-6
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory disease affecting the synovial joints also leading to extra-articular manifestations, characterized by persistent high-grade systemic inflammation. Classical clinical presentation consists of a symmetrical polyarthritis, related to intense leukocyte infiltration, hyperplasia, and neovascularization of the synovial tissue, resulting in an inflammatory destruction of cartilage and subchondral bone.1 The disease affects 0.5%–1% of adults in developed countries, with a global prevalence ~0.25%, thus representing an important cause of disability and preterm mortality worldwide.1,2 Among the extra-articular manifestations, accelerated cardiovascular disease (CVD) represents the main driver of the 2× higher risk of death observed in these patients when compared to age- and sex-matched non-RA subjects.3,4
Although the etiology of RA remains substantially unknown, it is well recognized that proinflammatory cytokines, particularly tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6), are of crucial importance in the pathogenesis of the disease, driving both joint inflammation and extra-articular comorbidities.5 This evidence has led in the last 15 years to the development of drugs specifically inhibiting these cytokines, thus kicking off to the era of biologic drugs that have revolutionized the therapeutic approach to RA. Starting from early 2000s, TNF-α inhibitors were the first class of cytokine-targeting drugs introduced for RA therapy, followed by the IL-1 receptor antagonist anakinra. In 2010, the first, and to date the only, IL-6 inhibitor tocilizumab (TCZ), a humanized IL-6 receptor (IL-6R)-inhibiting monoclonal antibody, was approved for the treatment of modest-to-severe RA in patients who have failed other disease-modifying antirheumatic drugs (DMARDs), including biologics. A large body of evidence demonstrated the potency and effectiveness of TCZ in reducing the signs and symptoms, as well as radiological disease progression of RA, thus pointing to this drug as a mainstay in the current treatment of the disease.6 In the last years, these successful results have encouraged the development of novel biologic DMARDs targeting IL-6 or IL-6R, among which sirukumab (SRK), a human anti-IL-6 monoclonal antibody currently under evaluation in Phase III studies in patients with RA, is promising.7
The aim of this paper is to review the evidence available to date supporting the future use of SRK in the treatment of RA in the light of the key role played by IL-6 in the pathogenesis of the disease, including both articular and extra-articular manifestations.
Biology of IL-6
IL-6 is a small (~25 kD) secreted glycoprotein composed of 184 amino acids and characterized by a four-helix bundle structure. It is produced by several cell types, including leukocytes (T- and B-lymphocytes, monocytes, macrophages), fibroblasts, osteoblasts, keratinocytes, endothelial cells, mesangial cells, adipocytes, skeletal myocytes, cardiomyocytes, brain cells (astroglia, microglia, neurons), and some tumor cells in response to various stimuli, such as lipopolysaccharide and other bacterial products, viruses, cytokines (TNF-α, IL-1, transforming growth factor [TGF]-β), adenosine triphosphate, parathormone, vitamin D3, homocysteine, and angiotensin II.8–19
Circulating IL-6 is found in the blood of healthy humans at low concentration (≤1 pg/mL), and significantly increases during inflammatory conditions, reaching concentrations in the range of μg/mL during sepsis.19 In fact, this cytokine critically contributes to host defense against infections and tissue injuries by stimulating acute-phase immune response and hematopoiesis, but it also regulates metabolic, regenerative, and neural processes under physiological conditions. Once released, IL-6 exerts its pleiotropic biological effects by activating a unique IL-6R signaling system, including the IL-6R and downstream signaling molecules.8,9,20
IL-6R
The IL-6R is constituted by two chains: 1) an IL-6 binding chain or IL-6Rα, which exists in two forms, ie, a 80 kD transmembrane IL-6R (mIL-6R), and a 50–55 kD soluble IL-6R (sIL-6R) and 2) a 130 kD signal-transducing chain, named gp130 or IL-6Rβ.8,9,19 The mIL-6R is expressed on the surface of a limited number of cell types, ie, hepatocytes, megakaryocytes, and leukocytes, including monocytes, macrophages, neutrophils, and T- and B-lymphocytes. The sIL-6R is present in human plasma (25–75 ng/mL) and tissue fluids and can be generated by proteolytic cleavage (shedding) of the mIL-6R by metalloproteases (A Disintegrin And Metalloproteinases [ADAM], 10 and 17), or, in minor part, via alternative splicing by omission of the transmembrane domain. Conversely, gp130 is ubiquitously expressed on all human cells.8,9,20,21
Upon biding to either mIL-6R or sIL-6R, IL-6 induces the homodimerization of the IL-6R/gp130 chains, resulting in the formation of a hexamer (comprising two IL-6, two IL-6R, and two gp130 proteins),22 which in turn triggers the downstream signaling cascade. Cellular activation via IL-6 binding to mIL-6R is named “classic signaling”. All other cells not expressing mIL-6R obtain their IL-6 signals by “trans-signaling”: IL-6 binds to the circulating sIL-6R, and this complex forms the signaling complex with gp130 on the cell surface. Trans-signaling can occur in a broad range of human cells, thus contributing to explain the pleiotropic activities of IL-6. Increasing evidence indicates that IL-6 trans-signaling, rather than classical signaling, is particularly involved in disease development. Although the picture is not completely clear, it is currently believed that homeostatic and regenerative activities of IL-6 are mediated by classical signaling, while proinflammatory effects mainly result from trans-signaling pathway activation.20 Interestingly, a soluble form of the gp130 (sgp130) was also detected in the circulation at relatively high concentrations, mainly produced by alternative splicing. Since sgp130 can bind to the IL-6/sIL-6R complex, it acts as a natural and specific inhibitor of IL-6 mediated trans-signaling. Indeed, classic signaling is not affected by sgp130. Recently, a fusion protein consisting of the extracellular portion of gp130 fused to the Fc region of human IgG1 (sgp130Fc) was developed. By forming dimers, sgp130Fc acts as a potent inhibitor of IL-6 trans-signaling (> tenfold more potent than monomeric natural occurring sgp130 form). Although preclinical studies showed efficacy of sgp130Fc treatment in animal models of inflammatory arthritis, peritonitis, inflammatory bowel disease, and colon cancer, it is not still currently clear whether this approach actually preserves mIL-6R-mediated anti-inflammatory responses.20
While IL-6R is a unique binding receptor for IL-6, the gp130 signal-transducing chain is shared by members of the IL-6 family, comprising leukemia inhibitory factor, oncostatin M, ciliary neurotrophic factor, IL-11, cardiotrophin-1, neuropoietin-1, IL-27, and IL-35.9
IL-6 signal transduction
After IL-6 binding, receptor homodimerization promotes the interaction between the gp130 chain with the tyrosine kinase JAK (Janus kinase), resulting in their mutual transactivation. In turn, JAK activation triggers three main intracellular signaling pathways, via phosphorylation of two key proteins, ie, 1) the Src Homology domain-containing protein thyrosin Phospatase-2 (SHP-2), and 2) the signal transducer and activator of transcription proteins (STAT1–STAT3). Once phosphorylated, SHP-2 can, on one hand, interact with Grb2 (growth factor receptor bound protein 2), leading to the activation of the Ras/ERK/MAPK (rat sarcoma protein/extracellular signal-regulated kinase/mitogen-activated protein kinase) cascade, and/or, on the other hand, can activate the PI3K/Akt (phosphoinositol-3 kinase/protein kinase B) pathway. On the contrary, phosphorylation of STATs proteins induces the formation of heterodimers (STAT1/STAT3) or homodimers (STAT1/STAT1 and/or STAT3/STAT3), which subsequently translocate into the nucleus. In all cases, the final result of the activation of these intracellular pathways is the induction of the transcription of multiple target genes accounting for the pleiotropic biological activities of IL-68,9,23 (Figure 1).
