")
Back to Journals » International Journal of Nanomedicine » Volume 14
Self-Assembly Of Retinoid Nanoparticles For Melanoma Therapy
Authors Liao H , Zhao S, Wang H, Liu Y, Zhang Y, Sun G
Received 5 December 2018
Accepted for publication 27 July 2019
Published 1 October 2019 Volume 2019:14 Pages 7963—7973
DOI https://doi.org/10.2147/IJN.S196974
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Han Liao,1,2 Shan Zhao,1,2 Huihui Wang,1,2 Yang Liu,1 Ying Zhang,1 Guangwei Sun1
1Scientific Research Center for Translational Medicine, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, People’s Republic of China; 2University of Chinese Academy of Sciences, Beijing 100049, People’s Republic of China
Correspondence: Guangwei Sun
Scientific Research Center for Translational Medicine, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, People’s Republic of China
Tel/Fax +86-411-82463027
Email [email protected]
Background: Amphiphilic fusion drugs are covalent conjugates of a lipophilic drug and a hydrophilic drug or their active fragments. These carrier-free self-assembly nanomaterials are helpful to co-deliver two synergic drugs to the same site regardless of pharmacokinetic properties of individual drugs. Retinoic hydroxamic acid (RHA) is a “fusion drug” of all-trans retinoic acid (ATRA) and vorinostat, a histone deacetylase (HDAC) inhibitor showing synergic effect with ATRA on cancer therapy. Although RHA was synthesized in 2005, its nanoscale self-assembly property, anticancer activity, and possible related mechanism are still unclear.
Methods: RHA nanoparticles were observed under transmission electron microscope (TEM). Both in vitro cell viability, colony formation assay, and in vivo xenograft mouse tumor model were employed here to study anticancer activity of RHA nanoparticles. The putative synergic anticancer mechanism of activating retinoic acid receptor (RAR) and inhibiting HDAC were investigated via receptor inhibitor rescue assay and in vitro enzyme activity assay, respectively.
Results: RHA could form nanoparticle formation by self-assembly and abrogates growth of several solid tumor cell lines even after RHA nanoparticles’ washout. However, opposite to our initial hypothesis, pre-treating the melanoma cells with RAR antagonists showed no impact on inhibitory effect of RHA nanoparticles, which suggested that the target of the molecule on melanoma cells is not RAR and retinoid X receptor (RXR). Importantly, RHA nanoparticles inhibited the growth of xenograft tumors without obvious impact on haematological indexes and hepatorenal function of these tumor-bearing mice.
Conclusion: Our findings demonstrate the promise of RHA nanoparticles in treating malignant melanoma tumors with high efficacy and low toxicity.
Keywords: nano-drugs, self-assembly, retinoid, cancer therapy, melanoma
Introduction
Since the 1980s, the medical drug all-trans retinoic acid (ATRA) has been clinically utilised for the treatment of acute promyelocytic leukemia.1 Unlike traditional chemotherapeutic agents, ATRA results in high complete remission rates, but without adverse effects, such as bone marrow hypoplasia.2 Previous in vitro studies demonstrated that ATRA can inhibit proliferation and induce differentiation of some solid tumor cell lines.3,4 However, the results of in vivo studies and clinical trials were not as compelling.5,6 Hence, there is significant scope to investigate methods to enhance the anticancer efficiency of ATRA on solid tumor cell lines.
It is known that nanoparticles tended to accumulate in solid tumors due to enhanced permeability and retention (EPR) effects.7,8 This effect has been exploited to enhance anticancer drug potency when the drugs are prepared as nanoparticles. Previous investigations have reported that ATRA-loaded nanoparticles can reverse ATRA resistance, including inhibiting cell proliferation and inducing cell differentiation.9,10 On the other hand, many drugs, such as histone deacetylase inhibitor vorinostat, showed substantial synergistic effect with ATRA on cancer therapy.11,12 Previous study presented a successful fusion drug strategy of structurally combining two synergistic drugs together to assemble nanoparticles for delivery.13 Here, we attempted to use this strategy on ATRA and vorinostat for providing a possible way for treating ATRA-resistant solid tumors. Because structurally modifying carboxylic acid group of ATRA to amide showed increased cytotoxicity against solid tumor cells,14,15 and hydroxamic group is the active centre of vorinostat, retinoic hydroxamic acid (RHA), a hydroxamic derivative of ATRA, was selected for this study.
RHA comprised of a lipophilic ATRA hydrocarbon backbone and a hydrophilic hydroxamic group. These functional groups may provide a potential synthetic avenue for the formation of nanoparticles by self-assembly. Although RHA was synthesized a couple of years ago, there is no previous investigation into the nanoscale characteristics and anticancer activity of RHA.16,17 In this report, we investigated whether RHA can form stable nanoparticles by self-assembly and thereafter investigate their anticancer effect on solid tumors.
Materials And Methods
Materials
ATRA was purchased from Meilunbio (Dalian, China). MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide), DCC (dicyclohexylcarbodiimide), NHS (N-hydroxylsuccinimide), DMAP (4-dimethylaminopyridine), DIPEA (N, N-diisopropylethylamine), and hydroxylamine hydrochloride were purchased from Sigma-Aldrich (Darmstadt, Germany). All the cell lines were obtained from Shanghai Institutes for Biological Science, CAS (Shanghai, China). Cell apoptosis and cell cycle detection kits were from Gefan Biotechnology (Shanghai, China). Poloxamer 184 and poloxamer 188 were purchased from Haian petrochemical technology (Nantong, China) and further purified by dialysis before use. AGN193109 and UVI3003 were purchased from Santa Cruz (California, USA). Histone deacetylase activity kit was purchased from Haling Biotechnology (Shanghai, China). Balb-c/nu mice were purchased from Beijing Vital River Laboratory Animal Technology (Beijing, China). β-actin and β-catenin antibody solutions were purchased from Proteintech (Rosemont, USA). SH-SY5Y and LN-229 cell lines were purchased from the American Type Culture Collection, while the rest of cell lines, U-118 MG, A-375, HL-60 and L-929 were purchased from Type Culture Collection of Chinese Academy of Science (Shanghai, China). Balb-c/nu mice used in this study were purchased from Vitalriver (Beijing, China) and raised in SPF centre of Dalian Medical University. All animal experiments were approved by the Institutional Animal Care and Use Committee of Dalian Medical University Laboratory Animal Center (China) and conformed to Chinese Ministry of Public Health (CMPH) and US National Institute of Health (NIH) guidelines.
