")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Sinomenine reduces neuronal cell apoptosis in mice after traumatic brain injury via its effect on mitochondrial pathway
Authors Fu C, Wang Q, Zhai X, Gao J
Received 18 October 2017
Accepted for publication 22 November 2017
Published 5 January 2018 Volume 2018:12 Pages 77—84
DOI https://doi.org/10.2147/DDDT.S154391
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Chuanjing Fu,1,* Qi Wang,2 Xiaofu Zhai,3,* Juemin Gao1
1Department of Neurosurgery, Jiangsu Hospital of Traditional Chinese Medicine, Nanjing, 2Department of Radiology, The Fourth People’s Hospital of Huai’an, 3Department of Neurosurgery, The Second People’s Hospital of Huai’an, Xuzhou Medical College, Huai’an, People’s Republic of China
*These authors contributed equally to this work
Background: Sinomenine (SIN) has been shown to have protective effects against brain damage following traumatic brain injury (TBI). However, the mechanisms and its role in these effects remain unclear. This study was conducted to investigate the potential mechanisms of the protective effects of SIN.
Methods: The weight-drop model of TBI in Institute of Cancer Research (ICR) mice were treated with SIN or a vehicle via intraperitoneal administration 30 min after TBI. All mice were euthanized 24 h after TBI and after neurological scoring, a series of tests were performed, including brain water content and neuronal cell death in the cerebral cortex.
Results: The level of cytochrome c (Cyt c), malondialdehyde (MDA), glutathione peroxidase (GPx) and superoxide dismutase 1 (SOD) were restored to some degree following the SIN treatment. The SIN treatment significantly decreased caspase-3 expression and reduced the number of positive cells by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay and improved the survival of neuronal cells. Additionally, the pretreatment levels of MDA were restored, while Bax translocation to mitochondria and Cyt c release into the cytosol were reduced by the SIN treatment.
Conclusion: SIN protected neuronal cells by protecting them against apoptosis via mechanisms that involve the mitochondria following TBI.
Keywords: sinomenine, traumatic brain injury, apoptosis, mitochondria
Introduction
Traumatic brain injury (TBI) is defined as a mechanical injury, which causes numerous deaths and has an adverse impact on families and society. The mechanism of TBI involves primary and secondary brain damage. Although the primary brain injury is the major factor, the secondary brain damage precipitates a complex pathological process, which can lead to a sequence of deleterious side effects near the center of the initial injury.1,2 One of the important components of the secondary injury is swelling and release of various chemicals, which promote inflammation and cell injury.3 In these processes, oxidative stress plays an important role in the secondary damage. Damaged mitochondria produce a massive amount of reactive oxygen species (ROS), and the dysfunctional mitochondria may be degraded by autophagy to protect neurons and reduce the oxidative stress.4 During this process, the membrane permeability of impaired mitochondria increases and mitochondrial apoptosis-associated proteins promote neuronal cell death.5,6 Accordingly, the protection of mitochondrial biogenesis can enhance neuronal cellular function and promote cellular recovery from TBI.
Sinomenine (7,8-didehydro-4-hydroxy3,7-dimethoxy-17-methyl-9a,13a,14a-morphinan-6-one; SIN) is an active alkaloid extracted from the plant Sinomenium acutum, which has a broad spectrum of pharmacological properties including antiallergenic, anti-inflammatory, and cytoprotective.7,8 SIN exerts neuroprotective effects in different central nervous system (CNS) diseases, including cerebral ischemia, intracerebral hemorrhage, and neurodegenerative diseases.9,10 However, the mechanisms of these effects are still not well understood. Besides, few studies have addressed the neuroprotective effect of SIN in TBI. Accordingly, the purpose of the present study was to investigate the neuroprotective effect of SIN on a TBI model and whether it was related to the activation of mitochondrial protection.
Methods
Animals
Male Institute of Cancer Research (ICR) mice weighing 28–32 g were obtained from the Experiment Animal Center of Nanjin Medical University. The mice were maintained under the conditions of controlled temperature (24°C±0.5°C) with ad libitum access to food and water for 7 days before experiment. Experiment protocols were approved by the Animal Care and Use Committee at Jiangsu Hospital of Traditional Chinese Medicine and conformed to the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health (Bethesda, MD, USA).
Models of TBI
The TBI model used in this study was a modified version of Marmarou’s weight-drop model.11,12 Briefly, ICR mice were anesthetized via an intraperitoneal injection of 10% chloral hydrate and placed in a stereotaxic frame. A midline longitudinal scalp incision ~1.5 cm was made, and then, fascia and skull were exposed. After locating the left anterior frontal area, a 200 g weight was dropped. The scalp wound was subsequently closed. The mice were returned to quondam cages. Sham animals underwent identical procedures except the weight drop.