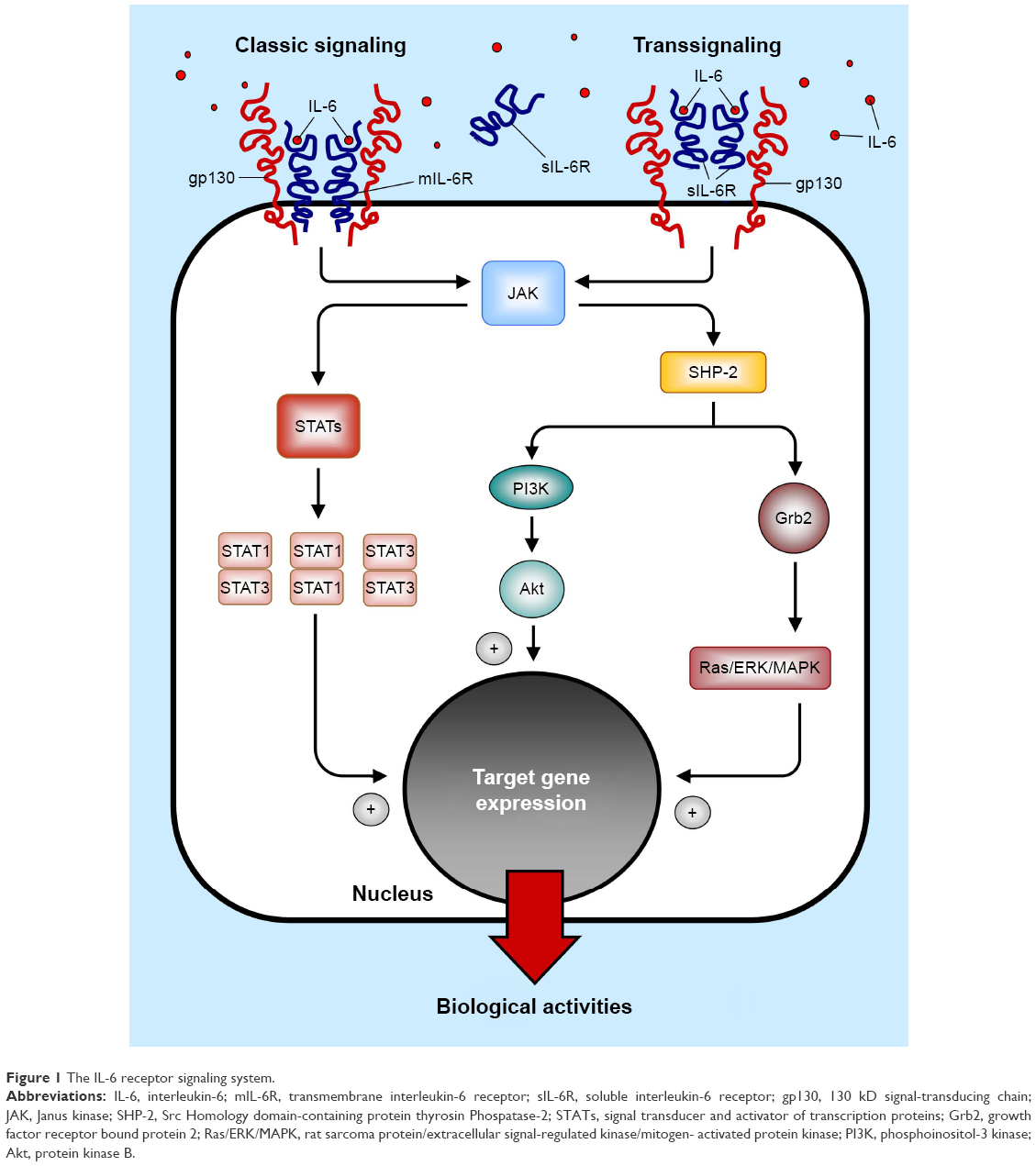 | Figure 1 The IL-6 receptor signaling system. |
Pleiotropic biological effects of IL-6
IL-6 exerts a wide range of biological activities, crucially implicated in the activation of the acute inflammatory response, as well as in the transition from innate to acquired immunity. Moreover, IL-6 has several additional roles in a variety of other processes, including metabolism, cognitive function, and embryonic development.
Effects on the immuno-inflammatory response
When infections or tissue injuries of various origins occur, a systemic reaction named acute-phase response is rapidly induced to neutralize pathogens and prevent their further invasion, minimize tissue damage, and promote wound healing. The acute-phase response, consisting of fever and production of acute-phase proteins by hepatocytes, is chiefly driven by IL-6. In fact, IL-6 increases body temperature by acting on the neurons of the preoptic hypothalamic region involved in thermoregulation24 and stimulates the liver to synthesize acute-phase proteins, such as C-reactive protein (CRP), fibrinogen, complement component C3, serum amyloid A, hepcidin, haptoglobin, α1-acid glycoprotein, α1-antitrypsin, α1-antichymotrypsin, and ceruloplasmin, while albumin, transferrin, fibronectin, transthyretin, and retinol-binding protein (“negative” acute-phase proteins) production is inhibited. In addition, IL-6 promotes monocyte-to-macrophage differentiation, stimulates the maturation of myeloid precursors and megakaryocytes leading to neutrophilia and thrombocytosis, induces angiogenesis via vascular endothelial growth factor production, enhances lymphocyte and neutrophil trafficking by upregulating adhesion molecule expression on endothelial cells (particularly, the intracellular adhesion molecule-1 and the vascular cell adhesion molecule), increases antibody production by B lymphocytes, and potentiates the proliferation of T helper (TH) lymphocytes promoting their differentiation toward TH2 or TH17 cells.8,9,16,20 In all cases, these changes contribute, via different but synergistic mechanisms, to realize an integrated response finalized to host defense.
Extra-immunoinflammatory effects
Besides its key involvement in the immune-inflammatory response, IL-6 also plays an important role under physiological conditions by modulating a number of multisystemic functions such as embryogenesis,25 glucose and lipid metabolism,26 bone remodeling,27,28 liver regeneration,29 neural tissue homeostasis, cognitive function, sleep, memory, pain, and emotional behavior.17,30,31 The knowledge of these extra-immunoinflammatory effects may help explain the pathogenesis of some systemic manifestations observed in RA and other chronic inflammatory diseases characterized by persistently elevated IL-6 levels.
In RA patients, the impact of this cytokine on metabolism and bone homeostasis may be of particular pathophysiological and clinical interest. Adipose tissue considerably contributes to IL-6 production under physiological conditions, accounting for ~35% of circulating IL-6 levels. Moreover, during prolonged exercise, contracting skeletal muscle becomes a major source of IL-6, increasing its plasma levels up to 100-fold.19,26 IL-6 stimulates lipolysis (and inhibits lipogenesis) in adipocytes, and increases cholesterol and trygliceride uptake by peripheral tissues via enhancement of the very low-density lipoprotein receptor expression, promoting body weight loss and serum lipid levels reduction. In addition, IL-6 enhances insulin sensitivity in hepatocytes and muscle cells, improving glucose utilization and tolerance. Although these effects suggest that this cytokine may be part of a physiological mechanism underlying the exercise-induced increase of insulin activity that enhances endurance, evidence indicates that chronic elevation of IL-6 conversely leads to insulin resistance in liver and adipose cells.19,26
With regard to the impact on bone tissue, IL-6 affects bone resorption and bone formation that are required for skeletal development, growth, and maintenance, by regulating differentiation and activity of osteoblasts, osteoclasts, and chondrocytes.27,28 In particular, of particular relevance is the role of IL-6 in enhancing the expression of receptor activator of nuclear factor-kB ligand (RANKL) on the surface of stromal/osteoblastic cells, which in turn stimulates osteoclast differentiation and bone resorption. Although this process can promote bone remodeling with potential positive effects for bone homeostasis under physiological conditions, in RA its exaggerated and long-lasting activation induces abnormal osteoclastogenesis, leading to osteoporosis and bone destruction.27,28,32
The role of IL-6 in RA
A large body of evidence from experimental and clinical studies has demonstrated the key role of IL-6 in the pathogenesis of RA, both in promoting basic immunopathologic mechanisms and mediating target organ damage.23,33 In particular, RA patients show elevated IL-6 levels in both synovial fluid and blood, which correlate with disease activity and structural damage progression.34–37 Moreover, IL-6-deficient mice did not develop experimental inflammatory arthritis, both in antigen- and collagen-induced models.38,39 Finally, selective IL-6 blockade with TCZ produces unequivocal clinical benefits in RA patients as a result of a marked reduction in joint involvement and systemic manifestations.6,40,41
Autoimmune process development
RA is an autoimmune disorder driven by both T- and B-lymphocyte activation. Increasing evidence indicates that IL-6 actively mediates the immunologic process underlying the disease by promoting the transition from innate to acquired immune response as well as autoantibody production. IL-6 is essential for CD4+ T-lymphocyte differentiation to TH17 cells, and it inhibits TGF-β-induced regulatory T-cell (Treg), leading to an increased TH17/Treg ratio that is thought to play a major role in RA development. Moreover, IL-6 induces B-cell differentiation through a direct action on plasmablasts, as well as by stimulating the development of T follicular helper cells, which secrete the B-cell differentiation factor IL-21.23,33 Accordingly, in RA patients, IL-6 and IL-21 serum levels are associated with increased autoantibody secretion (rheumatoid factor, RF, and anti–cyclic citrullinated protein, anti-CCP), and higher levels of markers of B-cell activation.37 Moreover, it has been demonstrated that TCZ treatment restored TH17/Treg imbalance,42,43 reduced IL-21 expression by CD4-positive T-cells,44 and inhibited RF and anti-CCP production.44,45
Articular damage
Activated synovial fibroblasts and macrophages represent a major source of IL-6 in the RA joint. The synovial fluid also contains high levels of sIL-6R, mainly deriving from monocytes and lymphocytes infiltrating into the synovium. Thus, via activation of classical signaling and, particularly, transsignaling pathway, IL-6 can target a wide range of joint cells in the synovium, cartilage, and subchondral bone, chiefly involved in the development of RA articular damage (Figure 2).8,23,33,41