Synthesis Of RHA
A mixture of 300 mg ATRA, 24.4 mg DMAP, 230 mg NHS, 247 mg DCC, and 150 μl DIPEA was dissolved in 3 mL tetrahydrofuran and then stirred at room temperature for 10 hrs. A filtered mixture of 20 mL 0.5 M hydroxylamine hydrochloride (ethanol solution) and 9 mL 1.0 M fresh sodium ethoxide (ethanol solution) was added, and then stirred at room temperature for 16 hrs. After solvent evaporated, the product was purified by D101 macroporous resin.
Preparation Of RHA And ATRA Nanoparticles
RHA and ATRA nanoparticle solutions were prepared using solvent diffusion methods: 100 μL of RHA or ATRA (ethanol solution) was injected into 900 μL of vigorously stirring water or PBS. Poloxamer-coated RHA nanoparticles solution were prepared by adding poloxamer 184 and poloxamer 188 in 10 mM RHA nanoparticles solution to a final concentration of 189 mg L−1 and 94.5 mg L−1, respectively.
Characterization
Mass spectrum of RHA was analyzed by a high-resolution mass spectrometer (6540 QTOF, Agilent) with negative charged mode. 1HNMR and 13CNMR of RHA were analyzed by a 700 MHz NMR spectrometer (Avance III HD, Bruker) using CDCl3 as solvent. Microscopic images of RHA nanoparticles were obtained from a high-resolution transmission electron microscopy (JEM-2100, JEOL). Size and polydispersity index (PDI) of RHA and ATRA nanoparticles were measured by dynamic light scattering assay with a Zetasizer (ZS90, Malvern).
Proliferation Analysis
A modified MTT colorimetric assay was employed here to determine the proliferation of the cells.18 Briefly, cells were seeded in 96-well plates with a density of 6000 cells per well. After attachment for 24 hrs, cells were treated with RHA nanoparticles or ATRA and incubated for 48 hrs (non-treated cells were used as control). A solution containing 100 g L−1 SDS, 50 g L−1 isobutanol, and 0.04 M HCl was utilized to dissolve formazan. IC50 values were calculated using regression probit assay with SPSS V19.0 (Chicago, USA), according to the results of proliferation analysis.
Flow Cytometry Analysis
Cells were seeded in 10-cm dishes with a density of 2 × 106 cells per dish. After attachment for 24 hrs, the cells were treated with variant concentrations of RHA nanoparticles for 48 hrs (non-treated cells as control). Apoptosis and cell cycle of these treated cells were analyzed using flow cytometry (Moflow XDP, Beckman Coulter) with the protocol provided by the manufacturer of the kits.
Long-Term Cytotoxicity
Cells were seeded in 6-well plates with a density of 2 × 105 cells per well. After attachment for 24 hrs, the cells were treated with 25.0 μM of RHA nanoparticles for 48 hrs and then maintained in RHA-free medium. Viable cell numbers and viability of the cells were analyzed daily thereafter using a cell counter (Vi-cell XR, Beckman Coulter) based on trypan blue staining assay.
Soft Agar Colony Forming Assay
Cells were treated with 25.0 μM of RHA nanoparticles for 0 (as control), 2, 3, and 4 days, and then seeded in 6-well plates with a density of 2500 cells per well as previously described.19 Liquid media was renewed every other day. After two weeks, the colonies were stained with 0.5 mg/mL MTT.
In Vivo Distribution Experiment
All the in vivo studies were permitted by local animal ethics committee. Distribution of the nanoparticles was determined consulting a fluorescent-dye-labelled assay described previously.13 Hexadecylamine-modified Cy7 dye (HA-Cy7), a near-infrared probe, was incorporated into RHA nanoparticles during preparation with a molar ratio of 0.5% in order to trace the RHA nanoparticles. HA-Cy7 was synthesized as we described previously.20 A single dose of HA-Cy7 or HA-Cy7 labelled nanoparticles was injected into tumor-bearing Balb-c/nu mice intravenously. Anesthetized by isoflurane, the mice were scanned using a fluorescence imaging system (Maestro EX, CRI) at scheduled time intervals. The excitation and filter wavelength were 605 nm and 675 nm, respectively. Images were obtained with an emission wavelength range from 680 to 850 nm and an exposure time of 1 s.
In Vivo Antitumor Experiment
A subcutaneous xenografted A-375 tumor model was built using Balb-c/nu mice. A-375 melanoma cells were inoculated subcutaneously into 12 Balb-c/nu mice with a dose of 4 × 106 cells per mouse. Fourteen days later, when the average tumor size reached approximately 0.6 cm3, the mice were randomly divided into 2 groups. A solution consisting of 10% (v/v) ethanol with 189 mg L−1 poloxamer 184 and 94.5 mg L−1 poloxamer 188 was used as vehicle control, and 10 mM poloxamer-coated RHA nanoparticles were used for treatment. These 2 solutions were injected into the mice intravenously with a dose of 10 mL kg−1 every other day for 6 times. Body weight and tumor sizes of the mice were measured weekly. The tumor volume was calculated using a formula described previously:21 volume = 0.5× length × width2. Two days after treatment was completed, blood samples from the mice were obtained and then analyzed using a haematology analyzer (BC5300, Mindray) or a biochemical parameter (BS380, Mindray).