Experimental design
All the ICR mice were divided into the following six groups (n=18 per group): sham, TBI, TBI + vehicle, and TBI + SIN (with three subgroups receiving different SIN doses: 10, 30, and 70 mg/kg). The mice in the three TBI + SIN subgroups were intraperitoneally injected with SIN (Sigma-Aldrich Co., St Louis, MO, USA) at 10, 30, and 70 mg/kg, respectively, 30 min after onset of TBI. At the corresponding time points, all mice in the TBI + vehicle group were administered isometric volume of vehicle (saline containing 1% DMSO). The mice from each group were anesthetized and sacrificed 24 h post-TBI for biochemical and histological analyses.
Isolation of mitochondria
Mitochondrial and cytosolic proteins were extracted using the Tissue Mitochondria Isolation Kit for Tissue Protocols (Beyotime Institute of Biotechnology, Shanghai, China). Fresh tissue was quickly homogenized using a Polytron homogenizer in ice-cold homogenization buffer and centrifuged at 600× g for 5 min at 4°C. The obtained supernatants were centrifuged at 1,500× g for 10 min at 4°C, and the sediment was mitochondria. The supernatants were collected and centrifuged at 11,000× g for 10 min at 4°C to obtain cytoplasmic proteins. The protein level in each sample was confirmed with a protein assay kit.
Brain water content
The brain water content was measured as previously described.13 Mouse brain was removed and placed on a cooled brain matrix. After that, the brain stem and cerebellum were removed, the left cerebral hemispheres were separated and harvested, and the wet weight (ww) of each hemisphere was measured immediately. The samples were then dried at 80°C for 72 h and the dry weight (dw) was weighed. Water content was calculated as a percentage by the following formula: (ww − dw)/ww ×100%.
Neurological deficit
Neurological deficit was evaluated by the grip test, which was developed on the basis of the test of gross vestibulomotor function as described elsewhere.14 Briefly, mice were placed on a thin, horizontal, metal wire (45 cm long) that was suspended between two vertical poles 45 cm above a foam pad. A score of 0 was given if the mouse was unable to remain on the wire for <30 s; one point was given if the mouse failed to hold on to the wire with both forepaws and hind paws together; two points were given if the mouse held on to the wire with both forepaws and hind paws but not the tail; three points were given if the mouse used its tail along with both forepaws and hind paws; four points were given if the mouse moved along the wire on all four paws plus tail; and five points were given if mouse that scored four points also ambulated down one of the posts used to support the wire. The grip test was performed in triplicate, and a total value was calculated for each mouse. The test was conducted by an investigator who was blinded to the experimental groups.
Nissl staining
Coronal sections of the brain tissue (5 μm thick) were stained with cresyl violet as previously described.15 Normal neurons had large cell bodies and cytoplasmic volume, with one or two large, round nuclei. In contrast, damaged neuronal cells were identified as those with shrunken cell bodies, condensed nuclei, and dark cytoplasm containing many empty vesicles. Histological examination was performed by two observers who were blind to the group assignment.
Western blot analysis
Mitochondrial, nuclear and cytosolic proteins were extracted from the cerebral cortex tissue and quantified following the instructions in the Protein Extraction Kit (Beyotime Institute of Biotechnology). Equal amounts of protein samples were subjected to electrophoresis on 10%–12% sodium dodecyl sulfate-polyacrylamide gel for 45 min at 80 V, followed by 100 min at 100 V, and then were transferred onto a polyvinylidene fluoride membrane. The membrane was blocked with 5% skimmed milk for 2 h at room temperature and then incubated with primary antibodies overnight at 4°C. For this step, the antibodies used were cytochrome c (Cyt c) (1:5,000 dilution; Abcam, Cambridge, UK), β-actin (1:5,000 diluted, rabbit; Bioworld Technology Inc., St Louis Park, MN, USA), Bax (1:400 diluted, rabbit; Abcam), Bcl-2 (1:1,000 diluted, rabbit; Cell Signaling Technology, Danvers, MA, USA), COX IV (1:1,000 diluted, mouse; Cell Signaling Technology), and cleaved caspase-3 (1:1,000 diluted, rabbit; Cell Signaling Technology). Subsequently, the membranes were incubated for 2 h with the corresponding secondary antibodies and stained, and the bands were visualized.