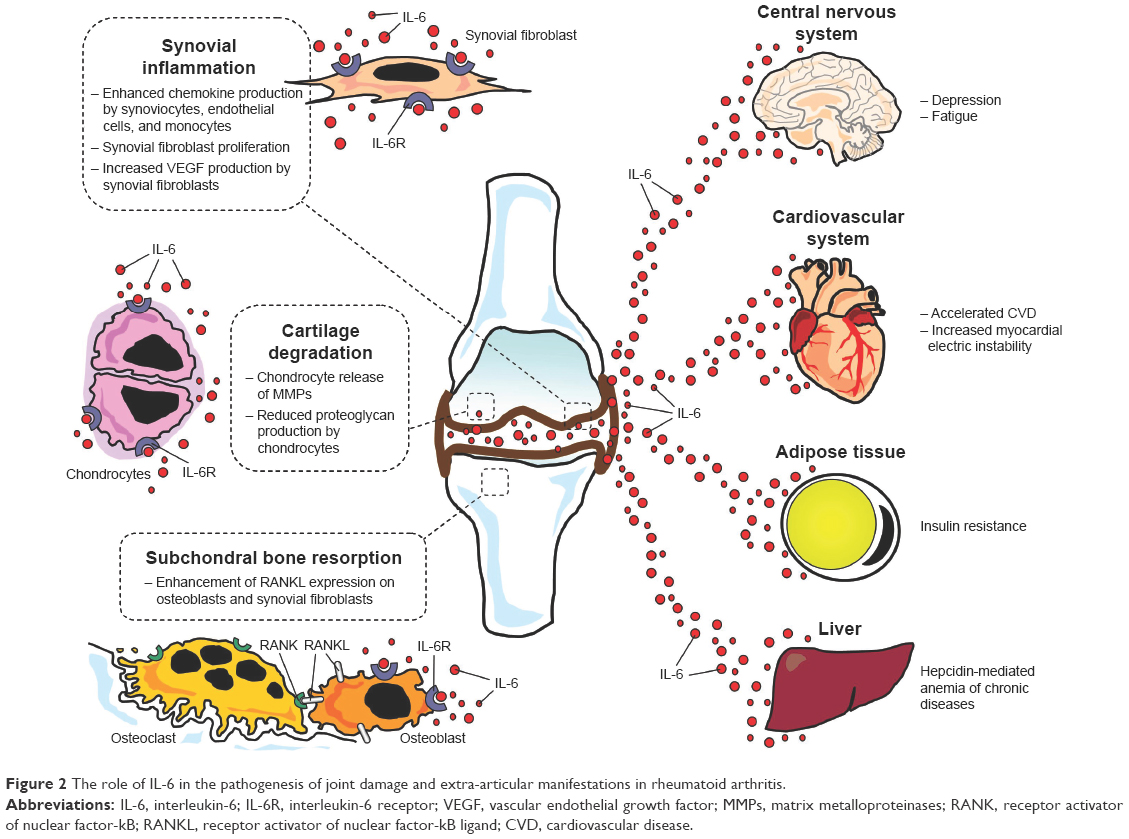 | Figure 2 The role of IL-6 in the pathogenesis of joint damage and extra-articular manifestations in rheumatoid arthritis. |
By amplifying inflammatory cell infiltration, neovascularization, and synoviocyte hyperplasia, IL-6 aggravates the synovitis process and favors the formation of the inflammatory vascular tissue known as “pannus”.8,41 In particular, IL-6 exerts potent stimulating effects on synovial fibroblastic cells, which result in cell proliferation,46 and vascular endothelial growth factor synthesis, which promotes angiogenesis.47 Moreover, IL-6 enhances chemokine production (monocyte chemotactic protein-1 and IL-8) from endothelial cells, mononuclear cells, and synovial fibroblasts, as well as adhesion molecules expression on endothelial cells, leading to increased monocyte accumulation in the synovium.16,33 Notably, these changes markedly reduced when IL-6 signaling was inhibited by using TCZ in both experimental models and clinical studies in RA patients.41
Once established, synovial inflammation drives joint structural damage by inducing cartilage degradation and bone resorption. Also in this case, IL-6 plays a key role as a result of specific effects on chondrocytes and bone cells of the subchondral bone. Indeed, IL-6 targets chondrocytes leading to production and release of matrix metalloproteinases (MMP-1, MMP-3, and MMP-13), a group of enzymes playing a crucial role in cartilage catabolism, as well as inhibition of cartilage matrix synthesis.41,48 Moreover, osteoclastogenesis is enhanced via stimulation of RANKL expression on osteoblasts and synovial fibroblasts (Figure 2).32,49 TCZ treatment reduced MMP-3 expression and increased cartilage synthesis in IL-6-stimulated cultured human chondrocytes, prevented proteoglycan loss in arthritic monkeys, and improved serum levels of cartilage turnover markers (N-terminal propeptide of Type II collagen, Type II collagen helical peptide, MMP-3) in RA patients41. Finally, TCZ significantly inhibited bone erosion and joint space narrowing, along with an improvement in bone resorption markers (C-terminal cross-linking telopeptide of Type I collagen) when administered to RA subjects for 52 weeks.33,41
Extra-articular manifestations
Besides stimulating acute-phase protein synthesis leading to elevation of circulating markers of inflammation, particularly CRP, IL-6 significantly contributes to a number of other systemic signs and symptoms frequently observed in RA patients, including fever, malaise, fatigue and muscle weakness, sleep disturbances, anxiety, and depression.41,50 Moreover, IL-6 is actively involved in the development of anemia and accelerated CVD, which represent two among the most important extra-articular manifestations of the disease (Figure 2).
Anemia occurs in 30%–60% of patients with RA. In the majority of cases, it is characterized by decreased serum iron and transferrin saturation, along with elevated serum ferritin levels. This form, termed anemia of chronic diseases, is chiefly driven by IL-6 via induction of hepcidin synthesis by the liver (Figure 2).41,50 Hepcidin is an iron-regulator hormone mediating evolutionary mechanisms contributing to host defense by decreasing iron availability to invading pathogens. However, by inhibiting iron absorption in the small intestine and the release of recycled iron from macrophages, hepcidin also leads to reduced iron delivery to maturing erythrocytes in the bone marrow.51 The severity of anemia of chronic diseases in RA strictly correlates with IL-6 levels, while TCZ treatment induces a rapid increase in hemoglobin levels as a result of an inhibitory effect on hepcidin production.41,52
In RA patients, the prevalence of CVD is significantly increased, accounting for ~50% of premature deaths observed.3 Evidence indicates that enduring systemic inflammation plays a key role in accelerating heart disease development and progression in these patients. In this scenario, inflammatory cytokines play a pivotal role through a myriad of pathogenic mechanisms. In particular, IL-6 enhances the immunoinflammatory process underlying atherogenesis either directly, affecting the cells of the plaque, or indirectly, promoting a number of metabolic changes, including insulin resistance, dyslipidemia, and prothrombotic/antifibrinolytic effects. This results in profound vascular changes including both endothelial dysfunction-dependent and endothelial-independent defects, in turn promoting atherosclerosis, as well as increased plaque inflammation leading to higher vulnerability to rupture and thrombosis.3,4 Accordingly, two large genetic studies demonstrated that the IL-6R pathway is causally implicated in the development of coronary artery disease in the general population.53 Moreover, a TCZ single dose administration reduced inflammatory response and myocardial injury in patients with acute myocardial infarction.54 In addition, in RA patients, TCZ treatment decreases insulin resistance,55 improves endothelial function and arterial stiffness,56,57 and reduces plasminogen activator inhibitor-1 serum levels.58 Furthermore, although cholesterol (total and LDL) and triglyceride levels increase, TCZ concomitantly increases HDL cholesterol with no changes in the atherogenic index, also reducing lipoprotein(a) levels, and improving HDL cholesterol efflux capacity.57,59
In RA patients, the risk of arrhythmic events is also significantly higher than in non-RA subjects, as indicated by the increased incidence of sudden cardiac death and atrial fibrillation. Although this is in part explained by accelerated structural heart disease, increasing evidence indicates that arrhythmogenicity in RA may be also the result of inflammation-driven nonstructural heart abnormalities of electrophysiological origin (Figure 2).4,60,61 In particular, it has been demonstrated that IL-6 prolongs action potential duration in pig ventricular cells by enhancing L-type calcium current,62 also increasing the propensity to develop atrial fibrillation in an in vitro model of rat atrial tissue.63 Moreover, in RA patients, circulating IL-6 levels correlated with the duration of ventricular repolarization as assessed by the corrected QT interval (QTc) on electrocardiogram,64 and TCZ therapy was associated with a rapid (within 3 months) QTc shortening, which correlated with the decrease in CRP levels.65 Finally, a recent study on a large cohort of women with RA demonstrated that inflammation, as assessed by IL-6 circulating levels, more strongly correlated with fatal than nonfatal cardiovascular events.66
Pharmacology of SRK
SRK is a human anti-IL-6 monoclonal antibody that binds to IL-6 with high affinity and specificity. It prevents IL-6 from interacting with IL-6Rs, both the transmembrane and the soluble form, thus resulting in an inhibition of IL-6-mediated signal pathways and related biological effects.