Anticancer Effect Of RHA Nanoparticles After RAR Or RXR Inhibition
Cells were seeded in 96-well plates with a density of 6000 cells per well and incubated for 24 hrs. A375 cells were treated with 6.25 μM of RHA nanoparticles alone, or together with either 25 μM of AGN193109 (pan-RAR antagonist) or 25 μM of UVI3003 (pan-RXR antagonist). HL-60 cells were also treated in the similar way with reduced final concentration of all compounds to half. Proliferation of the cells was determined using MTT assay after 48 hrs of treatment.
Determining Histone Deacetylase Activity
Activity of histone deacetylase was determined using a kit based on colorimetric assay. Briefly, histone deacetylase was extracted from 5 × 106 A375 cells and mixed with substance provided by the manufacturer. After aliquot, RHA, vorinostat, or vehicle was added and the mixtures were incubated at 37°C for 2 hrs. Absorbance of the samples was read after pre-treatment as described in the manufacturer’s instruction.
Statistics
IC50 values were calculated from the data of proliferation study (n = 4) by Probit regression with SPSS V19.0 (Chicago, USA). An average proliferation value, instead of individual ones of each treatment, was used for IC50 calculation to get better fitted curve. All other measurement data were shown as mean with SD error bars. Statistical analysis was carried out using independent sample t-test (2 groups) or LSD one-way ANOVA test (over 2 groups) with SPSS V19.0 (Chicago, USA). Statistical significant were marked as * (P < 0.05), ** (P < 0.01), and *** (P < 0.001).
Results
Preparing RHA And RHA Nanoparticles
RHA has previously been synthesized via an phosphoric anhydride intermediate, with a total yield of 45%.16 After purification by a D101 macroporous resin, this alternative method (Scheme 1) produced an increased total yield of 53% ± 6.6%. The successful synthesis of RHA was confirmed by high-resolution mass spectroscopy and NMR spectroscopy (Figures S1–S3, Tables S1 and S2).
After RHA synthesized, we moved on to investigating RHA nanoparticle formation by self-assembly. RHA could form stable nanoparticles using solvent diffusion assay while ATRA nanoparticles made in the same way aggregated 10 mins after preparation (Figure S4A). No obvious aggregation was detected while RHA nanoparticles were in serum (Figure S4) and PBS (Figure S5). The average size of RHA nanoparticles measured using dynamic light scattering assay was found to be 78.5 nm, which was significantly smaller than that of ATRA (Figure 1A, P<0.001). Moreover, the greater standard error of the polydispersity index (PDI) indicated poor reproductivity of ATRA nanoparticle formation (Figure 1B). Microscopic structure of RHA nanoparticles was observed using transmission electron microscopy, revealing solid spheres with smooth edges (Figure 1C). Overall, these features are similar to previously reported self-assembled nano-drugs,13 indicating that RHA forms nanoparticles by self-assembly.
 |
Figure 1 (A) Size and (B) polydispersity index of RHA nanoparticles analyzed by dynamic light scattering. Data are shown as mean with SD error bars (n = 3). Significance difference: *** (P < 0.001). (C) Transmission electron microscopy image of RHA nanoparticles. |
Anti-Tumor Proliferative Activity Of RHA Nanoparticles
Several high-malignancy solid tumor cell lines22 were selected for this study. We found that IC50 values of RHA nanoparticles against all tested cell lines were under 100 μM (Figure 2A), while the IC50 values of ATRA for most tested cells were greater than 100 μM (Figure S6), the maximum soluble concentration of ATRA in media. These findings indicated that ATRA-resistant solid tumor cell lines were still sensitive to RHA nanoparticles treatment. Among these tested cell lines, melanoma (A-375) cells were the most sensitive to RHA nanoparticles with an IC50 value of 7.5 μM (Figure 2A). Importantly, proliferation of A-375 cells was not inhibited when these cells were treated with 100 μM of ATRA. This finding indicated that A-375 is completely resistant to ATRA (Figure S6). To further compare anticancer potency of RHA nanoparticles and ATRA against A-375 cell line, the cells were treated with RHA nanoparticles or ATRA at the same final concentration. The result showed more significant proliferative inhibition activity of RHA nanoparticles than that of ATRA on A-375 cells (Figure 2B, P < 0.001). These data strongly suggest that RHA nanoparticles should be a more effective drug than ATRA for melanoma therapy. In order to learn the cytotoxicity of RHA nanoparticles against non-cancer cells, we used a normal cell line L-929 as reference in this cytotoxicity study. The IC50 value of RHA nanoparticles against mouse fibroblast L-929 cells was 36.9 μM which is 4.9 times higher than that of A-375 cells (Figure S7).
