")
Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 7
Spotlight on anagrelide hydrochloride for the treatment of essential thrombocythemia
Authors Sarma A, Mclornan DP, Harrison CN
Received 7 September 2016
Accepted for publication 7 October 2016
Published 31 January 2017 Volume 2017:7 Pages 11—23
DOI https://doi.org/10.2147/ODRR.S93451
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Lise Aagaard
Anita Sarma,1 Donal P Mclornan,1,2 Claire N Harrison2
1Department of Haematology, King’s College Hospital NHS Foundation Trust, 2Department of Haematology, Guy’s and St Thomas’ NHS Foundation Trust, London, UK
Abstract: Anagrelide (ANA) hydrochloride is an oral imidazoquinazoline registered as an orphan drug in Europe. It is indicated as a second-line agent for the reduction of thrombocytosis in high-risk essential thrombocythemia (ET) in Europe and in any myeloproliferative neoplasm-associated thrombocytosis and for the amelioration of thrombo-hemorrhagic events in the context of myeloproliferative neoplasm in the USA and Japan. The compound has been in clinical use for almost two decades with approval in the European Union (EU) for over a decade. The licensed indication encompasses the apparently specific action of the drug to reduce the platelet count; however, the precise mode of action of ANA remains unclear. Here, we review the current data from two large phase 3 studies, PT-1 and ANAHDRET, and a phase 4 post-approval observational study EXELS. All of these studies were conducted in the EU and therefore pertain to ET as the only licensed indication. Data from these studies suggest that ANA is on the whole as effective as the most commonly used agent hydroxycarbamide (HC). Although ANA, when compared to HC, appears to be slightly less effective in preventing arterial thrombosis and myelofibrotic transformation, it is associated with lower risk of venous thrombosis. Since the initial data from the PT-1 study, a caution has been recommended for the combined use of ANA and aspirin as this may provoke excess hemorrhage. ET is a clinically and biologically heterogeneous condition, and these biological variations may in part explain some of the clinical differences observed in various studies in response to specific treatments. No new toxicities of ANA have emerged in the past decade, which means that clinicians and patients can be reassured about the efficacy and safety of this agent, which is of particular importance in a chronic condition such as ET.
Keywords: anagrelide, essential thrombocythemia, PT-1, ANAHYDRET, EXELS
Introduction
Essential thrombocythemia (ET) is a clonal myeloproliferative neoplasm (MPN) with an incidence of 0.5–2.5 per 100,000 population per year and a slight female predominance. It is characterized by a sustained platelet count ≥450×109/L, without evidence of reactive causes, iron deficiency and exclusion of other MPNs, but equally of importance in making the diagnosis is the exclusion of myelodysplasia and chronic myeloid leukemia. The current diagnostic criteria for ET according to the 2016 World Health Organization (WHO) classification1 and recently updated British Committee for Standards in Haematology (BCSH) guidelines2 are shown in Table 1.
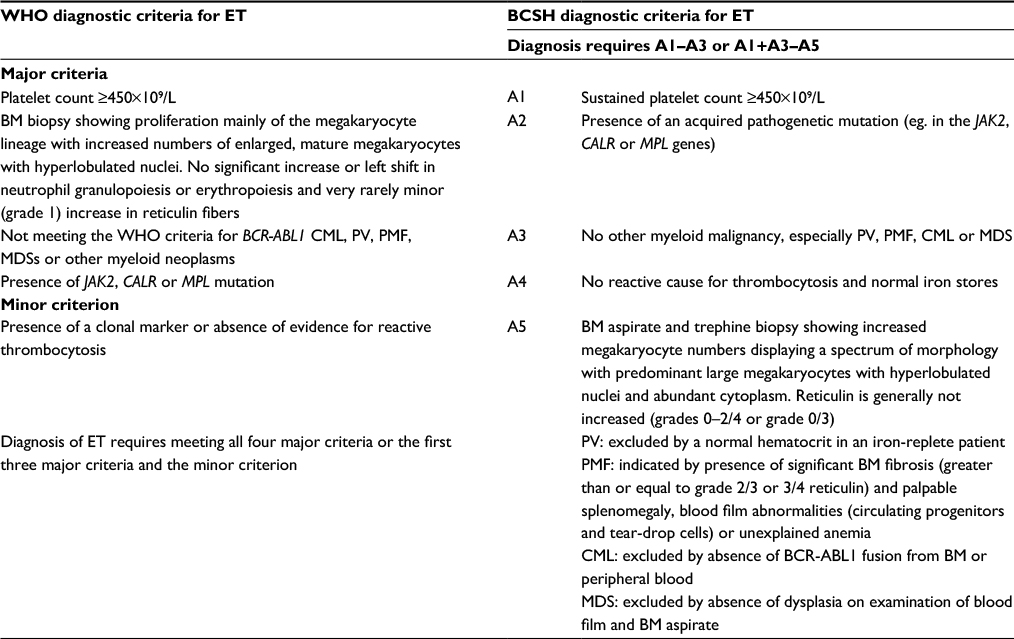 | Table 1 Comparison of the revised 2016 WHO1 and proposed 2014 BCSH diagnostic criteria2 for ET Note: Data from Arber et al1 and Harrison et al.2 Abbreviations: WHO, World Health Organization; BCSH, British Committee for Standards in Haematology; ET, essential thrombocythemia; BM, bone marrow; CML, chronic myeloid leukemia; PV, polycythemia vera; PMF, primary myelofibrosis; MDS, myelodysplastic syndrome. |
Approximately 50–60% of ET cases harbor a somatically acquired mutation in the JAK2 gene. The activating point mutation V617F in exon 14 is the most common mutation across all MPNs; more rarely, insertions or deletions are identified in exon 12 of JAK2, which are almost exclusively associated with a diagnosis of polycythemia vera (PV), a related MPN.3 Twenty to thirty percent of JAK2-unmutated ETs are associated with insertion/deletion mutations in exon 9 of the CALR gene. Overall, 3–5% of ET patients have mutations in the thrombopoietin receptor, also known as the MPL protein, and the remainder are considered triple-negative without a known genetic defect.4,5 Recent work suggests that these patients may either have hereditary thrombocytosis or harbor as yet less well-described mutations in MPL or JAK2.6–8
Patients with ET are commonly asymptomatic, but may throughout the course of the disease develop microvascular (headache, dizziness, erythromelalgia), thromboembolic or hemorrhagic complications. Disease progression to myelofibrosis (MF), termed post-ET myelofibrosis (PET-MF), in ET is thought to be uncommon particularly when strict interpretation of the WHO diagnostic criteria is applied, making the distinction between the so-called true ET and prefibrotic MF.9
This is a contentious issue, and experts in the field remain divided as to its utility. It is important to note, however, that the WHO classification places considerable emphasis on bone marrow morphology as a key feature. Training in the use of these criteria was shown in a Danish publication to increase reproducibility and interobserver variation in applying the WHO criteria, but even after such training, there is still significant lack of agreement.10 This is in contrast to the current BCSH criteria, which similar to the previous Polycythemia Vera Study Group (PVSG) classification has a greater focus on the exclusion of other subtypes of MPNs or myelodysplastic syndromes.11 In a recent analysis, Gisslinger et al12 demonstrated equivalence of the WHO and BCSH criteria when each were correctly applied in terms of predicting overall survival and progression to PET-MF.13 As noted in correspondence associated with this analysis, it is likely that improved understanding of the molecular disease-driving mechanisms will better improve risk stratification and effective management.13
As a clonal disorder, ET shows a weak propensity to transform to acute myeloid leukemia (AML).14 Exact figures are skewed by environmental factors and the use of potentially leukemogenic agents in the treatment of ET. However, according to data from the Swedish registry, 25% of patients whose disease ultimately transformed to AML were untreated, thus supporting the idea that the risk of transformation is at least in part intrinsic to the disease.15 It is likely that the observed transformation rate is not a true reflection of actual risk, as the lag period to transformation, of any kind, is usually long. This means that accurate data, regarding risk of PET-MF and secondary leukemia and any negative or positive treatment-related modifiers, are extremely difficult to accurately quantify and study.