After intravenous (IV, 0.3–10 mg/kg)67 or subcutaneous (SC, 25–100 mg)68 single administration of SRK in healthy subjects, maximum concentration and area under the concentration–time curve increase in an approximately dose-proportional manner. On the contrary, the clearance and half-life (estimated value 20.9 days) of the drug appeared to be independent of dose. The volume of distribution at steady state in a subject weighing 70 kg was 8.3 L, ie, ~2 times the plasma volume, suggesting that the molecule is primarily located in the circulatory system, with limited extravascular tissue distribution. These data, considered as a whole, indicate that SRK has a linear pharmacokinetic profile (Table 1),67,68 different from the nonlinear behavior showed by TCZ. Studies demonstrated that sex and race do not significantly affect these pharmacokinetic parameters. Conversely, to date, no studies have addressed the effect of hepatic or renal impairment on the pharmacokinetics of SRK.
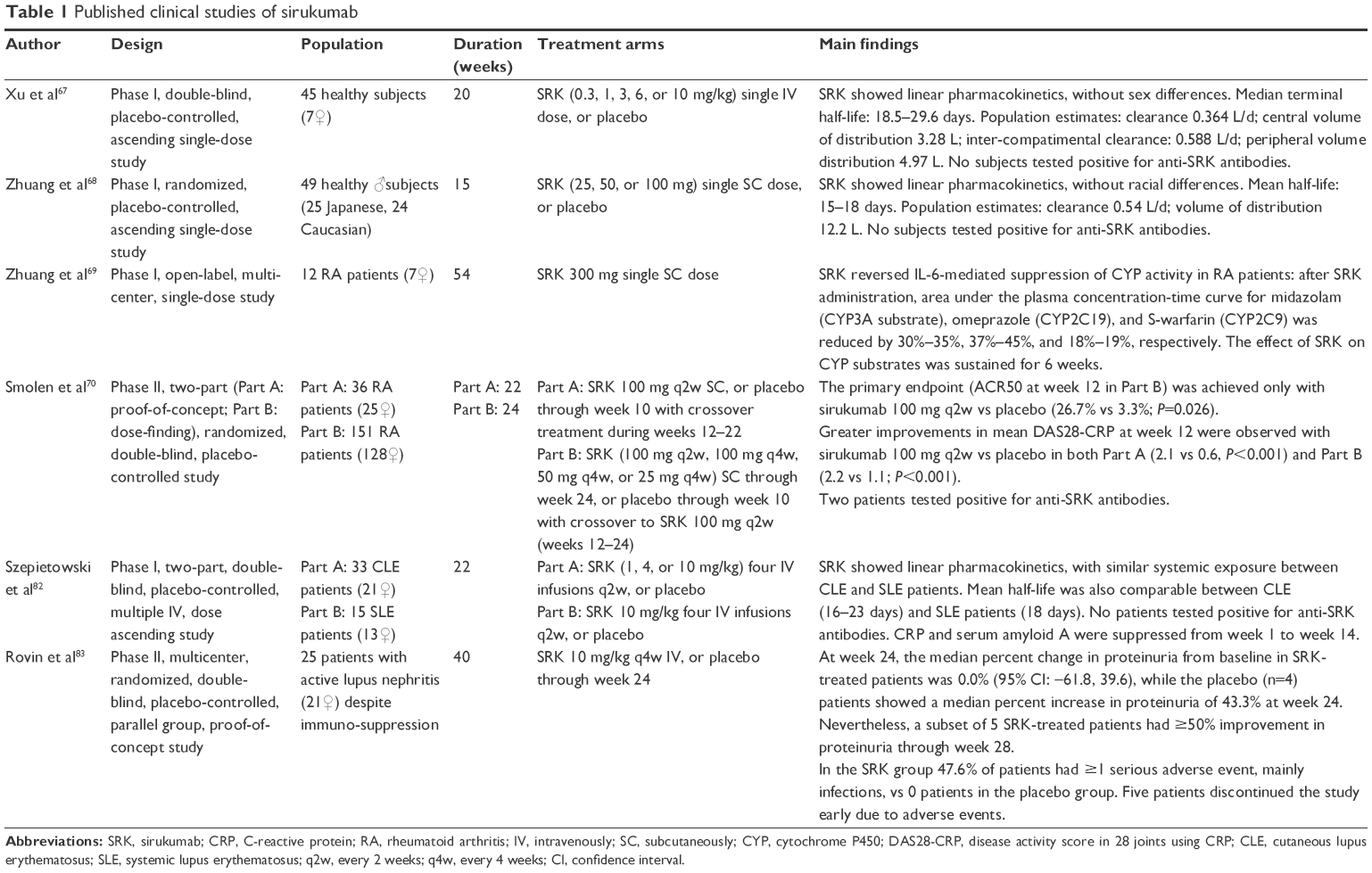 | Table 1 Published clinical studies of sirukumab |
Limited information also exists regarding the impact of this drug on cytochrome p450 (CYP) activity. Nevertheless, it is well documented that IL-6 induces inhibition of CYP enzymes, including CYP34, CYP2C19, CYP2C9, and CYP1A2, and available data suggest that SRK may reverse this effect. In particular, an in vivo study demonstrated that the levels of the CYP substrates midazolam, omeprazole, and warfarin reduced by 30%–35%, 37%–45%, and 18%–19%, respectively, 15 days after a single SC administration of SRK (300 mg) in patients with active RA. Notably, drug effect on CYP substrates was sustained for at least 6 weeks. As a result, drugs characterized by a narrow therapeutic range and profoundly influenced by CYP activity, such as warfarin, need to be closely monitored upon initiation or discontinuation of SRK.69
SRK in RA
In consideration of the aforementioned pivotal role played by IL-6 in the pathogenesis of RA, as well as of the large evidence of effectiveness demonstrated by TCZ in these patients in the clinical practice, in the past years marked attention has been focused on the potential of SRK as a new therapeutic tool in the treatment of RA.
To date, one Phase II clinical study was completed and published in 2014.70 Moreover, SRK has been or is being evaluated in five Phase III studies, among which three have been already completed or have estimated completion dates in late 2016 or early 2017 (Tables 1–3).
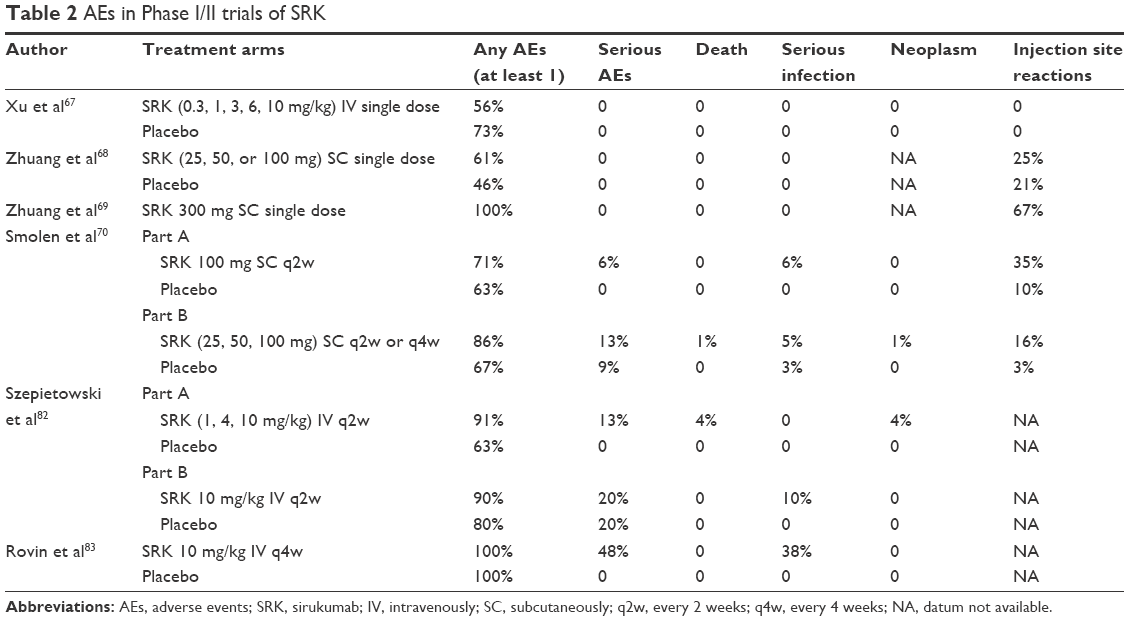 | Table 2 AEs in Phase I/II trials of SRK |
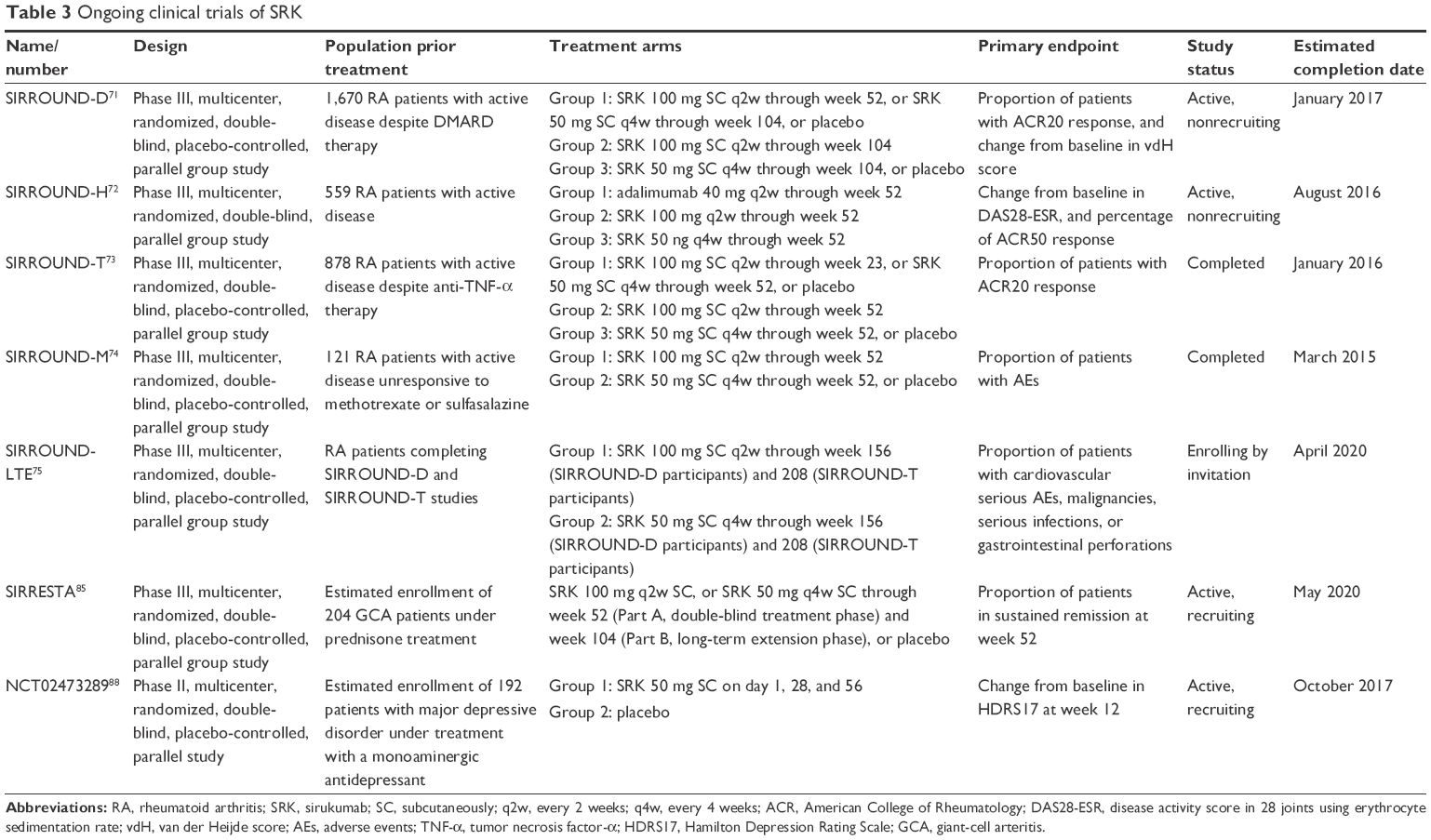 | Table 3 Ongoing clinical trials of SRK |
Phase II clinical study
Smolen et al70 conducted a randomized, two-part (proof-of-concept and dose-finding) Phase II study in patients with active RA despite methotrexate (MTX) therapy, with the primary objective represented by the evaluation of SRK safety and efficacy (Table 1).
In part A (proof of concept), 36 RA patients were randomly allocated to receive SC placebo or SRK 100 mg every 2 weeks (q2w) through week 10, with crossover treatment during weeks 12–22.
In part B (dose finding), 151 RA patients were randomized to receive SC SRK (100 mg q2w, 100 mg every 4 weeks (q4w), 50 mg q4w, or 25 mg q4w) for 24 weeks, or placebo through week 10, with crossover to SRK 100 mg q2w (weeks 12–24). The differences in the percentage of patients reaching an American College of Rheumatology 50 (ACR50) response and change from baseline in the 28-joint count disease activity score using CRP (DAS28-CRP) were determined.
The primary end point (ACR50 at week 12 in Part B) was achieved only with SRK 100 mg q2w vs placebo (26.7% vs 3.3%; P=0.026). Greater improvements in mean DAS28-CRP at week 12 were observed with SRK 100 mg q2w vs placebo in Parts A (2.1 vs 0.6, P<0.001) and B (2.2 vs 1.1; P<0.001). Clinical response to SRK occurred as early as week 2 and persisted through week 24 despite SRK crossover to placebo. SRK rapidly reduced CRP to or near the limit of detection (3.0 mg/L, within 2 weeks), also maintaining these levels through at least week 24. Changes in neutrophil and platelet counts consistent with the degree of CRP suppression over time were also observed. In both parts, transient elevations in alanine transferase were reported, more commonly within 4 weeks, with levels that declined or returned to normal without interruption of dosing. Moreover, increases in lipid levels were observed in the SRK group within 2 weeks, and sustained through week 12 (part A)–24 (part B). In particular, ~35% of SRK-treated patients with normal baseline LDL levels had abnormal values (>130 mg/dL) at week 24.