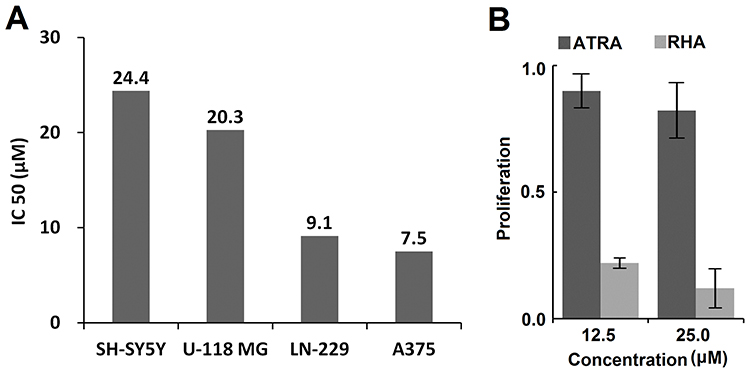 |
Figure 2 (A) IC50 values of RHA nanoparticles against 4 cancer cell lines. (B) Proliferation of A-375 cells after treatment with 12.5 μM and 25.0 μM of RHA nanoparticles or ATRA, respectively, for 48 hrs. Data are shown as mean with SD error bars (n = 4). Significance difference: *** (P < 0.001). |
Apoptosis And Cell Cycle Arrest Induced By RHA Nanoparticles
In general, decreased proliferation is usually considered as a result of cell cycle arrest and/or cell death. In order to clarify the mechanism of RHA nanoparticles-induced proliferative inhibition on A-375 cells, cell cycle alteration and apoptosis of RHA nanoparticles treated A-375 cells were determined. RHA nanoparticles demonstrate a dose-dependent cell cycle arrest (Figure 3A). Treated with 12.5 μM of RHA nanoparticles, A-375 cells were arrested in S phase; while treatment with 25.0 μM of RHA nanoparticles results in cell arrest in G0/G1 phase (Figure 3A). Therefore, we assume that RHA nanoparticles slow down DNA replication at lower concentrations and completely stop DNA replication at a higher concentration. To further verify whether RHA was DNA replication inhibitor, we treated thymidine double blocking synchronized A-375 cells with RHA nanoparticles (25 μM) and released at the same time. While non-treated (RHA-) A-375 cells entered S and G2/M phase 6 hrs after release, the cells with treatment (RHA+) failed to do so (Figure S8). This result confirmed that RHA nanoparticles were efficient DNA replication inhibitors. Besides cell cycle arrest, RHA nanoparticles also induced apoptosis of A-375 cells in a dose-dependent manner. The proportion of intact cells was decreased from 91.75% to 61.31% when the concentration of RHA nanoparticles increased from 6.25 μM to 25.0 μM (Figure 3B). Early apoptotic cells (AV+, PI-) increased after RHA nanoparticle treatment (Figure 3), specifically indicating that RHA nanoparticles could induce apoptotic cell death. These results suggested that both cell cycle arrest and apoptosis contributed to RHA nanoparticles-induced proliferative inhibition of A-375 cells.
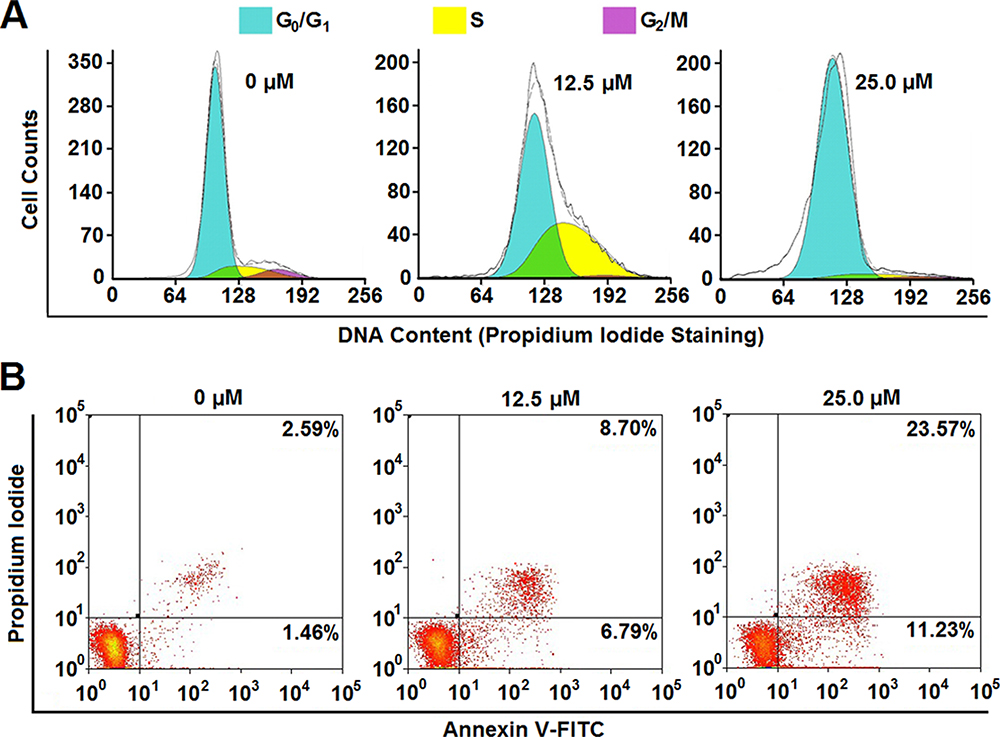 |
Figure 3 (A) Cell cycle analysis and (B) apoptotic cell proportion of A-375 cells treated with various concentrations of RHA nanoparticles for 48 hrs. |
Long-Lasting Anticancer Effect Of RHA Nanoparticles
To explore whether RHA nanoparticles have long-lasting anticancer effects after RHA withdrawal, A-375 cells were treated with 25.0 μM RHA nanoparticles for 2 days and then cultured in RHA-free media. At that time, 18.5% of the A-375 cells were killed (Figure 4A), which aligns well with that found in apoptotic cell analysis (Figure 3B). During the following 4 days, the viability of A-375 cells decreased continuously (Figure 4A) and the total counts of viable cells also reduced accordingly (Figure 4B). These data suggested that the cytotoxicity of RHA nanoparticles on A-375 cells was long lasting and there was no sign of “relapse” in this in vitro cell culture model.