Although at present ET is a disease that is regarded as incurable, life expectancy appears to be almost comparable to age-matched peers at least for the initial decade of disease, but beyond that, there is a degree of uncertainty. Risk stratification in ET is used to determine which patients should receive cytoreductive therapy (CRT) to prevent thrombosis; it should be noted that such therapy has not been shown to prolong survival. Here, older patients (age >60 years) and/or those who have already experienced thromboembolic events are deemed to be at high risk. Formal risk stratification tools such as the international prognostic score for the risk of thrombosis in ET (IPSET-thrombosis) lack prospective validation limiting their use.14 It is generally recommended that patients identified to be at high risk should receive platelet-reductive therapy and aspirin (ASA).16,17 Effective treatment is regarded as one that achieves platelet count reduction, minimizes thrombo-hemorrhagic events and controls microvascular symptoms without adversely increasing risk of disease progression or causing major side effects. Of note, recent attention has focused upon leukocytosis as a risk factor for thrombosis, although there is little consensus on reducing the leukocyte count. Response criteria for ET have been produced by the European LeukemiaNet (ELN),18 but these have not been designed for standard clinical use; instead, they are suggested for use in clinical trials. Moreover, the utility of some aspects such as molecular and histological responses has been questioned.
Therapy for ET must therefore include anti-aggregatory therapy, targeted treatment of cardiovascular risk factors and in selected patients cytoreduction to achieve platelet number normalization (<400×109/L), a normal spleen size and a white blood cell (WBC) count of <10×109/L with no disease-related symptoms.
Current evidence suggests that hydroxycarbamide (HC) in combination with ASA is the most efficacious and most cost-effective treatment for high-risk ET. However, in patients who are treatment refractory or are intolerant to HC, anagrelide (ANA) is licensed as second-line treatment in the European Union (EU); it is also licensed as first-line treatment in Japan and the USA. The number of other cytoreductive agents for patients with ET is limited to interferon alpha (IFN α), which is unlicensed, and a handful of older drugs with more significant leukemogenic potential such as busulfan, pipobroman or radioactive phosphorus. Experimental therapies currently being explored in ET include JAK inhibitors, histone deactylase inhibitors and the telomerase inhibitor imetelstat.19
This review is an update on studies evaluating the use of ANA in the treatment of thrombocytosis in the context of MPNs with particular emphasis on its use in high-risk ET. Current evidence suggests an important role of ANA in the treatment of individuals for whom HC is inappropriate or inadequate, or might be associated with long-term toxicity, such as younger patients.
ANA and ET
ANA hydrochloride monohydrate Xagrid® (Shire, Dublin, Ireland), Thromboreductin® (AOP Orphan Pharmaceuticals, Vienna, Austria) and Agrylin® (Shire) are oral imidazoquinazolines registered as orphan drugs in Europe. ANA is indicated for the reduction of thrombocytosis in high-risk ET, as a second-line agent, in Europe, and in any MPN-associated thrombocytosis and for the amelioration of thrombo-hemorrhagic events in the context of MPN in the USA. High-risk ET is defined as having one or more of the following risk factors: age >60 years, platelets >1500×109/L, previous thrombo-hemorrhagic events and additional cardiovascular risk factors.17
ANA received its primary marketing authorization in the USA in 1997 for ET and subsequently for other MPNs. It was initially developed as an inhibitor of platelet aggregation20 but found to have value as a platelet-lowering agent for the treatment of ET.21,22 Unlike many other treatments used in MPN, ANA is non-mutagenic, has no effect on angiogenesis and does not increase risks of leukemia, which make it a particularly useful drug for young patients with a longer natural history of ET.
ANA mechanism of action
The mechanism of action of ANA is not fully characterized. In vitro studies suggest that the platelet-lowering effect results from impaired megakaryocyte maturation and reduction in pro-platelet formation (PPF).23 ANA and its active metabolite 3-hydroxy-ANA specifically, reversibly and dose-dependently block the maturation of late-stage megakaryocytes. ET is associated with hyperproliferation of megakaryocytes, with increased cell size and ploidy. In ET patients, ANA induces left-shifted maturation and reduces both megakaryocyte size and DNA content.24–27 ANA’s effect on megakaryocytes has not been completely elucidated and appears to be specific to humans, limiting pharmacodynamic studies of the drug.28 A postulated route is through primary inhibition of TPO/c-mpl receptor-mediated events in megakaryocytes, including GATA 1 and FOG-1 expression.29 This might be consistent with the apparently platelet-selective effects of ANA without effect on erythropoiesis and granulopoiesis. ANA appears to have no effect on hemopoietic stem cells and does not cause DNA damage.30 Both ANA and its active metabolite 3-hydroxy-ANA elicit a marked effect on megakaryocyte growth with similar specificity and potency in vitro.31 ANA appears to normalize different aspects of platelet function in ET, including platelet coagulant activity and endothelial function.32,33 The inhibitory effect of ANA on platelet aggregation in ET occurs at significantly higher doses than required for platelet reduction, which may be important when considering patients with bleeding risk.
Aside from its effect on megakaryocytes, ANA also inhibits cyclic adenosine monophosphate (cAMP) phosphodiesterase (PDE) activity. 3-Hydroxy-ANA is nearly 40 times more potent at inhibiting PDE than the parent compound. The potent inhibition of PDE is likely to contribute significantly to the observed cardiovascular side effects of ANA.31
Pharmacodynamics and pharmacokinetics
ANA is very well absorbed independent of concurrent food intake (>70%) with a bioavailability of around 50%.34 The pharmacokinetic properties of ANA are linear in the dose range 0.5–2 mg, without plasma accumulation with repeated administration. ANA reaches peak plasma concentrations (Cmax) in 2 hours and has a terminal half-life of 1.7 hours in patients with ET.35 Case reports of intentional overdoses describe patients presenting with sinus tachycardia and vomiting, which resolve with conservative management.