From a pharmacokinetic point of view, mean half-life values ranged from 15 to 19 days, while serum SRK concentrations achieved steady state by week 12. The incidence of adverse events (AEs) was similar for SRK- and placebo-treated patients through week 12 in Part A (70.6% and 63.2%, respectively) and B (67.8% and 66.7%, respectively). Infections were the most common type of AE. Opportunistic infections were not reported, nor were gastrointestinal perforations. One death occurred, in Part B (SRK 100 mg q2w), related to brain aneurysm rupture. Injection site reactions occurred more frequently among patients receiving SRK, but in most cases they were considered mild (Table 2). Two patients (1.2%) tested positive for antibodies to SRK: neither patient had an injection site reaction. Although one patient was also positive for neutralizing anti-SRK antibodies, he remained a responder through week 30.70
Phase III clinical studies
SRK is currently being investigated in three main Phase III clinical trials for treating patients with moderate-to-severe active RA: SIRROUND-D, SIRROUND-H, and SIRROUND-T (Table 3).
SIRROUND-D (NCT01604343) is a multicenter, randomized, double-blind, placebo-controlled, parallel group study in 1,670 patients with active RA who were unresponsive to DMARDs. The primary objective was to assess the efficacy of SC SRK as measured by the reduction of the signs and symptoms of RA and inhibition of radiographic progression. Although the estimated study completion date is January 2017,71 preliminary results have been very recently presented (June 2016) at the Annual Congress of Rheumatology (EULAR 2016) in London, UK. Compared to placebo, both SRK 50 mg q4w and 100 mg q2w significantly reduced signs/symptoms of RA (ACR20 at week 16 and ACR50 at week 24 achieved by 26.4% vs 54.8%–53.5%, and 12.4% vs 30.2%–33.2%, respectively; P≤0.001), inhibited radiographic progression, and improved health-related quality of life, showing a safety profile consistent with the known safety profile of anti-IL-6 treatment.72
SIRROUND-H (NCT02019472) is a multicenter, randomized, double-blind, parallel-group study comparing SC SRK with the anti-TNF-α-monoclonal antibody adalimumab, each given as monotherapy in 559 biologic naïve patients with moderately to severely active RA who were intolerant to MTX, who were considered inappropriate for treatment with MTX, or who were unresponsive to MTX. The primary objective was to assess the efficacy of each treatment as monotherapy. The estimated study completion date is August 2016.73
SIRROUND-T (NCT01606761) is a multicenter, randomized, double-blind, placebo-controlled, parallel group study of SRK in 878 patients with active RA who were unresponsive or intolerant to anti-TNF-α agents. The primary objective was to assess the efficacy of SC SRK as measured by the reduction of the signs and symptoms of RA. The study has been completed in January 2016, but no results have been still published.74
Two additional trials include SIRROUND-M (NCT01689532), in 122 Japanese patients with moderately to severely RA who did not respond to treatment with MTX or sulfasalazine (completed in March 2015; results still not published),75 and SIRROUND-LTE (NCT01856309), the extension study for patients who complete the SIRROUND-T and SIRROUND-D studies to evaluate long-term safety and efficacy of SRK in RA (estimated completion date April 2020); (Table 3).76
SRK vs TCZ and other IL-6 inhibitors in RA
The clinical efficacy and safety shown by SRK in RA in the available clinical studies seems to be largely overlapping those reported for TCZ and other IL-6 inhibitors, regardless that the specific inhibitory mechanism is the direct targeting of the cytokine or its receptor. In fact, besides SRK, the other two anti-IL-6 monoclonal antibodies, ie, olokizumab and clazakizumab, and the one other anti-IL-6R monoclonal antibody, ie, sarilumab, have been recently evaluated in Phase II clinical studies in RA patients.77
In particular, the differences in ACR20 and ACR50 response rates between the SRK groups vs placebo at week 12 (27%–54% and 16%–23%, respectively) reported by Smolen et al70 were consistent with those observed with TCZ (week 16: 22%–33% and 3%–24%, respectively), as well as olokizumab (week 12: 32%–61% and 11%–33%, respectively), sarilumab (week 12: 3%–26% and 7%–25%, respectively), or clazakizumab (week 16: 29%–46% and 26%–35%, respectively).