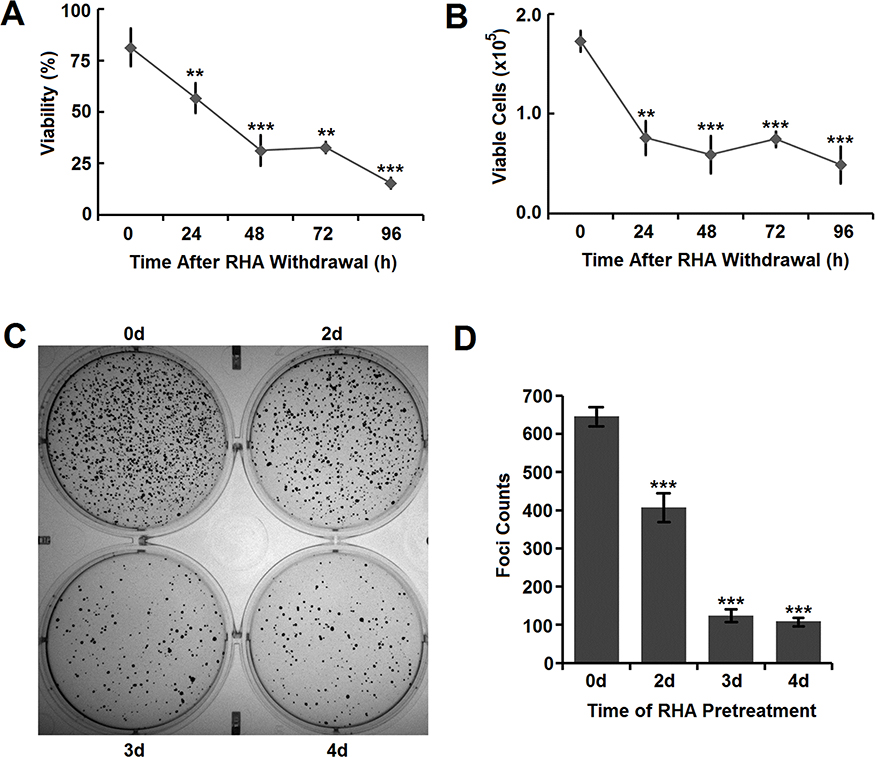 |
Figure 4 (A) Viability and (B) viable cells of A-375 cells after RHA withdrawal. Cells were pre-treated with 25 μM of RHA nanoparticles for 48 hrs. (C) Image and (D) foci counts of colony formation of A-375 cells pre-treated with 25 μM of RHA nanoparticles for various days. Data were shown as mean with SD error bars (n=3). Significance difference: ** (comparing with 0 hrs, P < 0.01) and *** (comparing with 0 hrs, P < 0.001). |
Anti-Tumorigenicity Of RHA Nanoparticles
Long-term proliferative inhibition is closely related to reducing tumorigenicity which could be indirectly determined using cell colony formation assay.23,24 RHA nanoparticles significantly prevented colony formation of A-375 cells in a time-dependent manner (Figure 4C and D), which meant that A-375 cells would lose their tumorigenicity with RHA nanoparticles treatment.
In Vivo Distribution And Optimization Of RHA Nanoparticles
Before in vivo RHA nanoparticles therapy, dynamic bio-distribution of RHA nanoparticles was confirmed via near-infrared imaging. We find that the naked RHA nanoparticles were mainly accumulated in the liver after administration (Figure 5). This undesired accumulation might be attributable to a nonspecific uptake by mononuclear phagocyte systems in liver, which is highly correlated with recognition and elimination of colloidal carriers.25 To remedy this defect, a mixture of poloxamer 188 and 184 was used as stealth material to coat RHA nanoparticles. While the poloxamer coating slightly enlarged the size of RHA nanoparticles, the impact on PDI and cytotoxicity of RHA nanoparticles was insignificant (Figure S9). In particular, as expected, poloxamer-coated RHA nanoparticles accumulate more in tumor and less in liver than the naked ones (Figures 5, S10). Therefore, poloxamer-coated RHA nanoparticles were used for in vivo administration thereafter.
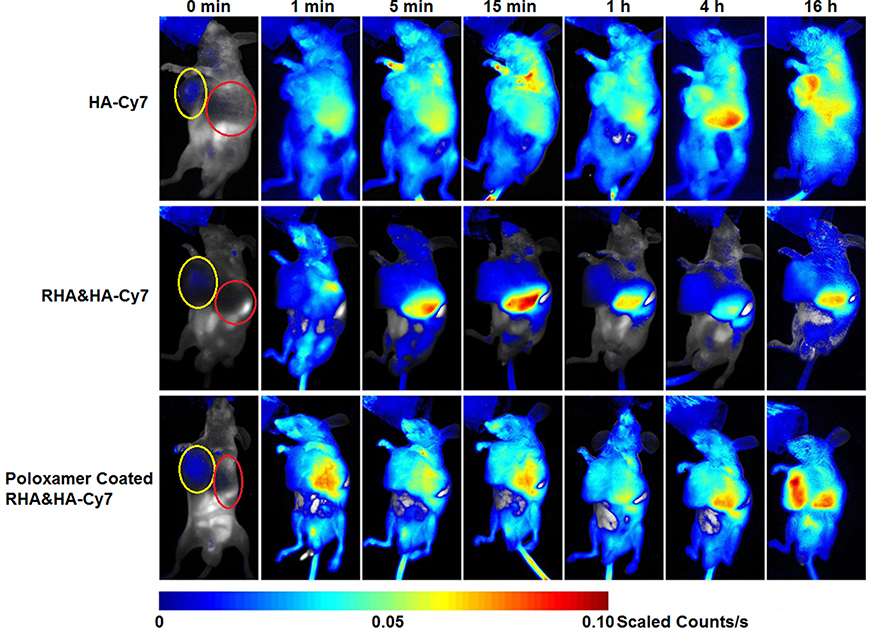 |
Figure 5 Dynamic in vivo distribution of RHA nanoparticles and poloxamer-coated RHA nanoparticles observed using 0.5% (mol/mol) hexadecylamine-modified Cy7 (HA-Cy7) as tracer. A single dose of nanoparticles or HA-Cy7 was administered intravenously. Yellow circle indicated the location of tumors, while red one indicated the location of livers. |
In Vivo Cancer Therapy With RHA Nanoparticles
While the volume of tumors on vehicle control (10% ethanol with poloxamer) mice increased rapidly (Figure 6A and B), the volume of tumors on RHA-nanoparticles-treated mice almost sustained in their initial sizes (Figure 6A and B). In addition, the volume of tumors on RHA-nanoparticles-treated mice was significantly smaller than that of control mice (Figure 6B, P < 0.01), which clearly illustrated those RHA nanoparticles effectively inhibited growth of xenograft melanoma.