Immunohistochemistry
Caspase-3 expression was evaluated by immunohistochemistry analysis. Brain tissue samples were fixed in formalin for 24 h and then embedded in paraffin. Sections were cut at 5 μm thickness, deparaffinized in xylene, dehydrated in a graded series of ethanol, subjected to antigen retrieval in citrate buffer (pH 6.0) for 30 min in a 37°C chamber, and then washed in PBS. The sections were quenched in 3% hydrogen peroxide and blocked with PBS containing 10% goat serum (Sigma-Aldrich Co.) for 1 h at 37°C, then incubated with a rabbit monoclonal anti-caspase-3 antibody at 1:300 (Cell Signaling Technology) overnight at 4°C, followed by three 15 min washes in PBS and incubation with horseradish peroxidase (HRP)-conjugated IgG at 1:500 dilution (Bioworld Technology Inc.) for 60 min at room temperature. After three washes in PBS, sections were counterstained with hematoxylin, dehydrated, and cleared with xylene before mounting. Control tissue was subjected to the same procedure but without the primary antibody step.
Mitochondrial malondialdehyde (MDA) content and superoxide dismutase (SOD) and glutathione peroxidase (GPx) activities
MDA content and SOD and GPx activities were measured using the appropriate kits (Nanjing Jiancheng Biochemistry Co, Nanjing, China) in accordance with the manufacturer’s instructions, using a spectrophotometer. Total protein concentration was determined by the Bradford method. The MDA content and SOD and GPx activities were expressed as nanomole per milligram of protein and unit per milligram of protein, respectively.
Statistical analyses
The SPSS 17.0 software (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. Data are expressed as the mean ± SEM and were evaluated by analysis of variance (ANOVA) and the Tukey’s post hoc tests. Statistical significance difference was set at P<0.05.
Results
SIN alleviated cerebral edema post-TBI
The brain water content was significantly increased after TBI, and there was no significant difference between the TBI group and the TBI + vehicle group (Figure 1).
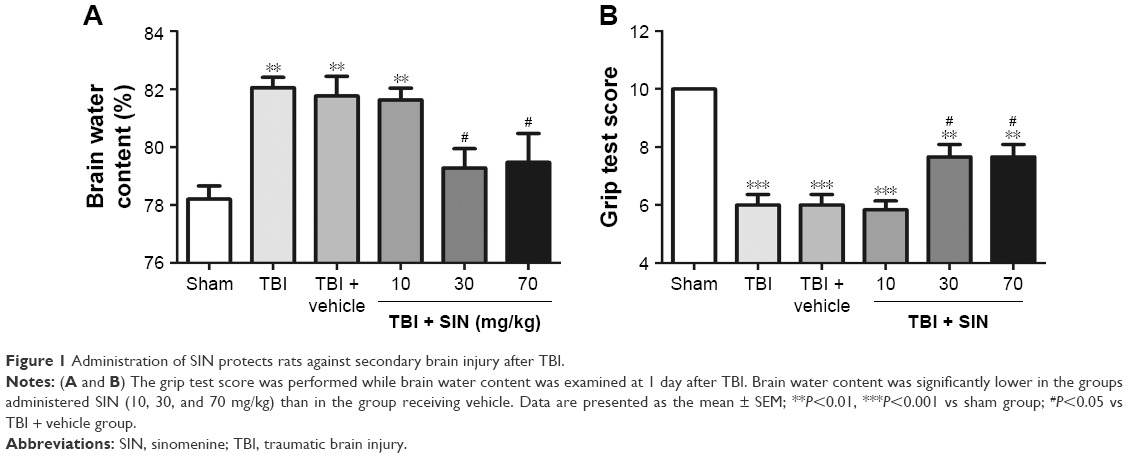 | Figure 1 Administration of SIN protects rats against secondary brain injury after TBI. |
In comparison, animals treated with SIN (30 or 70 mg/kg) had significantly lower brain water content than the TBI + vehicle group. However, larger doses (70 mg/kg) failed to provide additional neuroprotective effect. The results supported that SIN has protective effect after TBI. The dose of 30 mg/kg of SIN provided the best result and thus was chosen to be used in the subsequent experiments.
SIN decreased neuronal degeneration
Few positive cells were found in the brain tissue of the mice in the sham group, as shown in Figure 2. Also, the apoptotic index was clearly increased in the TBI + vehicle group relative to that in the sham group (Figure 2A–C, and E). No difference was detected between the TBI group and the vehicle-treated group (Figure 2B, C, and E). However, a decrease in the apoptotic index was observed in the TBI + SIN group (Figure 2D and E).