However, platelet reduction from ANA is dose-dependent in humans. Overdose could therefore induce spontaneous bleeding related to severe thrombocytopenia. ANA undergoes extensive first-pass metabolism via CYP1A2 to generate the active metabolite 3-hydroxy-ANA, which is further metabolized via CYP1A2 to the inactive metabolite, 2-amino-5,6-dichloro-3,4-dihydroquinazoline. The latter is mainly excreted in urine. Less than 1% of ANA is recovered in the urine, and even severe renal impairment (CrCl <30 ml/min) does not appear to have a significant impact on ANA pharmacokinetics.35,36
Exposure to ANA in moderate hepatic impairment is eightfold, and its use in patients with moderate–severe hepatic impairment is contradicted. Caution is advised in patients with mild hepatic impairment. ANA should only be used if the benefit of its treatment clearly outweighs the risk. In patients with mild hepatic impairment, dose reduction is required and careful monitoring for cardiovascular side effects is advised. ANA is not recommended in patients with elevated transaminases (>5 times the upper limit of normal).35
In patients with known or suspected cardiac disease, the use of ANA must be carefully considered. It is recommended that ANA should be used with caution in any patient with known risk factors for prolongation of the QT interval, such as congenital long QT syndrome, a known history of acquired QTc prolongation, use of medicinal products that can prolong QTc interval and hypokalemia.3
Since ANA is at least in part metabolized by CYP1A2, other substances that have an inhibitory effect on CYP1A2 such as grapefruit juice and fluvoxamine and enoxacin should be avoided. There is a theoretical risk that other inotropic PDE inhibitors such as theophylline and milranone could exacerbate the cardiovascular side effects of ANA.35,36
Clinical efficacy
ANA is a highly effective platelet-reducing agent with response rates ranging between 76% and 94%. The reported differences are most likely related to variable response criteria used in different studies.37–44 When used as a second-line agent after HC failure, complete response rates are around 50%.45
It is important to note however that response to CRT according to the ELN criteria does not seem to correlate with thrombotic risk reduction in ET.46,47 In that respect, Hernandez-Boluda et al did not observe any significant clinical benefit in ET patients achieving complete response with ANA.48
Perhaps not surprisingly, there are no placebo-controlled studies investigating the effect of ANA on thrombotic events. The ameliorating effect of ANA on thrombosis risk has been inferred from studies using historical controls and appears to be superior to no cytoreductive agent and similar to that of HC. There are however some important and clinically significant differences, which have been reported in the only two randomized studies comparing HC and ANA, and these studies are discussed as follows.
Primary Thrombocythemia 1 (PT-1) study
The United Kingdom Medical Research Council PT-1 study was an open-label randomized phase 3 study comparing HC and ASA against ANA and ASA in patients with high-risk ET.49 This independent academic study involved 809 patients meeting the PVSG diagnostic criteria for ET with high-risk features including at least one of the following: age >60 years, platelet count >1000×109/L, and history of thrombo-hemorrhagic events, hypertension or diabetes mellitus. It may be important to note that these patients were diagnosed using PVSG criteria and that the definition of high-risk disease in this study was slightly different from that currently used.
Patients received HC (initial dose 0.5–1 g/day; n=404) or ANA (initial dose 0.5 mg twice daily; n=405), with doses adjusted to maintain a platelet count <400×109/L. Treatment failure was defined as failure to achieve a platelet count of <600×109/L after 3 months of therapy. All patients received ASA 75–100 mg once daily, or in case of intolerance, clopidogrel or dipyridamole. At the time of the primary analysis, the median observation time was 39 months (range: 12–72 months). The composite end point of the study was thrombosis, major hemorrhage, transformation or death from thrombosis or serious hemorrhage. The secondary end points were time to first arterial/venous thrombosis, serious hemorrhage, time to death, incidence of fibrotic, leukemic and myelodysplastic transformation and transformation to PV and control of platelet count.
Although the platelet count control was quicker with HC than ANA, it was equivalent in the long term (ie. after 6 months). The primary composite end point occurred more frequently in patients treated with ANA (16% in the ANA group versus 11% in the HC group at 5 years), and the secondary end point arterial thrombosis was almost twice as common in the ANA group compared to the HC group. This difference was mainly related to an increased number of transient ischemic attacks (TIAs) in patients receiving ANA and ASA. The reported frequency of TIAs might have been related to known cardiac side effects. However, another theory suggests the possibility of microbleeds rather than actual ischemic events in patients receiving a combination of ANA and ASA.50 No evidence of the so-called microbleeds was seen on any scans in this study, so such conjecture seems unfounded. Furthermore, there were significantly higher rates of serious hemorrhage, with gastrointestinal hemorrhage being particularly common. This observation is thought to be due to synergistic anti-aggregatory activity of ANA and ASA, which is now cautioned against in the summary of product characterization (SPC) of ANA.35 The rate of death due to thrombo-hemorrhagic events was comparable in both groups. In contrast, the rate of venous thromboembolism in the ANA group was approximately one-quarter of that in the HC group, with a corresponding significantly lower rate of deep-vein thrombosis, but no increase in the occurrence of pulmonary emboli or hepatic vein thrombosis. Although the study reported an increase in arterial events in the ANA compared to the HC arm, the investigators reported that ANA had an ameliorating effect on arterial thrombotic risk. This is supported by evidence from studies comparing HC to placebo in the treatment of high-risk ET, where reported arterial thrombosis rates in the placebo group51 were significantly higher than in patients treated with ANA in the PT-1 study.
Interestingly, and controversially, the study observed a significant increase in transformation to PET-MF in patients treated with ANA compared to HC, which formed the basis for premature study closure. The estimated actuarial risk of MF at 5 years after trial entry was 2% for the HC group compared to 7% for the ANA group. There was no statistically significant difference in myelodysplastic or leukemic transformation between the study arms at the time of the primary analysis. Patients withdrew more frequently from ANA compared to HC therapy. This was related either due to side effects or less frequently due to the development of a study end point or serious adverse events.
The results from the PT-1 trial were clearly in favor of combination therapy of HC and ASA over ANA and ASA for patients with high-risk ET. However, there are some critical points that warrant further discussion. The reason for increased rate of MF in the ANA arm compared to HC was not conclusively explained.
The study used the PVSG criteria for the diagnosis of ET, which do not necessitate a bone marrow assessment. Therefore, patients may have had varying degrees of established reticulin/collagen fibrosis at baseline. This could have concealed the important distinction between true ET and prefibrotic MF, which is likely to have influenced the reported rate of myelofibrotic transformation. However, the randomized nature of the study should have mitigated against such bias. Stratified randomization did not achieve perfect balance between the two arms of the trial, since the published supplemental data showed an imbalance in the disease duration, favoring the HC arm possibly skewing the fibrotic transformation rate in the ANA arm; a higher proportion (6%) in the ANA arm also received prior busulfan (3%) than in the HC arm. Furthermore, a third of patients in the ANA arm had previously been treated with HC. It is possible that ANA does not actually affect progression of early fibrosis to classical MF, but that HC is a more effective agent at halting fibrotic progression. Therefore, the reported rate of myelofibrotic transformation in the ANA arm may not be treatment related. Subsequent studies and previously reported data support the notion that ANA does not hinder the rate of myelofibrotic progression.9 But there is lack of clarity regarding the potential of ANA to accelerate fibrosis. Finally, it can be argued that the statistical power of the PT-1 study should be protective against a randomly uneven distribution of patients with an inherently higher propensity for MF between treatment arms. However, since the event number was so low overall (n=21), there is a theoretical potential for problems with completeness of event reporting and a significant effect produced by even a minor flaw.