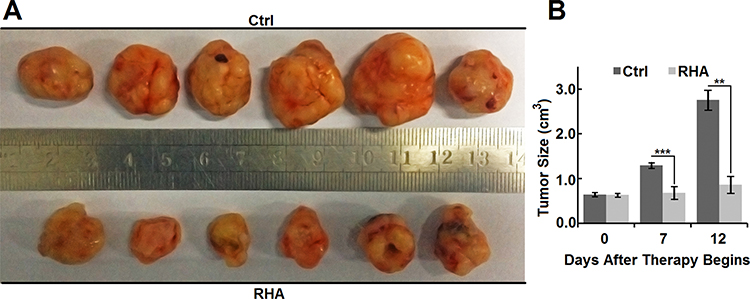 |
Figure 6 (A) Appearance and (B) size of Balb/c-nu mice tumors treated with vehicle or RHA nanoparticles. Vehicle or 10 mmol/kg RHA nanoparticles were administrated intravenously every 2 days from the 14th day to 24th day of A-375 cell presentation. Data are shown as mean with SD error bars (n = 6). Significance difference: ** (P < 0.01) and *** (P < 0.001). |
Net body weight is a general indicator for nutritional status and physical condition and chemotherapy-induced body weight loss is frequently attributed to toxicity. Our results show that RHA nanoparticles have no obvious effect on net body weight of the tumor-bearing mice during treatment (Figure S11A). Frequently reported adverse reactions of chemotherapeutic agents included haematopoietic,26 hepatic,27 and renal toxicity.28 No significant difference in red blood cell (P = 0.72), white blood cell (P = 0.30), and platelet counts (P = 0.57) between RHA nanoparticles treated and control mice indicated that RHA nanoparticles have negligible influence on the haematopoietic system (Figure S11B). RHA nanoparticles did not increase serum urea (P = 0.17) or uric acid, demonstrating its low toxicity to kidneys (Figure S11C). On the contrary, serum uric acid level decreased (P < 0.05) after RHA nanoparticles treatment (Figure S11C). As a metabolite of purine, high serum uric acid levels suggest poor prognosis in many malignant diseases.29,30 Therefore, the significant down-regulated serum uric acid observed here by RHA nanoparticles indicates a favourable prognosis. Moreover, as albumin (P = 0.76), serum aspartate aminotransferase (P = 0.24) and serum alkaline phosphatase (P = 0.98) levels were unaffected by RHA nanoparticles (Figure S11D), RHA nanoparticles may also be a drug without obvious hepatic toxicity. Only one mouse treated with RHA nanoparticles showed a higher serum alanine aminotransferase (ALT) level, but the overall ALT levels of RHA-nanoparticles-treated mice and control mice had no statistical significance (Figure S11D, P = 0.16). Since RHA nanoparticles were initially dissolved in alcohol, the abnormal increase of ALT might be caused by individual sensibility to alcohol.31 Overall, the toxicology data above demonstrate that RHA nanoparticles are safe with no obvious physiological adverse reactions detected.
Mechanism Study Of RHA
RHA contains the hydrocarbon backbone of ATRA, which is the key structural fragment for biological effects.32 Therefore, we investigated whether anticancer effect of RHA depended on RAR and RXR receptor like other retinoids. Interestingly, although RHA showed RAR- and RXR-receptor-dependent cytotoxicity against HL-60 cells (Figure S12A), its cytotoxicity against A-375 cells was independent to RAR and RXR receptor (Figure S12B). Furthermore, even though the structure of RHA is similar to vorinostat, in vitro studies demonstrate that histone deacetylase inhibition activity of RHA was only approximately 1% compared to that of vorinostat (Figure S13).
Discussion
RHA nanoparticles demonstrated several outstanding properties as a promising drug for cancer therapy. The first one is their long-lasting proliferative inhibition. Previous studies have shown that the high efficacy of ATRA on treating acute promyelocytic leukemia is related to its irreversible differentiation activity.33 Patients could benefit from this long-lasting cytotoxicity against cancer cells because that means lower possibility of recurrence and shorter treatment period. The second property is low systemic toxicity of RHA nanoparticles. One of the possible reason of this selective toxicity should be tumor-specific accumulation of poloxamer-coated RHA nanoparticles via EPR effect. Poloxamer is a type of inert amphiphilic block polymer with high biocompatibility.34 It is widely used for coating hydrophobic nanoparticles by adsorbing onto their surface, thus decreasing mononuclear phagocyte nanoparticle uptake.25,35 Another possible reason might be the selective toxicity of RHA nanoparticles themselves. Tumor cell lines from various origins demonstrated different sensitivity to RHA nanoparticles (Figure 2A). This difference indicated that the target of RHA nanoparticles should not be general cytobiological processes which are essential for the majority of cells.