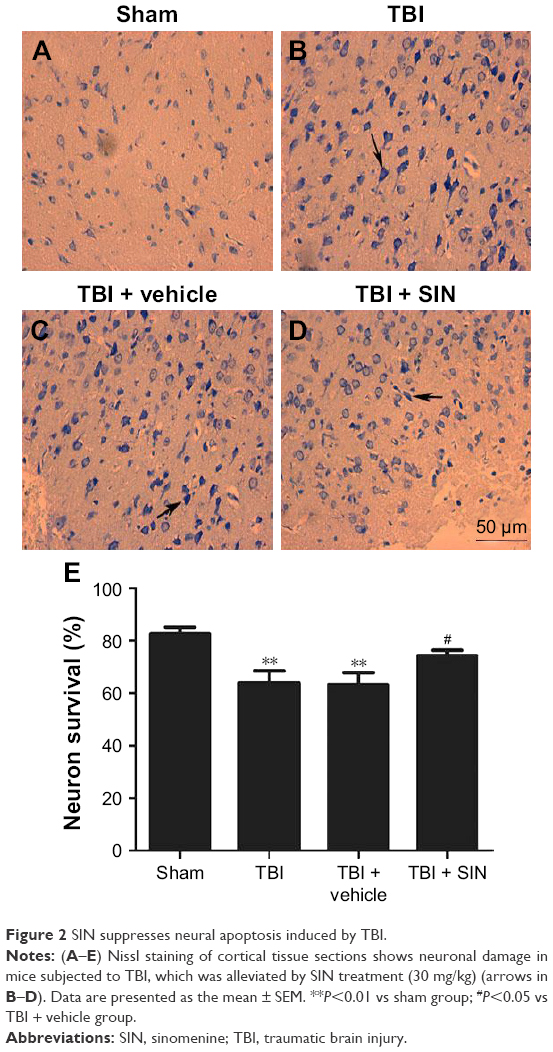 | Figure 2 SIN suppresses neural apoptosis induced by TBI. |
Expression of caspase-3 was downregulated by SIN administration
The protective effects of SIN against neuronal apoptosis after TBI were also detected by Western blot analysis and immunohistochemistry of mouse brain tissues. The number of caspase-3-positive cells in the group treated with SIN was fewer than in the group that received the vehicle treatment. In addition, the cleaved caspase-3 expression rose 24 h after TBI (Figure 3A–E and G). Also, compared with the sham group, in the SIN-treated group, the level of Bax protein was increased, whereas that in the cytosol was significantly decreased (Figure 4A–F). Except that, expression of the anti-apoptotic factor Bcl-2 decreased when compared with the sham group (Figure 4A, B, G, and H).
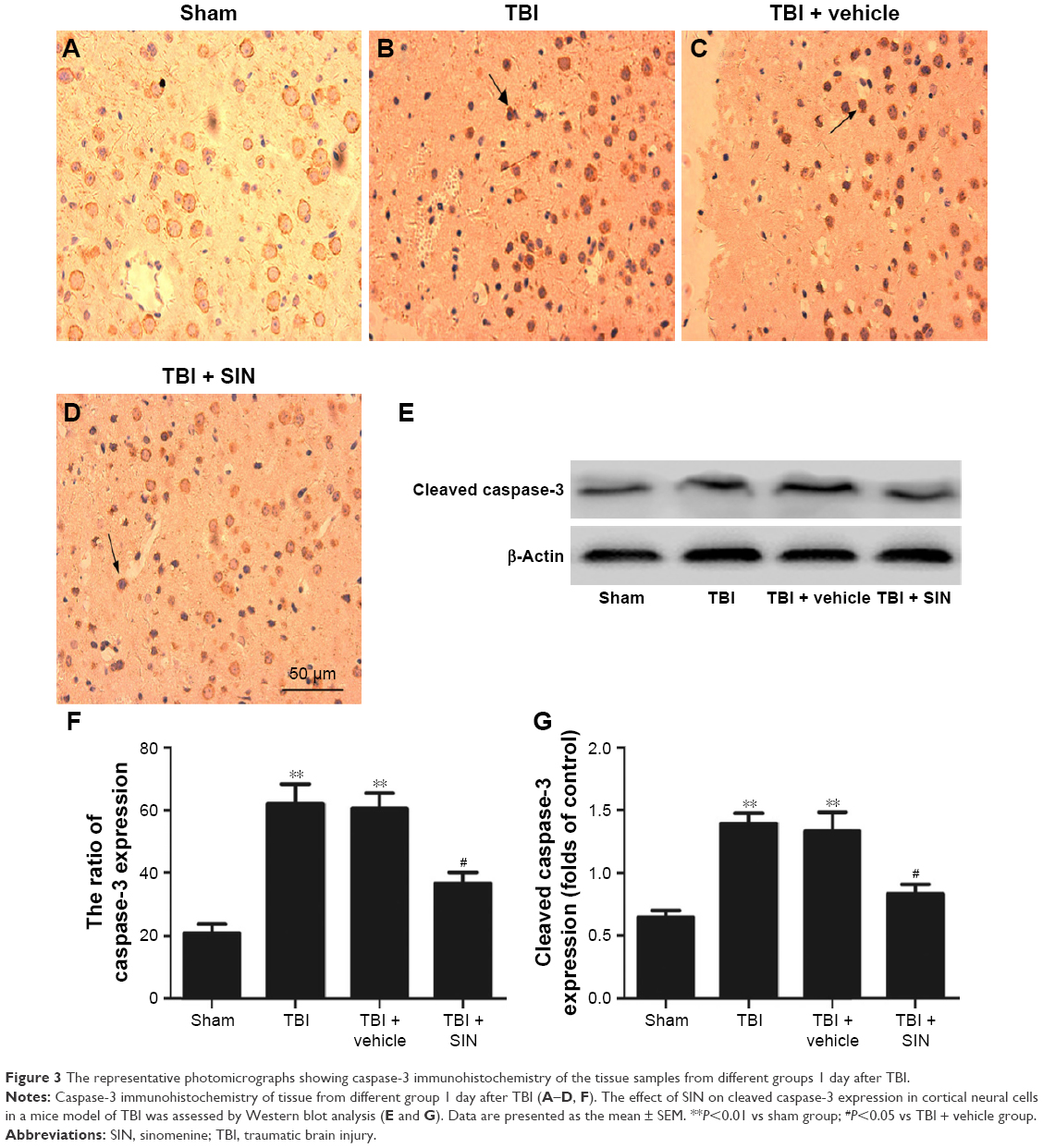 | Figure 3 The representative photomicrographs showing caspase-3 immunohistochemistry of the tissue samples from different groups 1 day after TBI. |
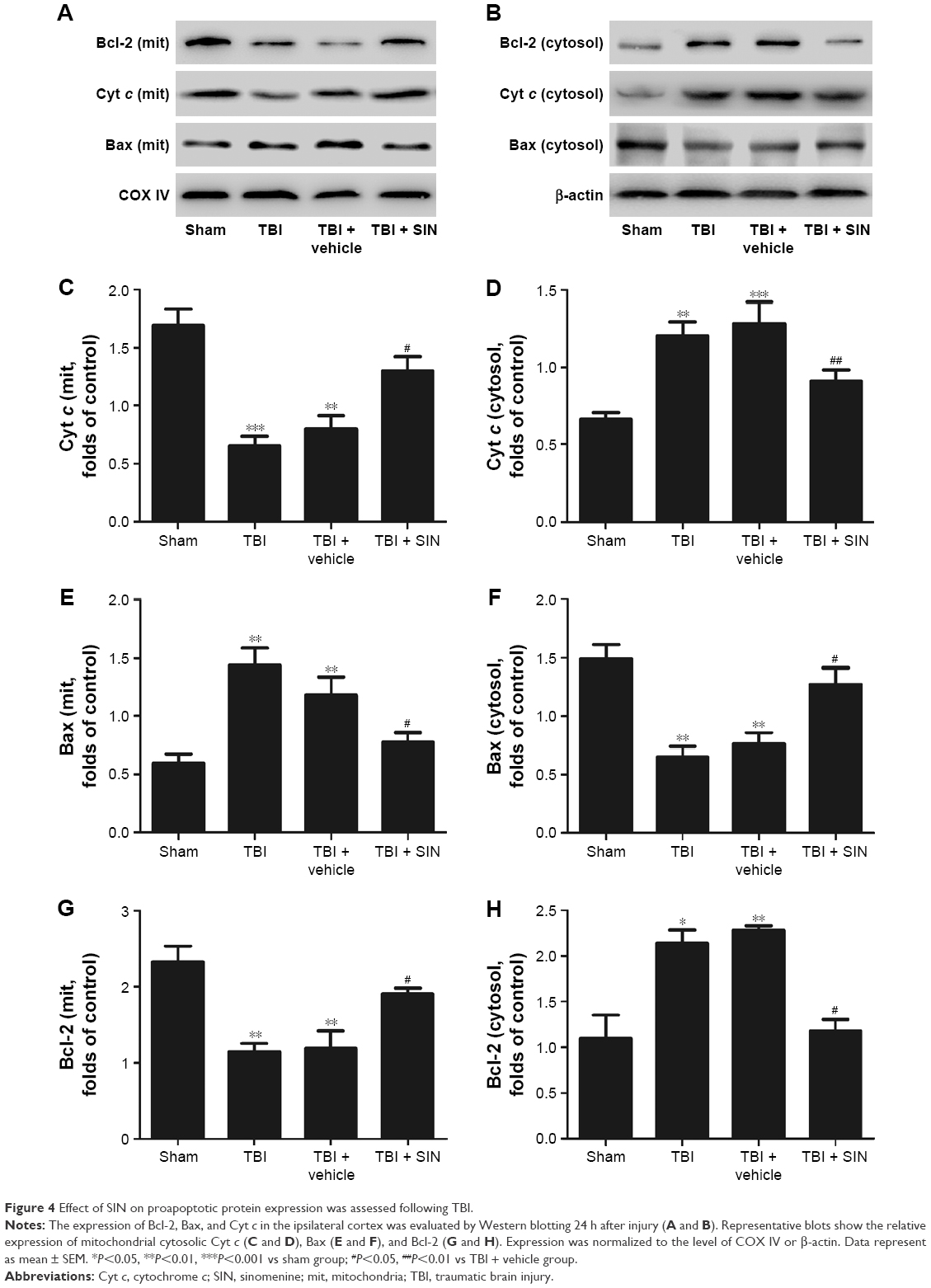 | Figure 4 Effect of SIN on proapoptotic protein expression was assessed following TBI. |