Similar considerations can be done for the drug safety profile. Also in this case, incidence and severity of commonly reported AEs, including respiratory and urinary tract infections, gastrointestinal, and nervous system disorders, showed no significant differences among different IL-6 inhibitors. Moreover, opportunistic infections, tuberculosis, or gastrointestinal perforations have not been reported so far with these new drugs, although safety data from daily clinical practice should be collected. Among laboratory changes, neutropenia, transient liver enzyme elevation, and increases in lipid levels were the most commonly observed. Exact clinical consequences and mechanisms of these abnormalities remain to be elucidated, but also in this case they appear broadly comparable in terms of frequency and magnitude among the five monoclonal antibodies.77
To exert its biological effects, IL-6 needs to bind the IL-6R: only when this complex is formed, signal pathways in target cell can be activated. For this reason, it is probable that indirect, rough comparisons show similar efficacy and safety among different IL-6 inhibitors, either anti-IL-6 or anti-IL-6R antibodies. Thus, it is currently unknown whether the inhibition of IL-6 rather than IL-6R has any advantages or disadvantages. A further, intriguing step forward will be to determine whether a specific block of IL-6 transsignaling by sgp130Fc can offer additional clinical benefits over monoclonal antibody therapy against IL-6 or IL-6R.
SRK beyond RA
As largely discussed in the first part of the review, IL-6 is a pleiotropic cytokine whose effects range from the activation of basic mechanisms of inflammation and immunity, critically involved in a number of systemic immuno-mediated diseases beyond RA, to the regulation of key physiological functions, including development, metabolism, and mental processes.
SRK is currently under investigation for other indications different to RA, including systemic lupus erythematosus (SLE), giant-cell arteritis (GCA), and major depressive disorder (Tables 1 and 3).
Systemic lupus erythematosus
Many data indicate that IL-6 is actively implicated in the pathogenesis of SLE. In particular, in SLE patients, IL-6 induces B-cell differentiation and increased production of antinuclear and anti-double-stranded DNA autoantibodies,78 while anti-IL-6 antibodies inhibited spontaneous polyclonal antibody production by SLE monocytes and lymphocytes in vitro.79 Moreover, in murine models, inhibiting IL-6 activity delayed the onset of lupus nephritis,80 and in lupus nephritis urine IL-6 levels increase with the severity of the renal involvement.81 On the basis of this rationale, two clinical studies evaluated the efficacy and/or safety of SRK administration in SLE patients. In a Phase I, randomized, double-blind, and placebo-controlled study, 46 patients with cutaneous lupus erythematosus or SLE underwent multiple SRK IV infusions (1–10 mg/kg, 4 times q2w). The drug was generally well tolerated and showed a linear pharmacokinetics as observed in previous studies in healthy subjects. Moreover, SRK suppressed CRP and serum amyloid A mean concentrations until week 14, and although the study was not designed for testing efficacy, SRK group showed a tendency to improve in the British Isles Lupus Assessment Group musculoskeletal domain score.82
These findings were not confirmed in a more recent Phase II, multicenter, randomized, double-blind, and placebo-controlled study aimed at evaluating the effects of a short-term (6 months) SRK treatment (10 mg/kg q4w) in 25 patients with active lupus nephritis. In fact, at week 24, SRK did not reduce proteinuria in the majority of patients (percent change from baseline: 0.0%). Moreover, in the SRK group, ~50% of patients had ≥1 serious AE, mostly infection related, in five cases leading to early study discontinuation83 (Tables 1 and 2).
Other diseases
Two further clinical trials evaluating SRK in GCA and major depressive disorder, respectively, are currently in progress (Table 3).
GCA is a granulomatous vasculitis affecting medium- and large-sized arteries responsible for cranial symptoms (ie, headaches, jaw claudication, and visual disturbance), polymyalgia rheumatica, and constitutional manifestations. IL-6 has been linked to GCA activity, and its blockade might abrogate signs and symptoms of the disease via a Treg/TH17 cell rebalance, critically involved in the GCA pathogenesis.84 The Phase III study SIRRESTA (NCT02531633) is a multicenter, randomized, double-blind, placebo-controlled trial, which evaluates the efficacy and safety of SC SRK with a prespecified tapering dose of prednisone for the treatment of GCA. This approach will specifically answer whether SRK treatment can reduce the duration of steroid treatment typical in clinical practice. The study started in October 2015, and estimated completion date is May 2020.85
IL-6 has been associated with major depression disorder. In particular, a recent meta-analysis showed that there was a significant elevation of IL-6 levels in the plasma of depressed patients.86 In addition, in a post hoc analysis of the data from the earlier reported Phase II clinical trial by Smolen et al,70 Hsu et al87 found that a significant percentage of RA patients (~25%) had prevalent depressed mood and anhedonia symptoms, which significantly improved upon SRK treatment, regardless of clinical response, but with a significant association with baseline sIL-6R levels. The specific molecular mechanism underlying the link between IL-6 and major depression disorder is unclear, but both stimulation of the activity of the hypothalamic–pituitary–adrenal axis and modulation of tryptophan and serotonin metabolism have been suggested.31 On this basis, a Phase II clinical trial (NCT02473289) aimed at evaluating the efficacy of SRK as adjunctive treatment to monoaminergic antidepressants in patients who have had a suboptimal response to standard therapy and a screening high sensitivity CRP ≥0.3 mg/dL started in July 2015 (estimated completion date in October 2017).88
Perspectives
IL-6 has been implicated in the pathogenesis of many autoimmune diseases different to RA and SLE, such as systemic juvenile idiopathic arthritis, adult-onset Still’s disease, Takayasu arteritis, sarcoidosis, as well as in a wide number of other disorders, ranging from malignancies to spinal disease, allograft rejection after organ transplantation, graft-versus host disease, and CVD.6 Although all these conditions may theoretically represent intriguing fields of application for SRK treatment, to date no clinical trial or preclinical study is available or in progress to test this possibility, perhaps with the only exception of CVD.
RA and atherosclerosis are chronic inflammatory diseases that share many pathological and molecular features. Indeed, having RA doubles the risk of CVD compared to non-RA subjects.3,4 In particular, elevated circulating IL-6 is an independent risk factor for CVD in the general population,53 and it also correlates with disease activity and progression in RA patients.35,36 On this basis, the inhibitory potential of SRK on vascular inflammation was recently evaluated in a human in vitro model of RA-associated CVD in comparison with other RA drugs including TCZ, the anti-TNF-α monoclonal antibody adalimumab, and the small-molecule JAK inhibitor tofacinib. By using a human endothelial cell and smooth muscle cell coculture system in which in vivo circulating concentrations of oxidized LDL, TNF-α, and sIL-6R were added, and atheroprone flow conditions applied, Feaver et al89,90 demonstrated a robust transcriptional response of inflammatory genes and oxidative stress pathways compared to control conditions. Anti-IL-6 treatment with SRK significantly improved the vascular health phenotype by significantly decreasing adhesion molecule and nuclear factor-kB gene expression, while simultaneously increasing vasculoprotective responses, such as endothelial nitric oxide synthase and krüppel-like factor expression, and promoting a contractile smooth muscle cell phenotype. Although broadly comparable, SRK was more potent than TCZ in suppressing vascular inflammation or promoting vascular health. Adalimumab showed a similar but weaker trend compared to IL-6 inhibitors, while tofacinib was not effective or exacerbated CVD pathways. These results, although preclinical, are consistent with preliminary clinical data showing improvement of endothelial function and arterial stiffness in small groups of RA patients after treatment with TCZ,56,57 and support the hypothesis that SRK-dependent IL-6 inhibition may provide vascular protection in RA, and possibly beyond. Large clinical trials are warranted to confirm this hypothesis that may open new intriguing avenues in the treatment of CVD.
Conclusion
The evidence to date indicates SRK as an effective and well-tolerated new therapeutic tool for patients with active RA, with some preliminary data suggesting a specific beneficial impact on relevant systemic complications associated with the disease, such as depression and CVD. Conversely, although pathophysiological considerations make plausible the hypothesis that IL-6 blockade with SRK may be also beneficial in the treatment of many diseases other than RA (either autoimmune or not), available clinical data in patients with SLE do not seem to support this view, giving rise to potentially relevant concerns about drug safety. If the large Phase III clinical trials currently in progress in patients with RA confirm the efficacy and tolerability of SRK in these patients, then in the long term, this drug could in the near future occupy a place in the treatment of the disease, potentially also opening the doors to a more extended use of SRK in a wide range of disorders in which IL-6 plays a key pathogenic role.
Disclosure
The authors report no conflicts of interests in this work.