This work also indicated that the anticancer mechanism of RHA nanoparticles is totally different from that of ATRA. First, anticancer effect of RHA nanoparticles against A-375 cells is fully independent on RAR and RXR, which are the major targets for ATRA and other retinoids. Second, ATRA only induced G1 phase arrest to both melanoma and other malignant cells without S phase arrest,36,37,38 while RHA nanoparticles clearly induced S phase arrest to A-375 cells. Although RHA nanoparticles can induce proliferative inhibition to ATRA sensitive HL-60 cells via RAR/RXR-dependent pathways, the RAR/RXR-independent cytotoxicity of RHA nanoparticles played a key role in anticancer effect against ATRA-resistant cell lines.
In this study, we first found that RHA could self-assemble into nanoparticles with a high anticancer efficacy. Coating RHA nanoparticles with poloxamer could obviously enhance their tumor-targeting efficiency. This investigation also revealed that RHA nanoparticles have potent and persistent proliferative inhibition effect against A-375 melanoma cells, leading to cell cycle arrest and apoptosis. Moreover, RHA nanoparticles were found to suppress the growth of xenograft tumors but importantly, without obvious adverse in vivo reaction. Even though RHA nanoparticle showed RAR/RXR-independent cytotoxicity and low histone deacetylase inhibition activity which is opposite to our initial expectation, their potent and long-lasting anticancer effect plus their low systemic toxicity made RHA nanoparticles a promising drug for melanoma therapy.
Acknowledgments
We thank DNL2001 and DNL2002 labs of Dalian Institute of Chemical Physics for providing technical supports of MS, NMR, and TEM. We also thank our present and past technicians, Xin Guo, Dan Sun, Luyao Deng, and Yi Zang, for routine technical supports and cell line maintaining.
Funding
This work is supported by the innovation program (DICP TMSR201601) of science and research from the Dalian Institute of Chemical Physics, Chinese Academy of Science, and National Natural Science Foundation of China (No.31470944).
Disclosure
Dr Han Liao reports a patent retinoic hydroxamic acids and their application in cacner therapy pending. All authors declare they have no other conflicts of interest in this work.
References
1. Daenen S, Vellenga E, van Dobbenburgh OA, Halie MR. Retinoic acid as antileukemic therapy in a patient with acute promyelocytic leukemia and aspergillus pneumonia. Blood. 1986;67(2):559–561.
2. Huang ME, Ye YC, Chen SR, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72(2):567–572.
3. Liang C, Yang L, Guo S. All-trans retinoic acid inhibits migration, invasion and proliferation, and promotes apoptosis in glioma cells in vitro. Oncol Lett. 2015;9(6):2833–2838. doi:10.3892/ol.2015.3120
4. Teppola H, Sarkanen JR, Jalonen TO, Linne ML. Morphological differentiation towards neuronal phenotype of SH-SY5Y neuroblastoma cells by estradiol, retinoic acid and cholesterol. Neurochem Res. 2016;41(4):731–747. doi:10.1007/s11064-015-1743-6
5. Freemantle SJ, Spinella MJ, Dmitrovsky E. Retinoids in cancer therapy and chemoprevention: promise meets resistance. Oncogene. 2003;22(47):7305–7315. doi:10.1038/sj.onc.1206936
6. Schenk T, Stengel S, Zelent A. Unlocking the potential of retinoic acid in anticancer therapy. Br J Cancer. 2014;111(11):2039–2045. doi:10.1038/bjc.2014.412
7. Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11(17–18):812–818. doi:10.1016/j.drudis.2006.07.005
8. Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3–6. doi:10.1016/j.addr.2015.01.002
9. Schultze E, Ourique A, Yurgel VC, et al. Encapsulation in lipid-core nanocapsules overcomes lung cancer cell resistance to tretinoin. Eur J Pharm Biopharm. 2014;87(1):55–63. doi:10.1016/j.ejpb.2014.02.003
10. Wang Y, Wang H, Lv X, et al. Enhancement of all-trans retinoic acid-induced differentiation by pH-sensitive nanoparticles for solid tumor cells. Macromol Biosci. 2014;14(3):369–379. doi:10.1002/mabi.201300295
11. Dummer R, Beyer M, Hymes K, et al. Vorinostat combined with bexarotene for treatment of cutaneous T-cell lymphoma: in vitro and phase I clinical evidence supporting augmentation of retinoic acid receptor/retinoid X receptor activation by histone deacetylase inhibition. Leuk Lymphoma. 2012;53(8):1501–1508. doi:10.3109/10428194.2012.656625
12. Fouladi M, Park JR, Stewart CF, et al. Pediatric phase I trial and pharmacokinetic study of vorinostat: a children’s oncology group phase I consortium report. J Clin Oncol. 2010;28(22):3623–3629. doi:10.1200/JCO.2009.25.9119
13. Huang P, Wang D, Su Y, et al. Combination of small molecule prodrug and nanodrug delivery: amphiphilic drug–drug conjugate for cancer therapy. J Am Chem Soc. 2014;136(33):11748–11756. doi:10.1021/ja505212y
14. Al-Sheddi ES, Al-Oqail MM, Saquib Q, et al. Novel all trans-retinoic acid derivatives: cytotoxicity, inhibition of cell cycle progression and induction of apoptosis in human cancer cell lines. Molecules. 2015;20(5):8181–8197. doi:10.3390/molecules20058181
15. Li HX, Zhao W, Shi Y, et al. Retinoic acid amide inhibits JAK/STAT pathway in lung cancer which leads to apoptosis. Tumour Biol. 2015;36(11):8671–8678. doi:10.1007/s13277-015-3534-8
16. Ech-Chahad A, Minassi A, Berton L, Appendino G. An expeditious hydroxyamidation of carboxylic acids. Tetrahedron Lett. 2005;46(31):5113–5115. doi:10.1016/j.tetlet.2005.05.131
17. Mai A, Rotili D, Tarantino D, et al. Small-molecule inhibitors of histone acetyltransferase activity: identification and biological properties. J Med Chem. 2006;49(23):6897–6907. doi:10.1021/jm060601m
18. van de Loosdrecht AA, Nennie E, Ossenkoppele GJ, Beelen RH, Langenhuijsen MM. Cell mediated cytotoxicity against U 937 cells by human monocytes and macrophages in a modified colorimetric MTT assay. A methodological study. J Immunol Methods. 1991;141(1):15–22. doi:10.1016/0022-1759(91)90205-t