Concerning thrombosis, the superior effect of HC over ANA on arterial thrombotic risk may be partly related to its overall myelosuppressive effect, with reduction in white cell count and reduction in hematocrit.52 HC also has a modulatory effect on endothelial function and acts as a nitrous oxide donor. All of these aspects may potentially explain the apparent superiority of HC.53,54
In a study, 806 ET patients including 776 from the original PT-1 study were tested for JAK2 mutation status. The purpose of the study was to determine a pattern between JAK2 mutation status, clinical phenotype and differential response to HC compared to ANA. Those with JAK2-positive ET had a greater resemblance with PV patients with features of panmyelosis and increased venous thrombosis.55 JAK2-positive ET patients responded better to HC than ANA, than those who were JAK2 negative. Similar data were also recently reported on the PT-1 cohort suggesting that features of PET-MF including anemia developed more frequently in patients with CALR or MPL mutations when treated with ANA versus HC.56 These data argue that in the future, ET therapies may be chosen on the basis of clinical and molecular features specific to the individual patient.
Anagrelide compared with Hydroxyurea in WHO-classified ET (ANAHYDRET) study
This prospective randomized, single-blind phase 3 trial was designed to demonstrate that the selective platelet-lowering therapy with ANA is non-inferior to HC in the prevention of thrombo-hemorrhagic events in high-risk ET diagnosed according to the WHO criteria (WHO-ET).57 The study included 259 patients with untreated high-risk WHO-ET for an observation time of 36 months. Patients were randomized to receive ANA (n=122) or HC (n=137) with or without the addition of antiplatelet therapy. The two treatment arms compared platelet-, hemoglobin- and white cell count-lowering effects, ET-related complications and safety and tolerability parameters. The diagnosis of WHO-ET was confirmed by histopathological examination of the bone marrow at study entry and later reexamined in a blinded fashion at a pathology reference coordinating center. A proportion of the cohort (n=199) were also assessed for the JAK2V617F mutation as an additional thrombophilic marker. ANA was commenced at 1 mg/day and HC started at 1.5 g/day, and titrated to a dose to achieve a target platelet count ≤450×109/L.
There was no relevant difference in platelet- or hemoglobin-lowering effect between the groups, but a significantly lower white cell count in the HC group. In contrast to the results from the PT-1 study, there was no apparent difference in major clinical events; specifically no significant difference in arterial or venous thrombosis. Subanalysis of the cohort for JAK2V617F mutation status showed no statistical difference in thrombotic events or event-free survival between patients with mutant or wild-type JAK2. There were also slightly more minor rather than major bleeding events in the ANA arm, half of which were associated with concomitant use of ASA. This discrepant result compared to the PT-1 study, which reported more major bleeding events in the ANA with ASA arm, may have been due to restrictive use of ASA with ANA and comparative lack of statistical power of the ANAHYDRET study.
Adverse events including serious adverse events and those leading to treatment discontinuation were equally distributed between the two study arms. However, there were relatively more cardiovascular events in the ANA group and more significant leukopenia and minor infections in the HC group.
The ANAHYDRET study also included a retrospective analysis for the development of PET-MF, mainly based on clinical and biochemical surrogate parameters (constitutional symptoms, anemia, leukoerythroblastosis, splenomegaly, high serum lactate dehydrogenase) rather than bone marrow assessment for reticulin/collagen fibrosis. According to these criteria, only one patient in the HC and two patients in the ANA arm developed PET-MF over a 9-year follow-up period, with none of the patients developing more than one of the surrogate markers. Clonal evolution with transformation to leukemia or myelodysplasia was not observed.
The authors concluded that in this homogeneous study population of WHO-ET, ANA was non-inferior to HC in the prevention of ET-related thrombo-hemorrhagic events with comparable tolerability and safety characteristics. The exceptions were bleeding risk and cardiovascular side effects with ANA. It was suggested that the former could be minimized by avoiding concomitant use of ASA, and the latter by dose reduction according to tolerance. Consistent with previous reports, patients with WHO-ET in this study had low JAK2V617F allele burden.
Contrary to the results from the PT-1 study, the smaller ANAHYDRET study did not show a significant difference in thrombosis risk reduction between the two study arms, while there was a marked reduction in arterial thrombosis in the HC and ASA arm when compared to the ANA and ASA arm in the PT-1 study. In the PT-1 study, patients were diagnosed according to the PVSG criteria (PVSG-ET). Consequently, the cohort was more heterogeneous and likely to have included both patients with true ET and patients with prefibrotic MF. It is possible that patients with prefibrotic MF, who appear to have an inherently higher arterial thrombosis risk, respond better to the unselective cytoreductive effect of HC, with superior arterial thrombosis risk reduction, than to megakaryocyte-selective ANA and that this may in part be the reason for the different results from PT-1 and ANAHYDRET.
The authors from the ANAHYDRET study suggest that the conclusions drawn from the PT-1 study may not be generally applicable to patients with WHO-ET and that distinction between WHO-ET and prefibrotic MF may play an important role in choosing an appropriate cytoreductive agent. But there are still considerable concerns regarding reproducibility and different histopathologists being able to clearly distinguish between the two entities, as discussed earlier.10,13,19 Nonetheless, the proposal that treatment recommendations for WHO-ET may not apply to patients with PVSG-ET is intriguing. However, the non-inferiority design of the ANAHYDRET study is a clear limitation with inadequate statistical power to permit universal treatment recommendations.
The Evaluation of Xagrid® Efficacy and Long-term Safety (EXELS) study
The EXELS study is the largest prospective study in ET so far. It is a phase 4 post-approval commitment observational multicenter study designed to monitor efficacy, safety and pregnancy outcomes in ET patients treated with ANA or other CRTs in routine clinical practice. The study sponsor was Shire Pharmaceuticals.
A total of 3649 high-risk ET patients were recruited from May 2005 to April 2009, in 13 European countries. Data including predefined clinical events (PDCEs) were collected at registration and every 6 months thereafter for 5 years. Efficacy assessment involved evaluation of platelet reduction, incidence of thrombo-hemorrhagic events and drug utilization patterns. The results presented here are from a scheduled datacut in September 2011, 2.5 years after registration of the last patient and the preliminary final data presented by Birgegard et al at the American Society of Hematology annual conference in 2014.58 Final results including data on platelet response and pregnancy outcomes are still awaited; the authors understand that a final analysis of the study including analyses for major events is underway.
ET patients, diagnosed according to the PVSG and WHO criteria, whichever was used by the recruiting center, with at least one high-risk feature (as described earlier) previously treated or commenced on CRT were eligible for inclusion in the study. As a non-interventional study, the choice of CRT was determined prior to study entry, and patients were managed according to local policy. This study gives valuable insight into the real-world management patterns of ET. As a result, there is considerable variation in treatment strategies, making analyses of differences in treatment-determined outcomes highly complex. The size of the EXELS study however means that it is extremely important and potentially provides very powerful data.
Most patients had already been treated with CRT (80.6%), and were therefore not treatment naïve. A large proportion (69.8%) received anti-aggregatory therapy at the time of study enrollment. The two most common cytoreductive monotherapies were HC (65.1%) and ANA (22.2%). The remaining patients received IFN α, busulfan, pipobroman, phosphorus-32 or a combination of HC with ANA. Many patients switched therapy during the course of the study.