SIN reduces mitochondrial oxidative stress following TBI
Mitochondrial MDA level was increased in the TBI + vehicle group compared with the sham-operated controls (Figure 5A); this effect was mitigated by the administration of 30 mg/kg SIN. Mitochondrial GPx and SOD are antioxidant enzymes responsible for scavenging oxidative metabolites generated by free radicals. GPx and SOD activities were decreased after TBI (Figure 5B and C), but SIN treatment at a dose of 30 mg/kg increased GPx and SOD activities.
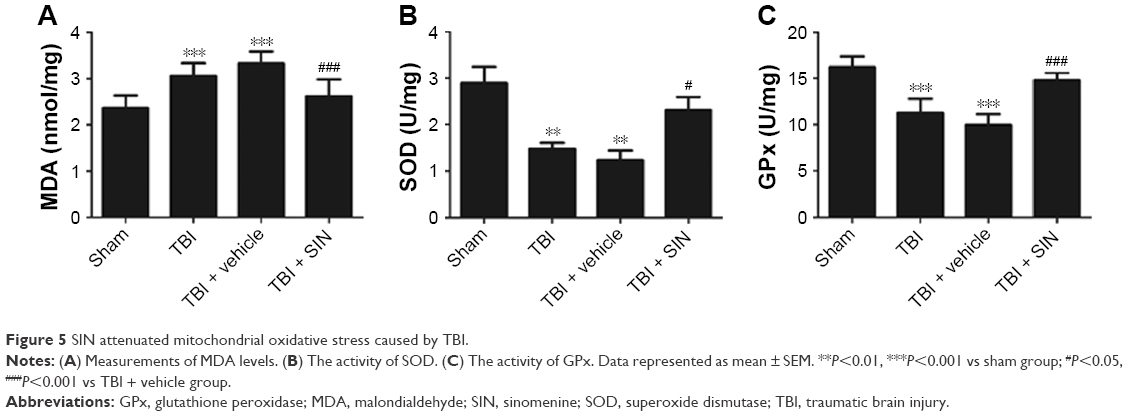 | Figure 5 SIN attenuated mitochondrial oxidative stress caused by TBI. |
Discussion
There is currently no effective strategy to alleviate brain damage caused by trauma, and the molecular mechanism of TBI-induced neurodegeneration remains unclear. SIN has been shown to exert neuroprotective effects in several CNS disease models.16–18 The result of this study also indicated that treatment with SIN post-TBI resulted in less cell apoptosis in the area surrounding the cortical contusion and could potentially ameliorate the secondary brain insult. However, studies on the molecular mechanisms of this activity have rarely been studied during the brain damage process after TBI.
Secondary brain injury after trauma may result from oxidative stress, excitotoxicity, or the inflammatory response.19,20 Oxidative stress is caused by ROS resulting from excitotoxicity and the failure of antioxidant mechanisms, which induce lipid peroxidation and DNA damage.21 Mitochondria play an important role in the injured brain by activating signaling pathways through ROS production or by inducing mitochondria-dependent apoptosis.22 Accordingly, neuroprotective approaches should involve limiting and reversing mitochondrial dysfunction. Lipid peroxidation is typically determined by the MDA content, which starts to increase immediately after injury and remains elevated 24 h later.23–25 Oxidative stress can also be evaluated by the activity of GPx or SOD, which converts peroxides into nontoxic forms. Mitochondrial metabolic impairment leads to oxidative modification of proteins, lipids, and DNA and leads to the concomitant increase in ROS.26,27 In the present study, SIN treatment reduced the mitochondrial MDA level and increased GPx and SOD activities compared to vehicle-treated mice, suggesting that SIN exerts antioxidant effects by protecting mitochondria.