References
Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376(9746):1094–1108. | ||
Cross M, Smith E, Hoy D, et al. The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73(7):1316–1322. | ||
Gabriel SE. Cardiovascular morbidity and mortality in rheumatoid arthritis. Am J Med. 2008;121(10 Suppl 1):S9–S14. | ||
Lazzerini PE, Capecchi PL, Laghi-Pasini F. Systemic inflammation and arrhythmic risk: lessons from rheumatoid arthritis. Eur Heart J. 2016. pii: ehw208. Epub June 1, 2016. | ||
Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118(11):3537–3545. | ||
Shetty A, Hanson R, Korsten P, et al. Tocilizumab in the treatment of rheumatoid arthritis and beyond. Drug Des Devel Ther. 2014;8:349–364. | ||
Reichert JM. Antibodies to watch in 2016. MAbs. 2016;8(2):197–204. | ||
Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (Lond). 2012;122(4):143–159. | ||
Tanaka T, Kishimoto T. The biology and medical implications of interleukin-6. Cancer Immunol Res. 2014;2(4):288–294. | ||
Lazzerini PE, Capperucci C, Spreafico A, et al. Rosuvastatin inhibits spontaneous and IL-1β-induced interleukin-6 production from human cultured osteoblastic cells. Joint Bone Spine. 2013;80(2):195–200. | ||
van Aken BE, Jansen J, van Deventer SJ, Reitsma PH. Elevated levels of homocysteine increase IL-6 production in monocytic Mono Mac 6 cells. Blood Coagul Fibrinolysis. 2000;11(2):159–164. | ||
Lazzerini PE, Selvi E, Lorenzini S, et al. Homocysteine enhances cytokine production in cultured synoviocytes from rheumatoid arthritis patients. Clin Exp Rheumatol. 2006;24(4):387–393. | ||
Inoue K, Hosoi J, Denda M. Extracellular ATP has stimulatory effects on the expression and release of IL-6 via purinergic receptors in normal human epidermal keratinocytes. J Invest Dermatol. 2007;127(2):362–371. | ||
Caporali F, Capecchi PL, Gamberucci A, et al. Human rheumatoid synoviocytes express functional P2X7 receptors. J Mol Med. 2008;86(8):937–949. | ||
Ancey C, Corbi P, Froger J, et al. Secretion of IL-6, IL-11 and LIF by human cardiomyocytes in primary culture. Cytokine. 2002;18(4):199–205. | ||
Hou T, Tieu BC, Ray S, et al. Roles of IL-6-gp130 signaling in vascular inflammation. Curr Cardiol Rev. 2008;4(3):179–192. | ||
Trapero I, Cauli O. Interleukin 6 and cognitive dysfunction. Metab Brain Dis. 2014;29(3):593–608. | ||
Fonseca JE, Santos MJ, Canhão H, Choy E. Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun Rev. 2009;8(7):538–542. | ||
Febbraio MA, Pedersen BK. Muscle-derived interleukin-6: mechanisms for activation and possible biological roles. FASEB J. 2002;16(11):1335–1347. | ||
Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813(5):878–888. | ||
Wolf J, Rose-John S, Garbers C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine. 2014;70(1):11–20. | ||
Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300(5628):2101–2104. | ||
Yoshida Y, Tanaka T. Interleukin 6 and rheumatoid arthritis. Biomed Res Int. 2014;2014:article 698313. | ||
Conti B, Tabarean I, Andrei C, Bartfai T. Cytokines and fever. Front Biosci. 2004;9:1433–1449. | ||
Onishi K, Zandstra PW. LIF signaling in stem cells and development. Development. 2015;142(13):2230–2236. | ||
Glund S, Krook A. Role of interleukin-6 signalling in glucose and lipid metabolism. Acta Physiol. 2008;192(1):37–48. | ||
Franchimont N, Wertz S, Malaise M. Interleukin-6: an osteotropic factor influencing bone formation? Bone. 2005;37(5):601–606. | ||
Sims NA, Walsh NC. GP130 cytokines and bone remodelling in health and disease. BMB Rep. 2010;43(8):513–523. | ||
Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: from physiopathology to therapy. J Hepatol. 2016;64(6):1403–1415. | ||
Rothaug M, Becker-Pauly C, Rose-John S. The role of interleukin-6 signaling in nervous tissue. Biochim Biophys Acta. 2016;1863(6 Pt A):1218–1227. | ||
Spooren A, Kolmus K, Laureys G, et al. Interleukin-6, a mental cytokine. Brain Res Rev. 2011;67(1–2):157–183. | ||
Edwards CJ, Williams E. The role of interleukin-6 in rheumatoid arthritis-associated osteoporosis. Osteoporos Int. 2010;21(8):1287–1293. | ||
Hashizume M, Mihara M. The roles of interleukin-6 in the pathogenesis of rheumatoid arthritis. Arthritis. 2011;2011:765624. | ||
Houssiau FA, Devogelaer JP, Van Damme J, de Deuxchaisnes CN, Van Snick J. Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum. 1988;31(6):784–788. | ||
Madhok R, Crilly A, Watson J, Capell HA. Serum interleukin 6 levels in rheumatoid arthritis: correlations with clinical and laboratory indices of disease activity. Ann Rheum Dis. 1993;52(3):232–234. | ||
Baillet A, Gossec L, Paternotte S, et al. Evaluation of serum interleukin-6 level as a surrogate marker of synovial inflammation and as a factor of structural progression in early rheumatoid arthritis: results from a French national multicenter cohort. Arthritis Care Res. 2015;67(7):905–912. | ||
Gottenberg JE, Dayer JM, Lukas C, et al. Serum IL-6 and IL-21 are associated with markers of B cell activation and structural progression in early rheumatoid arthritis: results from the ESPOIR cohort. Ann Rheum Dis. 2012;71(7):1243–1248. | ||
Alonzi T, Fattori E, Lazzaro D, et al. Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187(4):461–468. | ||
Ohshima S, Saeki Y, Mima T, et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci U S A. 1998;95(14):8222–8226. | ||
Patel AM, Moreland LW. Interleukin-6 inhibition for treatment of rheumatoid arthritis: a review of tocilizumab therapy. Drug Des Devel Ther. 2010;4:263–278. | ||
Hashizume M, Tan SL, Takano J, et al. Tocilizumab, a humanized anti-IL-6R antibody, as an emerging therapeutic option for rheumatoid arthritis: molecular and cellular mechanistic insights. Int Rev Immunol. 2015;34(3):265–279. | ||
Guggino G, Giardina AR, Raimondo S, et al. Targeting IL-6 signalling in early rheumatoid arthritis is followed by Th1 and Th17 suppression and Th2 expansion. Clin Exp Rheumatol. 2014;32(1):77–81. | ||
Thiolat A, Semerano L, Pers YM, et al. Interleukin-6 receptor blockade enhances CD39+ regulatory T cell development in rheumatoid arthritis and in experimental arthritis. Arthritis Rheumatol. 2014;66(2):273–283. | ||
Carbone G, Wilson A, Diehl SA, Bunn J, Cooper SM, Rincon M. Interleukin-6 receptor blockade selectively reduces IL-21 production by CD4 T cells and IgG4 autoantibodies in rheumatoid arthritis. Int J Biol Sci. 2013;9(3):279–288. | ||
Iannone F, Tampoia M, Giannini M, et al. Changes in anti-cyclic citrullinated peptide antibodies and rheumatoid factor isotypes serum levels in patients with rheumatoid arthritis following treatment with different biological drugs. Clin Exp Rheumatol. 2016;34(3):424–429. | ||
Mihara M, Moriya Y, Kishimoto T, Ohsugi Y. Interleukin-6 (IL-6) induces the proliferation of synovial fibroblastic cells in the presence of soluble IL-6 receptor. Br J Rheumatol. 1995;34(4):321–325. | ||
Nakahara H, Song J, Sugimoto M, et al. Anti-interleukin-6 receptor antibody therapy reduces vascular endothelial growth factor production in rheumatoid arthritis. Arthritis Rheum. 2003;48(6):1521–1529. | ||
Aida Y, Honda K, Tanigawa S, et al. IL-6 and soluble IL-6 receptor stimulate the production of MMPs and their inhibitors via JAK-STAT and ERK-MAPK signalling in human chondrocytes. Cell Biol Int. 2012;36(4):367–376. | ||
Hashizume M, Hayakawa N, Mihara M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology (oxford). 2008;47(11):1635–1640. | ||
Moreland LW, Curtis JR. Systemic nonarticular manifestations of rheumatoid arthritis: focus on inflammatory mechanisms. Semin Arthritis Rheum. 2009;39(2):132–143. | ||
Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011;117(17):4425–4433. | ||
Isaacs JD, Harari O, Kobold U, Lee JS, Bernasconi C. Effect of tocilizumab on haematological markers implicates interleukin-6 signalling in the anaemia of rheumatoid arthritis. Arthritis Res Ther. 2013;15(6):R204. | ||
Boekholdt SM, Stroes ES. The interleukin-6 pathway and atherosclerosis. Lancet. 2012;379(9822):1176–1178. | ||
Kleveland O, Kunszt G, Bratlie M, et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. Eur Heart J. pii:ehw171. Epub May 8, 2016. | ||
Chen DY, Chen YM, Hsieh TY, Hsieh CW, Lin CC, Lan JL. Significant effects of biologic therapy on lipid profiles and insulin resistance in patients with rheumatoid arthritis. Arthritis Res Ther. 2015;17:52. | ||