Importantly for analyzing the results of EXELS and reflecting real-life practice, here a significant number of younger patients (<60 years) received ANA (59.3%) or IFN with only a small proportion receiving HC (19.3%) or another CRT. This observation is in accordance with current international recommendations to avoid CRT with mutagenic potential in patients <40 years of age. Some country-specific differences did emerge reflected due to national guidance and also reimbursement patterns across the EU.59 In the older age group (>60 years), HC was by far the most commonly used agent, and only a few very elderly patients (>80) received ANA, most likely due to concern over cardiovascular side effects and potential bleeding risk. An analysis of ET therapy in the very elderly has been published already from the EXELS database.60 The most frequently reported PDCEs were cardiovascular symptoms including palpitations, tachycardia, headaches, hypotension, lightheadedness, dizziness, syncope, exertional dyspnea and peripheral edema in patients receiving ANA alone or in combination with another CRT. Concerning disease-related complications, the overall reported thrombotic events were low and partly in line with the results from the ANAHYDRET study. Arterial thrombosis rates were similar in the ANA and other CRT groups. Hemorrhagic events on the other hand were highest in the ANA group, especially in patients also receiving anti-aggregatory therapy, resonating the results from the PT-1 study. Thus, the EXELS study also supported the difference in venous versus arterial thrombosis previously seen in the PT-1 study.
Disease transformation to MF (n=105) and acute leukemia (n=62) was observed in all treatment groups and one of the more frequent PDCEs (3.5%), an important observation when determining disease treatment strategy. The rate of PET-MF was higher in patients receiving ANA compared to other CRTs, whereas the rate of leukemic transformation was higher in patients receiving CRTs other than ANA. This difference in fibrotic and leukemic transformation remained apparent when ANA and HC monotherapies were compared with each other. Interestingly, all patients who had ever received ANA and transformed to acute leukemia had previously also received HC. The incidence of death and transformation was higher in patients with a WBC count >15×109/L at any time during the study. Further analyses of these events and also other non-hematological malignancies which appear to be higher in the other CRT group than the ANA group are awaited, and an analysis with EU cancer registries is planned.
The EXELS study provides a real-world insight into common prescribing practices in the management of high-risk ET. Although it is not a standard recommendation, combination therapy with ANA and HC was being utilized in approximately 10% of patients, in order to improve cytoreductive efficacy without increasing toxicity, and data concerning this combination was published from the study and is in line with other experience.61 Further studies might help to determine for which patient group, combination therapy might be most appropriate. Similar to the PT-1 study, the patient cohort in the EXELS study was heterogeneous including patients with WHO-ET and PVSG-ET. Additional data are not expected with regard to molecular subtypes of ET since these were not collected in the EXELS study, but multivariate analyses allowing further dissection of the data and removing some of the age imbalance bias are planned.
Side effect profile and tolerability
The extended ANA study group described the tolerability profile of ANA at a mean dose of 2 mg/day in 942 patients and a further non-comparative study of 3660 patients. These are outlined in the manufacturers’ prescribing information and include headache (44% in ET), palpitations (26%), diarrhea (26%), fluid retention (21%), asthenia (22%), nausea (17%), generalized abdominal pain (16%), dizziness (15%), tachycardia and chest pain (8%).37 These side effects were also more frequently described in the EXELS study and in the ANA arm of the PT-1 study.49,61 It has been suggested that most side effects are mediated by ANA and the potent effect of its metabolite 3-hydroxy-ANA on PDE and cAMP, causing vasodilation and a positive inotropic effect. Patients treated with ANA may experience some of these effects in the first few weeks of therapy. There have been conflicting reports on whether side effects due to ANA resolve with time.62 Serious, but rare (0.1–0.01%) noncardiac adverse events include pleural effusions, pulmonary infiltrates, pulmonary fibrosis, hypersensitivity pneumonitis, renal tubular injury, pancreatitis, hepatitis, stroke, TIA, myelofibrotic transformation and bleeding, although these are unlikely to occur in patients on ANA monotherapy.
The reported rate of discontinuation from ANA therapy due to adverse events ranges between 20% and 40%. PT-1 study reported a drop-out rate due to adverse events of 22%.49 Adverse effects related to ANA are dose-related, and common side effects such as headaches can be managed with dose titration and simple analgesia and often subside with time. However, administration of high enough doses of ANA to achieve a target platelet count of <400×109/L may not be possible without causing excessive toxicity especially in older patients. Cardiovascular side effects have been a concern and are specifically addressed as follows.
Cardiovascular side effects
The most commonly reported non-thrombotic cardiovascular side effects of ANA are palpitations, tachycardia and fluid retention. More serious cardiovascular adverse events including congestive cardiac failure, hypertension and tachy-arrhythmia are uncommon (0.1–1%), and angina, myocardial infarction, pericardial effusion, postural hypotension and cardiac syncope are reassuringly very rare (0.1–0.01%). These results were also seen in the EXELS study where serious adverse cardiovascular events were rarely reported in patients treated with ANA.61 There have been case reports of patients with reversible ANA-associated cardiomyopathy with full restoration of left ventricular function after discontinuation of ANA.
The outcome of a prospective observational single-center study did not support in-depth cardiovascular risk factor assessment prior to commencement of ANA to help predict and prevent ANA-induced cardiovascular side effects.63 However, from a safety perspective in managing an individual patient, this is still recommended. The manufacturers’ SPC recommends that all patients commencing on ANA should have a baseline electrocardiogram and echocardiogram considered.35
Recently, promising data from a phase 2 pilot single-arm, open-label, multicenter study evaluating the efficacy and safety of ANA controlled-release (CR) GALE-401 (Galena Biopharma, San Ramon, CA, USA) in subjects with thrombocytosis secondary to ET and other MPNs has been published.64 According to the preliminary report, ANA-CR has superior tolerability characteristics with less cardiovascular side effects than ANA immediate release (IR). It has been suggested that the peak plasma concentration of ANA-IR may be too high, causing excessive PDE inhibition and leading to increased side effects. GALE-401 showed favorable pharmacokinetic characteristics of ANA and 3-hydroxy-ANA with a significantly reduced Cmax, while maintaining good overall plasma exposure. The compound shows promising platelet-reducing efficacy (overall response rate 70–80%) comparing favorably to previous reports on ANA-IR.49,57 GALE-401 is expected to decrease the frequency and severity of side effects in comparison to the IR formulation, improving tolerability and effective dose titration. AOP has also developed an ANA slow-release compound. Results from the two phase 3 trials conducted in Austria and eastern Europe assessing the treatment effect of ANA retard against placebo and against Thromboreductin (ANA-IR) are still pending.
Differential response of patients with ET according to gene mutation status
Improved understanding of the role of driver mutations in the pathophysiology of MPNs and their effect on clinical phenotype has prompted studies investigating the differential impact of ANA on ET according to MPN gene mutation status.
Several studies have observed the difference in clinical phenotype between JAK2 mutation-positive ET and JAK2 wild-type ET. Those with JAK2-positive ET show relatively higher hemoglobin levels, white cell counts, lower serum erythropoietin levels and lower platelet counts.55 The diagnosis of ET was not based on the WHO criteria resulting in a more heterogeneous cohort. Although the observed differences were statistically significant, they were clinically small. Since then a large retrospective study in patients with ET (n=867) with 20-year retrospective follow-up did not show a statistical difference in thrombosis rate between JAK2-positive and JAK2-negative ET patients;65 three meta-analyses have been published supporting the contrary.66–68 The level of JAK2 mutant allele burden in true ET is much lower than in PV with the majority of cells harboring a heterozygous JAK2 mutation. Furthermore, there is evidence supporting an increased risk of myelofibrotic progression in patients with ET or PV with an evolving or high stable JAK2 allele burden.