Mounting evidence indicates that dysfunctional mitochondria can promote cell death through the mitochondrial apoptotic pathway.28,29 Briefly, an increase in expression of proapoptotic factors relative to the expression of Bax is the downstream factor in the mitochondrial apoptosis pathway.30 Then, mitochondrial Bax forms a channel permeable to Cyt c, which only exists in the mitochondrial intermembrane space.31,32 Next, the Cyt c released from the mitochondria leads to the sequential activation of caspase-3. This study showed that the release of Cyt c into the cytosol and the translocation of Bax to the mitochondrial membrane were significantly increased after TBI.33 The results indicated that the mitochondria impairment in brain tissue was increased after TBI. All these changes led to the upregulation of the levels of cleaved caspase-3, which in turn led to neuronal apoptosis. However, administration of 30 mg/kg of SIN inhibits the translocation of Bax to the mitochondrial membrane and reduces the release of Cyt c from the mitochondria to the cytoplasm.
Conclusion
This study demonstrated that SIN exerts neuroprotective effects against TBI by mitigating oxidative stress and suppressing the mitochondrial apoptotic pathway via the inhibition of Bax translocation and Cyt c release from the mitochondria. These findings indicate that SIN has potential therapeutic applications for preventing secondary brain damage following TBI.
Author contributions
JG edited and proofread the manuscript. QW provided substantial contributions to interpretation of data. CF and XZ provided technical assistance for this work. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Vascak M, Jin X, Jacobs KM, Povlishock JT. Mild traumatic brain injury induces structural and functional disconnection of local neocortical inhibitory networks via parvalbumin interneuron diffuse axonal injury. Cereb Cortex. Epub 2017 Mar 04:1–20. | ||
Bigler ED, Tsao JW. Mild traumatic brain injury in soldiers returning from combat. Neurology. 2017;88(16):1490–1492. | ||
Abe H, Shimoji K, Nagamine Y, Fujiwara S, Izumi SI. Predictors of recovery from traumatic brain injury-induced prolonged consciousness disorder. Neural Plast. 2017;2017:9358092. | ||
Wang JW, Li JP, Song YL, Zhao QH. Humoral and cellular immunity changed after traumatic brain injury in human patients. Ann Clin Lab Sci. 2017;47(1):10–16. | ||
Li X, Wang H, Gao Y, et al. Quercetin induces mitochondrial biogenesis in experimental traumatic brain injury via the PGC-1α signaling pathway. Am J Transl Res. 2016;8(8):3558–3566. | ||
Gao Y, Zhuang Z, Gao S, et al. Tetrahydrocurcumin reduces oxidative stress-induced apoptosis via the mitochondrial apoptotic pathway by modulating autophagy in rats after traumatic brain injury. Am J Transl Res. 2017;9(3):887–899. | ||
Huang L, Dong Y, Wu J, et al. Sinomenine-induced histamine release-like anaphylactoid reactions are blocked by tranilast via inhibiting NF-κB signaling. Pharmacol Res. 2017;125(pt B):150–160. | ||
Jiang Y, Jiao Y, Wang Z, et al. Sinomenine hydrochloride inhibits human glioblastoma cell growth through reactive oxygen species generation and autophagy-lysosome pathway activation: an in vitro and in vivo study. Int J Mol Sci. 2017;18(9):E1945. | ||
Qiu J, Wang M, Zhang J, et al. The neuroprotection of Sinomenine against ischemic stroke in mice by suppressing NLRP3 inflammasome via AMPK signaling. Int Immunopharmacol. 2016;40:492–500. | ||
Yang Y, Wang H, Li L, et al. Sinomenine provides neuroprotection in model of traumatic brain injury via the Nrf2-ARE pathway. Front Neurosci. 2016;10:580. | ||
Xu J, Wang H, Ding K, et al. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic Biol Med. 2014;71:186–195. | ||
Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E. Mouse closed head injury model induced by a weight-drop device. Nat Protoc. 2009;4(9):1328–1337. | ||
Ding K, Wang H, Xu J, et al. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. | ||