19. Borowicz S, Van Scoyk M, Avasarala S, et al. The soft agar colony formation assay. J Vis Exp. 2014;92:e51998.
20. Wang H, Wu H, Shen H, et al. A bimodal MRI and NIR liposome nanoprobe for tumor targeted molecular imaging. J Mater Chem B. 2015;3(45):8832–8841. doi:10.1039/C5TB01160D
21. Zhang R, Xing R, Jiao T, et al. Carrier-free, chemophotodynamic dual nanodrugs via self-assembly for synergistic antitumor therapy. ACS Appl Mater Interfaces. 2016;8(21):13262–13269. doi:10.1021/acsami.6b02416
22. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi:10.3322/caac.21387
23. Cohen SM, Ellwein LB. Genetic errors, cell proliferation, and carcinogenesis. Cancer Res. 1991;51(24):6493–6505.
24. Sebastian C. Tracking down the origin of cancer: metabolic reprogramming as a driver of stemness and tumorigenesis. Crit Rev Oncog. 2014;19(5):363–382.
25. Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53(2):283–318.
26. Shabaruddin FH, Chen LC, Elliott RA, Payne K. A systematic review of utility values for chemotherapy-related adverse events. Pharmacoeconomics. 2013;31(4):277–288. doi:10.1007/s40273-013-0033-x
27. Grigorian A, O’Brien CB. Hepatotoxicity secondary to chemotherapy. J Clin Transl Hepatol. 2014;2(2):95–102. doi:10.14218/JCTH.2014.00011
28. Jones DP, Chesney RW. Renal toxicity of cancer chemotherapeutic agents in children: ifosfamide and cisplatin. Curr Opin Pediatr. 1995;7(2):208–213.
29. Szkandera J, Gerger A, Liegl-Atzwanger B, et al. Uric acid levels in blood are associated with clinical outcome in soft-tissue sarcoma patients. Clin Chem Lab Med. 2015;53(3):493–497. doi:10.1515/cclm-2014-0486
30. Yamauchi T, Negoro E, Lee S, et al. A high serum uric acid level is associated with poor prognosis in patients with acute myeloid leukemia. Anticancer Res. 2013;33(9):3947–3951.
31. Liu Z, Que S, Xu J, Peng T. Alanine aminotransferase-old biomarker and new concept: a review. Int J Med Sci. 2014;11(9):925–935. doi:10.7150/ijms.8951
32. Sporn MB, Dunlop NM, Newton DL, Henderson WR. Relationships between structure and activity of retinoids. Nature. 1976;263:110. doi:10.1038/263110a0
33. Kamimura T, Miyamoto T, Harada M, Akashi K. Advances in therapies for acute promyelocytic leukemia. Cancer Sci. 2011;102(11):1929–1937. doi:10.1111/j.1349-7006.2011.02045.x
34. Singh-Joy SD, McLain VC. Safety assessment of poloxamers 101, 105, 108, 122, 123, 124, 181, 182, 183, 184, 185, 188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 331, 333, 334, 335, 338, 401, 402, 403, and 407, poloxamer 105 benzoate, and poloxamer 182 dibenzoate as used in cosmetics. Int J Toxicol. 2008;27(Suppl 2):93–128. doi:10.1080/10915810802244595
35. Zhang W, Liu J, Li S, Chen M, Liu H. Preparation and evaluation of stealth tashinone IIA-loaded solid lipid nanoparticles: influence of poloxamer 188 coating on phagocytic uptake. J Microencapsul. 2008;25(3):203–209. doi:10.1080/02652040701852181
36. Dimberg A, Bahram F, Karlberg I, Larsson LG, Nilsson K, Oberg F. Retinoic acid-induced cell cycle arrest of human myeloid cell lines is associated with sequential down-regulation of c-Myc and cyclin E and posttranscriptional up-regulation of p27(Kip1). Blood. 2002;99(6):2199–2206. doi:10.1182/blood.v99.6.2199
37. Su B, Chen X, Zhong C, Guo N, He J, Fan Y. All-trans retinoic acid inhibits mesangial cell proliferation by up-regulating p21Waf1/Cip1 and p27Kip1 and down-regulating Skp2. J Nephrol. 2012;25(6):1031–1040. doi:10.5301/jn.5000090
38. Zhang H, Rosdahl I. Expression of p27 and MAPK proteins involved in all-trans retinoic acid-induced apoptosis and cell cycle arrest in matched primary and metastatic melanoma cells. Int J Oncol. 2004;25(5):1241–1248.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.