Calreticulin is a highly conserved multifunctional endoplasmic reticulum protein with partly unknown function. In 2013, two variant insertion/deletion mutations in the CALR gene were found to be present in approximately 20–30% patients with ET and primary MF.3,4 Although it does not aid distinction between ET and MF, the presence of a CALR mutation appears to be almost mutually exclusive with the presence of either a JAK2 or an MPL mutation. Clinically, of patients with CALR-mutated ET seem to have a distinct clinical phenotype from those with a JAK2 mutation. Such patients are younger and have a higher platelet count, lower white cell count, a relatively lower hemoglobin and a lower risk of thrombosis.69 This is consistent with the observation that both CALR-positive ET and MF patients have lower risk disease.4 It is conceivable that both a high white cell count and high hematocrit contribute to the increased thrombosis risk in JAK2-positive ET. Although evidence supporting the correlation between a higher white cell count and thrombosis risk in ET is conflicting, an association was found in the PT-1 study.55 It is reasonable to assume that a higher white cell count increases the risk of thrombosis, but not overwhelmingly so.
There have been a few small studies recently, investigating the impact of status of JAK2, CALR and MPL mutations on treatment response to ANA in patients with ET.
The first was a hypothesis-generating study of 47 (n=46 included in the analysis) patients with high-risk WHO-ET who were intolerant or refractory to first-line therapy.70 JAK2 mutation-positive and wild-type patients had a good overall platelet response (67–77%), with JAK2-positive ET patients being more responsive to ANA than wild-type patients; however, the numbers were too small to permit a conclusive statement. A more recent Italian study reviewed the impact of status of JAK2, CALR and MPL gene mutations on response to ANA in 21 consecutive patients, diagnosed between 1983 and 2014.71 The results suggest a high hematological response in patients with a JAK2 and triple-negative mutational status versus those with the CALR mutation. It has been demonstrated that the C-terminus of the CALR-mutated protein loses capacity to bind calcium, in turn affecting a variety of cellular functions.72 The authors hypothesize that because ANA exerts its effect on megakaryocyte adhesion and PPF in a calcium-dependent manner,24,73 patients with the CALR mutation are less sensitive to ANA platelet-reducing effect.74 The number of patients in this study was again too small, and the cohort was likely to have been fairly heterogeneous to draw a convincing conclusion. Additionally, and in conflict to the data discussed earlier, a different long-term follow-up (median 110 months) study of 47 WHO-ET patients with JAK2, CALR and MPL mutation frequencies of 46.3%, 28.3% and 1.5%, respectively, treated with ANA did not show a differential response to ANA among molecular subsets.75
Finally, prospective data on disease characteristics and treatment response of different molecular subtypes in the PT-1 study patient cohort was recently presented at the annual European Hematology Association congress.56 In this cohort, JAK2, MPL and CALR mutations were present in 53%, 4% and 26% of patients with ET, respectively. These were almost entirely mutually exclusive, but 1% of patients harbored more than one mutation and analysis of hemopoietic colonies showed that somatic mutations within these genes can co-occur in the same tumor subclone.
CALR and MPL mutations affected males and females equally, while JAK2 mutation and triple-negative mutation status predominated in females. In terms of age distribution, patients who were triple negative were youngest, followed by patients with a CALR and then patients with JAK2 or MPL mutations. Consistent with historical reports, CALR-mutated ET patients presented with lower hemoglobin levels and total white cell counts, and higher platelet counts. Additionally, venous thrombotic events occurred most frequently in the JAK2-mutated subgroup, while CALR-mutated ET patients suffered increased rates of myelofibrotic transformation. There was no suggestion that these events were precipitated or influenced by exposure to either ANA or HC. Platelet count control was equivalent after treatment with either HC and ASA or ANA and ASA among all molecular subgroups of patients with high-risk ET. However, CALR-mutated and MPL-mutated patients were more likely to develop a lower hemoglobin level in response to treatment with ANA, independent of ANA dose. These results support the notion that driver mutations cause distinct clinical subtypes.
The specific interaction of these mutations in triggering and leading to disease progression remains to be elucidated. Improved understanding of their specific role may help to guide management decisions in the future and may be more meaningful and reproducible than nuances of megakaryocyte morphology as the cornerstone of the WHO criteria.
Conclusion
ANA is a megakaryocyte-specific platelet-lowering agent used in the management of high-risk ET. Data from two significant prospective phase 3 randomized studies comparing ANA to the most commonly used first-line agent HC have provided valuable information on the efficacy, tolerability and safety of ANA. While ANA was shown to be a highly effective platelet-lowering agent with a considerable effect on overall thrombosis risk reduction, results also showed that ANA was less effective than HC in arterial thrombosis reduction, though it had a superior effect on venous thrombosis risk. The PT-1 study also showed a greater myelofibrotic transformation risk in patients treated with ANA compared to HC, which led to premature study closure. The latter observation may have been related to ANA’s inability to halt myelofibrotic transformation rather than promoting it, at least in a subset of patients with ET. ANA in combination with ASA also resulted in a greater major hemorrhage risk, which caused the manufacturer to caution against the use of ANA with ASA.
Another prominent feature observed in these studies was the frequency of cardiovascular side effects associated with ANA, which is thought to be mainly related to its potent PDE inhibition. Although side effects such has headache, palpitations and dizziness were common, more serious cardiovascular side effects were reported very rarely.
These results were also corroborated in the largest prospective study in ET so far. The EXELS phase 4 post-approval commitment observational multicenter study was designed to monitor efficacy, safety and pregnancy outcomes in ET patients treated with ANA or other CRTs in routine clinical practice. Minor cardiovascular side effects were by far the most commonly reported predefined clinical event in patients treated with ANA. Results from a recent phase 2 study assessing the efficacy of ANA-CR are interesting and promise greater tolerability due to improved pharmacokinetic characteristics.
Disease transformation to MF was also not an infrequent observation in the EXELS study, but was reported in all treatment groups. Similar to results from the ANAHYDRET study, EXELS did not confirm the difference seen in arterial thrombosis in patients treated with ANA compared to other CRTs. Bleeding risk with ANA appeared to be increased in this patient cohort, which like in the PT-1 study may have been confounded by the concurrent use of ASA.
These variations in outcome in patients with high-risk ET treated with ANA may have been influenced by the clinical and pathological heterogeneity of patients with ET, particularly in the PT-1 and EXELS study where patients were diagnosed according to the PVSG or WHO criteria for ET.
The study populations may have included patients with a high inherit propensity to fibrotic transformation and greater arterial thrombosis risk. However, considering the statistical power of these large studies, this observation remains speculative.
Recent studies have highlighted the importance of differentiating between patients with true ET and those with prefibrotic MF. It appears that patients from these different entities have a very different natural history with variation in clinical phenotype and the former patient group displaying a far more indolent disease course. There has been an increasing interest in the role of driver mutations in ET and other MPNs and their role in influencing clinical phenotype and response to treatment.