Protogerou AD, Zampeli E, Fragiadaki K, Stamatelopoulos K, Papamichael C, Sfikakis PP. A pilot study of endothelial dysfunction and aortic stiffness after interleukin-6 receptor inhibition in rheumatoid arthritis. Atherosclerosis. 2011;219(2):734–736. | ||
McInnes IB, Thompson L, Giles JT, et al. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann Rheum Dis. 2015;74(4):694–702. | ||
Makrilakis K, Fragiadaki K, Smith J, Sfikakis PP, Kitas GD. Interrelated reduction of chemerin and plasminogen activator inhibitor-1 serum levels in rheumatoid arthritis after interleukin-6 receptor blockade. Clin Rheumatol. 2015;34(3):419–427. | ||
Ormseth MJ, Yancey PG, Solus JF, et al. Effect of drug therapy on net cholesterol efflux capacity of HDL-enriched serum in rheumatoid arthritis. Arthritis Rheumatol. doi:10.1002/art.39675. Epub March 18, 2016. | ||
Lazzerini PE, Capecchi PL, Acampa M, Galeazzi M, Laghi-Pasini F. Arrhythmic risk in rheumatoid arthritis: the driving role of systemic inflammation. Autoimmun Rev. 2014;13(9):936–944. | ||
Lazzerini PE, Capecchi PL, Laghi-Pasini F. Long QT syndrome: an emerging role for inflammation and immunity. Front Cardiovasc Med. 2015;2:26. doi:10.3389/fcvm.2015.00026. | ||
Hagiwara Y, Miyoshi S, Fukuda K, et al. SHP2-mediated signaling cascade through gp130 is essential for LIF-dependent I CaL, [Ca2+]i transient, and APD increase in cardiomyocytes. J Mol Cell Cardiol. 2007;43(6):710–716. | ||
Mitrokhin VM, Mladenov MI, Kamkin AG. Effects of interleukin-6 on the bio-electric activity of rat atrial tissue under normal conditions and during gradual stretching. Immunobiology. 2015;220(9):1107–1112. | ||
Adlan AM, Panoulas VF, Smith JP, Fisher JP, Kitas GD. Association between corrected QT interval and inflammatory cytokines in rheumatoid arthritis. J Rheumatol. 2015;42(3):421–428. | ||
Lazzerini PE, Acampa M, Capecchi PL, et al. Antiarrhythmic potential of anti-cytokine therapy in rheumatoid arthritis: tocilizumab reduces QTc interval by controlling systemic inflammation. Arthritis Care Res (Hoboken). 2015;67(3):332–339. | ||
Mackey RH, Kuller LH, Deane KD, et al. Rheumatoid arthritis, anti-cyclic citrullinated peptide positivity, and cardiovascular disease risk in the women’s health initiative. Arthritis Rheumatol. 2015;67(9):2311–2322. | ||
Xu Z, Bouman-Thio E, Comisar C, et al. Pharmacokinetics, pharmacodynamics and safety of a human anti-IL-6 monoclonal antibody (sirukumab) in healthy subjects in a first-in-human study. Br J Clin Pharmacol. 2011;72(2):270–281. | ||
Zhuang Y, Xu Z, de Vries DE, et al. Pharmacokinetics and safety of sirukumab following a single subcutaneous administration to healthy Japanese and Caucasian subjects. Int J Clin Pharmacol Ther. 2013;51(3):187–199. | ||
Zhuang Y, de Vries DE, Xu Z, et al. Evaluation of disease-mediated therapeutic protein-drug interactions between an anti-interleukin-6 monoclonal antibody (sirukumab) and cytochrome P450 activities in a phase 1 study in patients with rheumatoid arthritis using a cocktail approach. J Clin Pharmacol. 2015;55(12):1386–1394. | ||
Smolen JS, Weinblatt ME, Sheng S, Zhuang Y, Hsu B. Sirukumab, a human anti-interleukin-6 monoclonal antibody: a randomised, 2-part (proof-of-concept and dose-finding), phase II study in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2014;73(9):1616–1625. | ||
Janssen Research & Development. A study of CNTO 136 (sirukumab), administered subcutaneously, in patients with active rheumatoid arthritis despite disease-modifying antirheumatic drug (DMARD) therapy (SIRROUND-D). Available from: https://clinicaltrials.gov/ct2/show/NCT01604343?term=NCT01604343&rank=1. NLM identifier: NCT01604343. Accessed August 9, 2016. | ||
Akeuchi T, Thorne C, Karpouzas G, et al. Efficacy and safety of sirukimab in patients with active rheumatoid arthritis despite disease-modifying anti-rheumatic drug treatment: results of a randomized, doubled-blind, placebo-controlled study. EULAR; London, UK; 2016: Abstract SAT0145. | ||
Janssen Research & Development. A study comparing sirukumab (CNTO 136) monotherapy with adalimumab (HUMIRA®) monotherapy in the treatment of active rheumatoid arthritis (SIRROUND-H). Available from: https://clinicaltrials.gov/ct2/show/NCT02019472?term=NCT02019472&rank=1. NLM identifier: NCT01604343. Accessed August 9, 2016. | ||
Janssen Research & Development. A study of CNTO 136 (sirukumab), a human anti-IL-6 monoclonal antibody, administered subcutaneously, in patients with active rheumatoid arthritis despite anti-TNF-alpha therapy (SIRROUND-T). Available from: https://clinicaltrials.gov/ct2/show/NCT01606761?term=NCT01606761&rank=1. NLM identifier: NCT01606761. Accessed August 9, 2016. | ||
Janssen Research & Development. A study of CNTO 136 (sirukumab) administered subcutaneously in Japanese patients with active rheumatoid arthritis unresponsive to methotrexate or sulfasalazine. Available from: https://clinicaltrials.gov/ct2/show/NCT01689532?term=NCT01689532&rank=1. NLM identifier: NCT01689532. Accessed August 9, 2016. | ||
Janssen Research & Development. Long-term safety and efficacy of sirukumab in participants With RA completing studies CNTO136ARA3002 or CNTO136ARA3003 (SIRROUND-LTE). Available from: https://clinicaltrials.gov/ct2/show/NCT01856309?term=NCT01856309&rank=1. NLM identifier: NCT01856309. Accessed August 9, 2016. | ||
Tanaka Y, Martin Mola E. IL-6 targeting compared to TNF targeting in rheumatoid arthritis: studies of olokizumab, sarilumab and sirukumab. Ann Rheum Dis. 2014;73(9):1595–1597. | ||
Kitani A, Hara M, Hirose T, et al. Autostimulatory effects of IL-6 on excessive B cell differentiation in patients with systemic lupus erythematosus: analysis of IL-6 production and IL-6R expression. Clin Exp Immunol. 1992;88(1):75–83. | ||
Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol. 1991;147(1):117–123. | ||
Liang B, Gardner DB, Griswold DE, Bugelski PJ, Song XY. Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology. 2006;119(3):296–305. | ||
Iwano M, Dohi K, Hirata E, et al. Urinary levels of IL-6 in patients with active lupus nephritis. Clin Nephrol. 1993;40(1):16–21. | ||
Szepietowski JC, Nilganuwong S, Wozniacka A, et al. Phase I, randomized, double-blind, placebo-controlled, multiple intravenous, dose-ascending study of sirukumab in cutaneous or systemic lupus erythematosus. Arthritis Rheum. 2013;65(10):2661–2671. | ||
Rovin B, van Vollenhoven R, Aranow C, et al. A multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of treatment with sirukumab (CNTO 136) in patients with active lupus nephritis. Arthritis Rheumatol. doi:10.1002/art.39722. Epub April 25, 2016. | ||
Guillevin L, Régent A. Treating giant-cell arteritis: is IL-6 the cytokine to target? Lancet. 2016;387(10031):1882–1883. | ||
GlaxoSmithKline. Efficacy and safety study of sirukumab in patients with giant cell arteritis. Available from: https://clinicaltrials.gov/ct2/show/NCT02531633?term=NCT02531633&rank=1. NLM identifier: NCT02531633. Accessed Aug 9, 2016. | ||
Dowlati Y, Herrmann N, Swardfager W, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67(5):446–457. | ||
Hsu B, Wang D, Sun Y, Chen G. Improvement in measures of depressed mood and anhedonia, and fatigue, in a randomized, placebo-controlled, phase 2 study of sirukumab, a human anti-interleukin-6 antibody, in patients with rheumatoid arthritis. Ann Rheum Dis. 2015;74(Suppl 2):720–721. doi:10.1136/annrheumdis-2015-eular.4081. | ||
Janssen Research & Development. An efficacy and safety study of sirukumab in participants with major depressive disorder. Available from: https://clinicaltrials.gov/ct2/show/NCT02473289?term=NCT02473289&rank=1. NLM identifier: NCT02473289. Accessed Aug 9, 2016. | ||
Feaver R, Collado S, Hoang S, et al. The anti-IL-6 antibody sirukumab inhibits vascular inflammation in a human surrogate model of atherosclerosis. 2014 ACR/ARHP Annual Meeting. Abstract number: 439. Available from: http://acrabstracts.org/abstract/the-anti-il-6-antibody-sirukumab-inhibits-vascular-inflammation-in-a-human-surrogate-model-of-atherosclerosis/. Accessed Aug 9, 2016. | ||
Feaver R, Collado S, Hoang S, et al. Neutralization of IL6 by sirukumab (SIR) inhibits inflammation and cellular stress in a human vascular surrogate system of atherosclerosis. Ann Rheum Dis. 2015;74:444–445. doi:10.1136/annrheumdis-2015-eular.5132. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.