Bermpohl D, You Z, Korsmeyer SJ, Moskowitz MA, Whalen MJ. Traumatic brain injury in mice deficient in Bid: effects on histopathology and functional outcome. J Cereb Blood Flow Metab. 2006;26(5):625–633. | ||
Wei W, Wang H, Wu Y, et al. Alpha lipoic acid inhibits neural apoptosis via a mitochondrial pathway in rats following traumatic brain injury. Neurochem Int. 2015;87:85–91. | ||
Yang H, Jiang C, Chen X, He K, Hu Y. Protective effects of sinomenine against LPS-induced inflammation in piglets. Microb Pathog. 2017;110:573–577. | ||
Rao S, Liu S, Zou L, et al. Erratum to: the effect of sinomenine in diabetic neuropathic pain mediated by the P2×3 receptor in dorsal root ganglia. Purinergic Signal. 2017;13(2):237. | ||
Qiu J, Yan Z, Tao K, et al. Sinomenine activates astrocytic dopamine D2 receptors and alleviates neuroinflammatory injury via the CRYAB/STAT3 pathway after ischemic stroke in mice. J Neuroinflammation. 2016;13:263. | ||
Vella MA, Crandall ML, Patel MB. Acute management of traumatic brain injury. Surg Clin North Am. 2017;97(5):1015–1030. | ||
Zwemer E, Bernson-Leung M, Rea C, et al. Education on the brain: a partnership between a pediatric primary care center and neurology residency. Clin Pediatr (Phila). Epub 2017 Feb 13. | ||
Wang JF, Li Y, Song JN, Pang HG. Role of hydrogen sulfide in secondary neuronal injury. Neurochem Int. 2014;64:37–47. | ||
Lu Y, Li Y, Xiang M, Zhou J, Chen J. Khat promotes human breast cancer MDA-MB-231 cell apoptosis via mitochondria and MAPK-associated pathways. Oncol Lett. 2017;14(4):3947–3952. | ||
Waseem M, Parvez S. Neuroprotective activities of curcumin and quercetin with potential relevance to mitochondrial dysfunction induced by oxaliplatin. Protoplasma. 2016;253(2):417–430. | ||
Ghosh A, Sarkar S, Mandal AK, Das N. Neuroprotective role of nanoencapsulated quercetin in combating ischemia-reperfusion induced neuronal damage in young and aged rats. PLoS One. 2013;8(4):e57735. | ||
Wan J, Deng L, Zhang C, et al. Chikusetsu saponin V attenuates H2O2-induced oxidative stress in human neuroblastoma SH-SY5Y cells through Sirt1/PGC-1α/Mn-SOD signaling pathways. Can J Physiol Pharmacol. 2016;94(9):919–928. | ||
Karuppagounder SS, Madathil SK, Pandey M, Haobam R, Rajamma U, Mohanakumar KP. Quercetin up-regulates mitochondrial complex-I activity to protect against programmed cell death in rotenone model of Parkinson’s disease in rats. Neuroscience. 2013;236:136–148. | ||
Panickar KS, Anderson RA. Mechanisms underlying the protective effects of myricetin and quercetin following oxygen-glucose deprivation-induced cell swelling and the reduction in glutamate uptake in glial cells. Neuroscience. 2011;183:1–14. | ||
Franco JL, Braga HC, Stringari J, et al. Mercurial-induced hydrogen peroxide generation in mouse brain mitochondria: protective effects of quercetin. Chem Res Toxicol. 2007;20(12):1919–1926. | ||
Ulbrich F, Kaufmann KB, Meske A, et al. The CORM ALF-186 mediates anti-apoptotic signaling via an activation of the p38 MAPK after ischemia and reperfusion injury in retinal ganglion cells. PLoS One. 2016;11(10):e0165182. | ||
Arredondo F, Echeverry C, Abin-Carriquiry JA, et al. After cellular internalization, quercetin causes Nrf2 nuclear translocation, increases glutathione levels, and prevents neuronal death against an oxidative insult. Free Radic Biol Med. 2010;49(5):738–747. | ||
Tian X, Gao L, An L, et al. Pretreatment of MQA, a caffeoylquinic acid derivative compound, protects against H2O2-induced oxidative stress in SH-SY5Y cells. Neurol Res. 2016;38(12):1079–1087. | ||
Gao Y, Chen T, Lei X, et al. Neuroprotective effects of polydatin against mitochondrial-dependent apoptosis in the rat cerebral cortex following ischemia/reperfusion injury. Mol Med Rep. 2016;14(6):5481–5488. | ||
Zhai X, Ding Y, Wang Q, Zhang H, Li F. Rutin acid ameliorates neural apoptosis induced by traumatic brain injury via mitochondrial pathways in mice. Neuroimmunomodulation. 2016;23(3):179–187. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.