The current evidence still supports HC as a highly effective first-line agent in the management of high-risk ET. However, ANA is a good second-line alternative, and its lack of mutagenic potential supports the use of ANA in younger patients with less cardiovascular risk and a prolonged disease course.
Disclosure
CN Harrison has previously received speaker’s fees from Shire Ltd. A Sarma and DP Mclornan declare that they have no conflicts of interest in this work.
References
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. | ||
Harrison CN, Butt N, Campbell P, et al. Modification of British Committee for Standards in Haematology diagnostic criteria for essential thrombocythaemia. Br J Haematol. 2014;167(3):421–423. | ||
Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. | ||
Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. | ||
James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. | ||
Cabagnols X, Favale F, Pasquier F, et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood. 2016;127(3):333–342. | ||
Milosevic Feenstra JD, Nivarthi H, Gisslinger H, et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood. 2016;127(3):325–332. | ||
Harrison CN, Vannucchi AM. Closing the gap: genetic landscape of MPN. Blood. 2016;127(3):276–278. | ||
Ejerblad E, Kvasnicka HM, Thiele J, et al. Diagnosis according to World Health Organization determines the long-term prognosis in patients with myeloproliferative neoplasms treated with anagrelide: results of a prospective long-term follow-up. Hematology. 2013;18(1):8–13. | ||
Madelung AB, Bondo H, Stamp I, et al. WHO classification 2008 of myeloproliferative neoplasms: a workshop learning effect – the Danish experience. APMIS. 2015;123(9):787–792. | ||
Murphy S, Peterson P, Iland H, Laszlo J. Experience of the Polycythemia Vera Study Group with essential thrombocythemia: a final report on diagnostic criteria, survival, and leukemic transition by treatment. Semin Hematol. 1997;34(1):29–39. | ||
Gisslinger H, Jeryczynski G, Gisslinger B, et al. Clinical impact of bone marrow morphology for the diagnosis of essential thrombocythemia: comparison between the BCSH and the WHO criteria. Leukemia. 2016;30(5):1126–1132. | ||
Harrison C. Equivalence of BCSH and WHO diagnostic criteria for ET. Leukemia. 2016. | ||
Barbui T, Thiele J, Carobbio A, et al. Disease characteristics and clinical outcome in young adults with essential thrombocythemia versus early/prefibrotic primary myelofibrosis. Blood. 2012;120(3):569–571. | ||
Björkholm M, Derolf ÅR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol. 2011;29(17):2410–2415. | ||
Barbui T, Finazzi G. When and how to treat essential thrombocythemia. N Engl J Med. 2005;353(1):85–86. | ||
Harrison CN, Bareford D, Butt N, et al. Guideline for investigation and management of adults and children presenting with a thrombocytosis. Br J Haematol. 2010;149(3):352–375. | ||
Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood. 2013;121(23):4778–4781. | ||
Alimam S, Wilkins BS, Harrison CN. How we diagnose and treat essential thrombocythaemia. Br J Haematol. 2015;171(3):306–321. | ||
Abe Andes W, Noveck RJ, Fleming JS. Inhibition of platelet production induced by an antiplatelet drug, anagrelide, in normal volunteers. Thromb Haemost. 1984;52(3):325–328. | ||
Oertel MD. Anagrelide, a selective thrombocytopenic agent. Am J Health Syst Pharm. 1998;55(19):1979–1986. | ||
Pescatore SL, Lindley C. Anagrelide: a novel agent for the treatment of myeloproliferative disorders. Expert Opin Pharmacother. 2000;1(3):537–546. | ||
Espasandin YR, Glembotsky AC, Grodzielski M, et al. Anagrelide platelet-lowering effect is due to inhibition of both megakaryocyte maturation and proplatelet formation: insight into potential mechanisms. J Thromb Haemost. 2015;13(4):631–642. | ||
Tomer A. Effects of anagrelide on in vivo megakaryocyte proliferation and maturation in essential thrombocythemia. Blood. 2002;99(5):1602–1609. | ||
Mazur EM, Rosmarin AG, Sohl PA, Newton JL, Narendran A. Analysis of the mechanism of anagrelide-induced thrombocytopenia in humans. Blood. 1992;79(8):1931–1937. | ||
Tefferi A, Silverstein MN, Petitt RM, Mesa RA, Solberg LA Jr. Anagrelide as a new platelet-lowering agent in essential thrombocythemia: mechanism of actin, efficacy, toxicity, current indications. Semin Thromb Hemost. 1997;23(4):379–383. | ||
Thiele J, Kvasnicka HM, Fuchs N, Brunnbauer K, Volkwein N, Schmitt-Graeff A. Anagrelide-induced bone marrow changes during therapy of chronic myeloproliferative disorders with thrombocytosis. An immunohistochemical and morphometric study of sequential trephine biopsies. Haematologica. 2003;88(10):1130–1138. | ||
Birgegard G, Bjorkholm M, Kutti J, et al. Adverse effects and benefits of two years of anagrelide treatment for thrombocythemia in chronic myeloproliferative disorders. Haematologica. 2004;89(5):520–527. | ||
Ahluwalia M, Donovan H, Singh N, Butcher L, Erusalimsky JD. Anagrelide represses GATA-1 and FOG-1 expression without interfering with thrombopoietin receptor signal transduction. J Thromb Haemost. 2010;8(10):2252–2261. | ||
Thiele J, Kvasnicka HM, Ollig S, Schmitt-Graff A. Anagrelide does not exert a myelodysplastic effect on megakaryopoiesis: a comparative immunohistochemical and morphometric study with hydroxyurea. Histol Histopathol. 2005;20(4):1071–1076. | ||
Wang G, Franklin R, Hong Y, Erusalimsky JD. Comparison of the biological activities of anagrelide and its major metabolites in haematopoietic cell cultures. Br J Pharmacol. 2005;146(3):324–332. | ||
Tomer A, Harker LA. Measurements of in vivo megakaryocytopoiesis: studies in nonhuman primates and patients. Stem Cells. 1996;14 Suppl 1:18–30. | ||
Cacciola RR, Cipolla A, Di Francesco E, Giustolisi R, Cacciola E. Treatment of symptomatic patients with essential thrombocythemia: effectiveness of anagrelide. Am J Hematol. 2005;80(1):81–83. | ||
Gaver RC, Deeb G, Pittman KA, Smyth RD. Disposition of anagrelide, an inhibitor of platelet aggregation. Clin Pharmacol Ther. 1981;29(3):381–386. | ||
SPC Ltd. Xagrid product specification leaflet. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000480/WC500056557.pdf2016. Accessed August 28, 2016. | ||
SPC Ltd. AGRYLIN® prescribing information. 2015. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/020333s023lbl.pdf. | ||
Petitt RM, Silverstein MN, Petrone ME. Anagrelide for control of thrombocythemia in polycythemia and other myeloproliferative disorders. Semin Hematol. 1997;34(1):51–54. | ||
Petrides PE, Beykirch MK, Trapp OM. Anagrelide, a novel platelet lowering option in essential thrombocythaemia: treatment experience in 48 patients in Germany. Eur J Haematol. 1998;61(2):71–76. | ||
Mills AK, Taylor KM, Wright SJ, et al. Efficacy, safety and tolerability of anagrelide in the treatment of essential thrombocythaemia. Aust N Z J Med. 1999;29(1):29–35. | ||
Laguna MS, Kornblihtt LI, Marta RF, Michiels JJ, Molinas FC. Effectiveness of anagrelide in the treatment of symptomatic patients with essential thrombocythemia. Clin Appl Thromb Hemost. 2000;6(3):157–161. | ||
Birgegard G. Anagrelide treatment in myeloproliferative disorders. Semin Thromb Hemost. 2006;32(3):260–266. | ||
Penninga E, Jensen BA, Hansen PB, et al. Anagrelide treatment in 52 patients with chronic myeloproliferative diseases. Clin Lab Haematol. 2004;26(5):335–340. | ||
Steurer M, Gastl G, Jedrzejczak WW, et al. Anagrelide for thrombocytosis in myeloproliferative disorders: a prospective study to assess efficacy and adverse event profile. Cancer. 2004;101(10):2239–2246. | ||
Fruchtman SM, Petitt RM, Gilbert HS, Fiddler G, Lyne A; Anagrelide Study Group. Anagrelide: analysis of long-term efficacy, safety and leukemogenic potential in myeloproliferative disorders. Leuk Res. 2005;29(5):481–491. | ||
Wagstaff AJ, Keating GM. Anagrelide: a review of its use in the management of essential thrombocythaemia. Drugs. 2006;66(1):111–131. | ||
Carobbio A, Finazzi G, Antonioli E, et al. Hydroxyurea in essential thrombocythemia: rate and clinical relevance of responses by European LeukemiaNet criteria. Blood. 2010;116(7):1051–1055. | ||
Hernandez-Boluda JC, Alvarez-Larran A, Gomez M, et al. Clinical evaluation of the European LeukaemiaNet criteria for clinicohaematological response and resistance/intolerance to hydroxycarbamide in essential thrombocythaemia. Br J Haematol. 2011;152(1):81–88. | ||
Hernandez-Boluda JC, Pereira A, Cervantes F, et al; Grupo Español de Enfermedades Mieloproliferativas Filadelfia Negativas (GEMFIN). Clinical evaluation of the European LeukemiaNet response criteria in patients with essential thrombocythemia treated with anagrelide. Ann Hematol. 2013;92(6):771–775. | ||
Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med. 2005;353(1):33–45. | ||
Petrides PE. Anagrelide: what was new in 2004 and 2005? Semin Thromb Hemost. 2006;32(4 Pt 2):399–408. | ||
Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med. 1995;332(17):1132–1136. | ||
Bouchard BA, Tracy PB. Platelets, leukocytes, and coagulation. Curr Opin Hematol. 2001;8(5):263–269. | ||
Brun M, Bourdoulous S, Couraud PO, Elion J, Krishnamoorthy R, Lapoumeroulie C. Hydroxyurea downregulates endothelin-1 gene expression and upregulates ICAM-1 gene expression in cultured human endothelial cells. Pharmacogenomics J. 2003;3(4):215–226. | ||
Nahavandi M, Tavakkoli F, Wyche MQ, Perlin E, Winter WP, Castro O. Nitric oxide and cyclic GMP levels in sickle cell patients receiving hydroxyurea. Br J Haematol. 2002;119(3):855–857. | ||
Campbell PJ, MacLean C, Beer PA, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood. 2012;120(7):1409–1411. | ||
Nangalia J. Phenotypic differences and treatment responses in molecular subgroups of essential thrombocythaemia from analysis of the PT-1 cohort. Paper presented at: 21st Congress of the European Hematology Association; June 12, 2016; Copenhagen. | ||
Gisslinger H, Gotic M, Holowiecki J, et al; ANAHYDRET Study Group. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood. 2013;121(10):1720–1728. | ||
Birgegard G, Besses C, Griesshammer M, et al. Treatment of essential thrombocythemia in Europe: an observational study of 3649 high-risk patients in EXELS. Blood. 2014;124(21):1846. | ||
Besses C, Kiladjian JJ, Griesshammer M, et al. Cytoreductive treatment patterns for essential thrombocythemia in Europe. Analysis of 3643 patients in the EXELS study. Leuk Res. 2013;37(2):162–168. | ||
Kiladjian JJ, Besses C, Griesshammer M, et al. Efficacy and safety of cytoreductive therapies in patients with essential thrombocythaemia aged >80 years: an interim analysis of the EXELS study. Clin Drug Investig. 2013;33(1):55–63. | ||
Gugliotta L, Besses C, Griesshammer M, et al. Combination therapy of hydroxycarbamide with anagrelide in patients with essential thrombocythemia in the evaluation of Xagrid(R) efficacy and long-term safety study. Haematologica. 2014;99(4):679–687. | ||
Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood. 2001;97(4):863–866. | ||
Tortorella G, Piccin A, Tieghi A, et al. Anagrelide treatment and cardiovascular monitoring in essential thrombocythemia. A prospective observational study. Leuk Res. 2015;39(6):592–598. | ||
Verstovsek S. Phase 2 study of a novel controlled-release formulation of anagrelide (GALE-401) in subjects with myeloproliferative neoplasm-associated thrombocytosis. Paper presented at: 20th Congress of the European Hematology Association; June 13, 2015; Vienna. | ||
Carobbio A, Finazzi G, Antonioli E, et al. JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol. 2009;37(9):1016–1021. | ||
Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica. 2008;93(9):1412–1414. | ||
Dahabreh IJ, Zoi K, Giannouli S, Zoi C, Loukopoulos D, Voulgarelis M. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res. 2009;33(1):67–73. | ||
Lussana F, Caberlon S, Pagani C, Kamphuisen PW, Buller HR, Cattaneo M. Association of V617F Jak2 mutation with the risk of thrombosis among patients with essential thrombocythaemia or idiopathic myelofibrosis: a systematic review. Thromb Res. 2009;124(4):409–417. | ||
Tefferi A, Pardanani A. Myeloproliferative neoplasms: a contemporary review. JAMA Oncol. 2015;1(1):97–105. | ||
Cascavilla N, De Stefano V, Pane F, et al. Impact of JAK2(V617F) mutation status on treatment response to anagrelide in essential thrombocythemia: an observational, hypothesis-generating study. Drug Des Devel Ther. 2015;9:2687–2694. | ||
Iurlo A, Cattaneo D, Orofino N, Bucelli C, Fabris S, Cortelezzi A. Anagrelide and mutational status in essential thrombocythemia. BioDrugs. 2016;30(3):219–223. | ||
Michalak M, Corbett EF, Mesaeli N, Nakamura K, Opas M. Calreticulin: one protein, one gene, many functions. Biochem J. 1999;344(Pt 2):281–292. | ||
Di Buduo CA, Moccia F, Battiston M, et al. The importance of calcium in the regulation of megakaryocyte function. Haematologica. 2014;99(4):769–778. | ||
Shivarov V, Ivanova M, Tiu RV. Mutated calreticulin retains structurally disordered C terminus that cannot bind Ca(2+): some mechanistic and therapeutic implications. Blood Cancer J. 2014;4(2):e185. | ||
Mela Osorio MJ, Ferrari L, Goette NP, et al. Long-term follow-up of essential thrombocythemia patients treated with anagrelide: subgroup analysis according to JAK2/CALR/MPL mutational status. Eur J Haematol. 2016;96(4):435–442. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.